目錄表
EYSUVIS和INVELTYS在2023年至2028年的全球淨銷售額合計為6,500萬美元,(2)EYSUVIS和INVELTYS在2023年至2028年的全球淨銷售額合計為1,000萬美元或以上,(3)EYSUVIS和INVELTYS在2023年至2029年的全球淨銷售額合計為1.75億美元或以上,以及(4)EYSUVIS和INVELTYS在2023年至2029年的全球淨銷售額合計為2.5億美元或以上時為1.6億美元。每個里程碑付款將只支付一次,如果有的話,在第一次達到這樣的里程碑,並且只有一次里程碑付款將支付關於一個日曆年。如果在一個日曆年實現了一個以上的里程碑,則只有在下一個日曆年再次實現相應的里程碑時,才會支付較高的里程碑付款和較低的里程碑付款。到目前為止,還沒有實現任何里程碑,我們也沒有收到愛爾康的任何里程碑付款。
下表描述了我們每個開發計劃的階段:
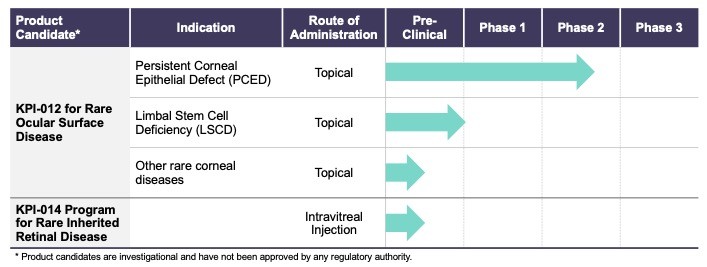
我們保留了我們的S平臺的全球商業權,包括KPI012和KPI014。我們擁有和/或獨家許可與此平臺相關的專利和專利申請,包括美國和外國已頒發的專利和未決的專利申請。我們控制的涵蓋KPI012的已頒發美國專利的到期日計劃不早於2040年到期,另外一系列涵蓋MSC-S平臺的額外美國和前美國專利申請目前正在起訴中。
戰略
我們的目標是成為一家領先的生物製藥公司,致力於眼前和眼後罕見和嚴重疾病的創新療法的研究、開發和商業化。我們戰略的關鍵要素包括:
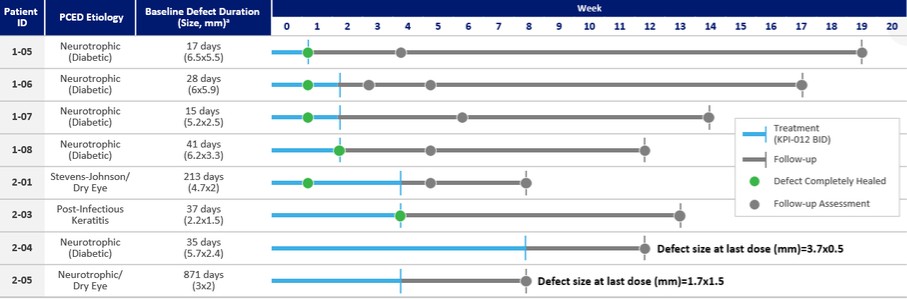
