美國
美國證券交易委員會
華盛頓特區,20549
表格:
(標記一)
截至本財年。
或
過渡期:_
佣金文件編號:

(註冊人的確切姓名載於其章程)
| (註冊成立或組織的州或其他司法管轄區) | (I.R.S.僱主 識別號) |
| (主要行政辦公室地址) | (郵政編碼) |
註冊人的電話號碼,包括
區號:
根據該法第12(B)款登記的證券:
| 每個班級的標題 | 交易代碼 | 註冊的每個交易所的名稱 | ||
| 這個 | ||||
| 這個 |
根據本法第12(g)條登記的證券:
沒有。
如果註冊人是著名的經驗豐富的發行人,則按《證券法》第405條的定義,用複選標記進行註冊。是的 ☐
如果註冊人不需要
根據《法案》第13條或第15(d)條提交報告,則使用複選標記進行標記。是的 ☐
用複選標記標出註冊人
(1)在
之前的12個月內(或註冊人被要求提交此類報告的較短期限內)是否提交了1934年證券交易法第13條或第15(d)條要求提交的所有報告,以及(2)在過去的90天內是否
遵守此類提交要求。
在過去12個月內(或在註冊人被要求提交和張貼此類
文件的較短時間內),通過勾選標記確認註冊人
是否已以電子方式提交了根據S—T法規第405條(本章第232.405節)要求提交的每個交互式數據文件。
通過複選標記來確定註冊人 是大型加速申報人、加速申報人、非加速申報人、小型申報公司還是新興增長型公司。 請參見"大型加速文件服務器,” “加速文件管理器“和”較小的報告公司 “和”新興成長型公司“在交易法第12B-2條中。
| 大型加速文件服務器 | ☐ | 加速文件管理器 | ☐ |
| ☒ | 規模較小的報告公司 | ||
| 新興成長 | |||
如果是新興增長型公司,請用複選標記 表示註冊人是否選擇不使用延長的過渡期來遵守根據《交易法》第13(a)條規定的任何新的或修訂的財務會計準則 。 ☐
用複選標記表示註冊人
是否已就其管理層根據《薩班斯-奧克斯利法案》(《美國法典》第15編第7262(B)節)第404(B)條對其財務報告進行內部控制的有效性提交了一份報告,並證明瞭編制或發佈其審計報告的註冊會計師事務所對其財務報告的內部控制的有效性。
如果證券是根據該法第(Br)12(B)節登記的,請用複選標記表示備案文件中包括的註冊人的財務報表是否反映了對以前發佈的財務報表的錯誤進行了更正。
用複選標記表示這些錯誤 更正中是否有任何重述需要根據§240.10D-1(B)對註冊人的任何高管在相關恢復期間收到的基於激勵的薪酬進行恢復分析。☐
用複選標記表示註冊人
是否是空殼公司(如《交易法》第12b-2條所定義)。是☐不,不是。
截至註冊人最近完成的第二
財政季度的最後一個營業日,註冊人的非關聯公司持有的有表決權和無表決權
普通股的總市值為美元
截至2024年3月22日,
以引用方式併入的文件
註冊人關於其2024年年度股東大會的最終 委託聲明的部分("2024年委託書”)以引用方式納入本年報第三部分,表格10—K(如有指明)。2024年委託聲明將在本報告所涉財政年度結束後120天內提交給美國證券交易委員會。
| 審計師事務所ID: | 審計師姓名: | 審計師地點:北京 |
目錄
| 詞彙表 | II |
| 關於前瞻性信息的警示聲明 | 六、 |
| 第一部分 | 1 |
| 項目1.業務 | 1 |
| 第1A項。風險因素。 | 51 |
| 項目1B。未解決的員工評論。 | 107 |
| 項目1C。網絡安全。 | 107 |
| 項目2.財產 | 108 |
| 項目3.法律訴訟 | 108 |
| 第4項礦山安全信息披露 | 108 |
| 第II部 | 109 |
| 項目5.登記人普通股的市場、相關股東事項和發行人購買股權證券 | 109 |
| 第六項。[已保留] | 110 |
| 第七項:管理層對財務狀況和經營成果的討論和分析。 | 111 |
| 第7A項。關於市場風險的定量和定性披露。 | 121 |
| 項目8.財務報表和補充數據 | 121 |
| 第九項會計和財務披露方面的變更和分歧。 | 121 |
| 第9A項。控制和程序。 | 122 |
| 項目9B。其他信息。 | 123 |
| 項目9C。披露妨礙檢查的外國司法管轄區。 | 123 |
| 第三部分 | 124 |
| 項目10.董事、行政人員和公司治理 | 124 |
| 第11項.行政人員薪酬 | 124 |
| 第12項:某些實益所有人的擔保所有權和管理層及相關股東事項。 | 124 |
| 第十三條某些關係和相關交易,以及董事的獨立性。 | 124 |
| 項目14.首席會計師費用和服務 | 124 |
| 第四部分 | 125 |
| 項目15.證物、財務報表和附表 | 125 |
| 項目16.表格10-K摘要 | 131 |
i
詞彙表
以下是本報告中使用的某些術語的縮寫和定義,這些術語通常在製藥和生物技術行業中使用:
“ACA“指 《患者保護和平價醫療法案》,通常簡稱為《平價醫療法案》,綽號”奧巴馬醫改“,這是一項美國聯邦 法規,提供了許多權利和保護,使醫療保險更公平,更容易理解,以及補貼 (通過“保險費税收抵免“和”分攤費用減少額“)以使其更容易負擔得起。這項法律還將醫療補助計劃擴大到覆蓋更多的低收入人羣。
“止痛藥“ 是一類專門用於緩解疼痛的藥物。
“安達“ 是指包含提交給FDA以供審查和潛在批准仿製藥產品的數據的簡化新藥申請。
“抗tnf“ 是一種抑制對TNF的生理反應的藥物。
“BLA“是指FDA的生物製品許可證申請,這是美國生物贊助商正式建議FDA批准一種新生物用於銷售和營銷的工具。
“BPCIA”指 生物製品價格競爭和創新法。
“大麻素“ 在中國發現的平均化合物大麻草..,在本報告中使用時,指的是在大麻植物中發現的不含THC的化合物。
“中央商務區“或大麻二酚是大麻植物中的活性成分。CBD是THC的非精神活性氧化降解產物。
“CBG“大麻酚是大麻植物中發現的一種化合物。
“CCMO" 是指De Centrale Commissie Mensgebonden Onderzoek(CMO),或指涉及人體受試者的研究中央委員會,該組織 負責審查和管理荷蘭涉及人體受試者的醫學研究。
“CHMP" 是指人用藥品委員會,以前稱為專利藥品委員會,是 歐洲藥品管理局的委員會,負責制定該機構對人用藥品 所有問題的意見。
“胞質"指 醫療保險和醫療補助服務中心,該中心是HHS內的一個聯邦機構,負責管理醫療保險計劃,並與州政府合作 管理醫療補助。
“皮質類固醇" 是一種降低體內炎症的藥物。
“CRO"指 合同研究組織,是一家以合同外包的研究服務形式向製藥、生物技術和醫療器械行業提供支持的公司。
“環孢素A"是指 《受控物質法》,該法令建立了美國聯邦藥物政策,根據該政策,某些物質的製造、進口、擁有、 使用和分銷受到管制。
“CTA"是指 臨牀試驗申請,是向主管國家監管機構提交的,以獲得在特定國家進行臨牀試驗的授權。這是一份包含試驗用藥品必要信息的申請。 CTA的目的是向衞生部門提供有關臨牀試驗的所有重要細節,以獲得 產品批准。
II
“DEA"是指 緝毒署,美國司法部下屬的美國聯邦執法機構,其任務是 打擊美國境內的毒品販運和分銷。
“EMA"指 歐洲藥品管理局,歐盟負責藥品評估和監督的機構。
“ETASU" 是指確保安全使用的要素,這是根據REMS降低藥物使用風險所需的幾種策略之一。
“歐盟“指歐洲聯盟。
“林業局"是指 美國食品和藥物管理局,它是美國衞生和公眾服務部的聯邦機構。FDA 負責通過確保人用藥品和獸藥、生物 產品和醫療器械的安全性、有效性和安全性,以及通過確保美國食品供應、化粧品和輻射產品的安全性來保護公眾健康。
“FDC法案" 指的是《聯邦食品、藥品和化粧品法案》,這是1938年由國會通過的一套美國法律,授權FDA監督 食品、藥品、醫療器械和化粧品的安全。
“FS指肩關節凍結,指的是肩關節僵硬和疼痛。
“GCP“意味着良好的臨牀實踐,這是一個國際質量標準,然後政府可以將其轉變為涉及人類受試者的臨牀 試驗的法規。GCP遵循國際人用藥品註冊技術要求協調委員會(ICH),並在臨牀研究的倫理方面執行嚴格的指導方針。
“普洛斯“指良好的實驗室操作規範,即與非臨牀健康和環境安全研究計劃、執行、監測、記錄、存檔和報告的組織過程和條件有關的質量體系。
“GMP"是指 FDA根據《食品和藥品監督管理局法》頒佈的藥品生產質量管理規範條例。這些具有法律效力的法規 要求藥品、醫療器械、某些食品和血液的製造商、加工商和包裝商採取積極措施 確保其產品安全、純淨和有效。
“HHS"是指 美國衞生和公眾服務部,也稱為衞生部,是美國聯邦政府的內閣級部門 ,其目標是保護所有美國人的健康並提供必要的公眾服務。
“HIPAA" 是指1996年的《健康保險攜帶和責任法案》,其目標是使人們更容易持有健康 保險,保護醫療保健信息的機密性和安全性,並幫助醫療保健行業控制行政 成本。
“HMGB1" 是指高遷移率組盒1,一種在人類中由HMGB1基因編碼的蛋白質。活化的巨噬細胞和單核細胞分泌 HMGB1作為炎症的細胞因子介質。
“IBD”指炎症性腸病,這是一個總括術語,用來描述涉及慢性消化道炎症的疾病。
“工業“是指正在研究的新藥申請。在開始臨牀試驗之前,研究必須得到批准。當研究人員想要在人體上研究藥物時,必須向FDA提交研究新藥或IND申請或請求。IND申請必須包含 某些信息,例如:研究結果,以便FDA可以決定該療法是否可用於人體測試; 藥物是如何製造的,由誰製造,裏面有什麼,它有多穩定,等等;審查計劃的臨牀研究的詳細大綱,稱為 研究方案,以確定人們是否可能面臨不必要的風險;以及關於臨牀試驗團隊的詳細信息,以確定他們是否擁有進行臨牀試驗的知識和技能。
三、
“可單獨識別的健康信息 "由HIPPA定義為健康信息子集的信息,包括從個人收集的人口統計信息,並且:(1)由醫療保健提供者、健康計劃、僱主或醫療保健信息交換所創建或接收;(2)與個人過去、現在或未來的身體或精神健康或狀況有關;向個人提供醫療服務;或向個人提供醫療服務的過去、現在或將來支付的費用;以及(a)識別個人的信息;或(b)有合理的依據相信信息可用於識別個人。
“IRB"是指 機構審查委員會,這是一個正式指定審查和監測涉及 人類受試者的生物醫學研究的小組。根據FDA法規,IRB有權批准、要求修改(以確保批准)、 或不批准研究。該小組在保護人類研究對象的權利和福利方面發揮着重要作用。
“醫療補助" 是美國聯邦和州醫療保險計劃,旨在幫助一些收入和資源有限的人支付醫療費用。 Medicaid還提供通常不在Medicare範圍內的福利,包括療養院護理和個人護理服務。
“醫療保險" 是美國的一項全國性醫療保險計劃,主要為65歲及以上的美國人提供醫療保險,但也為 一些由社會保障管理局確定的殘疾狀況的年輕人,以及患有終末期腎病和肌萎縮性側索硬化症(ALS或Lou Gehrig's disease)的人提供醫療保險。
“MHRA" 是指藥品和保健產品管理局,是 英國衞生和社會保健部的執行機構,負責確保藥品和醫療器械的有效性和可接受的安全性。
“物料需求計劃“是指相互承認程序,即在一個歐盟成員國授予並在其他歐盟成員國得到承認的市場授權。
“NCE“是否為不含FDA批准的任何其他用途的活性部分的藥物。
“NDA“或 ”完全保密協議“指FDA根據FDC法案第505(B)(1)條提交的新藥申請,這是美國的一個監管工具,藥品贊助商通過它正式建議FDA批准一種新藥用於銷售和營銷,要求申請人進行批准所需的所有調查。
“NIHR“ 意味着國家健康研究所是英國的一個政府機構,為健康和護理方面的研究提供資金,而 是歐洲最大的國家臨牀研究資助者。
“孤兒藥物名稱“ 是指為治療醫療疾病而開發的藥劑,由於這些疾病非常罕見,如果沒有政府的幫助,生產 將無利可圖。
“階段1 試驗通常是將藥物最初引入健康的人體受試者,並對安全性、劑量耐受性、吸收、代謝、分佈和消除進行測試。對於一些嚴重或危及生命的疾病的候選藥物,如癌症, 尤其是當候選藥物可能天生毒性太大而無法合乎道德地給健康志願者使用時,最初的人體試驗 通常在患者身上進行。
“第二階段 試驗通常是在有限的患者羣體中啟動臨牀試驗,目的是確定可能的不良反應和安全風險,初步評估候選藥物對特定目標疾病的療效,並確定劑量 耐受性和最佳劑量。第2階段試驗有時進一步分為:2a階段和2b階段試驗-2a階段專門側重於劑量要求。少數患者被給予不同數量的藥物,以評估是否存在劑量-反應關係,即與劑量增加相關的反應增加。此外,還探討了最佳劑量頻率;2b期試驗專門設計用於嚴格測試藥物在治療、預防或診斷疾病方面的療效。
四.
“第三階段 試驗是指在地理上分散的臨牀試驗地點進行臨牀試驗,以進一步評估在擴大的患者羣體中的劑量、臨牀療效和安全性。這些臨牀試驗旨在確定候選藥物的總體風險-收益比,併為監管批准和產品標籤提供充分的基礎。
“第四階段“ 試驗是需要作為批准條件進行的研究,以便收集有關該藥物在不同人羣中的效果以及與長期使用相關的任何副作用的更多信息。
“小靈通法案“ 指的是公共衞生服務法,這是1944年國會通過的一套美國法律,其中規定了FDA監管生物製品的法定權力。
“理療 是恢復、維持和最大限度地利用患者的活動能力、功能和福祉的治療。
“POCD" 是指術後認知功能障礙/譫妄。
“Ra“指類風濕性關節炎。
“REMS" 是指風險評估和緩解策略,是FDA可能要求某些具有嚴重 安全性問題的藥物實施的藥物安全性計劃,以幫助確保藥物的獲益超過其風險。
“政制事務局局長“是指合成大麻二醇類似物,這是一種合成的藥用級分子,與非精神活性大麻素 如CBD的近或遠類似物,用於治療炎症疾病和疼痛。
“第505(b)(2)條 保密協議“或”第505(b)(2)條申請"是指根據《食品和藥品監督管理法》第505(b)(2)條提交的新藥申請,該法案是美國的一種監管工具,藥物申辦者通過該工具正式提議FDA批准藥品的銷售和上市,允許申請人依賴於其他人之前進行的調查,而申辦者確實為此進行了調查 (b)沒有被調查的人或為其進行調查的人的引用或使用權。
“贊助商“ 指申請人或藥品贊助商,即對新藥的銷售承擔責任的個人或實體,包括對遵守FSC法案和相關法規適用條款的責任。”請注意,此處使用的 術語"保薦人"也可指我們IPO的保薦人KBL IV SponsorLLC,具體取決於使用該術語的上下文 。
“THC“指四氫大麻酚,它是大麻的主要精神活性成分。
“腫瘤壞死因子“是指腫瘤壞死因子,它是身體對炎症反應的一部分。
“英國“ 指聯合王國。
“美國“ 指的是美國。
v
有關前瞻性信息的警示聲明
本年度報告採用表格 10-K(以下簡稱“報告”),包含聯邦證券法規定的前瞻性陳述,包括1995年《私人證券訴訟改革法》所指的前瞻性陳述。在某些情況下,您可以通過以下詞語來識別前瞻性表述: “預期”、“相信”、“繼續”、“可能”、“估計”、“預期”、“打算”、“可能”、“正在進行”、“計劃”、“潛在”、“預測”、“項目”、“ ”或這些術語的否定或其他類似術語,儘管並非所有前瞻性表述都包含這些詞語。前瞻性陳述不是對未來業績或結果的保證,也不一定是對實現該業績或結果的時間或時間的準確指示。前瞻性陳述基於作出陳述時可獲得的信息,涉及已知和未知的風險、不確定因素和其他因素,這些風險、不確定因素和其他因素可能導致我們的結果、活動、業績或成就水平與本報告中的前瞻性陳述所明示或暗示的信息存在實質性差異 ,包括本報告第1A項中“風險因素”標題下所描述的信息。
具體來説,前瞻性 陳述包括但不限於任何不是當前或歷史事實的陳述,例如與我們對候選產品的臨牀和臨牀前開發、製造、監管批准和商業化的預期有關的陳述 ,我們對費用、未來收入和資本需求的估計的準確性,我們執行開發和營銷新藥產品計劃的能力以及這些開發計劃的時間和成本,以及對我們現有資本資源與未來預期現金流相結合的充足程度的估計。
前瞻性陳述 包括但不限於關於以下方面的陳述:
| ● | 對額外資金的需求,我們未來籌集資金的能力,這種資金的條款,以及由此造成的稀釋; | |
| ● | 對我們候選產品的臨牀和臨牀前開發、製造、監管批准和商業化的期望; |
| ● | 與該公司候選藥物的臨牀開發和監管批准相關的不確定性,包括臨牀試驗登記和完成的可能延遲,美國食品和藥物管理局(FDA)、歐洲藥品管理局(EMA)和英國藥品和保健產品監管機構(MHRA)提出的問題; | |
| ● | 美國和其他國家的監管動態; |
| ● | 我們成功地留住或招聘了我們的高級管理人員、關鍵員工或董事,或需要進行變動; |
| ● | 當期經營性現金為負; |
| ● | 新冠肺炎疫情對我們的業務運營和研發舉措的持續影響; |
| ● | 我們對費用、未來收入和資本需求的估計的準確性; |
| ● | 該公司依賴第三方進行臨牀試驗、招募患者以及生產其臨牀前和臨牀藥物供應; | |
| ● | 與此類第三方和合作夥伴達成相互同意的條款的能力,以及此類協議的條款、公司當前許可協議的條款以及與之相關的終止權; |
VI
| ● | 公司計劃產品的患者人數估計; | |
| ● | 候選藥物的非預期不良副作用或治療效果不足,可能限制批准和/或商業化,或可能導致召回或產品責任索賠; |
| ● | 公司有能力完全遵守適用於其產品開發活動的眾多聯邦、州和地方法律和法規要求,以及美國以外的規章制度; | |
| ● | 產品研發中固有的挑戰和不確定性,包括臨牀成功和獲得監管批准的不確定性;以及商業成功的不確定性; | |
| ● | 公司執行其開發和營銷新藥產品計劃的能力,以及這些開發計劃的時間和成本; |
| ● | 不斷變化的通貨膨脹和利率,經濟衰退,包括潛在的衰退,以及宏觀經濟、地緣政治、衞生和工業趨勢,流行病,戰爭行為(包括正在進行的烏克蘭/俄羅斯衝突和以色列/哈馬斯衝突)和其他大規模危機; | |
| ● | 估計我們現有的資本資源是否足夠,加上未來的預期現金流,以滿足我們的經營需求; |
| ● | 我們維持普通股和認股權證在納斯達克上市的能力,包括我們目前不遵守納斯達克的持續上市規則;以及 |
| ● | 其他風險和不確定性,包括下文“風險因素”下所述的風險和不確定性。 |
本報告中的任何前瞻性陳述 反映了我們對未來事件或未來財務表現的當前看法,並涉及已知和未知 風險、不確定性和其他因素,這些因素可能導致我們的實際結果、表現或成就與 這些前瞻性陳述中明示或暗示的任何未來結果、表現或成就存在重大差異。 鑑於這些不確定性, 您不應過分依賴這些前瞻性陳述。本文中包含的所有前瞻性陳述僅指提交本報告之日 。所有後續書面和口頭前瞻性聲明歸屬於公司或代表其行事的人員 ,均明確限定了上述警告性聲明的完整性。除法律要求外,我們 沒有義務以任何原因更新或修改這些前瞻性陳述,即使新的信息在未來變得可用。
第七章
第一部分
項目1.業務
引言
一般信息
本年報表格10—K中包含的信息應與 本報告結尾處包含的綜合財務報表和相關附註一併閲讀。
有關本報告中使用的生物技術行業縮略語和定義的列表,請參見上文的"術語表" 。
本報告使用了我們的徽標和部分商標 和商品名。本報告還包括屬於他人財產的商標、商品名和服務標誌。 僅為方便起見,本報告中提及的商標、商品名和服務標記可能不帶®、™和 SM符號。提及我們的商標、商品名和服務標記並不意味着我們 不會在適用法律下最大限度地主張我們的權利或適用許可人的權利(如有),也不會在適用法律下最大限度地主張 其他知識產權的各自所有人會在適用法律下最大限度地主張其權利。我們不打算 使用或展示其他公司的商標和商品名稱暗示與任何其他公司的關係,或由任何其他公司對我們的 認可或贊助。
The market data and certain other statistical information used throughout this Report are based on independent industry publications, reports by market research firms or other independent sources that we believe to be reliable sources; however, we have not commissioned any of the market or survey data that is presented in this Report. Industry publications and third-party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. We are responsible for all of the disclosures contained in this Report, and we believe these industry publications and third-party research, surveys and studies are reliable. While we are not aware of any misstatements regarding any third-party information presented in this Report, their estimates, in particular, as they relate to projections, involve numerous assumptions, are subject to risks and uncertainties, and are subject to change based on various factors, including those discussed under the section entitled “Risk Factors” of this Report. These and other factors could cause our future performance to differ materially from our assumptions and estimates. Some market and other data included herein, as well as the data of competitors as they relate to 180 Life Sciences Corp., is also based on our good faith estimates.
