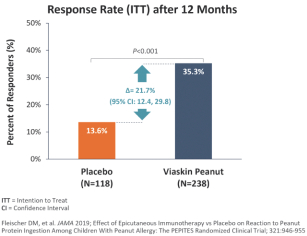
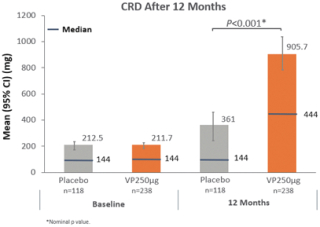
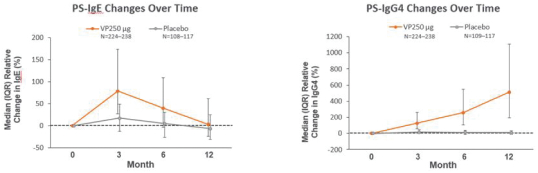
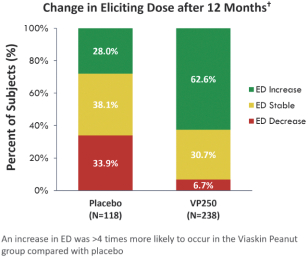
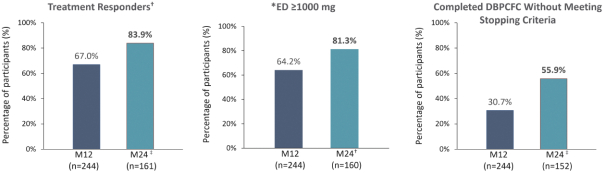
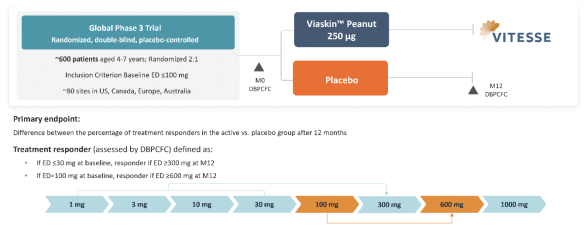
技術平臺,我們稱之為Viaskin,是一種潛在治療免疫疾病的創新方法,主要關注食物過敏。在EPIT中,完整的皮膚通過Viaskin技術使用含有微克量食物蛋白質的貼片暴露在過敏原中。通過EPIT施加的過敏原被特殊的抗原提呈細胞(表皮內的朗格漢斯細胞)以及真皮樹突狀細胞捕獲在皮膚淺層,從而限制接觸血液。在實驗模型中,EPIT誘導了一羣具有特定特性的調節性T細胞,即Tregs,從而抑制過敏反應。症狀和防止進一步敏感化的保護。EPIT誘導的表觀遺傳修飾有利於Treg介導的免疫反應和下調的Th2反應,並可能在效應的可持續性中發揮作用。根據我們的試驗和研究,我們相信EPIT有可能提供所有預期的過敏疾病修改治療的好處,同時避免嚴重或危及生命的過敏反應。
Viaskin補丁操作機制的關鍵元素如下所示:
| • | 該貼片的中心含有一層乾燥的過敏原,無需事先準備,即可放置在完整的皮膚上。 |
| • | 在皮膚和貼片中心之間形成的冷凝室會使皮膚過度水合,並積累水分。 |
| • | 水的積累溶解了過敏原。由於這個冷凝室,表皮變得更具滲透性,允許過敏原進入表皮。 |

一旦進入表皮,過敏原就會被一羣高度專業化的細胞捕獲:朗格漢斯細胞。這些細胞可以捕獲皮膚表面的蛋白質,對其進行處理,並將其表位呈現給淋巴結中的T淋巴細胞。
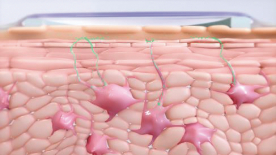
在塗抹Viaskin貼片後,表皮中的朗格漢斯細胞捕獲角質層(皮膚最外層)內的花生過敏原(以綠色表示),使過敏原溶解並滲透到皮膚中。
3