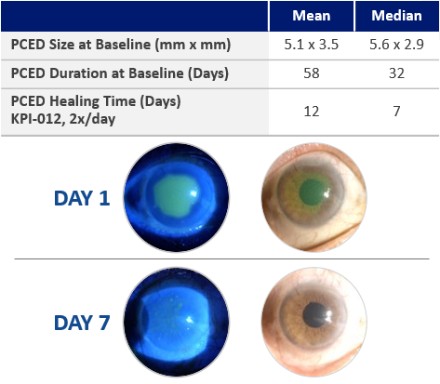
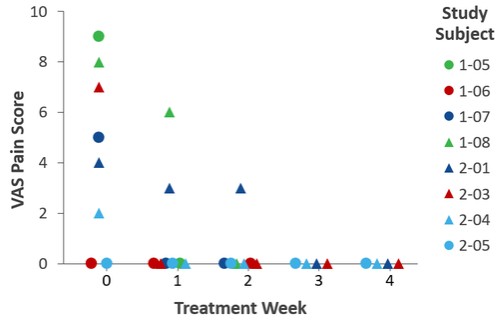
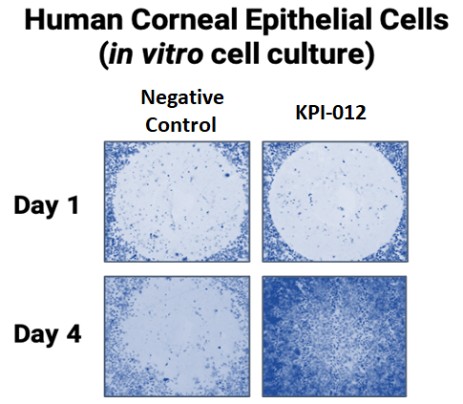
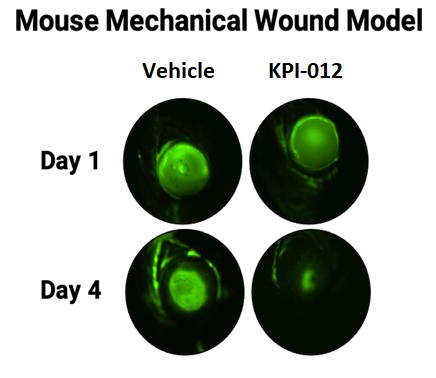
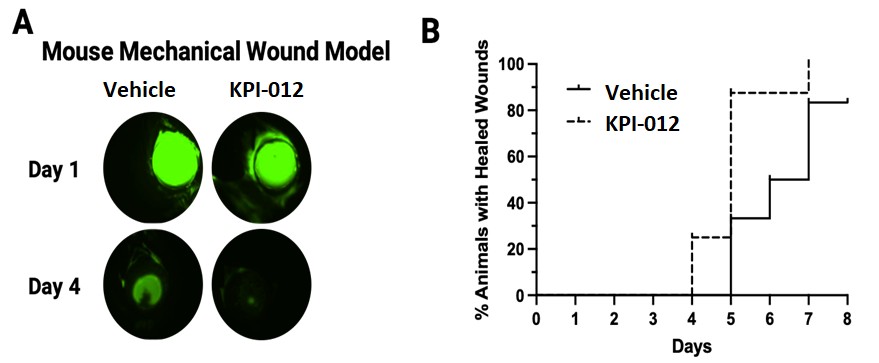
| ● | 推進治療PCED的KPI-012的臨牀開發,並尋求監管部門的批准。KPI-012是一種新型的、人骨髓來源的骨髓間充質幹細胞-S,目前正處於臨牀開發中,用於治療PCED。PCED是一種永久性的不可癒合的角膜缺損或傷口,對傳統治療方法來説是難以治癒的。2023年3月27日,我們宣佈了CHASE試驗的第一個隊列的陽性安全性數據,隨後啟動了第二個也是最後一個患者隊列。我們的目標是在2024年底之前為CHASE試驗報告TOPLINE安全性和有效性數據。如果結果是積極的,並取決於與監管機構的討論,我們相信這項試驗可能成為支持向FDA提交KPI-012的BLA所需的兩個關鍵試驗中的第一個。如果獲得批准,我們打算在美國通過一支小型的、有針對性的內部銷售隊伍將KPI-012商業化。我們還希望通過與一個或多個第三方的各種合作、分銷、聯合促銷和其他營銷安排,探索用於治療PCED的KPI-012在美國以外的某些市場的商業化。 |
| ● | 高級KPI-012用於其他罕見眼表疾病的適應症,KPI-014用於罕見的遺傳性視網膜退行性疾病。我們還在評估KPI-012治療其他罕見眼前疾病的潛力,如LSCD和其他威脅視力的罕見角膜疾病。此外,我們還有 |
7