我們的財政年度將於12月31日結束。中期業績按季度列報截至3月31日、6月30日和9月30日的季度,分別為第一季度、 第二季度和第三季度,其中截至12月31日的季度稱為我們的第四季度。2023財年指截至2023年12月31日的年度,而2022財年指截至2022年12月31日的年度。
反向拆分股票
2022年12月反向股票拆分
2022年12月15日,在180生命科學公司股東特別會議上,公司股東批准了對公司第二次修訂和重新修訂的公司註冊證書的修正案,對我們普通股的已發行和已發行股票進行反向股票拆分,每股面值0.0001美元,比例在四比一和二十比一之間,包括四比一和二十比一,具體比例將由我們的董事會或其正式授權的委員會自行決定。在修正案獲得批准後至2023年12月15日之前的任何時間(“股東管理局”)。2022年12月15日,公司董事會(“董事會”)與股東管理局一起批准了對我們第二次修訂和重新簽署的公司註冊證書的修正案,以影響我們普通股按20股1股的比例進行反向股票拆分(“反向 股票拆分”)。
緊隨特別會議及董事會批准後,吾等於2022年12月15日向特拉華州州務卿提交經修訂及重新修訂的第二份公司註冊證書(“修訂證書”),以實施股票反向拆分。
1
根據修訂證書 ,反向股票拆分於2022年12月19日凌晨12:01生效。美國東部時間(“生效時間”)。 公司普通股或公共認股權證股份的交易代號“ATNF”及“ATNFW”並無更改, 與股票反向拆分有關。
修訂證書 沒有減少我們普通股的授權股份數量,也沒有改變我們普通股的面值或修改任何投票權 或我們普通股的其他條款。
沒有發行與反向股票拆分相關的零碎股份,登記在冊的股東原本有權獲得零碎股份, 則有權將其零碎股份向上舍入至最接近的整體股份。
2024年2月反向股票拆分
2024年2月16日,在公司股東特別會議上,我們的股東批准了對我們的第二份經修訂並重新簽署的公司註冊證書,以實現我們普通股的已發行和流通股的反向股票拆分,每股面值0.0001美元,比例為四比一和四十比一,具體比例 將由我們的董事會確定。
2024年2月26日,公司董事會批准了對公司已發行普通股進行19年一股的首次反向股票拆分 ,並於2024年2月26日提交了公司註冊證書修正案證書,以影響此類反向股票拆分。反向股票拆分於2024年2月28日凌晨12:01生效。東部時間,股票於2024年2月28日開盤時開始在拆分調整的基礎上交易。關於反向股票拆分,截至生效時間,每19股公司已發行和已發行的普通股自動轉換為一股公司普通股 。
關於反向拆分,所有已發行的期權、認股權證、 和其他使其持有人有權購買或以其他方式獲得普通股股份的證券已根據每種證券的 條款進行了調整。根據本公司的股權激勵計劃可授予的股票數量也進行了適當調整 。反向拆分後,普通股的面值保持不變,每股面值為0.0001美元。反向 拆分並未改變普通股或優先股的核定股數。沒有發行與反向拆分相關的零碎股份,原本有權獲得零碎股份的股東獲得了一整股普通股 ,而不是該零碎股份。
19股1股反向拆分的影響已在本報告中追溯反映。
定義
除非上下文要求 另有規定,否則引用“公司,” “我們,” “我們,” “我們的,” “180人的生活”, “180ls“和”180生命科學公司“特指180生命科學公司及其合併的子公司。對“的引用”KBL線“指2020年11月6日之前的公司(以下討論和定義)。
此外,除上下文 另有要求外,僅為本報告的目的:
| ● | “計算機輔助設計“指加元; | |
| ● | “《交易所法案》“指經修訂的1934年《證券交易法》; | |
| ● | “£“或”英鎊“指英鎊; | |
| ● | “美國證券交易委員會“或”選委會“指美國證券交易委員會;及 | |
| ● | “證券法“指的是經修訂的1933年證券法。 |
在哪裏可以找到其他信息
我們向SEC提交年度、季度、 和當前報告、委託書和其他信息。我們提交的SEC文件可通過互聯網向公眾提供 ,網址為: Www.sec.gov 並可免費下載,在此類報告提交或提供給SEC後不久,在"投資者”—“美國證券交易委員會備案文件”—“所有SEC 文件"我們網站的頁面, Www.180lifesciences.com. 我們向SEC提交的文件的副本也可以免費向我們的祕書提供,請您通過本報告封面頁上列出的地址和電話聯繫 。我們的網站地址是 Www.180lifesciences.com.我們網站上的信息或可通過我們網站訪問的信息 不以引用的方式納入本報告,不應被視為本報告的一部分。
2
公司概述
我們是一家臨牀階段生物技術 公司,總部位於加利福尼亞州帕洛阿爾託,專注於通過採用創新研究和適當的聯合療法,開發治療慢性疼痛、炎症 和纖維化方面未滿足的醫療需求的療法。我們由生物技術和製藥領域的幾位世界領先的科學家創建。我們的世界知名科學家Marc Feldmann教授,Prof. Lawrence Steinman,Prof. Raphael Mechoulam,自去世以來,Jonathan Rothbard博士和Jagdeep Nanchahal教授在藥物發現方面擁有豐富的經驗和巨大的 成功。這些科學家來自牛津大學("牛津斯坦福大學和耶路撒冷希伯來大學(The "希伯來大學”),我們的管理團隊在融資和 發展早期醫療保健公司方面擁有豐富的經驗。Raphael Mechoulam教授於2023年3月去世,但他在希伯來大學的研究正在由其他科學家進行,正如本文件稍後在“SCAS平臺“部分。
我們有三個不同的產品 開發平臺,專注於不同的疾病或醫療條件,針對不同的因素、分子或蛋白質, 如下:
| ● | 抗腫瘤壞死因子平臺:專注於纖維化和抗腫瘤壞死因子(“抗腫瘤壞死因子”); |
| ● | SCA平臺:專注於合成大麻二酚(“中央商務區")或大麻酚("CBG“)和類似物(”政制事務局局長“);及 |
| ● | α7nAChR平臺:專注於α7煙鹼型乙酰膽鹼受體(“α7nAChR”). |
抗TNF平臺下的主要候選產品在英國("英國")完成了 2a期和2b期概念驗證臨牀試驗以及荷蘭的早期Dupuytren攣縮症,一種影響手掌纖維結締組織發育的疾病。
目前,我們正在計劃 或僅針對抗TNF平臺下的某些適應症進行臨牀試驗,例如計劃進行的針對術後 認知能力下降的II期試驗以及計劃進行的針對凍結肩的II期試驗。我們之前招募了患者進行 凍結肩的可行性試驗,由於英國的監管要求,我們已經停止了9名患者的招募。結束緩慢的招募 試驗。試驗結束的結果意味着將來可能需要進行另一項試驗以招募更多的 參與者。
我們最近獲得了一項關於阿達木單抗用於早期Dupuytren病的美國專利的 許可,如果獲得許可, 其有效期將不早於2037年到期。在我們的三個產品開發平臺中,只有SCA平臺涉及與大麻二酚(CBD)(而不是大麻或四氫大麻酚(THC))相關的產品,並且目前在美國或國外沒有針對SCA平臺下的適應症或產品進行臨牀試驗。我們目前正在開展SCA平臺的臨牀前 研發活動。由於公司資源有限,公司在α 7 nAChR平臺方面沒有取得 進展,同時暫停了進一步的研發活動。
公司目前正在評估 將現有資產貨幣化的所有選項,除了探索其他戰略替代方案,以最大限度地為其股東提供價值。 作為此過程的一部分,公司可能會探索或評估的潛在戰略替代方案包括但不限於 收購、合併、反向合併、其他業務合併、出售資產、許可或涉及 公司的其他戰略交易。
業務戰略
我們的目標是通過以下策略利用我們在慢性疼痛、炎症和纖維化方面的研究:
| ● | 將我們的臨牀階段候選產品從目前的後期開發階段推進到尋求並獲得英國批准,歐洲聯盟("歐盟”)和美國(”美國")對於此類候選產品,可能在英國將候選產品商業化,歐盟和美國,並確定全球其他市場的最佳商業途徑; |
| ● | 將我們的臨牀前候選產品轉移到臨牀試驗中,在英國尋求並獲得批准,歐盟和美國的此類未來候選產品,並可能在美國商業化此類未來候選產品,英國歐盟; |
| ● | 利用我們的專有產品開發平臺,發現、開發和商業化用於治療慢性疼痛、炎症和纖維化的新型一流產品;以及 |
| ● | 加強我們在慢性疼痛、炎症和纖維化方面的研究地位。 |
3
產品開發平臺概述
下表彙總了我們三個產品開發平臺的產品和適應症,包括目前處於臨牀試驗階段的產品和適應症。
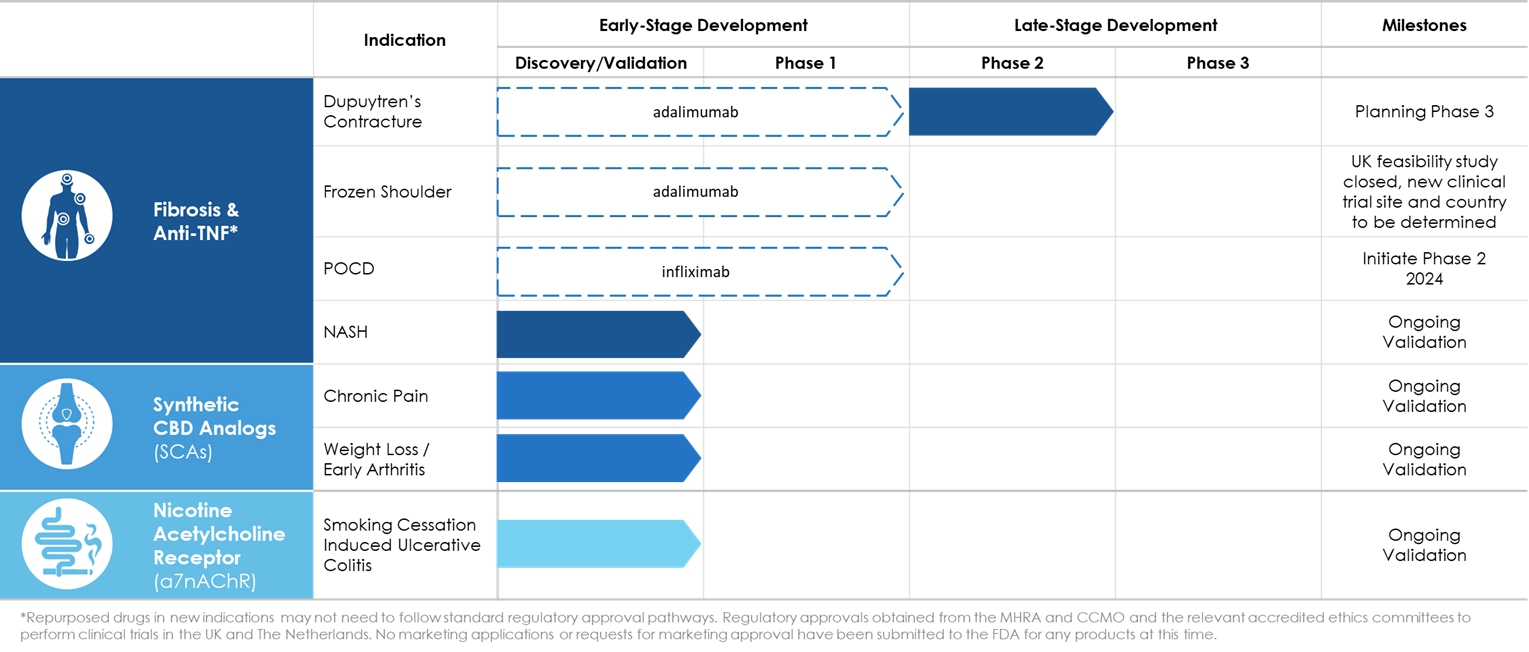
| * | 在英國進行臨牀試驗,獲得MHRA和CMO以及 相關經認可的倫理委員會的監管批准。以及荷蘭。尚未向美國食品藥品監督管理局("林業局"), 此時。 |
2021年12月1日,我們宣佈了Dupuytren‘s Constraint 2b期臨牀試驗的正面頂線數據。
2023年2月22日,我們宣佈 凍結肩關節可行性試驗的患者招募結束,由於英國的監管要求,我們已經結束了9名 患者的招募。結束緩慢的招募試驗試驗結束的結果意味着將來可能需要進行另一個 試驗以招募更多的參與者。
產品開發平臺 將在下面進行更詳細的描述。
纖維化與抗腫瘤壞死因子平臺
我們的抗腫瘤壞死因子(TNF)平臺始於我們的全資子公司180 Treateutics L.P.(180 LP“)。該平臺專注於研究炎症性疾病和纖維化的分子機制,以及發現腫瘤壞死因子作為纖維化的介質,以及其他免疫驅動的疾病。這項研究最早是在20世紀80年代由我們的執行聯席主席馬克·費爾德曼教授進行的, 基於對類風濕性關節炎患者組織的分析。Ra").我們正在將這種相同的方法應用於活動性纖維化患者的人類疾病組織的 分析,這是由牛津大學Jagdeep Nanchahal教授(他也是我們臨牀諮詢委員會的 主席)領導的研究,該研究導致了我們正在開發的新的治療靶點和方法的確定。 教授。Nanchahal和Feldmann與其他科學家合作,正在利用他們在開發抗炎藥方面的經驗和專業知識 來尋找抗TNF治療的新應用。我們正在尋求證明抗TNF藥物(如阿達木單抗)對新適應症(如Dupuytren攣縮、凍結肩和術後認知功能障礙/譫妄)有積極作用 ("POCD”).
4
我們臨牀開發的第一個候選產品 是用於治療手部早期纖維化的潛在藥物Dupuytren‘s Constraint,目前在英國或歐盟還沒有獲得批准的治療方法。來自溶組織梭菌的膠原酶在美國已被批准用於治療晚期Dupuytren肌攣縮症。建議的治療將通過將抗腫瘤壞死因子抗體阿達利單抗局部注射到早期疾病組織中來實施。由英國衞生部和美國惠康信託基金支持的Dupuytren收縮症2a期臨牀試驗的結果於2018年7月公佈。這項研究顯示了積極的組織反應,表明了抗纖維化的機制,並指導了後續試驗的劑量。在確定了最有效的劑量和製劑後,基於這些概念驗證數據,我們與惠康信託和英國衞生部一起,啟動了一項針對早期Dupuytren肌攣縮症患者的2b期試驗。最初的計劃是將138名患者按1:1的比例隨機分組,每隔3個月接受四次阿達利馬或安慰劑注射,並從基線開始跟蹤觀察總共18個月。在惠康信託的額外資金 下,2b期試驗於2019年4月完成了174名患者的招募,並於2017年2月開始服藥。 最後一名患者於2019年4月招募。治療早期Dupuytren肌攣縮症的2b期臨牀試驗已經完成。 2021年12月1日,我們公佈了試驗的頂線數據,表明超聲掃描的結節硬度的主要終點和結節大小的次級終點存在統計學意義上的差異。沒有相關的嚴重不良事件發生。正如之前報道的那樣,試驗的全部結果已經發表在同行評議的期刊上。通過這個纖維化和抗腫瘤壞死因子產品開發平臺,我們還在研究開發潛在的治療肩周炎、肝肺纖維化和POCD的方法。
我們已獲得英國MHRA和荷蘭Mensgebonden Onderzoek中心(CCMO)以及相關認可倫理委員會的監管批准,以便僅針對抗腫瘤壞死因子平臺下的適應症在英國和荷蘭進行臨牀試驗。 尚未向美國食品和藥物管理局(FDA)提交上市申請或上市批准請求(“林業局“)對於 目前抗腫瘤壞死因子平臺下的任何適應症或產品。2022年3月29日,我們向FDA提交了召開C型會議的請求,以討論抗腫瘤壞死因子平臺治療早期Dupuytren病臨牀試驗的臨牀結果評估。 2022年4月26日,FDA批准了會議請求,並同意提供書面答覆來代替會議。2022年6月9日,FDA提供了上述書面答覆,其中該機構質疑結節的硬度和大小是否會構成此類研究的合適終點。具體地説,FDA表示:“擬議的結節硬度和結節大小的結果指標似乎不是衡量患者感覺、功能或生存情況的臨牀結果指標,這將是在您未來的註冊研究中支持療效證明所必需的。”
在前面討論的2b期試驗之後, 證明瞭注射抗腫瘤壞死因子在減少或消除掌側結節的大小和硬度方面的有效性,我們認為這可能有助於預防手指殘疾和最終需要手術,我們的意圖仍然是 尋求有條件的營銷授權(“CMA“)在英國使用該療法。這項授權需要獲得英國MHRA的批准。隨後,我們計劃開始必要的程序,以尋求美國、歐盟和其他潛在國家的批准。
為支持我們的CMA申請, 我們與MHRA舉行了兩次科學建議會議。在這些會議期間,MHRA要求提供更多關於結節大小和硬度減少及其對最終需要對畸形手指進行手術的影響的信息。雖然我們提供了有限數量的患者的數據,表明與安慰劑組相比,接受抗腫瘤壞死因子注射治療的患者不需要早期手術,但樣本量太小,無法得出明確的結論。儘管如此,基於現有數據的優勢,我們認為,我們和我們在英國和美國的監管顧問認為,在啟動有限規模的第三階段結果試驗的同時,對英國市場進行CMA是合適的。必須在提交CMA審批請求文檔之前啟動此試驗 。我們的臨牀顧問已經設計了一項我們相信可以在三年內完成的試驗,預計 將滿足監管當局提出的要求,並允許考慮CMA。我們已經提交了與MHRA召開後續科學建議會議以討論這一試驗戰略的請求。由於該機構工作量大,人員有限,我們尚未收到本次會議的確認日期。
考慮到我們在美國和其他國家/地區尋求上市批准的計劃,我們還在尋求與美國FDA會面。這次會議被稱為C型會議,旨在探索一條通往IND(研究新藥)前申請和國際IND申請會議的途徑的潛力。我們認為,進行如上所述的後續3期試驗同樣是FDA接受該試驗的必要條件。我們已經向FDA提交了這次會議的申請,但尚未收到確認的日期。
隨着我們推行這一監管 戰略,我們將需要籌集額外資金。
2023年2月22日,我們宣佈 凍結肩關節可行性試驗的患者招募結束,由於英國的監管要求,我們已經結束了9名 患者的招募。結束緩慢的招募試驗試驗結束的結果意味着將來可能需要進行另一個 試驗以招募更多的參與者。
5
HMGB1計劃
我們的HMGB1:項目 於2021年11月2日獲得牛津大學的技術許可。我們的HMGB1計劃屬於纖維化和抗腫瘤壞死因子平臺。我們確定HMGB1是一種治療靶點,作用於多個內源性成體幹細胞,以加速對當前或未來損傷的生理再生反應。這些發現與幹細胞生物學和再生醫學領域具有廣泛的相關性,並建議了一種促進組織修復的治療方法,例如在NASH肝臟再生中。
由於這項研究計劃的持續成本以及公司需要將其資源集中在使用抗腫瘤壞死因子治療纖維化的公司的主要平臺上,公司董事會 於2023年9月22日決定終止公司與牛津公司的HMGB1許可協議,並於2023年9月22日,公司和牛津公司簽訂了終止函,正式終止許可證,從2023年9月22日起生效。解約信還澄清了我們在許可證終止後所欠的金額,包括在2023年12月支付的約20,000美元的 未開單費用。本公司並無因終止許可證而招致任何重大的提前終止罰款。
2022年10月25日,公司 簽訂了一項與HMGB1計劃相關的研究贊助協議,根據協議,公司同意資助牛津大學正在進行的與HMGB1計劃相關的研究 。根據這項協議,該公司有義務在2023年支付總計500,000英磅。到目前為止,公司還沒有為這一計劃支付任何款項,並正在與牛津大學談判,以減少這筆欠款。
SCAS平臺
我們的SCAS平臺始於我們的全資子公司CannBioRex PharmPharmticals Corp.(“CBR製藥“)與其創始人、已故的Mechoulam教授和Feldmann教授的合作工作。該平臺專注於開發用於治療炎症性疾病和疼痛的合成藥用級別分子 非精神活性大麻素類化合物的近似物或遠似物,如CBD。這些開發工作是費爾德曼教授和Mechoulam教授之間20年合作的結果,費爾德曼教授發現了抗腫瘤壞死因子療法並將其商業化,用於治療RA和隨後的一些炎症性疾病,這是目前世界上最暢銷的藥物類別。Mechoulam教授是世界領先的大麻化學專家,他成功地識別了THC、CBD和隨後的內源性大麻素。我們 正在與牛津肯尼迪研究所的研究團隊(由Feldmann教授、Richard Williams教授和其他人組成)和希伯來大學的研究團隊(由Avi Domb教授、Amnon Hoffman教授和其他人組成)合作,以生產新藥、測試它們,並優化它們的攝取和傳遞到疾病目標。其目的是開發基於合成化合物的新型口服活性止痛和消炎藥物,以治療慢性疾病。我們把這些合成化合物統稱為“人工合成的CBD類似物” (“政制事務局局長“)。我們的主要開發目標是關節炎以及慢性和複發性疼痛,而我們的次要開發目標是糖尿病/糖尿病神經病變、纖維肌痛、多發性硬化症、肥胖症和脂肪肝。 不幸的是,Mechoulam博士於2023年3月去世,雖然他將嚴重錯過預期,但他將根據需要繼續與同事們合作。
目前尚未就SCAS平臺下的任何產品或適應症尋求或獲得有關當局的監管批准。
α7nAChR平臺
我們的α7nAChR:平臺 始於我們的全資子公司KatExco,其創始人在那裏確定。α7nAChR:作為與阿爾茨海默氏症和帕金森氏症等疾病相關的澱粉樣蛋白的關鍵受體。α7nAChR:在大腦的神經細胞和免疫系統的重要細胞表面都有表達。喬納森·羅斯巴德博士和斯坦曼教授進行的研究表明,口服藥物的小分子可以與這種受體結合,潛在地減少炎症性疾病。羅斯巴德博士和斯坦曼教授也證明瞭。α7nAChR:在減少多發性硬化症和類風濕性關節炎以及心臟病發作和中風的 疾病動物模型中至關重要。我們 α7nAChR:產品開發 平臺目前專注於開發 α7nAChR:用於治療炎性疾病的激動劑,最初是戒煙後誘發的潰瘍性結腸炎。
6
目前尚未就任何產品或適應症尋求或獲得相關機構的監管批准, α7 nAChR 平臺。在我們能夠獲得足夠的資金來實施這一計劃之前,我們預計不會在我們的項目上取得重大進展。 α7nAChR:站臺。
候選產品
我們正在嘗試建立 一個廣泛而多樣化的產品線,用於治療慢性疼痛、炎症和纖維化。我們的候選產品已經並將 根據以下因素進行開發:解決未滿足的醫療需求的潛力;臨牀前 研究和開發工作確定的開發可行性;基於易於測量的經驗證監管終點快速實現概念驗證的潛力; 以及顯著的商業潛力。
抗TNF平臺Dupuytren攣縮
概述
Dupuytren攣縮, 也稱為手纖維化,是一種進行性的、無法治癒的疾病,其特徵是手掌中纖維索的發育,通常影響無名指和/或小指,通常影響多個關節,導致攣縮和無法伸直 受影響的手指。當向醫生提出症狀時,症狀範圍從手掌出現結節(可能是無痛的 或疼痛且常常使患者感到不安)到無法使用收縮手指。對於那些出現症狀性早期疾病的患者,目前還沒有批准的治療方案 。
手術仍然是掌腱膜攣縮症患者的標準 治療方法,但與恢復期延長和復發風險相關。
我們正在開發治療早期Dupuytren攣縮症的治療方法 ,該治療方法先前已批准並以商品名Humira用於治療多種自身免疫性疾病 。牛津大學的研究表明,抗TNF機制 可以減緩或阻止成肌細胞的增殖,導致手掌纖維結節/索的形成和生長 和可能的手指攣縮。我們已經通過2b期臨牀試驗推進了開發計劃,以評估多次病灶內注射對疾病進展和功能改善的影響。
患有晚期疾病的Dupuytren患者 主要由整形外科醫生或整形外科醫生治療,當攣縮 影響手部功能時,他們依賴侵入性幹預。目前的治療選擇包括開放性手術(筋膜切開術或筋膜切除術)和微創手術 針膜切開術(NA)或膠原酶注射。侵入性較低的手術旨在破壞收縮的 臍帶的完整性,從而可以拉直手指。不幸的是,這些選擇與高複發率有關。 醫療界和Dupuytren患者社區對晚期疾病結局的不滿以及缺乏早期/攣縮前階段幹預的選擇 表明早期幹預的醫療需求未得到滿足。
根據Dupuytren基金會,Dupuytren攣縮的患病率估計高達美國人口的7%。根據該基金會的 估計,約有300萬患者患有攣縮,應該得到治療,但這些患者中只有10%至20%得到治療。 缺乏治療的原因可能包括可用幹預措施的類型、不良的長期結局和報銷障礙。
在 2021年底與8名整形外科醫生進行的初次訪談中,由Red Sky Partners進行(一家獨立的第三方諮詢公司)代表我們 ,旨在更好地瞭解Dupuytren攣縮患者的未滿足需求,顯示了手外科醫生和患者強烈希望在發展到晚期攣縮之前儘早治療這種疾病,以非侵入性方式, 將限制進一步進展,保留功能並防止或延遲侵入性手術。外科醫生對 使用阿達木單抗解決這一未滿足需求的基本原理的反應總體上是積極的,並且認為抗TNF化合物的機制概念 令人信服。大多數接受調查的手外科醫生認為,阿達木單抗的無創、安全產品特徵可能會使其成為比目前通常治療範圍更廣的患者的重要治療選擇。假設臨牀療效和 安全性得到已發表數據的支持,我們認為阿達木單抗將成為手術、針式筋膜切開術 或膠原酶的一種有吸引力的替代藥物。此外,我們相信它在許多今天尚未接受治療的早期患者中有潛在的用途。
7
基於兩個主要的(來自這些醫生訪談的反饋 )和次要研究,Red Sky Partners得出結論,最初的標籤關注 明顯攣縮患者,阿達木單抗可以軟化結節並限制進展,這與當前的治療方法高度不同 ,在美國每年可產生3億至3.5億美元的收入。更重要的是,提供一種安全的、無創的治療方法以改善功能的機會可以顯著地擴大可治療的人羣,因為更多的患者尋求治療,更多的醫生有動力為他們的患者提供一種替代方法,而不是等待看看他們的疾病是否進展,而 他們現在無法做到這一點。
第二階段臨牀試驗
我們的全資子公司 180 LP與Wellcome Trust和 英國一起為Dupuytren攣縮症的2a期臨牀試驗提供了資金。衞生部,其使用實驗藥物臨牀試驗設計證明瞭積極的組織反應,以及 指導後續試驗的劑量和耐受性。該數據於2018年6月公佈。
在2a期試驗中,我們招募了28名患者,其中8名患者被分配到15毫克(Mg)組,12名患者被分配到35 mg組,8名患者被分配到40 mg組。ACTA2、COL1A1、COL3A1和CDH11.與安慰劑治療患者(1.51 ± 0.09 ng/μ g)相比,接受40 mg阿達木單抗治療患者(1.09 ± 0.09 ng/μ g總蛋白)的α—SMA蛋白表達水平 顯著降低(p = 0.006)。 與安慰劑組(817 ± 78 pg/μ g)相比,40 mg 阿達木單抗組(474 ± 84 pg/μ g總蛋白)中I型前膠原蛋白表達水平也顯著降低(p = 0. 019)。 有2起嚴重不良事件,均被認為與研究藥物無關。在這項劑量範圍研究中,注射40 mg 阿達木單抗(0.4 ml)導致肌成纖維細胞表型下調,2周時α—SMA 和I型前膠原蛋白表達降低證實。
在定義了最有效的 劑量和製劑後,根據這些積極的概念驗證數據,我們與惠康信託基金和英國一起, 衞生部啟動了一項針對早期Dupuytren攣縮患者的2b期試驗。最初的計劃是以1:1的比例隨機分配138例患者 ,接受阿達木單抗或安慰劑4次注射,間隔3個月,從 基線開始總共隨訪18個月。2b期試驗由惠康信託基金和英國的贈款資助。衞生部在180 LP的捐助下 購買該藥物,於2019年4月完成了174名患者的招募,並於2017年2月開始在英國 給藥。荷蘭格羅寧根。
早期Dupuytren攣縮的2b期臨牀試驗 已經完成。2021年12月1日,我們公佈了試驗的頂部數據, 表明超聲掃描結節硬度的主要終點和結節大小的次要終點存在統計學 顯著差異。無相關重度不良事件。完整結果已於2022年4月29日發表在《柳葉刀風濕病學》(Lancet Rheumology)上。根據2b期臨牀試驗的結果,該研究的研究人員分析了成本 和生活質量的數據,這是英國需要的。不錯的市場準入。他們使用患者水平模擬 模型推斷試驗結果,該模型估計阿達木單抗治療Dupuytren病的終身成本—效益。模擬模型還 評估了假設阿達木單抗每次結節重新激活(每三年一次)時重複給藥是否 治療進展性早期Dupuytren病可能具有成本效益。基於模型的外推表明, 一生中,阿達木單抗重複療程每獲得質量調整生命年可能會花費14,593英鎊,與目前的標準國民健康服務(NHS)實踐相比,這被認為是高度成本效益的。
其他候選產品 或適應症
除了潛在的治療方法,我們正在開發治療上述Dupuytren肌攣縮症的藥物,我們正在尋求改變抗腫瘤壞死因子的用途,將其用於治療其他纖維化疾病,如肩部凍結。費爾德曼教授在20世紀80年代的S的研究表明,抗腫瘤壞死因子是一種有效的抗炎藥物,具有多種可能的用途,隨後被批准用於各種形式的炎症性關節炎和炎症性腸病以及其他適應症。自那以後,產生了目前世界上最暢銷的藥物類別 抗腫瘤壞死因子療法,根據2022年11月3日發佈的一份研究報告世界報告,2022年該藥物的價值為427億美元。通過使用廣為人知和廣泛使用的治療藥物阿達利單抗,研究和開發過程可能會因為現有的與安全性有關的產品信息而被截斷,因為該藥物在過去20年中已在數百萬患者中廣泛使用。
8
凍結的肩部
凍結肩,也稱為粘連性關節囊炎,是一種極其痛苦和衰弱的疾病,影響個人的日常活動, 包括睡眠。根據美國國家衞生研究所的數據,凍結肩在40至60歲之間的人羣中最常見。 據估計,2%至5%的人口在某個時候會受到凍結肩的影響,女性中的這種情況比男性更常見 。患有糖尿病的人特別容易患上凍結肩:大約有10%到20%的人會患上凍結肩,但 目前還不知道為什麼會發生這種情況。此外,大約20%患有凍結肩的人會在他們的另一個肩膀上出現同樣的問題。根據2010年發表在Shoulder & Elbow上的一篇文章,估計高達30%的 糖尿病患者會發展為凍結肩,並且在這一組患者中症狀往往更持久和頑固。很大一部分冰凍肩患者也有Dupuytren綜合徵。我們的工作假設是,凍結肩涉及與Dupuytren類似的TNF驅動的纖維化過程 ,因此,抗TNF注射可能受益於我們在早期Dupuytren攣縮患者中進行的2b期試驗。
During the pain predominant inflammatory phase, patients are typically treated with analgesics, physiotherapy and corticosteroid injections. Patients with persistent stiffness may be referred to secondary care for capsular release by manipulation under anesthesia, hydrodilatation or surgical arthroscopy. To our knowledge, there is currently no approved targeted therapy, and in conjunction with the National Institute for Health Research (U.K.), we are investigating the feasibility of recruiting patients during the early pain-predominant inflammatory phase of the disease and delivery of a local injection of anti-TNF. The set-up stage for this Phase 2 clinical trial for the local injection of anti-TNF for frozen shoulder started in June 2021. A £250,000 grant has been awarded from NIHR to the University of Oxford to support execution and clinical trial sites are being identified. We are providing additional funding to support this trial. The recruitment of men and women across England with early-stage Frozen shoulder for a trial to determine the feasibility of conducting a large randomized controlled trial to assess whether an intra-articular injection of anti-TNF (Adalimumab) can reduce pain and improve function in people with pain predominant early-stage frozen shoulder, which was called the Anti-Freaze-F trial, began in May 2022. The Anti-Freaze-F trial was being run by the University of Oxford and originally sought to recruit 84 participants. Following delays in gaining approvals due to backlogs in the NIHR system due to COVID-19 and consequential staff vacancies, nine participants were recruited for participation in the trial through mid-February 2023. Subsequently, the NIHR’s Research Recovery and Reset program identified the trial as slow moving, due to the considerable challenges we faced to open recruitment sites and enroll sufficient participants during COVID-19. Therefore, the NIHR asked the chief investigators to close the trial for further recruitment. Our request for a no cost trial extension was denied. The participants enrolled to date have received their injections and follow up according to the established protocol. The result of the closure of the trial means that another trial will likely need to be undertaken at a future time to recruit additional participants.
