目錄表
正如下面概述的那樣,我們正在推進新型放射免疫療法和基於抗體的候選療法的重點臨牀流水線的開發,旨在改善各種癌症患者的生活:
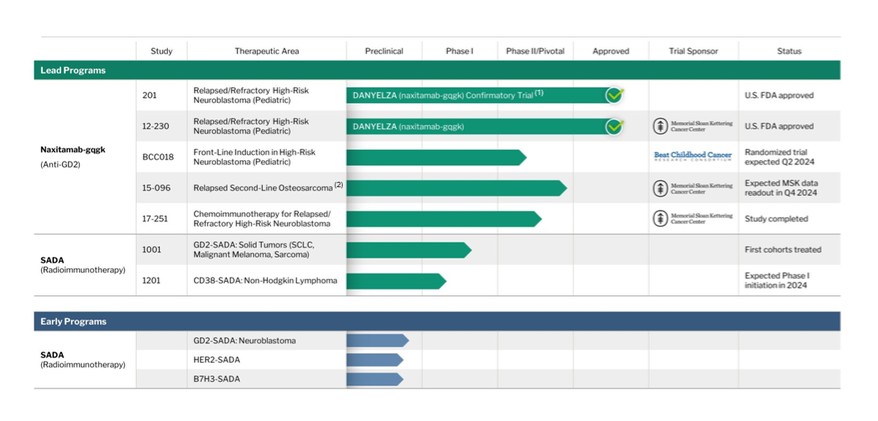
(1) | DANYELZA於2020年11月獲得FDA的加速批准。支持BLA提交的關鍵註冊研究,包括衡量藥代動力學、毒性和療效的研究12-230,以及另一項關鍵的第二階段多中心研究,研究201,旨在證明使用當前良好製造實踐或cGMP商業製造商的研究地點之間的可比性。研究201也是為了滿足FDA的驗證性研究和上市後要求而設計的。 |
(2) | 最初的研究代表了年齡從1歲到40歲的兒科和青年患者。 |
我們的使命是成為開發更好、更安全的放射免疫療法和基於抗體的腫瘤療法的全球領導者,以滿足明顯的未得到滿足的醫療需求,並因此對患者的生活產生革命性影響。我們打算獨立或與潛在合作伙伴合作,將我們的候選治療方案推進和擴大到某些成人癌症適應症。
丹尼爾扎
DANYELZA是美國食品和藥物管理局批准的第一個產品,是一種針對神經節苷脂GD2的重組人源化免疫球蛋白G,1K亞型,或IgG1mAb,針對神經節苷脂GD2,它在各種神經外胚層來源的腫瘤和肉瘤中高度表達。DANYELZA於2020年11月獲得FDA的加速批准,用於與GM CSF聯合治療1歲及以上的兒童患者和患有R/R高危NB的成人患者,這些患者對以前的治療表現出部分反應、輕微反應或病情穩定。我們正在將DANYELZA在美國商業化,並於2021年2月開始發貨。截至2023年12月31日和2022年12月31日的年度,DANYELZA的銷售額分別為8430萬美元和4930萬美元。2023年,SciClone製藥國際有限公司在中國推出了治療R/R高危NB患者的DANYELZA,並在以色列推出了武田以色列DANYELZA。此外,我們在2023年期間在巴西和墨西哥獲得了DANYELZA的監管批准。
DANYELZA的加速批准受到某些上市後要求和承諾的約束,包括必須完成的臨牀益處驗證性上市後試驗,以便將生物製品許可證申請(BLA)轉換為完全批准並防止FDA撤回許可證。FDA要求進行的驗證性上市後臨牀試驗是我們正在進行的201項研究,旨在招募至少80名可評估的患者,並報告總體應答率(ORR)、反應持續時間(DOR)、無進展生存期(PFS)和總生存期(OS)。ORR是主要的
7