目錄表
正在籌備中,包括進一步開發已批准的藥物
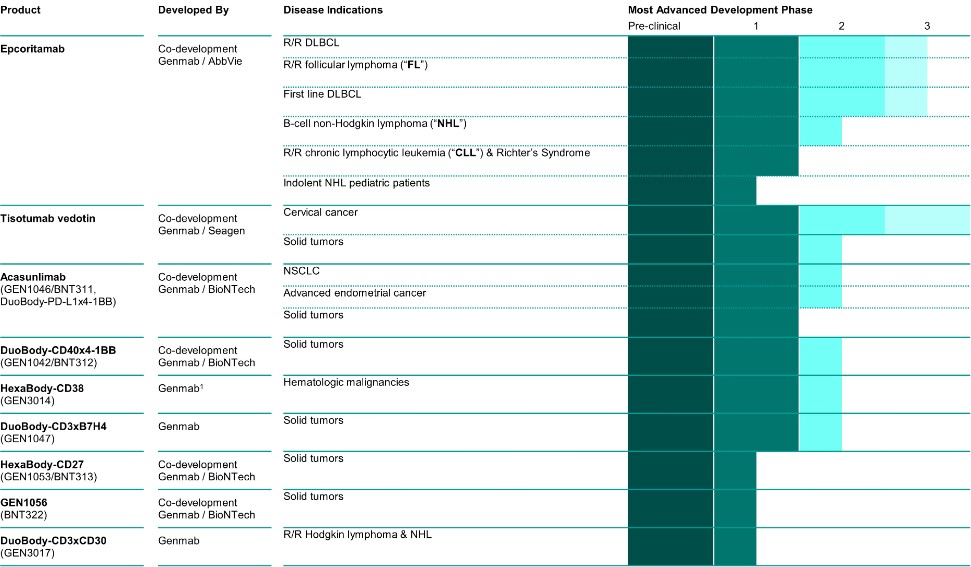
Epcoritamab
Epcoritamab是一種專有的雙特異性抗體療法,使用我們專有的DuoBody技術創建。Epcoritamab設計用於靶向CD3和CD20,CD3表達在所有T細胞亞型上,是T細胞受體的一部分,CD20是臨牀驗證的治療靶點。CD20在大多數B細胞惡性腫瘤中表達,包括CLL、DLBCL、FL和套細胞淋巴瘤(MCL)。我們使用了從Medarex,Inc.(“Medarex”)獲得許可的技術來產生形成epcoritamab一部分的CD20抗體。我們正在與AbbVie合作,在美國和日本共同開發和共同商業化epcoritamab。
Epcoritamab治療B細胞惡性腫瘤
DLBCL是全球最常見的B細胞非霍奇金淋巴瘤(B-NHL)類型,約佔所有NHL病例的30%,2022年美國估計有30,400例病例。DLBCL可出現在淋巴結和淋巴系統外的器官中,更常見於老年人,在男性中略多見。DLBCL是一種快速增長的非霍奇金淋巴瘤,是一種在淋巴系統發展的癌症,影響B細胞淋巴細胞,B細胞淋巴細胞是一種白細胞。對於許多患有DLBCL的人來説,他們的癌症要麼復發,這意味着它可能在治療後復發,要麼變得難以治癒,這意味着它對治療沒有反應。儘管已經有了新的治療方法,但治療管理仍然是一個挑戰。
FL是一種典型的惰性或生長緩慢的非霍奇金淋巴瘤,起源於B細胞淋巴細胞。淋巴瘤是第二種最常見的非霍奇金淋巴瘤,佔所有非霍奇金淋巴瘤病例的20%至30%,佔西方世界所有淋巴瘤的10%至20%。雖然FL是一種惰性淋巴瘤,但它被認為是傳統療法無法治癒的,而且獲得緩解的患者也經常復發。2022年3月和2023年11月,FDA分別批准了用於治療FL的Ecoritamab的孤兒藥物名稱和BTD。
42