人肝纖維化
肝纖維化的特徵是由於瘢痕組織替代正常肝組織而導致器官的長期損害。這種情況最常見的 是由非酒精性脂肪肝引起的("NAFLD),包括非酒精性脂肪肝(nfl“)和非酒精性脂肪性肝炎(”納什“)。根據《自然評論》2016年發表的一篇文章,非酒精性脂肪肝影響了大約30%的美國人口。大約2%的NFL患者和大約15%到20%的NASH患者進展為肝硬變、肝纖維化和主要的健康問題。
據我們所知, 目前還沒有批准的NASH患者治療方法。因此,我們認為,在創建有效的預防性治療方法方面,存在着巨大的潛在市場。根據Allied Market Research的數據,2018年肝纖維化治療市場約為130億美元,預計2022年將上升至約200億美元,複合年增長率(CAGR)超過11%。我們在2020年第二季度啟動了基於人類肝臟樣本的NASH臨牀前研究。我們已經進行了 臨牀前工作,但不打算在沒有籌集額外資金的情況下以重大方式向前推進。
9
術後認知功能下降(POCD)
POCD是一種常見的神經精神綜合徵 ,定義為注意力、意識和認知障礙,在短時間內發展並在一天中趨於波動 。髖部骨折患者發生POCD的風險特別高。英國第2018年的國家審計數據顯示,25%的髖部骨折患者患有譫妄。POCD與功能結局差、 生活質量降低和住院時間延長有關。患有髖部骨折的患者發生精神錯亂的可能性是住院患者的兩倍, 需要安置在療養院的可能性是住院患者的近四倍。POCD也與長期認知障礙密切相關。
髖部骨折是老年患者和醫療系統面臨的主要挑戰之一。根據2017年發表在《柳葉刀公共衞生》上的一篇文章,在美國和歐洲,髖骨骨折與中年和老年人平均損失2.7%的健康預期壽命有關。髖部骨折患者平均年齡為83歲,身體虛弱,三分之二為女性。他們的30天死亡率為7%,並且經歷了與健康相關的生活質量持續下降,類似於帕金森病或多發性硬化症的診斷。根據各種研究,13—40%的心臟手術患者發生POCD。在美國,每年有500,000例心臟直視手術和450,000例髖關節手術,對於老年患者,治療POCD的有益療法 對這些患者來説將是一個顯著的益處。我們計劃啟動一項使用抗TNF治療POCD的II期研究,並在2024年開始招募患者 。一項保護這種潛在用途的專利已獲肯尼迪風濕病學研究信託基金授權。我們的研究人員 發現,組織損傷手術會釋放TNF進入循環,進而打開大腦認知區域,使炎症介質流入 ,導致譫妄,手術時給予抗TNF可以預防。動物模型中的實驗 支持了這一假設。
SCAS平臺
概述
類大麻素是一種衍生自大麻植物的 化合物。大麻中含有的兩種主要大麻素是CBD和THC。雖然已知一種大麻素,THC, 引起與使用草藥大麻有關的精神活性效應,但已知沒有其他大麻素共享這些特性。 近幾十年來,有了重大的科學進展,導致發現了新的植物源性大麻素和 內源性大麻素系統。人體內源性大麻素系統中至少有兩種類型的大麻素受體,大麻素受體 1("CB1“)和大麻素受體2(”CB2"). CB1受體被認為是 大腦中表達最廣泛的G蛋白偶聯受體之一,並且在與運動和姿勢控制、疼痛和感覺感知、記憶、認知、情緒以及自主和內分泌功能有關的大腦區域中特別豐富。CB1受體 還存在於外周組織,包括外周神經和非神經元組織,如肌肉、肝組織和脂肪。CB2受體 主要在免疫系統的組織中表達,並被認為介導大麻素的免疫效應。CBD 不與CB 1受體相互作用,僅是CB 2受體的弱激動劑。CBD與人體內其他重要的神經遞質和神經調節系統相互作用,包括瞬時受體電位通道、腺苷攝取和血清素受體。多種大麻素的影響深遠 和多樣的藥理學為大麻素治療劑的開發提供了巨大的潛力,跨越 許多適應症和疾病領域,但也增加了研究的複雜性。
對於SCA計劃,我們已經與希伯來大學和牛津大學達成了 協議,根據這些協議,我們打算進行研究,以開發和表徵用於治療某些目標適應症的新型SCA,並進行早期臨牀試驗。通過與希伯來大學和牛津大學簽訂的研究協議,我們在希伯來大學和牛津大學建立了研究機構,將促進希伯來大學設計和合成的新型大麻素類化合物的開發和測試。希伯來大學的實驗室將合成這些化合物,並進行初步的療效和安全性研究。
一旦希伯來大學完成這些初步研究,化合物就會被送到牛津大學的理查德·威廉姆斯教授那裏,在那裏進行進一步的評估 以確定最有潛力進行臨牀療效和商業開發的候選藥物。隨後,我們 將支持先導化合物(S)的臨牀開發,最終進入第二階段臨牀試驗,以建立 慢性疼痛和炎症適應症的臨牀實用性。
10
研究的重點是開發安全和耐受性良好的化合物,具有止痛和免疫調節活性,並能夠與目前針對下游炎症過程的治療方法協同作用。在進行了初步研究和開發後,我們選擇了 最有希望的化學衍生物進入1/2期臨牀試驗,等待成功的毒性研究。此外,我們已經從動物研究中確定了CBD的兩種含鉛固體劑量口服制劑,目前正在進行準備工作,以促進健康人體志願者的藥代動力學分析。
候選產品或適應症
我們認為,對於口服的、相對安全的抗炎藥,尤其是具有鎮痛特性的抗炎藥,存在未得到滿足的需求。我們相信, SCA有潛力滿足這些需求,我們已經開始開發新型、口服和可申請專利的候選藥物,用於 治療某些疾病或病症,如關節炎、多發性硬化症、糖尿病、銀屑病、肥胖症和脂肪肝,以及各種 疼痛病症。我們在SCA方面的工作目前處於臨牀前開發階段。
由於醫用大麻是 200多種來自植物的化合物的複雜混合物,因此難以提供一致水平的感興趣的活性化合物或 控制其他天然化合物的水平。因此,我們正在研究口服可獲得的SCA,而不是來源於植物,以解決 上述醫用大麻的有害問題。如果成功,這些SCA可能成為批准的藥品,提供 穩定和安全的劑量,允許仔細控制患者的攝入量。
我們相信,SCA的開發和臨牀研究將揭示SCA相對於醫用大麻具有幾個關鍵優勢,包括:
| ● | 使用純化合物(>99.5%),而不是混合物; |
| ● | 能夠測試和控制劑量,進而控制療效和副作用水平; |
| ● | 創造可複製的產品;以及 |
| ● | 設計新的合成類似物以控制結合偏好以選擇受體、控制受體結合的激動劑或拮抗劑效應(藥代動力學和動力學)、改變藥物在體內的半衰期以及產生僅在特定組織中激活的前藥形式的能力,從而潛在地減少脱靶副作用。 |
除了上述優點外, 在科學的雙盲臨牀試驗中測試SCA將有助於緩解醫生對大麻基化合物治療用途的擔憂 。這一變化可能會增加獲得這些藥物治療的患者數量。如果臨牀 試驗成功,我們可以針對SCA的許多潛在市場和適應症,其中包括患有慢性和複發性疼痛、糖尿病、骨關節炎、肥胖症和脂肪肝的個體 。
α7nAChR平臺
概述
我們的兩位首席科學家,斯坦曼教授和羅斯巴德博士,之前發現了澱粉樣蛋白的一個關鍵受體,稱為α7nAChR,與阿爾茨海默氏症和帕金森氏症等疾病有關。α7nAChR在腦神經細胞和免疫系統的細胞表面都有表達。羅斯巴德博士和斯坦曼教授進行的研究表明,作為口服藥物的小分子可以與這種受體結合,從而有效地減少炎症性疾病。Rothbard博士和Steinman教授已經證明,這種受體在減少多發性硬化症和類風濕關節炎動物模型以及心臟病發作和中風的疾病方面至關重要。
我們努力瞭解高濃度的小分子熱休克蛋白的作用(“SHSP在多發性硬化症患者大腦病變中發現的這種蛋白使我們意識到,該蛋白在多發性硬化症、心臟和視網膜缺血以及中風的動物模型中具有(I)免疫抑制和(Ii)治療作用。一個重要的認識是,由蛋白質或小肽組成的澱粉樣纖維顯示出與sHSP相同的生物學反應。纖維和sHSPs專門結合並激活巨噬細胞(“MΦ“)和調節性B細胞。交聯和沉澱實驗表明,這兩個物種都與nAChR結合,並通過JAK2/STAT3信號轉導。我們意識到尼古丁治療實驗性自身免疫性腦脊髓炎 (“EAE“)誘導與我們的治療方法相同的免疫抑制模式,並顯示出臨牀前的療效 ,這與許多被批准用於多發性硬化症(MS)的藥物相當,當它們在EAE模型中進行測試時。總而言之,這些觀察為我們開發口服可用小分子α7nAChR激動劑治療炎症和自身免疫性疾病的戰略提供了依據。
11
α 7 nAChR的α 7亞基 是內源性免疫抑制途徑的一個組成部分,其中迷走神經的激活刺激乙酰膽鹼分泌, 進而結合M Φ s和調節性B淋巴細胞上的α 7 nAChR。M Φ s的激活啟動免疫抑制級聯反應 ,導致促炎細胞因子減少、B和T細胞活化抑制以及炎症控制。
在自身免疫性疾病中,如RA,有強烈的炎症破壞關節,多發性硬化症,其中大腦受到攻擊,關鍵的神經迴路受損,身體的免疫系統轉而對抗自身組織。從動脈粥樣硬化到痛風等其他疾病也顯示出不必要的自身免疫性攻擊的表現。
α7nAChR 的激活導致涉及JAK2和STAT3的信號下跌,導致巨噬細胞轉變為免疫抑制表型 併產生IL-10。已知IL-10可減少炎性細胞因子,尤其是腫瘤壞死因子、IL-1和IL-6。因此,α7nAChR激動劑應該補充抗腫瘤壞死因子治療,這為開發一類新的口服藥物開闢了可能性,這些藥物 是抗炎的,但比現有的藥物,如非類固醇、COX2抑制劑、甲氨蝶呤和JAK抑制劑要安全得多。這是因為α7nAChR激動劑激活了一條內源性調節途徑,而不是阻斷了各種過程所需的重要途徑。市場機會來自幾家大大小小的生物技術公司在開發專用於α7nAChR的口服部分激動劑頻譜方面所做的複雜而昂貴的努力。這些化合物經過了廣泛的臨牀前評估,並在包括2670名受試者的18項研究中使用。
這些藥物普遍被證明是安全的,但在神經和精神疾病(即阿爾茨海默病和精神分裂症)的試驗中無效。在針對阿爾茨海默病和精神分裂症認知障礙的隨機、安慰劑對照臨牀試驗中,化合物 未能達到其主要終點。
我們計劃以之前的這些研究為基礎,潛在地在該家族中開發一種可申請專利的α7nAChR類似物,用作免疫抑制藥,用於治療一系列炎症和自身免疫跡象,包括類風濕性關節炎、炎症性腸病、複發性和進展性多發性硬化症、動脈粥樣硬化、痛風和骨關節炎。我們的科學家發現,巨噬細胞上的α7受體和調節B淋巴細胞與迄今開發的藥物的靶點不同。
候選產品或適應症
我們打算通過篩選大量由製藥公司定義的已知激動劑的非專利類似物來鑑定、表徵、合成α7nAChR的口服小分子激動劑併為其申請專利。我們打算將這項工作外包給Evotec GmbH,這是一家集成的早期發現組織,我們過去曾與之合作,專門從事離子通道和轉運體,為客户提供從目標化合物到先導化合物的專業技術和科學專業知識。
在安全性和有效性 評估計劃之後,我們打算選擇臨牀前開發候選藥物,作為潛在啟動臨牀研究的前奏, 隨後可能進行新藥研究申請("工業”FDA。我們的α 7 nAChR開發平臺的第一個目標適應症是戒煙誘導的潰瘍性結腸炎。
在我們能夠獲得足夠的 資金來實施該計劃之前,我們不期望在我們的 α7nAChR:站臺。
12
外包與製造業
我們目前正在外包 我們的臨牀試驗,這些試驗在英國愛丁堡的牛津大學進行。和荷蘭格羅寧根,僅涉及抗TNF平臺下的某些適應症。我們希望繼續外包我們的臨牀試驗,並在(1)抗TNF平臺(牛津大學和荷蘭格羅寧根)的情況下進行,(2)SCA平臺(希伯來大學 和牛津大學)的情況下進行 α7nAChR平臺,待定。
我們還預計將外包我們的所有制造活動,包括那些處於研究或臨牀階段的活動,其中SCA將在希伯來語大學和α7nAChR將由Evotec GmbH和抗腫瘤壞死因子平臺利用現成的adalimumab生產。 此外,我們預計我們的產品將達到良好製造規範(GMP)級別,並由經認可的合同研究機構(CROS)生產。
材料協議
我們已簽訂材料 研究和許可協議(“研究協議")與各大學和各方合作,以便 進行研究,開發潛在的候選產品。我們還與多位科學家簽訂了其他材料諮詢和諮詢服務協議 (諮詢協議“),以協助這類研究。
研究協議概述 協議
研究協議包括與希伯來大學和牛津大學的 協議。對於抗腫瘤壞死因子平臺,我們與牛津大學簽訂的研究協議允許我們出資 資助針對抗腫瘤壞死因子平臺進行的研究。作為回報,我們將獲得獨家許可 研究協議產生的任何知識產權的選擇權。此外,我們還簽訂了許可協議,根據該協議,我們從牛津大學獲得了某些知識產權的獨家許可。
對於SCA計劃,我們已經與希伯來大學和牛津大學達成了 協議,根據這些協議,我們打算進行研究,以開發和表徵用於治療某些目標適應症的新型SCA,並進行早期臨牀試驗。通過與希伯來大學和牛津大學簽訂的研究協議,我們在希伯來大學和牛津大學建立了研究機構,將促進希伯來大學設計和合成的新型大麻素類化合物的開發和測試。
研究協議分別 如下所述。
與希伯來大學簽訂研究協議
2018年5月13日,我們的全資子公司CBR Pharma簽訂了一項研究和許可協議(The“2018年希伯來語協議")與耶路撒冷希伯來大學的Yissum研究開發公司("亞瑟姆),據此,Yissum授予CBR Pharma全球獨家許可(2018希伯來語許可證”)開發和商業化某些專利(“2018希伯來語許可專利”)、專門知識和研究成果(統稱為“2018年希伯來語許可技術), ,以便開發、生產、營銷、分銷或銷售產品,所有這些產品均使用2018年希伯來許可技術治療任何及所有獸醫和人類疾病,包括肥胖症、疼痛、炎症和關節炎(2018年字段”).
根據《2018年希伯來語協議》,儘管已授予2018年希伯來語許可,Yissm仍將代表希伯來大學保留以下權利:(I)將2018年希伯來語許可技術用於希伯來大學自身的研究和教育目的;(Ii)許可 或以其他方式將2018年希伯來語許可技術轉讓給其他學術和非營利研究組織用於非商業性研究;以及(Iii)將2018年希伯來語許可技術許可或以其他方式轉讓給任何第三方用於2018年領域之外的研究或商業應用 。
2018年希伯來語協議 進一步規定,CBR Pharma有權向2018年希伯來語許可技術授予一個或多個再許可,用於2018年領域的開發 。
2018年希伯來語許可技術的權利、所有權和權益 僅歸屬Yissm,CBR Pharma將僅根據2018年希伯來語協議的條款持有和使用根據 授予的2018年希伯來語許可的權利。
13
作為2018年希伯來許可證的對價,CBR Pharma向Yissum支付了75,000美元的許可證費用,並同意繼續支付年度許可證維護費(“許可證 維護費”)50,000美元,自2019年5月1日起,此後每年5月1日起。許可證維護費 不可退還,但每年可根據每年5月1日至4月30日期間的產品淨銷售額,從特許權使用費中扣除。
Yissm還同意進行研究和合成化合物,通過牛津大學和希伯來大學的額外研究,CBR Pharma將使用這些化合物來開發口服活性止痛和消炎藥物。化合物將從希伯來大學運往牛津,用於臨牀前研究,以確定對疼痛和炎症的療效。
在2018年希伯來語許可技術衍生的化合物方面達到某些里程碑後,CBR Pharma有義務向Yissm支付一定的 款項,包括但不限於以下內容:
| 里程碑 | 里程碑 費用 | ||||
| 為FDA提交首次IND測試 | $ | 75,000 | |||
| 開始與FDA進行一項1/2期試驗 | $ | 100,000 | |||
| 開始與FDA進行一項3期試驗 | $ | 150,000 | |||
| 每個產品的市場授權/許可(最多500,000美元) | $ | 100,000 | |||
| (最多50萬美元 | ) | ||||
| 每累計銷售2.5億美元的產品,直到達到10億美元的銷售額 | $ | 250,000 | |||
CBR Pharma將向Yissm 支付相當於(I)淨銷售額的3%的年淨銷售額,以及(Ii)淨銷售額達到或超過5億美元后淨銷售額的5%的版税。
如果 CBR Pharma股東出售其普通股或轉讓或轉讓2018年希伯來協議,CBR Pharma有義務 向Yissum支付CBR Pharma根據該公司交易收到的對價的5%的費用。在首次 公開發行或上市事件的情況下,CBR Pharma有義務在完成該交易的同時,在充分稀釋的基礎上向Yissum發行相當於已發行 和已發行普通股5%的註冊普通股。 於2020年11月6日完成的業務合併被視為上市事件,據此,我們於業務合併結束前向Yissum 發行了12,028股普通股。
CBR Pharma還同意 向Yissm償還專利費用(最高為30,000美元)。
Yissm和CBR Pharma還同意建立一個研究項目,CBR Pharma為截至2019年5月的12個月期間提供40萬美元的預算。該公司 計劃使用另一第三方進行研究,但尚未敲定協議。
發生下列情況時,2018年希伯來語協議將終止:(I)2018年希伯來語許可專利的最後一項到期; (Ii)任何監管機構或政府機構授予的任何產品的最後一次獨家經營權到期;(Iii)連續二十年期間在任何國家/地區沒有任何產品的商業銷售;或(Iv)如果我們根據2018希伯來語協議的條款選擇 獲取專有技術的獨家許可,則該獨家許可到期。
2019年11月11日,CBR Pharma簽訂了一項額外的研究和許可協議(2019年希伯來語協議)與Yissm合作,Yissm據此向CBR Pharma授予全球獨家和獨家許可(2019希伯來語許可證“)開發和商業化某些專利(”2019希伯來語許可專利)、技術訣竅和研究成果(統稱為2019希伯來語許可技術與2018年希伯來語許可技術一起,希伯來語 許可技術"),以便開發、製造、營銷、分銷、銷售、維修和翻新產品,所有這些都在 2019年希伯來許可技術的使用範圍內,用於(i)大麻酚鹽金屬鹽,包括一價、二價和三價金屬 ,如Li、Na、K、Ca、Mg、Zn、Fe和Al及其與天然或****的混合物,其藥物製劑, 包括口服和局部給藥;和(ii)藥物製劑,用於施用大麻素化學 衍生物,包括任何和所有獸醫和人類醫學病症,包括肥胖症、疼痛、炎症和關節炎("2019年現場”).
14
根據《2019年希伯來語協議》,儘管授予了2019年希伯來語許可,Yissm仍將代表希伯來語大學保留以下權利:(I)將2019年希伯來語許可技術用於希伯來語大學自己的研究和教育目的,但不用於商業目的,並對2019年希伯來語許可技術中包含的任何專有技術或未發佈的專利信息保密;(Ii)許可或以其他方式將2019年希伯來語許可技術轉讓給其他學術和非營利研究組織 2019年希伯來語許可技術用於非商業研究,並須對2019年希伯來語許可技術中包含的任何專有技術或未發佈的專利信息保密;以及(Iii)將2019年希伯來語許可技術許可或以其他方式轉讓給任何第三方用於2019年領域以外的研究或商業應用,但須對2019年希伯來語許可技術中包含的任何專有技術或未發佈的專利信息保密。
2019年希伯來語協議 進一步規定,CBR Pharma有權向2019年希伯來語許可技術授予一個或多個再許可,用於在2019年領域中進行開發 。
2019年希伯來語許可技術的所有權利、所有權和權益 僅歸屬Yissm,CBR Pharma將僅根據2019年希伯來語協議的條款持有和使用根據2019年希伯來語許可授予的權利。
2019年希伯來語許可技術 將在以下情況中較遲者發生時終止:(i)2019年希伯來語許可專利的最後一項到期; (ii)任何監管機構或政府機構授予的任何產品的最後一項排他性到期;(iii)連續20年的期限屆滿,加上任何適用的專利延長期,在此期間,任何國家均未進行任何產品的商業銷售。或(iv)如果我們選擇根據2019年希伯來協議的條款獲得專有技術的獨家許可, 該獨家許可到期。
2020年1月1日,CBR Pharma 和Yissm簽訂了2018年希伯來語協議的第一修正案(第一個希伯來語修正案”),這為Yissum公司對某些分子的新衍生物進行了額外的研究提供了條件。根據第一希伯來協議 修正案的條款,我們將支付Yissum每年200,000美元,加上35%額外的大學管理費用,為每個 教授在18個月期間進行的額外研究,從2019年5月1日開始。額外研究於2021年4月結束,預計 將在研究和開發一種潛在成功的給藥方法後進行進一步的臨牀前工作,該方法正處於開發後期。
與牛津大學簽訂研究協議
2013年11月1日,我們的全資子公司180 LP簽署了一份協議(第一個牛津協議”)與Oxford,根據該協議,180 LP將贊助Oxford的研究和開發,重新利用抗TNF治療Dupuytren攣縮症。
根據第一個牛津 協議,每筆款項將在項目的不同里程碑向ISIS創新公司(現為牛津大學創新公司)支付, 概述如下:
| 里程碑 | 里程碑 收費 | |||
| 已完成最小投資 | £ | 10,000 | ||
| 啟動許可產品的第二階段試驗 | £ | 10,000 | ||
| 啟動許可產品的第三階段試驗 | £ | 10,000 | ||
| 許可產品達到的可註冊III期試驗主要終點 | £ | 20,000 | ||
| 任何已發佈的美國專利的授權知識產權 | £ | 5,000 | ||
| FDA批准新藥申請("NDA“),由180 LP或其子許可人之一為許可產品提交 | £ | 30,000 | ||
| EMA批准由180 LP或其分許可人提交的許可產品的MAA | £ | 30,000 | ||
| 由180 LP或任何分授權人在美國首次商業銷售授權產品。 | £ | 50,000 | ||
| 180 LP或歐盟任何分許可方首次商業銷售授權產品 | £ | 50,000 | ||
15
ISIS Innovation也有資格 獲得特許權使用費,金額相當於在任何有有效索賠的國家/地區淨銷售額的0.5%,在其他國家/地區淨銷售額的0.25%,以及 根據或與180 LP授予的所有分許可證和 其他合同有關的所有前期、里程碑和其他一次性付款的費用收入特許權使用費率為7.5%。除非提前終止,否則第一個牛津協議有效, 只要指定專利申請作為已頒發專利、待審專利申請或補充保護 證書仍然有效,或有效期為20年,以較長者為準。
2018年8月15日,根據英格蘭和威爾士法律成立的公司CannBioRex Pharma Limited(“CannU.K.)與我們的全資子公司CBR Pharma的全資子公司 簽訂了研究協議(第二個牛津協議”)與 Oxford,據此CBR Pharma(通過CannU. K.)贊助了牛津大學從希伯來許可技術開發的SCA的研究和開發。在牛津,希伯來大學生成的SCA正在已建立的臨牀前模型中進行鎮痛和抗炎作用的測試。
根據第二個牛津協議,牛津大學開展了一項研究項目(研究項目")基於SCA的臨牀開發 已知具有抗炎和免疫調節特性。該研究項目的目的是開發 和表徵在希伯來大學合成的化合物,以創建治療慢性疼痛、RA和其他慢性 炎性疾病的方法,並最終獲得監管機構的批准,在2022年中至年底或 之後儘快啟動早期臨牀試驗。第二份牛津協議的初始期限為一年,自2019年3月22日開始,但經修訂延長 至2020年3月31日或雙方商定的任何較後日期,除非提前終止。第二個牛津協議 在2020年3月31日和Cannu. K之後沒有進一步延長。其與牛津的關係繼續存在,並與 牛津簽訂了其他協議,如下所述。
Cannu. K.,作為 研究項目的發起人,根據第二份牛津協議向牛津支付了以下款項:
| 里程碑 | 里程碑 收費 | |||
| 簽署《牛津協定》 | £ | 166,800 | ||
| 研究項目開始後6個月 | £ | 166,800 | ||
| 研究項目開始後9個月 | £ | 166,800 | ||
| 研究項目開始後12個月,在報告之後 | £ | 55,600 | ||
2020年9月18日,CannU.K. 與牛津大學簽訂了另一項研究協議(The“The”第三個牛津協議“),根據該計劃,英國政府贊助牛津大學南查哈爾教授領導的研究工作,以研究纖維化的機制。根據這項協議,加拿大政府最初提供了100,000美元,然後每隔6個月再提供一次資金,以支持林恩·威廉姆斯博士的工資和消耗品。
加拿大聯合王國作為發起國, 同意根據第三項牛津協議向牛津支付以下款項:
| 里程碑 | 金額 到期 (不包括 增值税) | |||
| 第三個牛津協定簽署後30天 | £ | 80,000 | ||
| 第三個牛津協定簽署後6個月 | £ | 178,867 | ||
| 簽署第三個牛津協定後12個月 | £ | 178,867 | ||
| 第三個牛津協定簽署後24個月 | £ | 178,867 | ||
| 第三個牛津協定簽署後36個月 | £ | 178,867 | ||
2020年9月21日,CannU.K. 與牛津大學簽訂了另一項研究協議(The“The”第四次牛津協定“),據此,CannUK同意贊助牛津大學的工作,開發和表徵新的大麻素衍生新化學實體(NCEs),用於治療炎症性疾病,並在3年內啟動患者的早期臨牀試驗。
16
加拿大聯合王國作為發起國, 同意根據第四項牛津協議向牛津支付以下款項:
| 里程碑 | 金額 到期 (不包括 增值税) | |||
| 第四次牛津協議簽署後30天 | £ | 101,778 | ||
| 簽署第四個牛津協議後6個月 | £ | 101,778 | ||
| 簽署第四個牛津協定後12個月 | £ | 101,778 | ||
| 《牛津第四協定》簽署後18個月 | £ | 101,778 | ||
| 《牛津第四協定》簽署後24個月 | £ | 101,778 | ||
2022年3月22日,CannU.K. 簽署了第四項牛津協議的修正案,將研究期延長至2023年12月31日,而不向 CannU.K.支付額外費用。
2021年5月24日,CannU.K. 與牛津大學簽訂了另一項研究協議(The“The”第五個牛津協定”),據此,CannU. K.將 在牛津大學贊助開展一項多中心、隨機、雙盲、平行組、抗TNF 注射治療疼痛主導期成人凍結肩的可行性研究。
CannU. K.,作為申辦方, 同意根據第五份牛津協議向牛津支付以下款項:
| 里程碑 | 金額 到期 (不包括 增值税) | |||
| 在簽署第五個牛津協定時 | £ | 70,546 | ||
| 《牛津第五協定》簽署後6個月 | £ | 70,546 | ||
| 《牛津第五協定》簽署後12個月 | £ | 70,546 | ||
| 《牛津第五協定》簽署後24個月 | £ | 70,546 | ||
牛津許可協議
2021年11月3日,我們 與牛津大學創新有限公司(“牛津許可協議“),據此 我們獲得了與用於肝再生的HMGB 1分子相關的某些專利權。
根據牛津許可協議,我們同意以下付款條款:
| 付款 | 應付金額 | |||
| 過去的專利成本 | £ | 49,207 | ||
| 許可費 | £ | 10,000 | ||
| 年度維護費 | £ | 3,000 | ||
17
| 里程碑 | 金額 到期 | |||
| 呈交IND | £ | 25,000 | ||
| 1聖何塞受試者在第一階段研究中為每種產品、每種適應症服用了藥物 | £ | 25,000 | ||
| 1聖何塞受試者在第二階段研究中對每種產品、每種適應症進行劑量測定 | £ | 100,000 | ||
| 1聖何塞受試者在第三階段研究中對每種產品、每種適應症進行劑量測定 | £ | 50,000 | ||
| 每種適應症的每種產品的新藥申請提交 | £ | 50,000 | ||
| 頒發的美國專利,每個專利 | £ | 5,000 | ||
| 在美國收到針對每種適應症的每種產品的監管批准 | £ | 1,250,000 | ||
| 在歐盟或英國收到監管批准。每種產品每種適應症 | £ | 550,000 | ||
| 在日本收到每種適應症的每種產品的監管批准 | £ | 150,000 | ||
| 總淨銷售額超過50億美元 | £ | 10,000,000 | ||
| 總淨銷售額超過100億美元 | £ | 50,000,000 | ||
| 淨銷售額(美元) | 版税 費率 | |||
| 1.00 | % | |||
| 2.5億-10億美元 | 2.00 | % | ||
| 10億至100億美元 | 3.00 | % | ||
| > $10B | 3.50 | % | ||
由於 該研究項目的持續成本,以及公司需要將資源集中在公司的主要平臺上,使用 抗TNF治療纖維化(腫瘤壞死因子),本公司董事會於2023年9月22日選擇終止本公司與Oxford的HMGB1許可協議 ,並於2023年9月22日,該公司和Oxford簽署了一份終止函,正式終止 許可證,於2023年9月22日生效。終止函還澄清了我們在許可證終止後欠下的金額, 包括約20,000美元的未計費費用。本公司未因 許可證終止而遭受重大提前終止處罰。
斯坦福大學許可協議
2018年5月8日,我們全資子公司Katexo的全資子公司Katexo PharmPharmticals Corp簽訂了一項期權協議(The“斯坦福大學選項“)與利蘭·斯坦福初級大學董事會合作(”史丹福“),據此,斯坦福大學授予Katexo 獲得某些發明的開發和商業化的獨家許可的選擇權。考慮到斯坦福大學的選項,Katexo向斯坦福大學支付了10,000美元(“期權付款“),可根據許可證發放費用協議記入貸方。
2018年7月25日(《斯坦福大學 生效日期),Katexo行使了斯坦福大學的選擇權,並簽訂了獨家許可協議(The斯坦福 許可協議與斯坦福大學合作,根據該協議,Katexo獲得了與以下方面有關的某些美國專利的權利:(Br)(I)用於治療自身免疫性脱髓鞘的α-B-晶體蛋白,以及(Ii)形成澱粉樣蛋白纖維的短小為六個氨基酸的多肽 可激活B-1細胞和巨噬細胞,對自身免疫和神經退行性疾病具有抗炎和治療作用 (斯坦福大學授權的專利").通過斯坦福許可協議,Katexco在斯坦福建立了研究設施 。我們將支持先導化合物的臨牀開發,最終在I期和II期臨牀試驗中建立 在潰瘍性結腸炎適應症中的潛在臨牀效用。
根據斯坦福許可協議,除根據斯坦福許可專利授予的權利外,不會將斯坦福的任何權利(包括知識產權)授予KatExco。
作為授予斯坦福大學許可專利的對價,Katexco向斯坦福大學支付了50,000美元的初始費用,包括期權付款。我們還向斯坦福大學發行了294股普通股,並提供了一封説明這些股票價值的信函。斯坦福大學發行的一部分股票後來 分配給了五個人,包括我們的首席科學官和聯席主席。
從斯坦福生效日期的一週年 開始,此後的每個週年,Katexco將在第一和第二週年向斯坦福支付20,000美元的年度許可證維護費,在隨後的每個週年向斯坦福支付40,000美元。此外,Katexco有義務支付以下 款項,包括(i)II期試驗啟動時支付100,000美元,(ii)FDA首次批准產品時支付500,000美元( "獲得許可的產品"),以及(iii)此後每件新的許可 產品支付250,000美元。特許權使用費,按淨銷售額的2.5%計算(計算方式為Katexco或其分許可人、其 分銷商或指定人根據斯坦福授權專利銷售、轉讓或其他處置產品所獲得的總收入減去5%), 將支付給斯坦福大學。此外,Katexco還向斯坦福大學償還了51,385美元,以抵消斯坦福大學許可專利的專利申請費用,並將償還斯坦福大學所有斯坦福大學許可專利申請費用,包括斯坦福大學在2018年3月3日之後發生的任何干擾和/或複審事宜。
18
我們可以通過提供30天的通知來無故終止斯坦福 許可協議。在控制權變更的情況下,在轉讓斯坦福許可協議後,Katexo有義務向斯坦福支付200,000美元的控制權變更費用。斯坦福許可協議還規定,斯坦福有權以現金購買最多(I)至10%或(Ii)斯坦福維持其在Katexo的按比例所有權權益所需的百分比,這些證券是以非公開發行的方式發行的。根據斯坦福許可協議向Stanford 發行的股份,使Stanford和五名獲得部分股份的個人 在2019年7月之前完成了180至180 LP、Katexo和CBR Pharma各自的公司重組,使Stanford和獲得部分股份的五名個人獲得了Katexo股票的2.11%的所有權,據此,180 LP、Katexo和CBR Pharma成為180LS(“重組“)在 協議下《商業公司法》。(不列顛哥倫比亞省)
《佩特卡納協定》
2018年8月20日,我們與Petcanna Pharma Corp.(“佩特卡納),這是一家由Marc Feldmann爵士(我們的執行聯席主席)和Yissm(The Yissm)教授創立的私人公司佩特卡納協定”).
