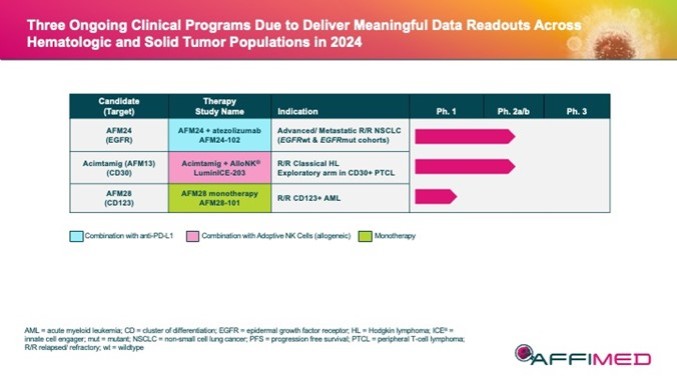
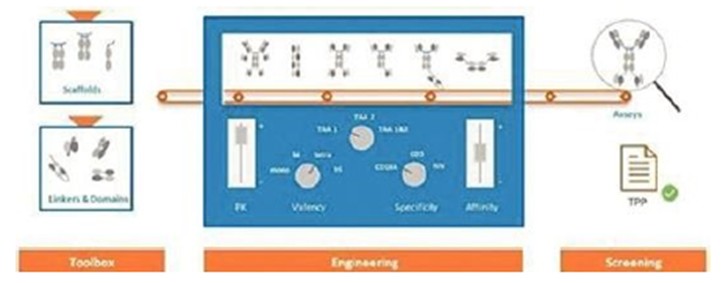
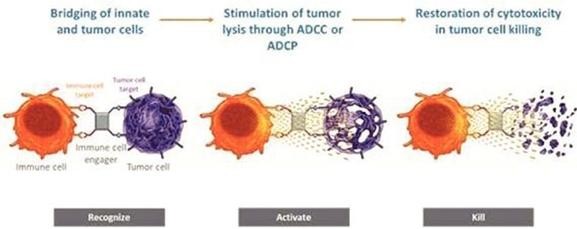
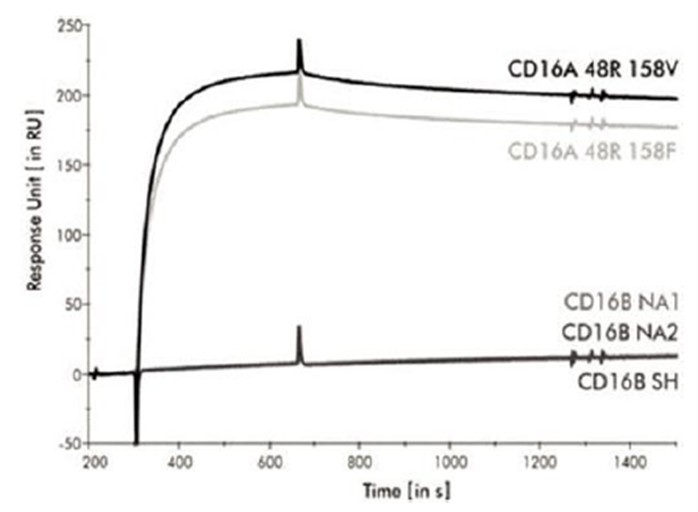
本附註所載的股份及每股資料追溯反映於2024年3月8日生效的反向股票分拆的影響(見附註12)。
帶服務條件的股份支付
大多數獎項都是以分期付款的形式發放的三年並且可以被行使到10年在授予之日之後。在2023年和2022年,集團授予822,175和491,560分別獎勵員工、管理委員會成員和監事會成員。這些獎勵在2023年授予日的公允價值為歐元。6.0百萬(美元)6.4百萬)。
在2023年,167,260由於與非僱員的僱傭關係終止或諮詢協議終止,員工持股2014年的獎勵被取消或沒收(2022:27,743),以及不是期權已被行使(2022年: 4,344期權的行使加權平均行權價為#美元。25.2).
截至2023年12月31日,2,181,8882014年ESOP獎項傑出(2022年12月31日: 1,526,973), 1,240,852獎項(2022年12月31日: 851,086)被授予。於2023年12月31日尚未行使的期權的行使價格在美元範圍內3.5至$134.7 (2022: $13.0至$134.7),加權平均剩餘合同期限為 7.3年份(2022年:7.4年),加權平均行使價為美元35.7 (2022: $49.1). 2023年和2022年,本集團預計年度沒收率約為 4未歸屬期權的%。
市場狀況下的股份支付
在2022年期間,公司發佈了282,500向管理委員會成員和員工提供基於市場的績效條件的期權。每筆贈款包括 三份額,由此 一- 當成交量加權平均股價高於之前時,授予總額的三分之一將歸屬 三十交易天數達到美元120, $150、和$180,分別。除控制權變更外,該等期權於授予日期一週年時歸屬。截至2023年12月31日, 20,000期權被取消。2022年授予日獎勵的公允價值為歐元2.9百萬(美元)3.2百萬),期權的合同期限為 兩年.授予日期後兩年內任何未授予的獎勵將失效。
基於股份的支付費用
2023年,費用為歐元10,714被認為影響研究和開發費用(歐元6,014)以及一般和行政費用(歐元4,700). 2022年,費用為歐元19,110被認為影響研究和開發費用(歐元10,351)以及一般和行政費用(歐元8,759). 2021年,費用為歐元11,820被認為影響研究和開發費用(歐元5,892)以及一般和行政費用(歐元5,928).
公允價值計量
Affimed N.V.發行的帶有服務條件的股票期權的公允價值是使用Black-Scholes-Merton公式估計的。該公式根據標的工具的價值、行權價格、股價回報的預期波動率、股息、無風險利率和期權到期時間等輸入參數來確定期權的價值。
在市場條件下,股票期權的公允價值是通過使用蒙特卡羅模擬法確定的,該模擬法將需要達到的障礙(或障礙)作為額外的輸入參數。股份權益結算補償計劃的公允價值於授出日計量,補償成本於歸屬期間確認,並相應增加權益。預計將授予的股票期權數量是在每個衡量日期估計的。