目錄表
分子的ETB平臺技術
雖然化療仍然是大多數癌症治療的基石,但新的和有針對性的治療類別的出現極大地改變了疾病治療的結果。單抗、信號轉導抑制劑和最近的免疫腫瘤學的出現在復發和難治性環境中都提供了實質性的臨牀益處,當聯合使用時,在早期的治療路線中。分子認為,ETB代表了一類具有不同生物學特性的新型靶向製劑,它們非常適合改善癌症患者的預後。
ETB似乎能誘導非或不良內化靶標的內化,具有不同的作用機制(酶促和不可逆轉的核糖體失活),具有相對可預測的PK譜,並且可以很容易地按照cGMP標準制造。從抗體樣靶向結構域的庫中,分子的研究和設計平臺允許全面的體外培養根據親和力和特異性、效力和表達來選擇針對給定靶標的前導ETB。鉛的選擇是通過使用動物模型來證實的,以驗證PK、吸收、分佈、代謝和排泄以及效力。ETB通過一種分化的作用機制具有強大的直接細胞殺傷作用,可以迫使受體內化,並可用於將外源I類抗原等有效載荷輸送到胞漿。
在所有臨牀階段的ETB中,分子公司使用一種高度有效和適當去免疫的SLTA支架,臨牀前和動物研究表明,該支架可顯著降低先天和獲得性免疫原性反應。對於腫瘤已被證明對T細胞參與敏感的適應症,分子公司開發了ETBS,它可以遞送外來的I類病毒抗原,呈現在腫瘤表面:分子的抗原種子技術,這是一種免疫腫瘤學的差異化方法。分子已經將其抗原種子技術集成到針對ETB的PD-L1中,MT-6402,並繼續建立動物模型,以進一步驗證和篩選更多的ETB候選對象來支持這一方法。
分子公司認為,其專有的ETB技術平臺代表了腫瘤學領域的一種差異化方法。EBCs具有基於抗體的治療方法的靶向特異性,但提供高度有效的有效載荷,以一種不受傳統化療耐藥機制或靶點內化限制的方式擾亂蛋白質合成,而不是ADC。分子還在尋求擴大藥物治療的潛在靶標的宇宙,方法是利用ETB的能力,針對通常不內化的受體強制內化。
腫瘤學治療需要新的作用機制,分子公司認為,其ETB平臺技術的不同作用機制可能提供超過現有治療模式的獨特好處。
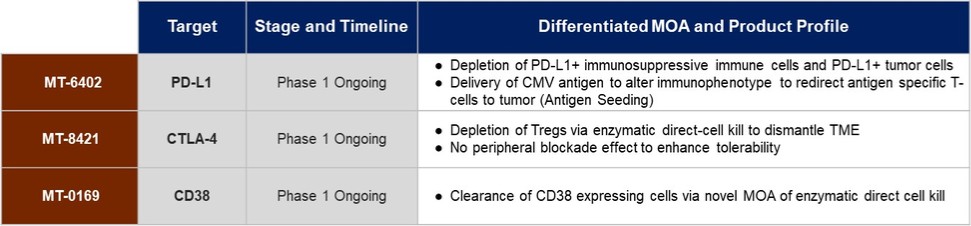
ETB產品線
分子公司正在開發一條ETB管道,分子公司相信這將有能力為癌症患者提供有意義和長期的好處。分子計劃將每一種藥物作為單一藥物和/或與其他療法聯合開發,如果適用的話。下表描述了分子公司目前的流水線:
9