根據《佩特卡納協議》, 我們授予佩特卡納獨家、全球範圍內、不可轉讓、不可再許可的再許可,以便將《佩特卡納協議》(《佩特卡納協議》)中所列與環己烯類化合物相關的某些專利用於商業用途。佩特卡納IP“)-為了開發、製造、營銷、分銷或銷售在用於治療獸醫疾病(最初是骨關節炎)的產品中加入Petcanna IP的產品。
作為 再授權的對價,Petcanna同意在2018年第四季度向我們發行約9,000,000股Petcanna普通股。截至本報告日期 ,Petcanna尚未向任何股東發行股份,且尚未開始運營。我們打算保留85%的股份,並將15%的股份轉讓給Yissum。如果Yissum不接受該等股份,我們將有義務 向Yissum支付該等股份當時公允市值的15%。Petcanna還將支付1%的版税給我們Petcanna的 淨銷售的產品,包含Petcanna知識產權。
好的,Petcanna IP的所有權和權益 ,包括對Petcanna IP的任何改進,將完全屬於我們公司。
除非 Petcanna協議訂約方另有書面約定,否則Petcanna協議將在以下情況發生時終止:(I)最後一項Petcanna IP到期之日,(Ii)任何監管或政府機構授予的任何產品的獨家經營權最終到期之日,以及(Iii)連續二十(20)年內沒有任何產品的首次商業銷售的期限屆滿之日。術語“產品”和“首次商業銷售”適用於《佩特卡納協議》中的定義。我們向Petcanna授予此再許可的能力取決於(I)Yissm擁有從所有適用各方向其轉讓的希伯來語專利申請所需的 權利,(Ii)Yissm能夠根據希伯來語協議的條款向我們授予許可,以及(Iii)希伯來語專利申請和任何相關的由此產生的專利有效 ,並根據希伯來語許可協議和Petcanna協議的各自條款保持良好。
肯尼迪許可協議
2019年9月27日,我們的全資子公司180 LP簽訂了獨家許可協議(肯尼迪許可協議“)與 肯尼迪風濕病研究信託基金(”肯尼迪”),據此,肯尼迪授予180 LP在美國的獨家 許可證,日本和歐盟成員國(包括英國),某些特許專利(“Kennedy 許可專利"),包括授予分許可的權利,以及研究、開發、銷售或製造任何藥物 產品的權利(i)如果沒有 肯尼迪許可協議授予的許可,其研究、開發、生產、使用、進口或銷售將侵犯肯尼迪許可專利,或(ii)含有抗體片段或衍生自 抗體的抗體,其研究、開發、生產、使用、進口或銷售,如果沒有根據《肯尼迪許可協議》授予的 許可,製造、使用、進口或銷售將侵犯肯尼迪許可專利,用於所有人類用途,包括疾病和病症的診斷、預防和治療。
19
根據肯尼迪許可協議,肯尼迪保留在全球範圍內永久、不可撤銷、非排他性、免版税、可再許可的權利,讓肯尼迪許可的專利及其附屬公司、員工、學生和其他研究人員為教學和進行研究開發的目的實施任何可能侵犯肯尼迪許可專利的行為,包括接受用於此類研究和開發的外部贊助的權利,以及為相同目的授予次級許可的權利。
作為授予肯尼迪許可專利的代價,180 LP向肯尼迪支付了60,000 GB的預付費用,還將向肯尼迪支付相當於淨銷售額的 至(I)第一年淨銷售額10億美元的淨銷售額的1%,以及(Ii)淨銷售額達到或超過10億美元后淨銷售額的2%,以及所有分許可收入的25%。但該等再許可或其附屬公司銷售的產品的首個累計淨銷售額 不得低於該等再許可或其附屬公司銷售的產品累計淨銷售額的1%,以及該等再許可或其附屬公司銷售的產品累計淨銷售額超過10億美元的部分的2%。
支付給肯尼迪的專利權使用費的有效期將在(I)肯尼迪許可專利中包括的涵蓋或聲稱產品在適用國家/地區的開發的專利的最後有效主張;(Ii)該產品在該國的監管排他性到期;或(Iii)自該產品在該國首次商業銷售起10年內到期。
我們可以通過提供90天的通知來無故終止肯尼迪許可協議。
Kinexum協議
2023年1月13日,我們在正常業務過程中與Kinexum簽訂了合同(“MSA“)。根據MSA,Kinexum將在有條件上市許可(CMA)和上市批准申請(MAA)方面向我們提供 協助,我們預計將向MHRA提交與我們計劃使用阿達利單抗治療進展性早期Dupuytren病相關的申請。 我們預計2024年不會有與Kinexum協議相關的重大成本,因為公司現在被要求在提交MAA之前完成針對Dupuytren病的第三階段試驗。
諮詢協議
諮詢協議 如下所述。
賈格迪普·南查哈爾教授諮詢協議
2021年2月25日,我們(和後來加入為協議一方的CannBioRex Pharma Limited)與JagDeep Nanchahal教授簽訂了一項諮詢協議,日期為2021年2月22日,並於2020年12月1日生效(經修訂,諮詢協議").教授 Nanchahal自2014年以來一直為我們和/或我們的子公司提供服務,之前是公司5%以上的股東 ,目前是我們的臨牀諮詢委員會主席。
2021年3月31日,我們與賈格迪普·南查哈爾教授簽訂了諮詢協議的第一修正案。第一個南恰哈爾修正案),修訂了與南查哈爾教授於2021年2月25日簽訂的諮詢協議,將在英格蘭和威爾士註冊成立並註冊的公司CannBioRex Pharma Limited(“CannBioRex公司“)及本公司的間接全資附屬公司, 作為訂約方,並更新先前的顧問協議,以規定應由CannBioRex支付應付Nanchahal教授的現金款項, 為税務目的,規定CannBioRex為協議的若干其他條文的訂約方,並就根據協議條款應付的若干 現金紅利的時間作出規定。
Prof. Nanchahal is a surgeon scientist focusing on defining the molecular mechanisms of common diseases and translating his findings through to early phase clinical trials. He undertook his Ph.D., funded by the U.K. Medical Research Council, whilst a medical student in London and led a lab group funded by external grants throughout his surgical training. After completing fellowships in microsurgery and hand surgery in the USA and Australia, he was appointed as a senior lecturer at Imperial College. His research is focused on promoting tissue regeneration by targeting endogenous stem cells and reducing fibrosis. In 2013 his group identified anti-tumor necrosis factor (TNF) as therapeutic target for Dupuytren’s Contracture, a common fibrotic condition of the hand. He previously lead a Phase 2b clinical trial funded by the Wellcome Trust and Department of Health to assess the efficacy of local administration of anti-TNF in patients with early-stage Dupuytren’s Contracture and a clinical trial for patients with early-stage frozen shoulder. He is a proponent of evidence-based medicine and was the only plastic surgery member of the NICE Guidance Development Groups on complex and non-complex fractures. He was a member of the group that wrote the Standards for the Management of Open Fractures published in 2020. This is an open-source publication to facilitate the care of patients with these severe injuries.
20
根據諮詢協議,Nanchahal教授同意在協議期限內擔任我們的顧問,並提供首席執行官和/或我們的董事會不時要求的服務,包括但不限於:(1)在Dupuytren肌攣縮症、肩部凍結和術後精神障礙/認知下降領域進行臨牀 試驗;以及(2)對包括肝和肺纖維化在內的其他纖維化疾病進行 實驗室研究。服務”).
作為提供服務的代價,我們(通過CannBioRex Pharma Limited)已同意在協議期限內每月向Nanchahal教授支付15,000英鎊(約20,800美元),自(A)Dupuytren‘s Constraint(RIDD)2b期臨牀試驗數據發表 之日起至23,000英鎊(約32,000美元),以及(B)我們成功籌集超過1,500萬美元資本之日。此後,費用將每年增加,以反映董事會批准的其他臨牀試驗和實驗室研究的進展 。我們還同意付給南查哈爾教授一筆獎金。獎金1")在提交Dupuytren攣縮臨牀試驗數據以供在同行評審期刊上發表時,總計100,000英鎊 ,該提交 發生於2021年12月,獎金於2021年12月支付。此外,對於之前完成的工作,包括完成RIDD(Dupuytren)試驗的招募,我們同意向Nanchahal教授支付434,673英鎊(約605,000美元)("獎金 2").根據Nanchahal教授的選擇,獎金2應至少50%(百分之五十)或更多(視Nanchahal教授的選擇)以我們普通股股份支付,每股1,140.00美元的股價或授予日期的股價(以較低者為準),其餘部分以英鎊支付。如果我們在2020年12月1日之後通過出售債務或股權籌集至少1500萬美元的額外資金,獎金2應被視為賺取和支付("歸屬日期“),在該歸屬日期之前不得應計、到期或支付。獎金2應由我們在歸屬日期後30個日曆日內支付。最後,南查哈爾教授將獲得另一筆一次性獎金(“獎金35,000英鎊(約合7,000美元),用於招募第一名患者參加2期冷凍肩部試驗,以及另一筆一次性獎金(“獎金4”)5,000英鎊(約7,000美元),用於招募第一名患者進入II期譫妄/POCD試驗。2021年3月30日,我們發行了Prof. Nanchahal 265股普通股以代替217,337英鎊,2021年4月15日,我們向Nanchahal教授發行了99股普通股以代替82,588英鎊。我們還放棄了公司必須籌集1500萬美元的要求,以便Nanchahal教授 同意通過發行股份獲得總計300,000英鎊。Nanchahal教授同意,根據獎金2到期的剩餘134,673英鎊將在我們籌集至少1500萬美元的額外資金後支付。2021年8月23日,應Nanchahal教授的要求,我們同意按每股1,140.00美元的價格向Nanchahal教授發行161股普通股,以換取紅利2剩餘的31%(或134,749英鎊,或184,606美元)。該等股份是根據我們的2020年綜合激勵計劃發行的, 該計劃已獲股東批准。
從2022年4月27日起,我們和CannBioRex與JagDeep Nanchahal教授簽訂了《諮詢協議第二修正案》。南查哈爾第二修正案 ").根據第二次Nanchahal修正案,Nanchahal教授同意在接受Dupuytren病2b期臨牀試驗數據以供發表後,(發生在2022年3月1日,須經編輯和最終批准), 他的月費增加到23,000英鎊,但其中4,000英鎊應累計,19英鎊,自2022年3月1日起,我們應根據我們的工資單慣例以現金支付每月000美元的此類費用,直至(a)2022年11月1日 或(b)董事會確定我們手頭有足夠現金支付此類應計金額的時間(以較早者為準),我們預計 在我們籌集了至少15,000,000美元("資金確定日期“),屆時所有應計款項均應到期。
2022年12月28日,我們 和CannBioRex與Nanchahal教授簽訂了諮詢協議第三修正案(The第三次南恰哈爾修正案“)。 《南恰哈爾修正案》第三修正案修訂了諮詢協議,規定根據該協議向南查哈爾教授支付的每月現金費用將保持當時的每月23,000 GB/月,直至2022年12月31日,然後在諮詢協議有效期內從2023年1月1日起增加到每月35,000 GB(統稱為 諮詢協議期限結束前)。收費").第三次Nanchahal修訂案亦規定,費用將根據董事會或本公司薪酬委員會的建議每年進行調整,而董事會或本公司薪酬委員會在釐定該等增加金額時將考慮 英國。消費者價格指數和Nanchahal教授對推進我們使命的貢獻等等。第三次Nanchahal 修訂案還規定,如果我方因非原因而終止諮詢協議,則Nanchahal 教授有權從終止日期起一次性支付12個月的月費。
21
儘管有上述規定, 本公司董事會或薪酬委員會可酌情不時以現金、股票或期權形式向Nanchahal教授發放額外獎金。
諮詢協議 的初始期限為三年,之後續簽三年,直至按照協議的規定終止為止。 目前的期限至2026年12月1日。諮詢協議可由任何一方提前12個月書面通知終止(前提是我們終止協議的權利只能在南查哈爾教授未能履行諮詢協議規定的職責的情況下行使),或在以下情況下由我們立即終止:(A)南查哈爾教授未能或忽視高效和勤奮地履行服務,或違反協議規定的任何義務(包括根據協議授予的任何同意);(B)Nanchahal教授犯有任何欺詐或不誠實行為,或其行為(無論是在履行服務或其他方面)已經或可能使Nanchahal教授、本公司或其任何附屬公司名譽受損,或 被判犯有可逮捕罪行(非監禁處罰的道路交通罪行除外);或(C)Nanchahal教授破產或與債權人達成任何安排或和解。如果我們 因任何其他原因終止諮詢協議,南查哈爾教授有權獲得終止之日起12個月費用的一次性付款。
諮詢協議 包括南查哈爾教授12個月的競業禁止和非招標義務,禁止他在他積極從事我們業務的任何國家/地區的任何地方與我們競爭,但某些例外情況除外,包括在牛津大學進行的研究。諮詢協議還包括慣常的保密和發明轉讓條款,在每一種情況下,均以我們與包括牛津大學在內的多所大學之前達成的協議為前提,南查哈爾教授在牛津大學擔任手、整形和重建外科教授。
與馬克·費爾德曼爵士簽訂服務協議
2018年6月1日,CannBioRex Pharma Limited(“CannBioRex公司)和我們的執行聯席主席Marc Feldmann Ph.D.教授簽訂了一項服務協議(費爾德曼僱傭協議“)。根據費爾德曼僱傭協議,費爾德曼爵士將擔任CannBioRex董事長、首席執行官兼董事首席執行官,或擔任與其身份相符的其他職位。費爾德曼爵士的職責包括他所擔任的角色的慣常職責。費爾德曼教授每年獲得115,000英磅的薪酬,年度薪酬由董事會審查,並由董事會決定是否有資格獲得酌情獎金。CannBioRex 還報銷費爾德曼爵士的旅費和其他商務費用。
根據費爾德曼僱傭協議,費爾德曼爵士創造的或與其僱傭相關的所有知識產權均屬於並歸屬於CannBioRex。
費爾德曼僱傭協議 包含一項慣例的非競爭條款,禁止費爾德曼爵士在任職期間為任何競爭企業工作,或持有其他企業的股權,但如果他及其家族的實益權益合計不超過該類別證券的5%,則他可以持有或實益擁有上市公司的證券。
費爾德曼爵士也被禁止在解聘後12個月內(“終止合同後期間")以任何身份 參與英國或任何其他國家的競爭業務或潛在合資企業。在終止後期間, 他不得向CannBioRex及其關聯公司的客户招攬業務;或他在工作期間積極參與的任何公司;或他持有保密信息的任何公司。Feldmann爵士進一步承諾,在終止後 期間,不得通過誘導或試圖誘導供應商採取不利行動來幹擾CannBioRex的業務關係。他還同意不誘導或試圖誘導任何CannBioRex員工在終止後期間離開公司。 Feldmann僱傭協議包含慣例的保密義務、病假和休假時間。
費爾德曼僱傭協議 沒有固定期限。任何一方均可提前9個月發出書面通知終止本協議。CannBioRex也可通過書面通知,隨時終止費爾德曼僱傭協議,立即生效。如果CannBioRex在沒有提供9個月書面通知的情況下終止了費爾德曼爵士的聘用,他將有權獲得相當於如果給予9個月通知他有權獲得的基本工資的付款。費爾德曼就業協議的適用法律 為英格蘭法律。
22
董事會亦可根據本公司薪酬委員會(及/或薪酬委員會)的建議或個別建議,不時(以股票、期權、現金或其他形式的代價)向費爾德曼教授發放獎金。
2021年11月17日,董事會根據薪酬委員會的建議,將費爾德曼教授的年薪提高到22.5萬美元。
自2022年4月27日起,CannBioRex和費爾德曼爵士簽訂了一項諮詢協議修正案,根據該協議,雙方同意從2022年3月1日起,費爾德曼爵士的工資將減少225,000美元(100%),並將在資金確定日應計並支付減少的金額。
2024年1月10日,並於2024年1月1日生效,公司與Feldmann爵士簽署了諮詢協議的第二次修訂。根據修正案,Feldmann爵士自2024年1月1日起生效,同意將其諮詢協議中規定的基本工資減少100%,至每年0英鎊,該工資減少額為每月14,167英鎊或每年170,000英鎊), 每月應計拖欠,將在公司籌集至少5美元后支付,在修正案日期之後的資助 ("資助日期"),前提是,如果資金日期未在2025年3月15日之前發生,則 應計金額將全部免除。
2024年3月7日,Marc Feldmann爵士博士。向董事會發出辭職通知,通知於同日 生效;見附註13—後續事項, 董事會成員的辭職,以獲取更多信息。
與勞倫斯·斯坦曼教授簽訂的諮詢協議
2021年11月17日和2021年11月1日生效,我們與執行聯席主席Lawrence Steinman醫學博士簽訂了諮詢協議(諮詢 協議“)。根據諮詢協議,Steinman博士同意向我們提供某些諮詢服務,包括但不限於,參與定義和設定公司的戰略目標;積極尋找收購和合並候選者;以及對我們的α7nAChR平臺(統稱為服務“)。 協議期限為一年(”初始項“);前提是協議在初始期限後自動延長 一年(每個期限均為”自動續約期限和初始條款一起 以及所有自動續訂條款(如果有)術語“),在符合續期要求(如下所述)的情況下,如果任何一方在初始期限或任何自動續期期限結束前至少30天向另一方發出書面通知,表明其不打算自動延長協議期限。任期只能延長, 條件是:(I)斯坦曼博士在緊接該自動續期開始日期之前的股東年會上再次當選為董事會成員;(Ii)董事會確認他被任命為適用的自動續期的聯席主席 (或沒有在該適用的自動續期之前任命其他人為聯席主席);以及(Iii)施泰因曼博士繼續負責公司α7nAChR平臺的科學發展( “續期要求“)。諮詢協議也將在以下較早的日期立即到期:(I)斯坦曼博士不再擔任聯席主席並不再對我們的α7nAChR平臺承擔主要科學責任的日期;以及(Ii)在以下任一方要求的任何較早日期:(1)我們(由董事會多數成員(不包括斯坦曼博士)在董事會會議上投票證明),或(2)斯坦曼博士(由斯坦曼博士向董事會發出的書面通知證明)。此外,如果斯坦曼博士不能或拒絕執行服務,我們可以立即終止諮詢協議,而無需事先通知。如果另一方 違反了諮詢協議的任何重大條款,則任何一方均可立即終止諮詢協議,而無需事先通知。
我們同意在協議期限內每年向Steinman博士支付225,000美元,並一次性支付43,750美元,這是自2021年4月1日起他的舊補償和新補償之間的差額。根據諮詢協議,Steinman博士同意在協議期限內不與我們競爭,除非獲得董事會的書面批准,並同意某些慣常的保密條款和發明轉讓要求。諮詢協議在終止後還有12個月的非徵集禁令。
23
2021年12月8日,Steinman博士 還被授予購買65股我們普通股的股票期權,期限為10年;行使價等於 我們普通股在授出日期的公平市值,每股1,501.00美元,並受我們2020年綜合激勵計劃的約束。 此外,自2022年日曆年開始,在諮詢協議期限內的每一年,我們將根據董事會的未來批准,向Steinman博士授予價值125,000美元的股權補償。未來股權授予將在48個月期間內歸屬,並 符合本計劃。未來補助金的時間、股權補助金的性質(例如,RSU、PSU、限制性股票等)且 未來股權價值的任何變動將由我們的薪酬委員會和/或審計委員會提出建議,並由 董事會批准。
自2022年4月27日起生效 本公司與Steinman博士對諮詢協議進行了修訂,據此雙方同意自2022年3月1日起,Steinman博士的工資將減少56,250美元(25%),並將於資金確定日期 計提並支付這筆減少的金額。
2024年1月10日,並於2024年1月1日生效,公司與勞倫斯·斯坦曼簽署了諮詢協議第三次修訂案。根據 修正案,Steinman博士自2024年1月1日起同意將其諮詢協議中規定的基本工資 減少100%,至每年0美元,並減少了工資數額(每月18,750美元或每年225,000美元),每月應計欠款, 將在資助日期支付,但如果資助日期不在3月15日之前,2025年,累計金額 將全部免除。
近期及重大事件
2023年1月13日, 公司在正常業務過程中與Kinexum簽訂了合同(MSA").根據MSA, Kinexum將就公司預期向英國提交的有條件上市許可(CMA)和上市批准申請(MAA)向公司提供協助。美國藥品和保健產品管理局(MHRA)與公司計劃使用阿達木單抗治療進展性早期Dupuytren病有關。我們預計2024年Kinexum協議不會產生重大 成本,因為公司現在需要在提交MAA之前完成Dupuytren病的III期試驗 。
如前面所討論的,公司的HMGB1計劃是與牛津大學創新有限公司(“University Innovation Limited”)簽訂獨家全球許可協議而形成的。牛津)於2021年11月開發和商業化HMGB1,這是一種促進肝臟修復和再生的再生分子(許可證“)。當時,由HMGB1分子激活的一條生理途徑已被證明通過靶向內源性幹細胞和祖細胞而導致組織再生。 然而,經過近兩年的研究,發現分子間的相互作用比最初設想的要複雜得多 ,至今仍未解決。因此,我們無法將這項研究進展到識別鉛分子以進行擴大和良好製造規範(GMP)生產以及安全和毒性測試的地步。
由於這項研究計劃的持續成本 ,以及公司需要將其資源集中在公司使用抗腫瘤壞死因子治療纖維化的主要平臺上 ,公司董事會於2023年9月22日決定終止公司與牛津的HMGB1許可協議,並於2023年9月22日,公司和牛津簽訂了終止函,正式 終止了2023年9月22日生效的許可。解約信還澄清了許可證終止後我們所欠的金額,包括大約20,000美元的未開單費用。本公司並無因終止許可證而招致任何重大的提前終止罰款。
2023年10月12日,公司收到了英國藥品和保健品監管機構(MHRA)關於2023年8月17日與MHRA舉行的會議的正式書面科學回復。該公司的管理和監管團隊與MHRA會面,提出了一條未來的道路,以批准該公司使用阿達利單抗作為抗腫瘤壞死因子(TNF)治療 ,以潛在地預防Dupuytren的肌攣縮殘疾。
在迴應中,MHRA(I)認識到Dupuytren‘s Constraint的衰弱性質;(Ii)同意公司為擬議的3期臨牀試驗(3期研究)提出的主要和次要終點;(Iii)同意如果觀察到令人信服的有效性和安全性證據,單個3期研究就足以支持營銷授權;(Iv)已確認MHRA認為 公司2b期試驗的結果導致太多不確定性,無法支持有條件營銷授權(CMA),因為試驗參與者數量較少,並且MHRA將要求第三階段研究的結果考慮 營銷授權;及(V)已就潛在的第三階段研究向公司提供指導,包括由每隔3個月進行四次注射的療程 是可接受的。
24
公司目前還在 與FDA進行互動,並準備與歐洲藥品管理局(EMA)聯絡,試圖就我們擬議的 臨牀開發計劃(如上文針對MHRA指南所述)達成協議,並努力尋求在所有這些司法管轄區批准阿達木單抗作為 一種抗TNF治療,用於潛在預防Dupuytren攣縮殘疾。
為了支持我們目前與FDA的互動,一家領先的製藥生物相似產品製造商已同意與該公司一起參加FDA關於阿達利瑪單抗建議生物類似物的製造和安全性的建議討論 。此外,該製造商已表示希望 供應將在第三階段研究中使用的抗腫瘤壞死因子生物相似藥物;但到目前為止,尚未與供應商 達成最終協議。預計與該供應商的任何協議將取決於上述FDA討論的結果,我們可能無法與該供應商達成雙方同意的最終條款。
本公司目前正在 在與FDA的討論中考慮MHRA的指導意見,並計劃在必要時進行潛在的第三階段研究,條件是此類研究的資金可用。
2023年12月,我們聘請了AG.P./Alliance Global Partners擔任財務顧問,以探索和評估提升股東價值的戰略選擇。作為這一過程的一部分,公司可能探索或評估的潛在戰略選擇包括但不限於收購、合併、反向合併、其他業務合併、出售資產、許可或涉及公司的其他戰略交易。 公司不打算在此過程中討論或披露進一步的發展,除非董事會已批准具體行動或以其他方式確定進一步披露是適當的。不能保證戰略審查流程將批准或完成任何特定交易或結果。
我們的普通股和公共認股權證在納斯達克上交易,代碼為“ATNF”和“ATNFW,“這兩個字分別是。儘管有這樣的上市, 不能保證任何經紀商都有興趣交易我們的證券。因此,可能很難公開出售我們的證券 。也不能保證通過永久滿足納斯達克的持續上市要求,我們能夠在任何時間內保持納斯達克上的上市。
2023年9月7日,公司收到納斯達克上市資格部的書面通知(“納斯達克“)並通知 本公司未遵守納斯達克上市規則第5550(A)(2)條關於 繼續在納斯達克資本市場上市的最低投標價格要求。納斯達克上市規則第5550(A)(2)條要求上市證券維持每股1.00美元的最低買入價,而上市規則第5810(C)(3)(A)條規定,如果未能達到最低買入價要求持續連續三十(30)個工作日,則存在未能達到最低買入價要求的情況。根據公司普通股在2023年7月26日至2023年9月6日連續三十(30)個工作日的收盤投標價格,公司不再滿足最低投標價格要求。
通知函指出,公司有180個歷日或至2024年3月5日,以重新遵守納斯達克上市規則第5550(A)(2)條。要重新獲得合規, 公司普通股的出價必須在至少連續10個工作日內至少達到每股1.00美元的收盤出價。如果公司在2024年3月5日之前仍未恢復合規,則只要公司滿足納斯達克資本市場初始上市標準(投標價格要求除外),並在必要時通過反向股票拆分的方式在第二合規期內書面通知納斯達克,就可以額外給予180天來恢復合規。如果本公司不符合第二個合規期的要求或未能在第二個180天的合規期內恢復合規,則本公司的普通股將被摘牌,屆時本公司將有機會向聽證小組就退市決定提出上訴。
25
The Company intends to monitor the closing bid price of its common stock and may, if appropriate, consider implementing available options to regain compliance with the minimum bid price requirement under the Nasdaq Listing Rules, including affecting a reverse stock split. The Company plans to hold a special stockholders’ meeting on February 16, 2024, to seek approval, for among other things, an amendment to our Second Amended and Restated Certificate of Incorporation, as amended, to effect a reverse stock split of our issued and outstanding shares of our common stock, by a ratio of between one-for-four to one-for-forty, inclusive, with the exact ratio to be set at a whole number to be determined by our Board of Directors or a duly authorized committee thereof in its discretion, at any time after approval of the amendment and prior to February 16, 2025. On February 16, 2024, the Company’s Board of Directors authorized a reverse stock split of our issued and outstanding shares of common stock in the amount of one-for-nineteen, which was effective on February 28, 2024. On March 13, 2024, the Company received a letter from Nasdaq notifying the Company that it has regained full compliance with the minimum bid price for continued listing on Nasdaq, pursuant to Nasdaq Listing Rule 5550(a)(2), because Nasdaq has determined that for 10 consecutive business days, the closing bid price of the Company’s common stock was at or above $1.00 per share.
於2023年10月11日,本公司收到納斯達克的書面通知,通知本公司,本公司不符合納斯達克上市規則第5635(D)條所載的股東批准要求 公開招股以外的交易涉及 以低於適用最低價格(定義見上市規則5635(D)(1)(A))的價格發行20%或以上的交易前已發行股份。
員工根據《上市規則》第5635(d)條作出的決定 涉及本公司發售及發行下列各項的合計:(i)35,102股 公司普通股,價格為每股12.35美元,(ii)購買最多207,814股普通股 的預融資認股權證,以每份預融資認股權證12.3481美元的價格及(iii)購買最多242,915股普通股的認股權證。每股 及相關普通認股權證的發行價為12.35美元,每份預存資金認股權證及相關普通認股權證的發行價為 12.3481美元。
工作人員確定, 本次發行不是納斯達克股東批准規則的“公開發行”,原因是 發行的類型、根據配售代理協議進行的盡力而為發行,以及一名投資者購買了本次發行的98%。 因此,由於發行量佔已發行普通股的20%以上,且定價低於最低價格, 員工決定公司須根據上市規則第5635(d)條事先獲得股東批准。
2023年10月11日的信函 要求公司在45天內提交重新獲得合規的計劃。隨後,公司 於2023年11月9日向納斯達克提交了合規計劃,並於2023年11月14日,納斯達克批准公司將合規計劃中規定的某些交易延期至2023年12月15日,以糾正其先前違反納斯達克規則的行為,如納斯達克2023年10月11日的信函中所述。
2023年8月14日,我們發行了 並出售給某些投資者,包括某個機構投資者(The“採購商(i)35,102股股份(“2023年8月股票)公司普通股;(Ii)預融資權證(2023年8月 預籌資權證“)購買最多207,814股普通股,及(Iii)認股權證(”2023年8月普通認股權證 “)就買方而言,根據本公司與買方於2023年8月9日訂立的證券購買協議購買最多242,915股普通股(2023年8月SPA”).
於2023年10月11日, 本公司收到納斯達克上市資格部的書面通知,通知本公司不符合《納斯達克上市規則》第5635(D)條所載的股東批准要求,即除公開發行外,涉及以低於適用最低價格(定義見上市規則第5635(D)(1)(A)條)的方式發行20%或以上的交易前已發行股份。
納斯達克根據上市規則第5635(D)條就2023年8月招股(“2023年8月提供“)。2023年8月普通股及相關普通權證的發行價為每股12.35美元,2023年8月預融資權證及相關普通權證的發行價為12.3481美元。
納斯達克認定,就納斯達克的股東批准規則而言,2023年8月的發售不是“公開發售”,原因是 發售的類型、根據配售代理協議進行的盡力發售以及一個投資者購買了2023年8月發售的98%的股份的事實。因此,由於2023年8月的發行佔已發行普通股的20%以上,並且定價低於最低價格,納斯達克決定,根據上市規則 第5635(D)條,公司必須事先獲得股東批准。於2023年11月及12月,本公司採取各種行動修訂2023年8月要約的條款,以遵守上市規則第5635(D)條,如下所述。
26
2023年11月28日, 公司與買方簽訂了2023年8月SPA的第1號修正案(2023年11月SPA修正案),據此,(I)買方同意額外支付830,769.30美元,用於重新定價2023年8月的股份 和2023年8月的預資金權證(“重新定價金額“),(Ii)本公司同意向買方(X) 發行預資金權證,以購買最多257,205股普通股,行使價為每股0.0019美元(”2023年12月預籌資金認股權證“),及(Y)認股權證購買最多477,058股普通股,行使價為每股3.23美元(”2023年12月普通權證此外,連同2023年12月的預付資權證,認股權證“)、 及(Iii)本公司與買方同意訂立權證修訂協議(定義及説明如下)。
認股權證將不可行使 ,直到公司獲得股東批准,在認股權證行使時發行734,262股普通股( "認股權證股份”)(“股東批准,以及股東批准的日期,股東 審批日期“),屆時2023年12月的預資資權證將繼續可行使,直至所有2023年12月的預資資權證全部行使,而2023年12月的普通權證將繼續行使,直至股東批准日期(定義見下文)五週年為止。
2023年11月SPA修訂案 包含公司的某些慣例陳述、保證和協議、關閉的慣例條件、公司的賠償 義務、各方的其他義務以及終止條款。根據2023年11月SPA修訂案, 本公司已同意,除某些例外情況外,自截止日期起至股東批准日期後15天內,將不發行任何普通股(或其等價物) 。2023年11月的SPA修正案還要求公司 向SEC提交登記聲明,以在 股東批准日期後六十(60)天內登記買方轉售權證股份。
根據《2023年11月SPA修正案》,本公司於2023年11月28日與買方訂立權證修訂協議(“授權書 修訂協議”),據此,本公司同意修訂買方持有的以下尚未行使的認股權證:(i)購買最多135,339股普通股的認股權證,於2022年12月22日發行,並於2023年1月、2023年4月和2023年8月修訂(“2022年12月認股權證(ii)購買最多16,138股普通股的認股權證,於2022年7月20日發行,並於2023年4月和2023年8月修訂(“2022年7月認股權證);(iii)購買最多82,668股普通股的認股權證,於2023年4月10日和2023年8月發行(2023年4月和8月的認股權證”,連同 2022年12月權證和2022年7月權證,2022年12月至2023年8月);及(iv)購買最多242,915股2023年8月普通認股權證(統稱為“現有常見 認股權證“)。根據《權證修訂協議》,現有的共同權證將被修訂(“授權書 修訂),因此,在公司獲得股東批准後,在行使現有普通認股權證(即"現有普通權證股份"). 現有普通認股權證的行使價為每股3.23美元,而現有普通認股權證將於股東批准日期("重新定價和延期“)。現有普通權證的其他條款保持不變。
上述交易已於2023年12月1日完成(“截止日期“)。於交易完成後,本公司 恢復遵守納斯達克上市規則第5635(D)條。
為取得 股東批准發行認股權證股份及現有普通權證股份,本公司同意於截止日期後九十(90)日或之前召開股東 會議(定義見2023年11月SPA修正案)(而 已安排於2024年2月16日召開該會議)。如本公司未能在首次股東大會上獲得股東批准,則本公司將於其後每九十(90)天召開一次股東大會,直至(I)獲得股東批准之日或(Ii)認股權證及現有普通權證不再有效之日(以較早者為準)。
在交易完成的同時,本公司簽訂了認股權證代理協議(“認股權證代理協議“)與大陸 股票轉讓與信託公司(”大陸航空公司“),據此,大陸航空將作為認股權證的 代理。
27
由於這些交易,納斯達克於2023年12月14日向本公司發出書面通知,表示本公司已遵守先前延期的條款; 本公司遵守上市規則第5635(D)(1)(A)條);以及此事現已結束。
2023年11月15日, 公司收到納斯達克的函,通知公司不符合在納斯達克資本市場繼續上市的最低股東權益要求 。納斯達克上市規則第5550(B)(1)條(“規則“)要求 在納斯達克資本市場上市的公司保持至少250萬美元的股東權益。在公司截至2023年9月30日的季度報告10-Q表中,公司報告股東虧損(149,327美元),低於根據規則繼續上市所需的最低股東權益。此外,本公司並不符合納斯達克上市規則下的另類納斯達克持續上市標準。
納斯達克要求公司在2024年1月2日之前向納斯達克提交重新獲得合規的計劃。我們及時提交了恢復合規的計劃, 並且在2024年1月11日,納斯達克通知公司,它已決定批准公司延期,以重新遵守規則 。
The terms of the extension are as follows: on or before May 13, 2024, the Company must complete certain transactions described in greater detail in the compliance plan, contemplated to result in the Company increasing its stockholders’ equity to more than $2.5 million, and opt for one of the two following alternatives to evidence compliance with the Rule: Alternative 1: The Company must furnish to the SEC and Nasdaq a publicly available report (e.g., a Form 8-K) including: 1. A disclosure of Staff’s deficiency letter and the specific deficiency(ies) cited; 2. A description of the completed transaction or event that enabled the Company to satisfy the stockholders’ equity requirement for continued listing; and 3. An affirmative statement that, as of the date of the report, the Company believes it has regained compliance with the stockholders’ equity requirement based upon the specific transaction or event referenced in Step 2; or Alternative 2: The Company must furnish to the SEC and Nasdaq a publicly available report including: 1. Steps 1 & 2 set forth above; 2. A balance sheet no older than 60 days with pro forma adjustments for any significant transactions or event occurring on or before the report date; and 3. That the Company believes it satisfies the stockholders’ equity requirement as of the report date. The pro forma balance sheet must evidence compliance with the stockholders’ equity requirement.
此外,在任何一種情況下,公司都必須披露納斯達克將繼續監督公司對股東權益要求的持續遵守情況,如果在下次定期報告時公司沒有證明遵守要求,公司可能會被摘牌 。
無論本公司選擇哪種方式 ,如果本公司在合規期結束後向美國證券交易委員會提交下一份定期報告(即截至2024年6月30日的季度報告)時未能證明其遵守情況,則本公司可能被摘牌。 如果本公司不滿足這些條款,納斯達克將以書面通知其證券將被摘牌。 屆時,本公司可就納斯達克的裁決向聽證會小組提出上訴。
該公司目前正在評估恢復合規的各種行動方案,並希望能夠在合規期內重新符合納斯達克的最低股東權益標準。然而,不能保證公司能夠完成合規計劃中預期的交易,公司希望這些交易能夠使公司重新遵守規則,也不能保證此類交易 將導致公司在納斯達克授予的合規期內重新遵守規則。如果我們未能及時糾正我們對此類適用要求的遵守,我們的普通股和公共認股權證可能會被摘牌。
自2023年12月17日起,唐納德·A。麥戈文,本公司董事會成員。麥戈文先生的辭職是由於健康原因。自2023年12月17日起,Francis Knuettel II、Pam Marrone、Teresa DeLuca、Larry Gold和Russell Ray辭去公司董事會成員職務;他們的辭職與成本削減舉措的意見分歧有關。
McGovern先生、Knuettel先生、DeLuca博士、Gold博士和Ray先生代表了我們的所有獨立董事,因此,截至2023年12月17日,我們沒有獨立 董事,也沒有審計委員會、薪酬委員會、提名和公司治理委員會以及風險、安全和監管委員會的任何成員。我們目前正在尋求合格人士任命為董事會的獨立成員; 參見附註13—後續事項, 委任新董事會成員和董事會的撤銷.
28
Due to recent financial constraints, the Company has been unable to timely pay amounts due to the University of Oxford, the licensor of the majority of the Company’s licenses and patents and the Company’s research partner. Oxford alleges that an aggregate of approximately £929,030 is owed from the Company and one of its subsidiaries to Oxford under the terms of licenses and agreements with Oxford and related parties. The Company is currently in ongoing discussion with Oxford to reduce that amount and enter into a payment plan with regards to the amounts owed, and has received preliminary acceptance of a payment plan; however, no definitive terms or extensions have been agreed to date. Oxford has also notified the Company that it is not willing to discuss any new projects or arrangements until all outstanding invoices have been paid or a payment plan has been agreed to; has engaged a law firm to seek the collection of the amounts owed, together with interest; and has threatened legal proceedings against us. While we are hopeful that we can come to mutually agreeable terms regarding a settlement, payment plan, and/or extension, with Oxford, we may not have sufficient funds to pay amounts due to Oxford in the near term, if at all, and Oxford may take action against us, including filing legal proceedings against us seeking amounts due and interest, attempting to terminate their relationship with us, and/or filing a wind-up petition against one of the Company’s subsidiaries in the U.K. If Oxford were to take legal action against us or terminate their relationship with us, we may be forced to scale back our business plan and/or seek bankruptcy protection. We may be subject to litigation and damages for our failure to pay amounts due to Oxford, and may be forced to pay interest and penalties, which funds we do not currently have. We plan to seek to raise funding in the future to support our operations, and to pay amounts due to Oxford, through a combination of equity offerings, debt financing or other capital sources, including potentially collaborations, licenses and other similar arrangements, which may not be available on favorable terms, if at all. The sale of additional equity or debt securities, if accomplished, may result in dilution to our then stockholders. Additionally, in December 2023, we engaged A.G.P./Alliance Global Partners as financial advisor to explore and evaluate strategic alternatives to enhance shareholder value. Potential strategic alternatives that may be explored or evaluated by the Company as part of this process include, but are not limited to, an acquisition, merger, reverse merger, other business combination, sale of assets, licensing or other strategic transactions involving the Company. The Company does not intend to discuss or disclose further developments during this process unless and until its Board of Directors has approved a specific action or otherwise determined that further disclosure is appropriate. There is no assurance that the strategic review process will result in the approval or completion of any specific transaction or outcome.
知識產權
我們的成功在很大程度上取決於我們是否有能力保護我們的候選產品、技術和專有技術的專有元素,在不侵犯他人專有權利的情況下運營,以及抵禦他人的挑戰和反對並防止其他人侵犯我們的專有 權利。我們已經並將繼續在美國、英國、歐洲和其他國家/地區為我們的專有技術尋求專利保護。
截至2024年3月20日,我們的知識產權組合包括已發佈和/或懸而未決的權利要求、藥物配方、藥物輸送和SCA的治療用途的11個專利系列, 以及專有技術和商業祕密,如果包括我們擁有獨家權利的合作伙伴持有的專利。
在美國境內,我們和/或我們的合作伙伴已經或已經授權了14項已發佈的專利和9項正在積極起訴的未決專利申請。在美國之外,假設歐盟是一個單一司法管轄區,還有另外15項已頒發的專利和13項待決的專利申請正在積極起訴中。
我們的政策是為我們認為對業務發展重要的技術、發明和改進尋求專利 保護,但僅在我們認為獲得專利保護的成本因技術的商業潛力而合理的情況下, 通常僅在我們認為存在重大商業機會的司法管轄區。我們還依靠商標、交易祕密、技術訣竅和持續創新來發展和保持我們的競爭地位。
個別專利的期限 取決於獲得專利的國家/地區。在我們提交申請的大多數國家/地區,專利期為自提交非臨時專利申請的最早日期起計20年。在美國,專利期限的延長可以通過專利期限調整來延長,專利期限調整可以補償專利權人因美國專利商標局(“USPTO”)在授予專利時的行政延誤而造成的損失, 如果一項專利被最終放棄,則可以縮短專利期限。
29
涵蓋FDA批准的藥物的專利期限也有資格延長,這允許恢復期限,作為對在FDA監管審查過程中丟失的期限的補償。1984年的《藥品價格競爭和專利期限恢復法》(《哈奇-瓦克斯曼法》)允許在專利到期後延長最多五年。專利期延長的時間長短與藥物接受監管審查的時間長短有關。延期不能將專利的剩餘期限從產品批准之日起延長14年以上,並且只能延長一項適用於批准藥物的專利。歐洲和其他非美國司法管轄區也有類似的條款,以延長涵蓋批准藥物的專利的有效期。
為了保護我們獲得 任何已發佈專利和專有信息的權利,我們可能需要對侵權第三方提起訴訟,利用 法院或參加聽證會來確定這些專利或其他專有權利的範圍和有效性。
我們還依靠商業祕密保護我們的機密和專有信息。我們的政策要求我們的員工、顧問、外部科學合作者、受贊助的研究人員和其他顧問在與我們開始僱傭或諮詢關係時執行保密協議 。
在我們正常的運營過程中,我們將不時地成為與知識產權相關的訴訟和其他爭議事項和索賠的一方。
180LS的研究、開發和許可協議
180LS已經與包括耶路撒冷希伯來大學和牛津希伯來大學在內的各方簽訂了研究和許可協議。有關這些 協議的信息,請參閲“材料協議“,上圖。
競爭
以下是對我們每個候選產品開發平臺和潛在候選產品的競爭環境的説明。
杜普伊特倫肌攣縮症
我們的治療是早期的Dupuytren攣縮,據我們所知,目前尚無批准的治療方法。現有的治療方法集中在晚期Dupuytren攣縮,當手指不可逆轉地捲曲到手掌。手術仍然是典型的標準治療方法,但相對 較長的術後康復已經推動了侵入性較低的技術。Xiaaflex是一種由Alcilium開發的藥物, 已經顯示出對嚴重攣縮患者有效的治療,儘管許多患者的副作用相對較輕。另一種 方法是用針破壞晚期脊髓,2018年發表在《骨 和關節外科雜誌》(美國)上的一項比較臨牀試驗數據顯示,膠原酶和經皮針筋膜切開術在2年時的複發率相似。由英國國家衞生研究所(National Institute for Health Research Health Technology Assessment Program)資助的臨牀試驗目前正在英國進行比較手術治療Dupuytren攣縮症與膠原酶治療的成本效果。 研究的目的是確定(i)膠原酶注射是否與手術治療這種疾病一樣有效和安全 和(ii)兩種治療的費用。
政制事務局局長
在收購之後 GW製藥公司及其Epidiolex(大麻二醇)和Sativex(THC&CBD)特許經營權,截至2010年Jazz 製藥(愛爾蘭),Jazz Pharma已成為大麻二醇領域的重要參與者.Epidiolex是一種口服大麻二醇溶液,被批准用於治療一系列兒童癲癇疾病的癲癇發作,包括Draves綜合徵(以前稱為嬰兒期嚴重肌陣攣癲癇)、Rett綜合徵和Lennox-Gastaut綜合徵。Jazz Pharma正在探索Epidiolex是否對Sturge-Weber綜合徵有效,這種綜合徵是指血管發育異常會導致出生時大腦、皮膚和眼睛出現缺陷,更廣泛地説,Epidiolex對自閉症譜系障礙有效。由Jazz Pharma贊助的臨牀試驗正在測試Epidiolex對多發性硬化症、潰瘍性結腸炎和克羅恩病等自身免疫性疾病的有效性。總而言之,這些努力代表了最廣泛的大麻二醇臨牀計劃。
30
據我們所知,多家公司在大麻治療領域開展工作,並正在尋求對其候選產品進行監管批准,包括:
| ● | Cardiol Therapeutics(加拿大)正在評估其口服CBD液體制劑對急性心肌炎患者心肌恢復的有效性。 |
| ● | Zynerba製藥公司(賓夕法尼亞州),專注於藥物生產的經皮大麻類藥物治療罕見和近乎罕見的神經精神疾病。Zynerba目前正在評估ZyglTM,這是一種受專利保護的CBD透皮凝膠,用於治療脆性X綜合徵,該公司向FDA提交了一份保密協議,涉及發育和癲癇腦病、22q缺失綜合徵和自閉症譜系障礙。 |
| ● | 奧科薩,(新澤西州),正在測試他們的CBD,Oravexx (口腔崩解片),以控制疼痛和炎症,希望減少臨牀對阿片類藥物的依賴。在第二階段試驗中,特殊的適應症是與膝關節骨關節炎相關的疼痛。 |
| ● | Stero Biotechs(以色列),它贊助了GVHD的第二階段試驗 表明CBD給藥(橄欖油中合成的CBD)要麼增強了類固醇的治療效果,要麼減少了類固醇的劑量,同時保持或改善了類固醇的原始治療效果。其他臨牀試驗包括IIa期試驗、類固醇依賴型克羅恩病的多中心試驗、慢性蕁麻疹(Hives)的IIa期試驗和嚴重新冠肺炎的I/II期試驗。 |
A7nAChR
選擇TNFa(Humira)或TNFa受體(Remicade)的抗體和核酸適配子是可注射的試劑,其固有性質 有臨牀侷限性。口服生物利用的TNFa抑制劑是抗體或適配子策略的天然補充試劑。 如果不同的治療試劑的作用模式不同,這一點尤其有吸引力。抗體和適配子都旨在幹擾腫瘤壞死因子α信號,而口服生物可用藥物是一種已知能激活迷走神經和大腦免疫系統接口的α7煙鹼型乙酰膽鹼受體激動劑。
180LS計劃開發一種口服生物可用的TNFa分泌抑制劑,這一計劃面臨着巨大的競爭。最突出的是被批准用於治療類風濕性關節炎、牛皮癬關節炎、幼年特發性關節炎、軸性脊柱性關節炎、潰瘍性結腸炎、特應性皮炎和斑禿的JAK抑制劑(Xeljanz、Cibinqo、OLumant、Rvoq和Jyeleca)的集合。作用模式是抑制Jak-Stat途徑,主要是在已知的分泌多種促炎細胞因子的巨噬細胞系的細胞中,包括 TNFa。這些試劑的商業成功為開發口服生物利用型產品的重要性提供了實際支持。 這些藥物不參與乙酰膽鹼受體途徑。Attenua Pharma,一家小型生物技術公司,正在使用一種α7nAChR激動劑Bradanicline在慢性咳嗽的第二階段臨牀試驗中由Coda Treateutics獲得,該計劃已停止。
電子公司 可以被視為競爭,或者是一個巨大的概念驗證。因為在許多方面,α7nAChR計劃可以被認為是對迷走神經的化學刺激,而受益於電刺激的每一個適應症都應該服從化學 刺激。據我們所知,最接近被批准用於炎症性適應症的產品的公司是SetPoint Medical Corporation,它具有治療炎症性腸病和類風濕性關節炎的活性。他們的設備是一個微型刺激器植入物,在手術中通過頸部左側的一個小切口在全身麻醉下放置在迷走神經上。一個意想不到的結果是,短的電脈衝導致炎性細胞因子的減少延長,約為8-10小時。
最後一個考慮是,最初開發α7nAChR激動劑的每一家大型製藥公司都可以重振他們的計劃,並將他們的藥物用於炎症適應症的臨牀試驗。
電子公司 可以被視為競爭,或者是一個巨大的概念驗證。因為在許多方面,α7nAChR計劃可以被視為對迷走神經的化學刺激,而受益於電刺激的每一個適應症都應該服從於化學刺激。
31
最後,最初開發α7nAChR激動劑的每一家大型製藥公司都可以重振其計劃,並將其藥物用於臨牀 炎症適應症試驗。
政府監管
我們已獲得英國藥品和保健品監管機構(MHRA)和荷蘭中央委員會Mensgebonden(CCMO)以及相關認可道德委員會的監管批准,以便僅針對抗腫瘤壞死因子平臺下的適應症在英國和荷蘭進行臨牀試驗。根據2b期Dupuytren‘s Contracture 臨牀試驗的成功結果,我們正在準備向MHRA提交的有條件營銷授權申請。我們沒有與美國食品和藥物管理局 舉行任何會議,也沒有向美國食品和藥物管理局提交任何申請或批准請求(“林業局“)對於 目前抗腫瘤壞死因子平臺下的任何適應症或產品。
FDA審批流程
在美國,包括藥品和生物製品在內的製藥產品受到FDA的廣泛監管。根據美國聯邦食品、藥品和化粧品法案(“FDC法案”), a “藥物“定義為包括”用於診斷、治癒、緩解、治療或預防人類或其他動物疾病的物品“和”物品(食物除外)旨在影響人或其他動物的身體結構或任何功能。“21 USC 321(G).與所有藥物一樣,生物製品也用於治療、預防或治癒人類疾病。然而,與化學合成的小分子藥物不同,小分子藥物具有明確的結構並可以進行徹底的表徵,而生物製品通常是從活體材料(如人、動物或微生物)中提取的,結構複雜,因此總是得到充分的表徵。小靈通法案“)將生物製品定義為”病毒、治療性血清、毒素、抗毒素、疫苗、血液、血液成分或衍生物、致敏產品或類似產品…用於預防、治療或治癒人類的一種疾病或狀況。“42 USC 262(I).FDA法規和政策已確定生物製品包括血液衍生產品、疫苗、體內診斷過敏製品、免疫球蛋白製品、含有細胞或微生物的製品以及大多數蛋白質製品。受《小靈通法案》約束的生物製品也符合以下定義:毒品問題根據FDC法案。生物製品是藥品的一個子集,因此兩者都受《食品和藥物管制法》條款的監管。然而,只有生物製品根據PHS法案第351條獲得許可,儘管一些治療性蛋白質產品是根據FDC法案第505條而不是PHS法案獲得批准的。
FDC法案、PHS法案和其他聯邦和州法規管理藥品和生物製品的研究、開發、測試、製造、儲存、記錄保存、審批、標籤、推廣和營銷、分銷、審批後監測和報告、抽樣和進口。不遵守適用的美國要求可能會使公司受到各種行政或司法制裁,例如實施臨牀封存、FDA拒絕批准未決的NDA或FDA的生物製品許可證申請(BLAS)或已批准的NDA/BLAS的補充、撤回批准、警告信、產品召回、產品扣押、全部或部分暫停生產或分銷、禁令、罰款、民事處罰和刑事起訴。
在美國,藥物和生物產品開發 通常涉及臨牀前實驗室和動物試驗,即向FDA提交IND,該IND必須在臨牀試驗開始前生效 。對於商業批准,申辦者必須通過所有合理適用的方法提交充分的檢測 ,以證明藥物/生物製劑在擬定標籤中規定、推薦或建議的條件下使用是安全的。 申辦者還必須提交實質性證據,通常包括充分、控制良好的臨牀試驗,以確定 藥物/生物製品在擬定標籤中規定、推薦或建議的使用條件下將具有其聲稱或聲稱具有的效果 ,包括符合FDA關於藥物安全性和有效性或生物製品純度和效力的標準。滿足FDA上市前批准要求 通常需要多年時間,實際所需時間可能因候選產品或疾病的 類型、複雜性和新穎性而有很大差異。
32
臨牀前測試包括對候選產品的化學成分、配方和毒性進行實驗室評估,以及評估候選產品的特性和潛在安全性和有效性的動物試驗。臨牀前試驗的進行必須符合聯邦法規和要求,包括FDA的良好實驗室操作規範(“普洛斯)、良好的臨牀實踐(GCP“)、 和藥品生產質量管理規範(“GMP“)法規和美國農業部 實施1996年《動物福利法》的法規。臨牀前試驗的結果作為IND 的一部分與其他信息一起提交給FDA,包括關於候選產品化學、生產和控制的信息,以及擬議的臨牀 試驗方案。長期臨牀前試驗,如生殖毒性和致癌性的動物試驗,可在 IND提交後繼續進行。
在開始人體臨牀試驗之前,需要在 提交每個IND後的30天等待期。如果FDA在這30天內未對IND實施臨牀 擱置或以其他方式對IND提出評論或質疑,則IND被視為已發佈,IND中提議的臨牀試驗 可以開始。
臨牀試驗涉及 在合格研究者的監督下,對健康志願者或患者進行試驗用新藥/生物製劑給藥。 必須進行臨牀試驗:(i)符合GCP(一項旨在保護患者權利和健康並定義臨牀試驗申辦者、管理者和監查員角色的國際標準和美國法律要求),(ii)符合其他聯邦法規,(iii)符合詳述試驗目標的方案,用於監測 安全性的參數和待評價的有效性標準。涉及對美國患者進行試驗的每個方案以及後續方案修訂 必須作為IND的一部分提交給FDA。
如果FDA認為臨牀試驗未按照FDA要求進行或對臨牀試驗患者構成不可接受的風險,則FDA可隨時下令暫時或永久停止臨牀試驗,或實施其他制裁。臨牀試驗中患者的試驗 方案和知情同意信息也必須提交給機構審查委員會(“IRB“), 供審批。IRB還可以因不符合IRB要求而阻止臨牀試驗開始或要求臨牀試驗機構暫時或永久停止臨牀試驗,或者可以施加其他條件。
Clinical trials to support NDAs/BLAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap or otherwise vary in particular circumstances. In Phase 1, the initial introduction of the drug/biologic into healthy human subjects or patients, the drug/biologic is tested to assess metabolism, pharmacokinetics, pharmacological actions, side effects associated with increasing doses and, if possible, early evidence on effectiveness. Phase 2 usually involves trials in a limited patient population to determine the effectiveness of the drug/biologic for a particular indication, dosage tolerance and optimum dosage, and to identify common adverse effects and safety risks. If a compound demonstrates evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to obtain the additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug/biologic and to provide adequate information for the labeling of the drug/biologic. In most cases, the FDA requires two adequate and well-controlled Phase 3 clinical trials to demonstrate the efficacy of the drug/biologic. The FDA may, however, determine that a single Phase 3 trial with other confirmatory evidence may be sufficient in some instances. In some cases, the FDA may require post-market studies, known as Phase 4 studies, to be conducted as a condition of approval in order to gather additional information on the drug’s/biologic’s effect in various populations and any side effects associated with long-term use. Depending on the risks posed by the drugs/biologics, other post-market requirements may be imposed.
為了響應《21世紀治癒法案》中提出的具體要求, 滿足患者更多地參與藥物開發和評價的需求, FDA已發佈了《重點藥物開發指南發佈計劃》,根據該計劃,FDA將發佈一系列指南, 旨在逐步解決以下問題:利益相關者如何從患者和護理人員處收集和提交患者體驗數據和其他相關信息 ,以用於醫療產品開發和監管決策。這些指南預計將促進 系統方法的發展和使用,以收集和使用可靠且有意義的患者和護理人員輸入,從而 更好地為醫療產品開發和監管決策提供信息。到目前為止,FDA已經發布了計劃中的四個指南中的三個。我們預計以患者為中心的藥物開發和評價問題將得到優先考慮,並在臨牀試驗設計中成為更多的因素。
完成所需的 臨牀試驗後,新藥申請(“NDA")/BLA已準備好並提交給FDA。候選產品在美國上市之前,需要獲得FDA對 NDA/BLA的批准。NDA/BLA必須包括所有臨牀前、 臨牀和其他測試的結果,以及與候選產品的藥理學、化學、生產、 和控制相關的數據彙編。編寫和提交國家發展援助/BLA的費用是巨大的。根據聯邦法律,大多數NDA/BLA的提交也需要 應用程序用户費,2023財年的費用約為400萬美元(其中需要臨牀 數據)。
33
FDA自收到NDA/BLA之日起有60天的時間根據該機構關於申請是否足夠完整以允許進行實質性審查的門檻確定是否接受申請備案。一旦提交的申請被接受,FDA就開始進行深入的審查。根據《處方藥使用費法案》,FDA已同意在審查非處方藥/BLAS時的某些績效目標。FDA目前的績效目標要求FDA在收到後10個月內完成對90%的標準(非優先)NDA/BLAS的審查,對於優先NDA/BLAS在6個月內完成審查,但對於新的分子實體/參考生物,標準和優先NDA/BLAS將額外增加兩個月的審查。如果一種藥物/生物藥物解決了嚴重疾病或危及生命的疾病或狀況中未得到滿足的醫療需求,則有資格優先審查。FDA可以將標準審查和優先審查的審查流程再延長三個月,以考慮某些遲提交的信息或旨在澄清提交中已提供的信息的信息。 這些時間表對FDA沒有法律約束力。
FDA還可以將新藥/生物製品或提出安全性或有效性難題的藥物/生物製品的申請提交給諮詢委員會,該委員會通常是一個由臨牀醫生和其他專家組成的小組,以審查、評估和建議是否應批准該申請。FDA不受諮詢委員會建議的約束,但它通常遵循此類建議。 在批准NDA/BLA之前,FDA通常會檢查一個或多個臨牀站點,以確保符合GCP。
此外,FDA將 檢查製造藥物的一個或多個設施。FDA將不會批准候選產品,除非符合GMPs,令人滿意,並且NDA/BLA包含提供大量證據的數據,證明該藥物是安全有效的,或者 生物符合所研究適應症的安全性、純度和效力標準。
在FDA對NDA/BLA和製造設施進行評估後,FDA會出具批准信或完整的回覆信。一封完整的回覆函 通常會概述提交中的不足之處,並可能需要大量的額外測試或信息,以便FDA重新考慮申請。如果或當FDA在重新提交NDA/BLA時對這些缺陷進行了滿意的處理,FDA將簽發批准信。FDA承諾在兩到六個月內審查90%的重新提交,具體取決於所包括的信息類型。儘管提交了任何要求的附加信息,FDA 最終可能會決定該申請不符合審批的監管標準。
批准函授權 藥物/生物製劑的商業銷售,並提供特定適應症的特定處方信息。作為NDA/BLA批准的一個條件, FDA可能要求風險評估和緩解策略("REMS"),以幫助確保藥物/生物製劑的獲益大於潛在風險 。REMS可以包括藥物指南、醫療保健專業人員的溝通計劃和 確保安全使用的要素(ETASU)。ETASU可以包括但不限於處方或配藥的特殊培訓或認證、 僅在某些情況下配藥、特殊監測和使用患者登記。REMS的要求可能 嚴重影響藥物/生物製劑的潛在市場和盈利能力。此外,候選產品批准可能需要大量的 批准後檢測和監督,以監測藥物/生物製劑的安全性或有效性。一旦獲得批准,如果未保持符合監管標準或在首次上市後發現問題,則可能撤回候選產品批准 。
在宣佈 COVID公共衞生緊急事件期間,FDA一直在對某些類別的產品行使執法自由裁量權, 已規定發放緊急使用授權(EUA),這使得許多產品在未經FDA正式常規批准或批准的情況下進入市場, 還將FDA資源從非COVID相關產品中抽走。美國 政府已宣佈自2023年5月11日起終止突發公共衞生事件。
34
臨牀 試驗信息的披露
某些FDA監管產品(包括處方藥/生物製劑)的臨牀試驗 申辦者必須在美國國立衞生研究院維護的公共網站上註冊和披露某些臨牀試驗 信息。與候選產品、患者 人羣、研究階段、研究中心和研究者以及臨牀試驗的其他方面相關的信息作為 註冊的一部分予以公開。申辦者也有義務在完成後披露這些試驗的結果。如果申辦者證明其正在尋求批准未批准的產品或 將在一年內提交批准已批准產品新適應症的申請,則提交這些試驗結果的截止日期 可延長至兩年。競爭對手可能會使用公開 信息來獲取有關我們開發計劃的設計和進度的信息。
快速通道指定 和加速批准
If our drug/biologic candidate meets the requirements of the FDA’s fast track program, we would seek to have our drug/biologic candidate expedited through this program. The FDA has programs to facilitate the development, and expedite the review of drugs/biologics that are intended for the treatment of a serious or life-threatening disease or condition for which there is no effective treatment and which demonstrate the potential to address unmet medical needs for the condition. Under the fast-track program, the sponsor of a new drug/biologic candidate may request that the FDA designate the drug/biologic candidate for a specific indication as a fast track drug/biologic concurrent with, or after, the filing of the IND for the drug/biologic candidate. The FDA must determine if the drug/biologic candidate qualifies for fast track designation within 60 days of receipt of the sponsor’s request. In addition to other benefits such as the ability to engage in more frequent interactions with the FDA, the FDA may initiate review of sections of a fast track drug’s/biologic’s NDA/BLA before the application is complete. This rolling review is available if the applicant provides, and the FDA approves, a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, the FDA’s time period goal for reviewing an application does not begin until the last section of the NDA/BLA is submitted. Additionally, the fast track designation may be withdrawn by the FDA if it believes that the designation is no longer supported by data emerging in the clinical trial process.
根據FDA的加速 批准法規,FDA可以批准一種藥物/生物製劑用於嚴重或危及生命的疾病,該藥物/生物製劑可為患者提供比現有治療更有意義的治療 獲益,其依據是合理可能預測臨牀獲益的替代終點,或 可在不可逆發病率或死亡率之前測量的臨牀終點,考慮到 病症的嚴重程度、罕見性或流行性以及替代治療的可用性或缺乏性,合理可能預測 對不可逆發病率或死亡率或其他臨牀獲益的影響。
In clinical trials, a surrogate endpoint is a measurement of laboratory or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient feels, functions or survives. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A drug/biologic candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to confirm clinical benefit. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, will allow the FDA to withdraw the drug/biologic from the market on an expedited basis. Unless otherwise informed by the FDA, for an accelerated approval product/biologic, an applicant must submit to the FDA for consideration during the preapproval review period copies of all promotional materials, including promotional labeling as well as advertisements, intended for dissemination or publication within 120 days following marketing approval. After 120 days following marketing approval, unless otherwise informed by the FDA, the applicant must submit promotional materials at least 30 days prior to the intended time of initial dissemination of the labeling or initial publication of the advertisement.
突破性治療指定
As with the FDA’s fast track program, if our drug/biologic candidate meets the requirements to receive the FDA’s Breakthrough Therapy designation, we would seek to have our drug/biologic candidate expedited through this program. The FDA’s Breakthrough Therapy designation program is intended to expedite the development and review of products that treat serious or life-threatening diseases or conditions. A Breakthrough Therapy is defined, under the Food and Drug Administration Safety and Innovation Act, as a drug/biologic that is intended, alone or in combination with one or more other drugs/biologics, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug/biologic may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The designation includes all of the features of fast-track designation, as well as more intensive FDA interaction and guidance. The Breakthrough Therapy designation is a distinct status from both accelerated approval and priority review, but these can also be granted to the same product candidate if the relevant criteria are met. The FDA must take certain actions, such as holding timely meetings and providing advice, intended to expedite the development and review of an application for approval of a breakthrough therapy. All requests for breakthrough therapy designation will be reviewed within 60 days of receipt, and the FDA will either grant or deny the request.
35
此外,《21世紀治癒法》創建了再生醫學高級治療(RMAT)名稱。RMAT作為一個類別適用於再生醫學。 某些細胞療法、治療性組織工程產品、人類細胞和組織產品以及某些組合 產品的申辦方可獲得其製劑的RMAT認證,如果該藥物預期用於治療嚴重或危及生命的疾病 或病症,並且如果有初步臨牀證據表明該藥物有可能滿足該疾病或病症的醫療需求 。申辦者可以在提交研究新藥申請時或之後提出此類請求。
RMAT指定 產品的申辦者有資格與FDA進行更多和更早的互動,類似於突破性指定 療法的申辦者可獲得的互動。此外,它們可能有資格獲得優先審查和加速批准。與RMAT指定 產品的申辦者的會議可能包括討論基於替代或中間終點是否適合加速批准,這些替代或中間終點合理 可能預測長期臨牀獲益,或依賴於從大量臨牀試驗機構獲得的數據。
一旦獲得批准,在適當情況下, FDA可以通過提交臨牀證據、 臨牀研究、患者登記或其他真實世界證據來源(如電子健康記錄);通過收集 更大的確證性數據集;或通過批准前對接受治療的所有患者進行批准後監測,允許在加速批准下滿足批准後要求。
快速通道認定、加速 審批、優先審評和突破性治療認定不會改變審批標準,但可能會加快開發 或審批流程。即使FDA批准了其中一項指定,FDA也可能在以後決定該藥物/生物製品不再符合資格條件。
橙皮書列表和 專利認證
根據1984年《藥品價格競爭和創新法案》(通常稱為Hatch—Waxman)對 FDC法案的修正案,在通過NDA尋求藥物批准時,申請人需要向FDA列出每項專利,其權利要求涵蓋申請人的候選產品或 候選產品的使用方法。藥物獲得批准後, 該藥物申請中列出的每項符合條件的專利均會在FDA的《具有治療等效性評估的批准藥品》(通常稱為 Orange Book)中公佈。奧蘭治書中列出的藥物,反過來,必須由簡化的 新藥申請的申請人進行特殊認證("安達"),用於藥物的仿製藥,或由混合申請的申請人(稱為505(b)(2)申請。ANDA規定,候選製劑具有與參考上市的創新藥物相同的活性成分( 規格和劑型)的上市,並且已證明與參考上市的創新藥物具有生物等效性 。除生物等效性試驗要求(無豁免)外,ANDA申請人無需進行臨牀前或臨牀試驗或提交 結果以證明其候選製劑的安全性或有效性。以這種方式 批准的藥物通常被稱為上市藥物的"仿製等同物",被認為在治療上與 上市藥物等同,並且通常可以由藥劑師根據針對原始上市藥物的處方進行替代。
要求ANDA申請人 向FDA證明FDA的奧蘭治書中列出的已批准候選產品的任何專利。具體而言, 申請人必須證明:(i)尚未提交所需的專利信息;(ii)所列專利已經過期; (iii)所列專利尚未過期,但將在特定日期過期,並在專利過期後尋求批准;或 (iv)所列專利無效或不會被新候選產品侵犯。ANDA申請人還可以選擇提交 一份"第八節聲明",證明其擬議的ANDA標籤不包含(或刻出)關於專利使用方法的任何語言 ,而不是證明列出的使用方法專利。
如果申請人未 對所列專利提出異議,則在所有聲稱引用產品的所列專利到期之前,ANDA申請將不會獲得批准 。
36
證明 新候選產品不會侵犯已獲批准的候選產品的列出專利,或證明此類專利無效 或不可執行,稱為第IV段證明。如果ANDA申請人已向FDA提供了第IV段認證, 申請人還必須在FDA接受ANDA申請後向NDA和專利持有人發送第IV段認證的通知。NDA和專利持有人隨後可以根據第 IV段證明的通知提起專利侵權訴訟。在收到第IV段認證通知後45天內提出專利侵權訴訟 自動阻止FDA批准ANDA,直到30個月、專利到期、訴訟解決、 侵權案件中對ANDA申請人有利的裁決或法院的其他命令中較早者。
申辦者還可以通過第505(b)(2)條申請尋求 藥品的上市版本,這是一種NDA途徑,允許申請人根據完整的安全性和有效性文件(其中一些文件可能來自文獻或由其他人進行)尋求藥品的批准, 申請人沒有引用權。對於代表先前獲批藥物的修改(如新適應症或新劑型)的藥品,可以提交NDA第505(b)(2)節申請 。第505(b)(2)條申請 可依賴FDA先前關於先前獲批藥物安全性和有效性的研究結果,以及505(b)(2)條申請人獲得的信息 ,以支持先前獲批藥物的修改。準備第505(b)(2)條申請 可能比完全基於新數據和信息準備保密協議的成本和時間更低。第505(b)(2)條的申請 與ANDA一樣,須遵守相同的專利證明程序。
新的化學實體排他性和臨牀研究排他性
在NDA批准新的 化學實體(“NCE”),即一種不含活性部分且已獲FDA在任何其他 NDA中批准的藥物,在此期間,FDA無法收到任何ANDA或505(b)(2)申請 尋求批准引用NCE藥物版本的藥物。藥物的某些變更,例如在藥品説明書中添加新適應症 ,需要批准新的臨牀研究,與三年的獨佔期有關, 在此期間,FDA無法批准包含該變更的ANDA或505(b)(2)申請。
如果提交了第四段認證,可在NCE排他性到期前一年提交ANDA或505(B)(2)申請 。如果Orange Book中沒有列出的專利,則可能沒有第四款認證,因此在 專營期到期之前不能提交ANDA或505(B)(2)。
對於植物藥, FDA可以確定活性部分是一種或多種主要成分或作為整體的複雜混合物。此決定 將影響任何五年獨家經營權的效用,以及任何潛在仿製藥競爭對手證明 其與原始植物藥是同一種藥物的能力。
5年和3年的獨家 不妨礙FDA在獨家期內批准一種藥物的505(b)(1)申請, 條件是505(b)(1)進行或獲得所有臨牀前研究以及證明安全性和有效性所需的充分且受控良好的臨牀試驗的參考權。
孤立指定和 排他性
FDA可能會授予孤兒藥指定給旨在治療罕見疾病或病症的藥物,這些疾病或病症影響美國少於20萬人,或者如果 影響美國超過20萬人,並且沒有合理的預期,開發和生產用於此類疾病或病症的 藥物的成本將從美國的銷售中收回。
孤兒藥指定 是一個財政激勵措施的一方,如臨牀研究費用、税收優惠和用户費用豁免的機會。 孤兒藥指定不會在監管審查和批准過程中帶來任何好處或縮短其持續時間。 此外,第一個獲得特定藥物的孤兒藥指定的NDA或BLA申請人有權獲得孤兒藥排他性, 這意味着FDA不得批准在美國針對相同適應症上市的任何其他申請 ,除非在有限的情況下。孤兒藥的排他性並不妨礙FDA批准不同的藥物用於 相同的疾病或條件,或相同的藥物用於不同的疾病或條件。
37
兒科研究和 排他性
NDA和BLA必須包含 數據,以評估試驗用新藥在所有相關兒科人羣中用於聲明適應症的安全性和有效性 ,以支持藥物安全有效的每個兒科亞羣的給藥和給藥。 FDA可主動或應申請人的要求,批准推遲提交部分或所有兒科數據,直至 產品獲準用於成人或全部或部分豁免(如果符合某些標準)。有關兒科開發 計劃的討論可隨時與FDA討論,但通常在II期會議結束和提交 NDA或BLA之間的任何時間進行。除非法規另有要求,否則兒科數據要求不適用於 已授予孤兒稱號的任何適應症藥物。
兒科排他性是 美國另一種非專利排他性,如果滿足FDA的某些要求,例如FDA確定 與兒科人羣中使用新藥相關的信息可能產生健康益處,申請人同意 在一定時間範圍內執行和報告FDA要求的研究,則可授予該專利。兒科排他性在申辦者為該活性成分持有的所有現有市場排他性和專利結束後增加了6個月的排他性 。這不是專利期限延長, 但它有效地延長了監管期限,在此期間,FDA無法接受或批准依賴NDA 或BLA申辦者數據的另一項申請。
生物製劑專屬性 和生物仿製藥
《2010年患者保護和 平價醫療法案》或《平價醫療法案》包括一個名為 2009年生物製品價格競爭和創新法案或BPCI法案的副標題,該法案對PHS法案進行了修訂,以根據PHS法案第351(k)節為生物製品創建一個簡化的批准途徑,證明與之類似或互換,FDA許可的參考生物製品,最初根據PHS法案第 351(a)節獲得許可。
參比生物製劑自首次獲得上市許可之日起獲得 12年的市場獨佔權,在此期間,參比製劑生物類似藥的351(k)申請 可能不會獲得批准。參比生物製劑還被授予四年的所謂數據排他性, 在此期間,參比產品的生物類似藥的351(k)申請不得提交審查。根據簡化批准途徑提交的第一個生物 產品被確定為可與參比產品互換,相對於根據簡化批准途徑提交的其他生物製劑,該生物 產品具有排他性 ,期限以較短者為準:(i)首次商業 上市後一年,(ii)批准後十八個月(如果沒有法律質疑),(iii)如果已提交申請,則在 申請人對挑戰生物製品專利的訴訟做出有利的決議後18個月,或(iv)如果訴訟在42個月期間內正在進行,則在申請獲得批准後42個月。
生物專利信息
與小分子藥物不同的是,申請者需要提交專利信息和他們的NDA和某些補充劑,而尋求生物製品許可證的申請者不需要在其BLA或補充劑中提交專利信息。此外,與小分子藥物不同的是,ANDA和505(B)(2)NDA的批准受到參考NDA藥物產品上市專利狀況的影響,而第351(K)條關於生物相似產品申請的批准目前與解決專利糾紛的各種程序脱鈎。生物相似申請人可以選擇是否參與生物製品價格競爭和創新法案(BPCIA)的專利訴訟條款,俗稱為“專利舞蹈”,以識別和提起一系列明確的專利。然而,與《橙色手冊》中列出的小分子參考上市藥物和專利不同,FDA的《許可生物製品清單》(俗稱《紫皮書》)中沒有列出專利的流程。然而,2020年12月,國會通過了生物製品專利透明法(“BPPT“)(最初提出的紫色圖書連續性法案創建了小靈通法案第351(K)(9)節)。該條款要求生物製品參考贊助商 在BPCIA專利訴訟程序中向生物相似申請人提供專利清單,現在必須在30天內將這些清單提交給FDA,此外,自2021年6月起,FDA必須在Purple Book數據庫中公開這些清單(以及任何 修訂或更新)。
38
專利期延長
在NDA或BLA批准後, 相關藥物或生物專利的所有者可以申請最長五年的專利延期。允許的專利期延長時間為產品測試階段(IND提交和NDA或BLA提交之間的時間)和所有審查階段(NDA提交和批准之間的時間,最長為五年)的一半。如果FDA確定申請人沒有經過盡職調查尋求批准,則可以縮短時間。展期後的總專利期不得超過14年。
對於在申請階段可能到期的專利,專利所有人可以申請臨時專利延期。臨時專利延期可將專利期限延長一年,最多可續訂四次。每授予一次臨時專利延期,批准後專利延期將減少 一年。美國專利商標局的董事必須確定,正在申請專利延期的專利所涵蓋的產品很可能獲得批准。對於尚未提交NDA或BLA的藥物,臨時專利延期不可用。
廣告與促銷
一旦NDA或BLA獲得批准, 候選產品將受到某些審批後要求的約束。例如,FDA密切監管藥品和生物製品的審批後銷售和促銷,包括直接面向消費者的廣告、標籤外促銷、行業贊助的科學和教育活動以及涉及互聯網的促銷活動的標準和法規。
藥品和生物製品只能根據批准的適應症和批准的標籤的規定進行銷售。
審批後變更
更改已批准申請中確定的某些條件,包括適應症、標籤或生產工藝或設施的更改,需要 提交和FDA批准新的NDA/BLA或NDA/BLA補充物才能實施。針對新的 適應症的NDA補充劑通常需要與原始申請中類似的臨牀數據,FDA在審查NDA補充劑時使用與審查NDA相同的程序和行動 。
不良事件報告和GMP合規性
在FDA批准NDA或BLA後,要求加快不良事件報告並提交定期不良事件報告。FDA還可能要求進行上市後測試,即所謂的第四階段測試、REMS和監督,以監控批准產品的效果,或者 FDA可能會在批准時附加條件,限制該產品的分銷或使用。此外,質量控制、藥品製造、包裝和標籤程序在獲得批准後必須繼續符合GMP。藥品製造商和他們的某些分包商被要求向FDA和某些州機構登記他們的工廠。在FDA註冊後,實體將接受FDA的定期突擊檢查,在此期間,FDA會檢查生產設施,以評估是否符合GMP。因此,製造商必須繼續在生產和質量控制方面投入時間、金錢和精力,以保持對GMP的遵守。如果一家公司未能遵守監管標準,如果它在初始營銷後遇到問題,或者如果後來發現了以前沒有意識到的問題,監管機構可以撤回產品批准、發佈警告信、請求產品召回或採取其他執法行動。
特殊協議評估
贊助商可根據特別協議評估與FDA達成協議(“水療中心“),關於旨在形成療效聲明的主要依據的臨牀試驗的所需設計和規模的過程。根據其績效目標,FDA承諾在收到評估擬議試驗是否充分的請求後45天內對90%的方案進行評估, 評估可能會導致討論和要求提供更多信息。必須在提議的試驗開始之前提出SPA請求 ,並且必須在試驗開始之前解決所有未決問題。如果達成書面協議,它將被記錄下來,併成為行政記錄的一部分。根據FDC法案和FDA實施法定要求的指南,SPA通常對FDA在試驗設計方面具有約束力,但在有限情況下除外,例如,如果FDA在研究開始後發現對確定安全性或有效性至關重要的重大科學問題,出現在方案評估時未意識到的公共衞生問題,贊助商和FDA以書面形式同意更改,或者研究贊助商未能遵循與FDA商定的方案 。
39
受管制物質
經修訂的《管制物質進出口法》(“環孢素A)和實施條例對受管制物質進行登記、安全、記錄保存和報告、儲存、製造、分銷、分發、進口和其他要求,由美國禁毒署監督(br})DEA“)。DEA是聯邦機構,負責監管受控物質,並要求生產、進口、出口、分銷、研究或分發受控物質的個人或實體遵守監管要求,以防止受控物質的轉移和濫用。
DEA將受管制的 物質列為附表I、II、III、IV或V物質,具體取決於該物質的醫療效力和濫用潛力。 目前已被認可可用於醫療用途且已獲準上市的藥品可被列為附表II、III、IV或V物質,其中附表II物質濫用的可能性最大,身體或心理依賴的可能性最大,而附表V物質濫用和依賴的相對可能性最低。DEA已將包括大麻二酚在內的某些藥品 列入附表V。
在國家藥品監督管理局批准含有附表I受控物質的藥物後,該物質必須重新安排為附表II、III、IV或V物質,然後才能上市。2015年11月25日頒佈的《提高新醫療療法監管透明度法》及其實施條例消除了與NDA批准後DEA重新安排流程的時間相關的不確定性,根據該規定,製造商 可以在以下較晚的時間內銷售其產品:(1)DEA從FDA收到科學 以及醫療評估和計劃建議的日期;或(2)DEA從FDA收到FDA通知 FDA已批准該藥物的日期。該法案還澄清,七年的孤兒排他期從NDA或DEA時間表的批准開始,以較晚的為準。這改變了以前的情況,即孤兒“鍾開始根據FDA的NDA批准進行操作,即使該產品在DEA計劃完成後才能上市。
CSA要求製造、分銷、分發、進口或出口任何受控物質的設施必須每年向DEA登記。進口和製造活動需要單獨註冊,每個註冊授權註冊人可以處理的受控物質的具體時間表 。在頒發受控物質註冊之前,DEA檢查所有生產設施,以審查受控物質的安全、記錄保存、報告和處理。具體的安全要求因所從事的商業活動的類型以及所處理的受控物質的類型、形式和數量而異。
此外,各州 有自己獨特的受控物質法律法規,包括對受控物質的許可、分配、分配、記錄保存和報告要求。州藥房委員會或類似當局對每個州的受控物質的使用進行管理。不遵守適用的要求,如受管制物質的損失或轉移,可能會導致行政罰款、暫停或吊銷許可證,以及民事和刑事責任。
英國/歐洲/世界其他地區 政府法規
除了美國的法規 ,我們現在和將來都將直接或通過我們的分銷合作伙伴遵守其他司法管轄區的各種法規,其中包括臨牀試驗和任何商業銷售(包括定價和報銷)以及我們 產品的分銷(如果獲得批准)。
無論產品是否獲得FDA批准,我們都必須在非美國國家/地區的監管機構開始 在這些國家/地區進行臨牀試驗或營銷之前獲得必要的批准。
40
在歐盟,醫藥產品 在上市前和上市後都受到歐盟和國家監管機構的廣泛監管。國家一級的其他規則也適用於受管制物質的製造、進口、出口、儲存、分銷和銷售。在許多歐盟成員國,負責醫藥產品的監管當局也對管制物質負責。然而,責任 在一些成員國是分開的。通常,任何在歐盟生產或分銷含有受控物質的醫藥產品的公司都需要持有國家主管機構頒發的受控物質許可證,並受具體的 記錄保存和安全義務的約束。進出成員 國的每批貨物都需要單獨的進口或出口證明。
在英國,藥品 受到藥品和保健產品監管局的廣泛監管(“MHRA“),這是一個執行機構,由衞生和社會保健部贊助。MHRA除了支持有益於公眾健康的創新和研發外,還通過確保藥品、醫療器械和用於輸血的血液成分符合適用的安全、質量和療效標準來進行監管。
臨牀試驗和營銷 批准
美國 以外的某些國家/地區的流程要求在開始 人體臨牀試驗之前提交臨牀試驗申請,這與IND非常相似。在歐盟/歐洲經濟區(“歐洲經濟區例如,臨牀試驗申請 (aCTA“),必須通過單一入口網站提交所有在歐盟/歐洲經濟區進行的臨牀試驗(”CTIS“)。 一旦CTA根據一個國家的要求獲得批准,並且一家公司獲得了有利的道德委員會的批准, 臨牀試驗開發就可以在該國進行。
2014年4月,《臨牀試驗條例》(歐盟)第536/2014號(“新法規“)被採納以取代臨牀試驗 指令2001/20/EC(”先行指令“)。為確保臨牀試驗規則在整個歐盟/歐洲經濟區內保持一致,歐盟通過了新的臨牀試驗立法,作為一項直接適用於歐盟/歐洲經濟區成員國的“規定”。新條例要求贊助商提交一份針對所有歐盟/歐洲經濟區成員國計劃的單一CTA,將通過CTIS這一簡化授權流程的在線門户網站提交。CTIS授權程序由兩部分組成:成員國在第一部分評估中共同合作,而第二部分由每個成員國單獨評估。這是一個重大變化, 因為根據之前的指令,贊助商必須尋求進行試驗的每個國家/地區的國家當局的單獨批准。新法規於2022年1月31日生效-從同一天起廢除之前的指令-並引入了一年的過渡期(至2023年1月31日),在此期間,贊助商可以選擇是根據之前指令管轄的舊制度還是根據新的CTI提交新的CTA。在過渡期之後,從2023年1月31日起,贊助商必須 根據新法規申請臨牀試驗,到2025年1月31日,根據先前指令 批准的所有正在進行的臨牀試驗將需要過渡到新法規。
在英國,之前的指令 (由人用藥物(臨牀試驗)條例2004/1031實施)仍然適用。2020年1月31日,英國根據《2018年歐盟和歐盟(退出)法案》(TheEUWA“)它開始生效。EUWA第1節廢除了之前允許歐盟法律適用於英國的《1972年歐洲共同體法》(ECA 1972)。然而,EUWA第1節立即保留了《ECA 1972》(經修改的形式)在過渡期內的大部分效力,而第1B節保留了實施歐盟要求的英國立法。因此,EUWA的實際效果是廢除了尚未適用於英國的新法規 ,但保留了先前的指令。本節中提及的英國主要指的是大不列顛。由於《北愛爾蘭議定書》第182條的規定,北愛爾蘭的某些歐盟/歐洲經濟區衍生的管理標準沒有被廢除,因此在某些情況下仍然適用。《北愛爾蘭議定書》於2023年被《温莎框架》取代。在英國,當計劃進行臨牀試驗時,必須獲得MHRA的批准。根據《人用藥物(臨牀試驗)條例2004/1031》和MHRA提供的其他適用指南文件,CTA必須 提交研究用藥品檔案,並由研究用藥品檔案和其他支持信息提供。此外,只有在主管倫理委員會對英國的臨牀試驗申請發表了積極的意見後,才能開始臨牀試驗。
管理臨牀試驗、產品許可、定價和報銷的要求和流程因國家/地區的不同而有所不同,儘管由於歐盟基本法規的國家實施,歐盟成員國已經有了一定程度的法律協調。 在所有情況下,臨牀試驗都必須按照國際協調會議關於GCP的指南和其他適用的法規要求進行。
41
要獲得監管部門批准 將藥物投放到英國或歐盟/歐洲經濟區國家/地區的市場,我們必須提交上市授權申請。此應用程序 類似於美國的保密協議,不同之處在於特定於國家/地區的文檔要求。所有申請程序都需要採用通用技術文檔格式的申請,其中包括提交有關產品製造和質量的詳細信息,以及非臨牀和臨牀試驗信息。除了使用國家授權程序(導致在相關歐盟/歐洲經濟區成員國有效的藥品上市授權)外,還可以使用(I)集中式授權程序、(Ii)相互認可程序或(Iii)分散 程序在歐盟/歐洲經濟區 授權藥品。歐盟/歐洲經濟區提供的這三種授權方法不再適用於英國。MHRA可以通過使用(I)創新許可和准入途徑,(Ii)150天評估路線,(Iii)新產品和生物技術產品評估的滾動審查路線,(Iv)歐洲委員會(EC)決定依賴程序,(V)分散和相互承認的依賴程序,在英國授權藥物。或(Vi)在北愛爾蘭獲得批准的營銷授權申請的“自由訪問”路線 。
歐盟委員會制定了集中審批人類藥物的程序,以促進在整個歐盟以及冰島、列支敦士登和挪威(在國家實施決定後)有效的銷售授權,這三個國家與歐盟成員國一起構成了歐洲經濟區。申請者向歐洲藥品管理局(EMA)提交營銷授權申請,在那裏 它們由相關的科學委員會審查,在大多數情況下是人用藥品委員會(“CHMP“)。 歐洲藥品管理局(”EMA“)將CHMP意見轉發給歐盟委員會,歐盟委員會將其用作決定是否授予營銷授權的 基礎。此程序導致由歐盟委員會授予的單一營銷授權在整個歐盟以及冰島、列支敦士登和挪威都有效。對於以下人類藥物來説,集中程序是強制性的:(I)源自生物技術過程,如基因工程;(Ii)含有一種新的活性物質,指示用於治療某些疾病,如艾滋病毒/艾滋病、癌症、糖尿病、神經退行性疾病、自身免疫性疾病和其他免疫功能障礙和病毒性疾病;(Iii)被官方指定孤兒藥物"(用於罕見人類疾病的藥物)和(iv)先進治療藥物,如基因治療、體細胞治療或組織工程藥物。 如果CHMP同意人用藥物(a)含有2004年11月20日尚未批准的新活性物質,則集中程序也可根據申請人的自願請求用於不屬於上述 類別的人用藥物;(b)構成一種重要的治療方法,科學或技術創新或(c)集中程序下的授權符合歐盟層面患者的利益 。儘管英國。在2023年12月31日之前,MHRA 可以使用分散和相互承認依賴程序,通過依賴歐盟 集中程序下的EC批准來授權藥物。在2023年12月31日之後,MHRA打算建立一個新的國際藥物依賴框架制度。
根據歐盟的集中程序 ,EMA評估上市許可申請的最長時限為210天(不包括時鐘 停止,當申請人在回答CHMP提出的問題時,申請人將提供額外的書面或口頭信息), 此後歐盟委員會採用實際上市許可。
在例外情況下,當從治療創新的角度來看,預期某種藥品具有重大公共衞生利益時,CHMP可能 批准加速評價,定義為三個累積標準:待治療疾病的嚴重性; 沒有適當的替代治療方法,以及預期具有特殊的高治療獲益。在這種情況下,EMA 確保在150天內完成對CHMP意見的評估,並在隨後發佈意見。
對於那些沒有集中程序的藥品 ,申請人必須通過以下三種程序之一向國家 藥品監管機構提交上市許可申請:(i)相互承認程序(如果產品已 在至少一個其他歐盟/歐洲經濟區成員國獲得授權,其中要求歐盟/歐洲經濟區成員國授予授權 承認其他歐盟/歐洲經濟區成員國的現有授權,除非它們發現對公共健康構成嚴重風險),(ii) 分散程序(在兩個或多個歐盟/歐洲經濟區成員國同時提交申請)或(iii)國家 授權程序(在單個歐盟/歐洲經濟區成員國獲得上市許可)。
42
有條件營銷 授權
歐盟委員會可能會對滿足未得到滿足的醫療需求的藥品授予有條件的上市授權。此營銷授權是在執行授權時強加的某些活動(例如,完成正在進行的或新的研究或收集額外的 數據)的前提下進行的 。這適用於申請人因客觀、可核實的原因和基於特定理由而無法提供有關藥品在正常使用條件下的有效性和安全性的全面數據,並且符合以下所有標準:
| ● | 藥物的獲益—風險平衡是積極的; |
| ● | 申請人在獲得授權後可能能夠提供全面的數據; |
| ● | 藥物滿足未滿足的醫療需求;以及 |
| ● | 患者可立即獲得藥物的益處更大 而不是仍然需要額外數據這一事實所固有的風險。 |
有條件營銷授權 有效期為一年,可每年續訂。
在英國(英格蘭、蘇格蘭、威爾士),MHRA引入並規範了國家有條件營銷授權,從2021年1月1日起生效;在北愛爾蘭,有條件營銷授權的申請必須提交給EMA。英國方案具有與歐盟方案相同的 資格標準。
互認程序
互認程序 (“物料需求計劃“),批准人類藥物是促進歐盟/歐洲經濟區內個別國家銷售授權的另一種方法。基本上,MRP可以適用於所有不是強制性的集中程序的人類藥物。MRP 適用於大多數傳統醫藥產品,如果該產品已在一個或多個成員國獲得授權,則必須使用該產品。
MRP的特點是,該程序建立在成員國已存在的上市許可的基礎上,該許可用作 在其他歐盟/歐洲經濟區成員國獲得上市許可的參考。在MRP中, 歐盟/歐洲經濟區的一個或多個成員國已經存在藥物的上市許可,隨後通過參考 初始上市許可在其他成員國提出上市許可申請。首先授予上市許可的成員國隨後將作為 參考成員國。隨後申請上市許可的成員國作為相關成員國行事。 相關成員國必須批准一項授權,以承認參考成員國現有的授權, 除非它們發現對公共健康構成嚴重風險。
MRP基於EU/EEA成員國相互承認各自國家上市許可的原則 。根據參考成員國的上市許可 ,申請人可以在其他成員國申請上市許可。在這種情況下,參考 成員國需要在90天內更新其關於該藥物的現有評估報告。評估完成後,報告副本 連同已批准的產品特性摘要、標籤和包裝 説明書一起發送給所有相關成員國。相關成員國隨後有90天的時間來認可參考成員國的決定以及產品特性、標籤和包裝説明摘要 。應在確認協議後30天內授予全國上市許可 。
如果任何成員國以潛在的嚴重公共健康風險為由拒絕承認參考成員國的銷售授權,則將把問題提交協調小組處理。在60天的時限內,成員國應在協調小組內作出一切努力,以達成共識。如果失敗,程序將提交給EMA科學委員會進行仲裁。然後將EMA委員會的意見 轉發給歐盟委員會,以開始決策過程。與集中式程序一樣,這一過程需要諮詢各歐盟委員會總幹事和人類醫藥產品常設委員會。
43
數據獨佔性
在英國和歐盟,仿製藥的上市許可申請不需要包括臨牀前和臨牀試驗的結果,但 可以參考參考產品的上市許可中包含的數據,該參考產品的監管數據獨佔性 到期。如果為含有新活性物質的藥品授予上市許可,則該產品將享受 八年的數據獨佔權,在此期間,涉及該產品數據的仿製藥上市許可申請 可能不被監管機構接受;以及另外兩年的市場獨佔權,在此期間,此類仿製藥 不會投放市場。如果在前8年內,批准了比現有療法具有顯著臨牀獲益的新治療適應症 ,則兩年期可延長至三年。
孤兒醫藥產品
EMA的孤兒藥品委員會(“COMP”), may recommend orphan medicinal product designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than five in 10,000 persons in the EU. Additionally, orphan designation is granted for products intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the product in the EU would be sufficient to justify the necessary investment in developing the medicinal product. The COMP may only recommend orphan medicinal product designation when the product in question offers a significant clinical benefit over existing approved products for the relevant indication. Following a positive opinion by the COMP, the European Commission generally grants orphan status within 30 days. When the draft decision of the European Commission is not aligned with the COMP opinion, the COMP will reassess orphan status in parallel with EMA review of a marketing authorization application and orphan status may be withdrawn if the drug no longer fulfills the orphan criteria (for instance, because a new product was approved for the indication and no data is available to demonstrate a significant benefit over that new product). Orphan medicinal product designation entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity is granted following marketing authorization. During this period, the competent authorities may not accept or approve any similar medicinal product for the same therapeutic indication, unless it offers a significant clinical benefit or if the holder of the marketing authorization for the original orphan drug is unable to supply sufficient quantities of the drug. This period may be reduced to six years if the orphan medicinal product designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity.
歐盟/歐洲經濟區孤兒指定 延伸至北愛爾蘭。否則在英國。英國退歐後,將孤兒藥產品指定的申請提交給MHRA,該申請採用與歐盟相同的實質性標準,並具有與歐盟相同的實質性效力(但與歐盟程序不同的是,不可能獲得早期孤兒指定;指定申請必須與 英國申請同時提交。銷售授權)。如果一種藥品在歐盟被指定為孤兒藥,那麼英國 孤兒藥上市許可申請可以向MHRA申請;如果沒有歐盟孤兒藥指定,英國—廣泛(包括北愛爾蘭)的孤兒 上市許可申請可以向MHRA提出。
兒科發展
在歐盟和英國,開發新藥品的公司 必須同意兒科研究計劃("皮普”),與EMA或MHRA 一起進行兒科臨牀試驗,除非豁免適用,例如,因為相關疾病 或病症僅發生在成人中。產品的上市許可申請必須包括根據PIP進行的兒科臨牀 試驗的結果,除非適用豁免或已批准延期,在這種情況下,兒科臨牀 試驗必須在稍後日期完成。根據PIP進行的兒科臨牀 試驗獲得上市許可的產品,有資格獲得補充保護 證書下的六個月的保護延期(如果該證書涵蓋的產品在批准時符合資格)。此兒科獎勵受特定條件的約束 ,當開發並提交符合PIP的數據時,該獎勵不會自動獲得。
如果我們未能遵守適用的外國監管要求,我們可能會受到罰款、暫停臨牀試驗、暫停 或撤銷監管批准、產品召回、產品扣押、經營限制和刑事起訴等處罰。
44
此外,大多數國家都是《1961年麻醉品單一公約》和《1971年精神藥物公約》的締約國,這兩項公約管理着包括大麻提取物在內的麻醉物質的國際貿易和國內管制。國家/地區可能會解釋和履行其條約義務 ,這可能會對我們在這些國家/地區獲得我們的候選產品的營銷批准造成法律障礙。這些國家/地區 可能不願意或無法修改其法律法規以允許我們的候選產品上市,或者 對法律法規進行此類修改可能需要很長一段時間。在這種情況下,我們將無法在不久的將來或根本無法在這些國家/地區銷售我們的產品。
報銷
醫藥產品在美國的銷售 將部分取決於第三方支付者(如政府 醫療計劃、商業保險和管理醫療保健組織)支付產品成本的程度。這些第三方付款人對醫療產品和服務的收費價格提出了越來越多的挑戰。此外,控制醫療保健費用已成為聯邦政府和州政府的優先事項,藥品價格也是這項工作的重點。美國政府、州立法機構和外國 政府對實施成本控制計劃表現出極大興趣,包括價格控制、使用管理 和仿製藥替代要求。採取價格控制和成本控制措施,以及在現有控制和措施的司法管轄區採取更多 限制性政策,可能會進一步限制我們的淨收入和業績。如果這些 第三方付款人不認為我們未來的產品與其他可用療法相比具有成本效益,則他們可能不會在批准後將 我們的產品作為其計劃中的一項福利支付給 ,或者如果他們這樣做,支付水平可能不足以讓我們 在盈利的基礎上銷售我們的產品。
2003年《醫療保險處方藥品、改進和現代化法案》(“MMA”), imposed requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries and included a major expansion of the prescription drug benefit under Medicare Part D. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities that provide coverage of outpatient prescription drugs. Part D is available through both stand-alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans. Unlike Medicare Parts A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payers often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from nongovernmental payers.
2009年2月17日,奧巴馬總統簽署了《2009年美國復甦和再投資法案》。這項法律為聯邦政府提供資金,以比較 不同治療方法對同一疾病的有效性。這項研究由衞生和公眾服務部、醫療保健研究和質量機構和國家衞生研究院監督,並且必須向國會提交關於研究狀況和相關支出的定期報告。雖然比較有效性研究的結果並不旨在 強制執行公共或私人支付者的覆蓋政策,但尚不清楚如何避免此類結果,以及如果任何此類產品或預期治療的病症是研究的主題, 研究將對我們候選產品的銷售產生什麼影響。也有可能是,證明競爭對手產品受益的比較有效性研究可能 對我們候選產品的銷售產生不利影響。我們候選產品的第三方報銷減少或第三方付款人決定 不支付我們候選產品的費用可能會減少醫生對候選產品的使用,並對我們的銷售、運營結果和財務狀況造成嚴重的 不利影響。
45
患者保護和平價醫療法案,經2010年《醫療保健和教育負擔能力協調法案》修訂(統稱為ACA)於2010年3月頒佈。ACA的制定旨在擴大未投保人羣的覆蓋範圍,同時控制總體 醫療保健費用。關於藥品,除其他外,ACA擴大並增加了醫療補助計劃下藥品的行業回****r},並對醫療保險D計劃下的覆蓋要求進行了修改。我們仍然無法完全預測 ACA對製藥公司的影響,因為許多ACA改革需要頒佈實施 法定條款的詳細法規,但尚未完成,醫療保險和醫療補助服務中心(Centers for Medicare & Medicaid Services)已公開宣佈 ,它正在分析已經頒佈的ACA法規和政策,以確定是否應該進行修改。此外,雖然 美國最高法院維持了ACA大部分內容的合憲性,但一些州表示他們打算不執行ACA的某些部分,一些國會議員仍在努力廢除ACA。這些挑戰增加了作為ACA一部分而頒佈的變更的不確定性 。此外,目前對ACA的法律挑戰,以及國會 廢除ACA的努力,增加了作為ACA一部分而頒佈的立法修改的不確定性。
此外,在一些外國 國家/地區,藥品的建議定價必須獲得批准,才能合法上市。管理藥品定價的要求因國家而異 。例如,一些歐盟司法管轄區實行正面清單和負面清單制度,在這種制度下,產品 只有在商定報銷價格後才能銷售。要獲得報銷或定價批准,其中一些國家/地區 可能需要完成臨牀試驗,將特定候選產品的成本效益與當前可用的療法進行比較。其他成員國允許公司固定自己的藥品價格,但監控公司的利潤。各國定價制度的這種差異 可能會造成歐盟成員國之間的價格差異。不能保證 對藥品實行價格控制或報銷限制的任何國家/地區會允許對我們的任何產品進行優惠的報銷和定價安排 。從歷史上看,在歐盟推出的產品並不遵循美國的價格結構。在歐盟,總體醫療成本,尤其是處方藥的下行壓力變得很大。因此,新產品的進入門檻越來越高,患者不太可能使用沒有得到政府或其他公共或私人支付者報銷的藥品。
其他醫療保健法律 和合規要求
在美國,除FDA外,我們的活動 還可能受到各種聯邦、州和地方當局的監管,包括CMS、美國衞生與公眾服務部的其他 部門(例如,監察長辦公室)、美國司法部 和司法部內的個別美國檢察官辦公室,以及州和地方政府。例如,銷售、營銷和科學/教育補助計劃必須符合《社會保障法案》、《虛假索賠法案》、《健康保險可攜性和責任法案》的隱私條款以及類似的州法律的反欺詐和濫用條款。 定價和返點計劃必須符合1990年《綜合預算調節法》和1992年《退伍軍人醫療法案》的醫療補助退税要求(“VHCA”,每一個都經過修改。如果未來的產品提供給聯邦供應計劃的授權用户 ,則適用其他法律和要求。根據VHCA,製藥公司必須以較低的價格向多個聯邦機構提供某些 藥物,包括美國退伍軍人事務部和美國國防部、 公共衞生服務和某些私營公共衞生服務指定實體,以參與其他聯邦資助 計劃,包括醫療保險和醫療補助。此外,美國國防部為TRICARE計劃採購的某些產品 必須通過折扣系統提供折扣價格。參與VHCA要求提交定價數據,並根據複雜的法定公式計算折****r}和回扣,以及簽訂受聯邦 採購法規約束的政府採購合同。
為了以商業方式分銷產品,我們必須遵守州法律,該法律要求在一個州註冊藥品 產品的製造商和批發商,包括在某些州將產品運往該州的製造商和經銷商,即使此類製造商或批發商在該州沒有營業地點。幾個州已頒佈立法,要求製藥公司 建立營銷合規計劃,向州政府提交定期報告,定期公開銷售和營銷活動 或註冊其銷售代表。某些州還頒佈了其他法律,禁止某些其他銷售和營銷行為。我們的所有活動都可能受到聯邦和州消費者保護以及不正當競爭法律的約束。 同樣,根據歐盟或歐盟成員國的法律或我們運營、製造或分銷產品的其他國家/地區的法律,這些活動受授權或許可證要求或其他法律要求的約束。
46
遵守環境法的成本
我們的運營受各種聯邦、州、地方和外國環境法律的監管,包括向空氣和水中排放污染物的法律,危險物質和廢物的管理和處置,以及受污染場地的清理。如果我們將來違反環境法或根據環境法承擔責任,我們可能會招致鉅額成本,包括清理費用、罰款和民事或刑事制裁,以及第三方損害或人身傷害索賠。我們不知道遵守環境法的任何成本或影響 。
與氣候變化相關的法規
我們的業務專注於醫藥產品的研究和開發,其中很大一部分研究和開發是在我們的設施之外進行的,並由外包合同研究機構或大學進行。因此,我們預計任何有關氣候變化的法規都不會影響我們的運營。
但是, 可能會出現更頻繁和更嚴重的天氣事件和水資源供應挑戰,這可能會影響我們合作伙伴和 未來供應商的設施。我們無法保證未來不會因氣候變化而對我們的合作伙伴和未來供應商和供應鏈造成的物理風險 。我們定期審查我們在潛在的天氣相關風險和其他 自然災害中的脆弱性,並相應地更新我們的評估。根據我們的審查,我們認為這些潛在風險 目前對我們的運營沒有重大影響。
員工與人力資本管理
截至2024年3月20日,我們和 我們的子公司有四名全職員工。其中一名僱員在英國,其中三家位於美國。
此外,我們還臨時聘用 數量有限的兼職員工,以及科學顧問、顧問和服務提供商,主要是 通過學術機構和合同研究組織。
我們從未發生過停工事件,我們的員工也沒有受到集體談判協議的保護,也沒有工會代表。我們相信我們 與員工有良好的關係。
我們的人力資本資源 目標包括(如適用)識別、招聘、留住、激勵和整合我們的現有和新員工、 顧問和顧問。我們的股權和現金激勵計劃的主要目的是通過授予基於股票和基於現金的薪酬獎勵來吸引、留住和獎勵員工 ,以通過激勵員工發揮最大能力並實現我們的目標來增加股東價值和 公司的成功。
企業歷史
形成
我們成立於2016年9月7日,是根據特拉華州法律成立的空白支票公司。我們成立的目的是為了實現合併、資本股票交換、股票購買、資產收購或其他類似業務與一家或多家運營企業的合併。 自成立以來,我們專注於收購醫療保健及相關健康行業的運營公司,儘管我們在確定潛在目標企業方面的努力並不侷限於特定行業。
首次公開募股
2017年6月7日,根據我們的首次公開募股(“首次公開募股(IPO)"), 我們以每單位10.00美元的購買價出售了11,500,000個單位,包括2017年6月23日在承銷商選擇完全行使其超額配售權後出售給承銷商的1,500,000個單位,產生的總收益為115,000,000美元。每個"單位 包括我們普通股的380分之一,一項在企業合併完成後獲得1-3800分之一我們普通股的權利(正確的),以及一份可贖回認股權證,購買1股普通股的1 760股 (公開認股權證“)。每份公共認股權證使持有人有權以每1/760股5.75美元的行使價購買1股普通股這是每股收益(每股4,370.00美元), 可調整。於行使公開認股權證時,將不會發行零碎股份。公開認股權證自首次公開招股結束起計12個月可行使,並於業務合併完成後五年(2025年11月6日)屆滿。
47
我們可以在提前30天發出通知的情況下, 以每份公開認股權證0.01美元的價格贖回全部而非部分公開認股權證(“30天的贖回期“), 僅當普通股在發出贖回通知日期前的第三個交易日結束的30個交易日內的任何20個交易日內的最後售價等於或超過每股6,840.00美元,但前提是有關於該等公開認股權證的普通股的有效 登記聲明,以及與該等普通股有關的當前招股説明書在整個30天的贖回期內可用。如果我們如上所述要求贖回公共認股權證 ,我們的管理層將有權要求所有希望行使公共認股權證的持有人在無現金 基礎。“在決定是否要求所有持有人行使其公共認股權證時”無現金基礎,“除其他因素外,管理層將考慮我們的現金狀況、已發行的公開認股權證的數量以及在行使公開認股權證時發行最大數量普通股對我們股東的稀釋影響。 在完成業務合併時,每個權利持有人將獲得1股普通股的1/3,800股。 權利交換時不會發行任何零碎股份。
私募
在IPO結束 的同時,KBL IV Sponsor LLC(“贊助商“)和承銷商購買了總計450,000個未註冊 單位(“私人單位“),每單位10.00美元,在私人配售中產生450萬美元的總收益。 此外,在2017年6月23日,我們完成了另外52,500個私人單位的銷售,每單位價格為10.00美元,由贊助商和承銷商購買,產生的總收益為525,000美元。其中,377,500個私人單位由保薦人購買,125,000個私人單位由承銷商購買。私人單位所得款項已加入 首次公開招股所得款項淨額,並存放於信託賬户(“信託帳户“).私人單位(包括其組成證券)在業務合併(定義見下文)和私人單位中包含的認股權證(“私募認股權證“)只要由保薦人、承銷商或其獲準受讓人持有,則不可贖回。如果私募認股權證由保薦人、承銷商或其獲準受讓人以外的其他人持有,私募認股權證將可由本公司贖回,並可由 該等持有人按與首次公開發售單位所包括的認股權證相同的基準行使。此外,只要私募認股權證由承銷商或其指定人或聯營公司持有,則自與IPO有關的註冊聲明生效日期起計五年後不得行使。否則,私募認股權證的條款和條款與作為IPO單位一部分出售的認股權證的條款和條款相同,並且沒有淨現金結算條款。
業務合併
2019年7月25日,我們簽訂了《企業合併協議》(經不時修訂)。企業合併協議“),與KBL 合併子公司(”合併子)、180生命科學公司(f/k/a 180生命科學公司)。180“)、Katexo 製藥公司(”Katexo)、CannBioRex製藥公司(CBR製藥)、180 Treateutics(br}L.P.(“180 LP與Katexo和CBR Pharma一起,180個子公司並與180生命科學公司一起,180個締約方),勞倫斯·彭布爾(Lawrence Pemble)以180個締約方股東代表的身份 (股東代表").業務合併 協議中描述的業務合併("業務合併),關閉並於2020年11月6日生效(結業"). 根據業務合併協議,除其他事項外,合併子公司與180合併,180繼續作為存續的 實體和本公司的全資子公司(“合併").在關閉之前, 180 Life Sciences Corp.提交了其在特拉華州註冊證書的修正證書,將其名稱改為180 Life Corp.,我們的公司(在我們進入業務合併時稱為KBL Merger Corp. IV,更名為180 Life Sciences Corp.)。
180於2019年1月28日在特拉華州註冊成立。在業務合併結束之前,180通過三家子公司運營:180 LP,一家特拉華州有限合夥企業,成立於2013年9月6日;Katexo,一家於2018年3月7日在加拿大不列顛哥倫比亞省註冊成立的公司;以及CBR Pharma,一家於2018年3月8日在加拿大不列顛哥倫比亞省註冊成立的公司。
48
2019年7月,180以及180 LP、Katexo和CBR Pharma各自完成了一項公司重組,據此,180 LP、Katexo和CBR Pharma成為180LS(重組“)。有關Katexo和CBR Pharma的公司重組安排已根據協議完成。《商業公司法》。(不列顛哥倫比亞省)
2020年11月6日( "截止日期”),我們在2020年11月5日舉行的股東特別大會後完成了業務合併,公司股東在會上審議並批准了(其中包括)採納業務合併的建議。 根據業務合併協議,除其他事項外,合併子公司與180合併,180繼續作為尚存的 實體和本公司的全資子公司。合併於二零二零年十一月六日生效(屆時,“生效時間 ,而合併的結束在本文中被稱為結業“)。在交易結束前,180公司在特拉華州提交了公司註冊證書修訂證書,更名為180生命公司,KBL合併公司第四公司更名為180生命科學公司。
於生效時間,於生效時間前發行及發行之普通股每股 自動轉換為收取本公司約0.44310股普通股的權利,每股面值0.0001美元(該等普通股根據企業合併協議可向180名普通股股東發行)。合併對價股份“)。迄今已向180名普通股股東發行了總計46,039股普通股,作為合併對價股份,包括託管 股份(定義如下)。同樣在生效時間,在生效時間之前發行和發行的180股優先股的每股基礎股份被轉換為有權獲得一股公司的C類特別投票權股份或一股公司的K類特別投票權股份(該等股份、“特別表決權股份“)。特別投票權股份使其持有人有權就任何特定事項、命題或問題,就任何特定事項、命題或問題,享有相等於不時發行的加拿大子公司CannBioRex Puracheco ULC及Katexo Puracheco ULC各自的可交換 股份數目(定義見下文)的總投票數 。
作為合併的結果, 現有可交換股份(統稱為“可交換股份“)CannBioRex Puracheco ULC及/或KatExco Puracheco ULC的股份已根據CannBioRex Puracheco ULC或Katexo Puracheco ULC(視何者適用而定)管理可交換股份的章程細則中的股份條文作出調整,使其乘以合併的交換比率 ,成為可交換普通股。可交換股份使持有人有權獲得與普通股持有人在經濟上實質上相等的股息和其他權利,而可交換股份持有人有權在本公司股東大會上投票。預留共14股普通股,以供在可交換股份交換時向可交換股份持有人發行。
根據企業合併協議,2,764股合併代價股份(該等股份、“託管份額“)被存入一個 託管帳户(”第三方託管賬户“)作為我們在企業合併協議項下的賠償權利的擔保和獨家付款來源,所有這些權利原計劃在企業合併結束後12個月發放給相同的股東,但克勞斯博士對這些股份的索賠仍在進行中。
由於業務合併,180的前股東成為本公司的控股股東,180成為本公司的全資子公司 。該業務合併被視為反向合併,因此180在會計和財務報告方面被視為收購方。
由於交易結束,我們從信託賬户(定義見下文)中提取了9,006,493美元資金,用於贖回2,149股股票。
反向股票拆分
2022年12月反向股票拆分
2022年12月15日,在180生命科學公司股東特別會議上,我們的股東批准了對我們的公司註冊證書的修正案 ,以實現對我們的已發行和流通股(每股面值0.0001美元)的反向股票分割,比例為 一比四比一比二十(含),具體比例為由我們的董事會或其正式授權的 委員會酌情決定的整數,在修訂案批准後和2023年12月15日之前的任何時間。2022年12月15日, 我們的董事會與股東管理局一起批准了對我們第二次修訂和重述的公司註冊證書的修訂,以影響 我們的普通股的反向股票分割,比例為1:20。
在特別 會議和董事會批准後,2022年12月15日,我們向特拉華州務卿提交了對我們的第二次修訂和重述 公司註冊證書的修訂證書,以實現反向股票分割。
49
根據修訂證書 ,反向股票分割於2022年12月19日東部時間上午12:01生效。與 反向股票分割有關,我們的普通股或公共認股權證股票的交易 代碼分別為"ATNF"和"ATNFW"。
修正證書 沒有減少我們普通股的授權股份數量,也沒有改變我們普通股的面值或修改我們普通股的任何投票權 或其他條款。
沒有發行與反向股票拆分相關的零碎股份,登記在冊的股東原本有權獲得零碎股份, 則有權將其零碎股份向上舍入至最接近的整體股份。
2024年2月反向股票拆分
2024年2月16日,在公司股東特別會議上,我們的股東批准了對我們的第二份經修訂並重新簽署的公司註冊證書,以實現我們普通股的已發行和流通股的反向股票拆分,每股面值0.0001美元,比例為四比一和四十比一,具體比例 將由我們的董事會確定。
2024年2月26日,公司董事會批准了對公司發行在外的普通股進行一股19股反向股票分割,並批准了對公司註冊證書進行修改的證書的備案,以影響此類反向股票分割,該證書是在2024年2月26日提交的。反向股票拆分於2024年2月28日東部時間上午12:01生效,股票 在2024年2月28日開市時開始交易。與反向股票分割有關,截至生效時間,每19股已發行和流通的公司普通股將自動轉換為一股公司普通股 。
與反向拆分有關,所有未行使的期權、認股權證、 和其他賦予其持有人購買或以其他方式接收普通股股份的證券都按照每種證券的 條款的要求進行了調整。根據公司股權激勵計劃可授予的股份數量也進行了適當的 調整。在反向拆分之後,普通股的面值保持不變,為每股面值0.0001美元。反向 拆分沒有改變普通股或優先股的授權股數。沒有發行與反向拆分有關的零碎股份,而原本有權獲得零碎股份的股東則獲得一整股普通股 ,以代替此類零碎股份。
19反向股票分割1的影響已追溯反映在本報告中。
公司結構
下圖顯示了我們當前的組織結構:

50
關於我們
我們的主要行政辦公室 位於3000 El Camino Real,Bldg. 4,Suite 200,Palo Alto,CA 94306,我們的電話號碼是(650)507—0669。我們維護 網站:www.example.com.我們未通過引用將我們網站中的信息或 可通過我們網站訪問的信息納入本報告,您不應將其視為本報告的一部分。
項目1a.風險 因素。
彙總風險因素
我們面臨與我們的業務相關的風險和不確定性,其中許多風險和不確定性是我們無法控制的。特別是,與我們的業務相關的風險包括:
| ● | 我們是一家臨牀階段的生物技術公司,截至2023年和2022年12月31日止年度沒有收入,預計在不久的將來不會產生收入; |
| ● | 我們需要額外的近期和長期融資,以支持我們的 運營、我們根據需要籌集此類融資的能力、此類融資的條款(如有)、潛在的重大稀釋 與此相關的契約和限制,以及我們可能需要遵守的與該等資金有關的契約和限制; |
| ● | 我們對未來候選產品成功的依賴,其中一些產品可能 未獲得監管部門批准或成功商業化;我們新產品的製造過程中存在問題和/或 我們未能遵守制造法規,或製造成本意外增加;分銷問題 我們的產品;以及未能充分營銷我們的產品; |
| ● | 與我們業務增長相關的風險,我們保持這種增長的能力, 管理我們的增長和執行我們的增長戰略的困難; |
| ● | 先前重報財務報表的負債,並與無效相關 控制和程序,以及與現任和前任管理人員和董事的賠償有關的成本和開支; |
| ● | 我們對關鍵人員的依賴,以及我們吸引和留住員工和顧問的能力; |
| ● | 來自擁有比我們更豐富資源和經驗的公司的激烈競爭的風險; |
| ● | 我們為候選產品獲得監管部門批准的能力,以及時間軸和成本 與此相關的,包括與我們候選藥物的臨牀開發和監管批准相關的不確定性, 包括臨牀試驗入組和完成的潛在延遲、FDA和MHRA提出的問題; | |
| ● | 我們未來的候選產品(如果獲得監管機構的批准)可能無法 實現預期的市場接受度,從而限制我們從新產品中產生收入的能力; |
| ● | 當前和未來的索賠和訴訟的結果,未來的政府調查, 及其他程序可能對我們的業務及經營業績造成不利影響; |
| ● | 我們的大多數許可協議為許可方和/或交易對手方提供的事實 使用、擁有和/或利用此類許可知識產權的權利; |
| ● | 臨牀前研究和早期臨牀試驗不一定能預測未來結果 並且可能不會取得有利的結果;我們的營銷經驗有限,未來成功商業化任何 我們的候選產品的數量,即使它們在未來獲得批准也是未知的;業務中斷可能會延誤我們的進程 開發我們未來的候選產品,並可能擾亂我們的產品銷售; |
| ● | 第三方付款人可能不為任何未來產品提供保險和適當的報銷水平; |
| ● | 訴訟責任(包括產品責任訴訟、股東訴訟和監管 事項),包括判決、損害賠償、罰款和處罰,以及目前未決訴訟的結果,潛在 未來的政府調查,以及可能對我們的業務和經營業績造成不利影響的其他訴訟; |
| ● | 安全漏洞、數據丟失和其他中斷可能阻止我們訪問關鍵 或使我們承擔責任或損害賠償; |
51
| ● | 與昂貴、耗時、不確定且易受影響的臨牀試驗相關的風險 變更、延遲或終止,且在試驗、測試、申請或批准中存在不同解釋、延遲 候選藥物的過程和/或我們獲得有前途候選藥物批准的能力,以及與之相關的成本; |
| ● | 我們遵守現有和未來的規則和法規,包括聯邦、州和 外國醫療保健法律法規及其實施或變更; |
| ● | 我們有能力充分保護我們未來的候選產品或我們的專有技術, 市場、第三方就我們涉嫌侵犯其知識產權提出的索賠和責任; |
| ● | 國家和其他司法管轄區之間的法律法規差異以及法律的變更 不對任何資料作商業性利用,包括但不限於在未經批准的情況下,複製在未經批准的情況下使用的任何資料並用於商業用途。 |
| ● | 我們的管理人員、董事、顧問和科學家之間的利益衝突; |
| ● | 與我們未能遵守某些事先約定的合同義務相關的處罰,以及 限制; |
| ● | 未來籌資、轉換/行使已發行的可轉換證券造成的攤薄,以及此類未來發行/出售對我們證券價值造成的下行壓力; |
| ● | 新冠肺炎疫情和其他未來可能的流行病對我們業務的負面影響; |
| ● | 我們證券的波動性極大,可能缺乏流動性; |
| ● | 我們的公司註冊證書規定了對高級管理人員和董事的賠償, 限制了高級管理人員和董事的責任,允許在沒有股東批准的情況下授權優先股,並且 包括某些其他反收購條款和排他性論壇條款; |
| ● | 我們有能力保持我們的普通股和認股權證在納斯達克上上市,以及遵守美國證券交易委員會和納斯達克規則和要求的成本 ; |
| ● | 我們的信息技術系統出現故障,包括網絡安全攻擊或其他數據安全事件,可能會嚴重擾亂我們的業務運營; |
| ● | 我們可能會收購其他公司,這可能會轉移我們管理層的注意力, 這會對我們的股東造成額外的稀釋,否則會擾亂我們的運營,損害我們的經營業績,如果我們進行任何收購,它們可能會擾亂我們的業務或對我們的業務產生負面影響; | |
| ● | 通貨膨脹和利率變化以及經濟衰退的影響,包括潛在的衰退,以及宏觀經濟、地緣政治、衞生和行業趨勢、流行病、戰爭行為(包括持續的烏克蘭/俄羅斯和哈馬斯/以色列衝突)和其他大規模危機的影響,以及國會在美國債務上限問題上陷入僵局或未來美國政府可能因預算分歧而停擺的潛在影響; | |
| ● | 我們目前沒有250萬美元或更多的股東權益,因此,我們不符合納斯達克資本市場的持續上市要求,我們的普通股和公募認股權證將被退市; |
| ● | 我們可能會將營運資本和未來資金用於最終不會改善我們的經營業績或增加我們證券價值的用途;以及 |
| ● | 我們的增長在一定程度上取決於我們與第三方戰略關係的成功。 |
52
您應該知道 投資我們普通股有很大的風險。在決定投資我們的普通股之前,您應該仔細考慮這些風險因素。
如果發生以下任何 風險,我們的業務、財務狀況、運營結果或其他前景可能會受到重大不利影響, 任何這些風險的發生都可能對我們的成功可能性產生重大影響。如果發生這種情況,我們 普通股的市場價格(如果有的話)可能會下跌,潛在投資者將失去對我們普通股的全部或部分投資。
我們的業務、財務狀況和經營結果會受到各種風險和不確定性的影響,包括下文所述的風險和不確定因素。本節討論的因素 可能導致我們的實際結果與預期和歷史結果大相徑庭的因素,無論是單獨的還是綜合的。我們的業務、財務狀況或運營結果可能會受到任何這些風險的重大不利影響。不可能預測或識別所有這些因素。因此,以下對風險因素的描述並不是對適用於我們業務的所有潛在風險或不確定性的完整討論。
與我們的業務運營相關的風險
我們目前的現金餘額 預計僅足以為我們計劃的業務運營提供資金,直至2024年5月左右。如果沒有額外資本 ,我們可能無法繼續我們計劃的業務運營,可能被迫更改我們計劃的業務運營,或者 可能採取其他可能對我們的股東造成不利影響的行動,包括尋求破產保護。
我們是一家臨牀階段生物技術公司,目前沒有收入。因此,我們的業務不會產生為我們計劃的業務運營提供資金所需的現金。 我們將需要大量額外資本來:(I)開發FDA和/或MHRA批准的產品並將此類產品商業化; (Ii)為與我們的候選產品相關的研究和開發活動提供資金,並獲得監管部門的批准;(Iii)保護我們的知識產權;(Iv)吸引和留住高素質人才;(V)有效應對競爭壓力; 和(Vi)獲取補充業務或技術。
我們未來的資本需求 取決於許多因素,包括:(I)與我們的研究、開發和商業化努力相關的範圍、持續時間和支出 ;(Ii)我們項目的持續科學進步;(Iii)潛在合作或許可交易的結果, 如果有的話;(Iv)競爭技術發展;(V)我們的專利地位;以及(Vi)我們產品的監管批准 過程。
我們將需要通過公開或私募股權發行、債務融資或戰略聯盟以及許可安排籌集大量 額外資金,為我們計劃中的業務運營提供資金。我們可能無法以對我們有利的條款獲得額外的融資,如果有的話。一般市場狀況、不斷上升的利率和通脹,以及全球衝突,如烏克蘭和俄羅斯、以色列和哈馬斯之間的持續衝突,可能會使我們難以從資本市場尋求融資,任何融資條款都可能對我們股東的持股或權利產生不利影響。例如,如果我們通過發行股權證券籌集額外資金,將導致我們的股東進一步稀釋,這可能會極大地稀釋他們的投資價值。任何股權融資也可能 降低我們已發行的可轉換或可行使證券的轉換或行使價格,這可能導致我們的普通股發行(或潛在發行)大量額外股票。此外,作為向我們提供額外資金的條件,未來的投資者可以要求並可能被授予高於現有股東的權利。 債務融資可能涉及限制性契約,可能會限制我們開展未來業務活動的靈活性 ,如果發生破產,可以在股權證券持有人收到我們資產的任何分配之前支付。我們可能被要求 放棄對我們的技術或候選產品的權利,或以對我們不利的條款通過聯盟、合資企業或協議授予許可證 ,以籌集額外資金。如果沒有足夠的資金,我們可能不得不推遲、 減少或取消與我們的業務相關的一項或多項計劃活動,或者終止我們的運營,或者可能被迫 尋求破產保護。這些行動可能會降低我們普通股的市場價格。
我們將需要額外的 資本,這可能無法以商業上可接受的條款獲得,這會引發對我們是否有能力繼續 作為持續經營的企業的質疑。
截至2023年12月31日,我們的累計赤字為127,343,657美元,營運資本赤字為1,422,710美元,截至2023年12月31日止年度,淨虧損為19,935,112美元,截至2023年12月31日止年度經營活動所用現金為10,922,223美元。 截至2024年3月18日,我們手頭現金約為80萬美元。 隨附的合併財務報表是假設我們將繼續作為一個持續經營企業編制的。由於我們沒有創造收入,我們需要籌集大量資金,以償還債務和支付運營成本。雖然我們最近 在2022年7月通過出售股權籌集資金,(約650萬美元總收益),2022年12月(約600萬美元的總收益),2023年4月(約300萬美元總收益),2023年8月(約300萬美元)和 2023年11月(約80萬美元),無法保證我們將能夠籌集額外所需資本,或此類資本 將以優惠條款提供。
53
在一個競爭激烈的行業中,我們面臨着發展新企業所固有的所有重大風險。由於缺乏長期的運營歷史和我們所在市場的新興性質,我們預計將出現運營虧損,直到 我們能夠成功實施我們的業務戰略,其中包括所有相關的收入流。我們可能永遠無法實現盈利 運營或產生可觀的收入。
我們目前每月 現金需求支出約為350,000美元。我們認為,總體而言,我們將需要大量額外的資本資金 來支持和擴大我們產品的研發和營銷,為未來的臨牀試驗提供資金,償還債務, 為額外設備和開發成本提供資本支出,支付義務,辦公空間和管理業務的系統,並支付其他運營成本,直到我們計劃的產品收入流完全實現,並開始抵消 我們的運營成本(如果有的話)。
自我們成立以來,我們 用股權和債務融資的收益為我們的運營提供資金。我們遇到了流動性問題,除其他原因外, 我們以可接受的條件籌集足夠資本的能力有限。我們過去一直依賴出售可轉換為普通股股份的股權和債務融資 來為我們的運營提供資金,並投入了大量努力來減少這種風險。 我們預計,在可預見的將來,我們將需要發行股本,為我們的運營提供資金,併為我們的運營開支提供資金。如果 我們無法實現運營盈利或無法成功獲得其他形式的融資,我們將不得不評估 其他措施,以減少運營費用並節約現金。
這些情況使人對我們作為持續經營企業的持續經營能力產生了極大的懷疑。本報告所載綜合財務報表乃根據美國公認的持續經營會計原則編制,該等會計原則考慮在正常業務過程中變現資產及清償負債。因此,本文所載的綜合財務報表 不包括任何與資產的可回收性和負債分類有關的調整,如果我們無法繼續經營下去的話 可能需要這樣做。本文中的綜合財務報表還包括一個持續經營的腳註。
此外,只要有可能, 我們的董事會(“董事會“或”衝浪板")將試圖使用非現金對價 來履行債務。在許多情況下,我們認為非現金代價將包括我們普通股的限制性股份、優先股或購買我們普通股股份的認股權證。我們的董事會擁有權力,無需股東採取行動或投票, 但須遵守納斯達克規則和法規(通常要求股東批准任何交易,如果交易導致 發行超過我們當時發行的普通股的20%,或佔我們當時發行的 股票的20%以上的投票權,但某些例外情況除外),發行全部或部分授權但未發行的普通股、優先股或購買該等普通股股份的認股權證。此外,我們可能會嘗試通過出售我們的普通股股票來籌集資金,可能在未來以低於市價的價格出售。這些行為將導致現有股東的所有權權益被稀釋, 可能進一步稀釋普通股賬面價值,並且這種稀釋可能是重大的。此類發行還可能有助於增強現有管理層 維持對我們控制的能力,因為這些股份可能會發行給承諾支持現有管理層的各方或實體。
我們將需要籌集 額外的資本,這些資本可能無法以優惠的條件獲得,如果有的話,將導致我們的股東股權稀釋、限制我們的運營 或對我們的業務運營能力產生不利影響。
我們可能無法以對我們有利的條件獲得 額外融資(如果有的話),包括由於宏觀經濟條件,例如嚴重或長期 經濟衰退。資本市場的中斷、不確定性或波動可能會增加我們的資本成本或限制我們籌集業務所需資金的能力 。中斷可能是由美聯儲的政策和行動、貨幣擔憂、 通貨膨脹、經濟下滑或不確定性、貨幣政策、金融機構倒閉、美國債務管理擔憂以及 美國債務限額和預算爭議(包括政府關門、歐洲和全球主權債務擔憂、其他全球或 地緣政治事件或其他因素)引起的。當前的宏觀經濟狀況對美國銀行業產生了負面影響,例如最近硅谷銀行和Signature銀行的倒閉和FDIC接管。雖然我們在 沒有任何賬户,也沒有與這些銀行的業務關係,但我們可能會受到這些 或類似事態發展導致的美國銀行系統其他中斷的負面影響。
54
我們有很大的流動性需求,而且還在不斷增加,需要額外的資金。
研發、 管理和行政費用(包括法律費用)以及用於運營的現金將繼續很大,並且在未來 與新的研發計劃、臨牀試驗、持續的產品商業化努力以及我們未來候選產品的推出有關, 可能大幅增加。我們將需要籌集額外資金以資助我們的運營, 繼續開展臨牀試驗以支持潛在的監管部門批准上市申請,併為我們未來候選產品的商業化提供資金 。
我們未來資金需求的數額和時間取決於許多因素,包括但不限於:
| ● | FDA和/或MHRA批准的時間(如有)以及其他國際批准的時間 我們未來候選產品的市場,如果有的話; |
| ● | 銷售我們產品的收入的時間和金額,或贈款收入,或 其他來源; |
| ● | 我們的臨牀試驗和其他產品開發計劃的進展率和成本; |
| ● | 建立或外包銷售、營銷和分銷能力的費用; |
| ● | 任何外包種植和商業製造供應安排的成本和時間 為我們未來的候選產品; |
| ● | 申請、起訴、辯護和執行任何專利權利要求和其他知識產權的費用 與我們未來候選產品相關的產權; |
| ● | 競爭的技術和市場發展的影響; |
| ● | 人員、設施和設備要求;以及 |
| ● | 任何其他合作、許可、聯合推廣或其他的條款和時間 我們可以建立的安排。 |
55
雖然我們預計未來的資本需求將來自多個來源,如運營現金流和進一步公開和/或非公開發行的收益,但我們不能向您保證這些資金來源中的任何一個都將以優惠的條款提供給我們,或者根本不會。此外,即使 如果我們能夠從上述所有來源籌集資金,籌集的金額也可能不足以滿足我們未來的資本需求。
我們與牛津大學和其他許可方的許可協議 在某些情況下可能會在未經我們同意的情況下終止。
我們與牛津大學和其他許可方簽訂的所有許可協議 仍受各種條件和約定的約束,併為許可方規定了某些終止權 。這些協議通常允許許可方因我們未能及時支付到期款項、 未能根據適用許可協議的條款糾正重大違約行為以及我們的破產而終止協議。因此,如果我們被視為 資不抵債,或在我們尋求破產保護的情況下,我們許可協議的許可方可以終止其與我們的許可協議 。如果此類許可協議終止,我們可能會失去開發我們所有平臺和技術的權利, 可能會失去為開發此類平臺和技術所做的任何投資,並且可能會失去任何知識產權、 產品途徑或開發機會。此類終止可能導致我們的證券價值下降或 變得毫無價值,需要我們改變我們的業務計劃,並可能導致公司尋求破產保護。
我們欠牛津大學一大筆錢,但我們沒有這筆錢。大學可能會對我們採取行動,以強制執行他們在未來的付款權利 ,這可能會對我們和我們的運營產生重大不利影響。
由於最近的財政拮据, 公司一直無法及時支付應付牛津大學的款項("牛津”), the licensor of the majority of the Company’s licenses and patents and the Company’s research partner. Oxford alleges that an aggregate of approximately £929,030 is owed from the Company and one of its subsidiaries to Oxford under the terms of licenses and agreements with Oxford and related parties. The Company is currently in ongoing discussions with Oxford to reduce that amount and enter into a payment plan with regards to the amounts owed; however, no definitive terms or extensions have been agreed to date. Oxford has also notified the Company that it is not willing to discuss any new projects or arrangements until all outstanding invoices have been paid or a payment plan has been agreed to; has engaged a law firm to seek the collection of the amounts owed, together with interest; and has threatened legal proceedings against us. While we are hopeful that we can come to mutually agreeable terms regarding a settlement, payment plan, and/or extension, with Oxford, we may not have sufficient funds to pay amounts due to Oxford in the near term, if at all, and Oxford may take action against us, including filing legal proceedings against us seeking amounts due and interest, attempting to terminate their relationship with us, and/or filing a wind-up petition against one of the Company’s subsidiaries in the U.K.. If Oxford were to take legal action against us or terminate their relationship with us, we may be forced to scale back our business plan and/or seek bankruptcy protection. We may be subject to litigation and damages for our failure to pay amounts due to Oxford, and may be forced to pay interest and penalties, which funds we do not currently have. We plan to seek to raise funding in the future to support our operations, and to pay amounts due to Oxford, through a combination of equity offerings, debt financing or other capital sources, including potentially collaborations, licenses and other similar arrangements, which may not be available on favorable terms, if at all. The sale of additional equity or debt securities, if accomplished, may result in dilution to our then stockholders. Additionally, in December 2023, we engaged A.G.P./Alliance Global Partners as financial advisor to explore and evaluate strategic alternatives to enhance shareholder value. Potential strategic alternatives that may be explored or evaluated by the Company as part of this process include, but are not limited to, an acquisition, merger, reverse merger, other business combination, sale of assets, licensing or other strategic transactions involving the Company. The Company does not intend to discuss or disclose further developments during this process unless and until its Board of Directors has approved a specific action or otherwise determined that further disclosure is appropriate. There is no assurance that the strategic review process will result in the approval or completion of any specific transaction or outcome.
我們的運營結果 可能會受到幣值波動的不利影響。
我們以美元以外的貨幣支出 。我們的報告貨幣是美元。某些子公司的本位幣為 加元(“計算機輔助設計“)或英鎊(”£“或”英鎊").由此產生的 換算調整在股東權益中確認為累計其他全面收益的組成部分。綜合 收入定義為實體所有來源(所有者投資或向所有者分配除外)的權益變動, 包括上述外幣換算調整。截至2023年及2022年12月31日止年度,由於外幣換算調整,我們錄得 其他全面(虧損)分別為15,816美元及3,702,963美元。
56
我們支付費用(並在未來獲得收入)的貨幣與美元之間的價值變化,可能會導致不利的 費用被記錄到我們的損益表中。
全球經濟狀況 可能對我們的業務、運營結果、財務狀況和增長產生重大不利影響。
不利的宏觀經濟狀況,包括通貨膨脹、增長放緩或衰退、新的關税或增加的關税、財政和貨幣政策的變化、信貸收緊、更高的利率、高失業率和匯率波動,以及國會在美國債務上限問題上陷入僵局或未來美國政府可能因預算分歧而停擺的潛在影響,可能會對我們的運營、 費用、獲得資本的渠道以及我們計劃中的未來產品的市場產生重大不利影響。此外,消費者信心和支出可能會因金融市場波動、負面金融消息、房地產和抵押貸款市場狀況、收入或資產價值下降、燃料和其他能源成本、勞動力和醫療成本以及其他經濟因素的變化而受到不利影響。
此外,全球或區域經濟狀況的不確定性 或衰退可能對我們的資金來源、供應商和 合作伙伴產生重大影響。潛在影響包括財務不穩定;無法獲得信貸以資助運營和購買我們未來 計劃產品;以及破產。
經濟環境的低迷 也可能導致我們出售股權或發行新債的能力受到限制;流動性降低;並導致我們金融工具的公允價值下降 。這些和其他經濟因素可能會對我們的業務、經營業績、財務狀況和增長造成重大不利影響。
我們的行業和更廣泛的美國經濟在2022至2023年間經歷了高於預期的通脹壓力,這與持續的供應鏈中斷、勞動力短缺和地緣政治不穩定有關。如果這些情況持續下去,未來的運營結果和現金流可能會受到實質性的不利影響。
在2022年和2023年初,由於供應受限、供應鏈中斷、需求增加、與充分就業的美國勞動力相關的勞動力短缺、高通脹和其他因素,某些材料、產品和運輸成本大幅上升。多個地緣政治事件造成的全球能源供應中斷進一步加劇了供需基本面,包括俄羅斯和烏克蘭之間的持續衝突,以及以色列和哈馬斯之間的衝突,這有可能蔓延到其他中東國家。服務、材料和運輸成本也相應增加,美國各地普遍存在的供應鏈和通脹問題導致運營成本增加。供應鏈限制和通脹壓力在過去和未來可能對我們的運營成本產生不利影響,並可能對我們未來的產品成本、諮詢成本和支出產生負面影響,這可能會對我們的業務、財務狀況、運營結果和現金流產生重大不利影響。
經濟不確定性 可能會影響我們獲得資本的機會和/或增加此類資本的成本。
全球經濟狀況繼續動盪和不確定,原因包括消費者對未來經濟狀況的信心,對衰退和貿易戰的擔憂,能源價格,利率波動,消費信貸的可用性和成本,政府刺激計劃的可用性和時機 ,失業率,通貨膨脹上升,税率,烏克蘭和俄羅斯之間的戰爭,這場戰爭於2022年2月開始,以色列和哈馬斯之間的戰爭,這場戰爭始於2023年10月,有可能蔓延到其他中東國家。 這些情況仍然不可預測,給我們未來籌集資金的能力帶來了不確定性。如果所需的資金在未來變得不可用或成本更高,可能會對我們的業務、未來的運營結果和財務狀況產生重大不利影響。
我們可能不會收到 合併前董事和高級管理人員保險單下與某些訴訟事項有關的任何金額。
2022年6月29日,AmTrust 國際承銷商DAC(“AmTrust“),它是KBL合併前董事和高級職員保險的保單承保人,向美國加州北區地區法院提起了針對我們的聲明性救濟訴訟 (宣告性救濟行動“)尋求申報AmTrust在董事 和高級人員保險單下的義務。在聲明救濟行動中,AmTrust聲稱,由於合併,我們不再是標的保險單下的被保險人,儘管我們尋求向AmTrust追回的費用與合併前發生的事項 有關。
57
2023年4月21日,法院 發佈命令,部分批准並部分拒絕公司的部分簡易判決動議。具體而言,法院 就以下問題作出了有利於公司的簡易裁決:(a)公司事實上是 AmTrust和Freedom保單下的被保險人;(b)某些SEC傳票相關的費用被告Marlene Krauss博士,公司的前首席執行官和董事,喬治霍尼格,前董事會主席,在AmTrust和Freedom保單的 基本承保範圍內;以及(c)AmTrust和Freedom所依賴的被保險人與被保險人除外 不適用于禁止任何此類承保。
法院還發現, 保單中包含的控制權變更除外條款存在爭議的事實,因此 法院無法批准公司剩餘的簡易裁決請求作為法律事項。因此,法院 此時駁回了公司進一步的簡易判決請求,並認為目前, 控制權變更問題將在審判時確定,以便發現保單提供(i)對 公司已經預付並將預付給Marlene Krauss博士和George Hornig博士的費用的承保;(ii)AmTrust違反了政策;(iii) AmTrust必須支付公司的此類費用;並且,一旦AmTrust政策用盡,(iv)Freedom將有義務 根據其政策支付公司的此類費用。
2023年8月4日,法院 批准了公司的請求,提出了第二項動議,要求在本案中進行部分即決判決,這一動議涉及 是否應要求AmTrust向公司預付Marlene Krauss博士和George Hornig 在案件未決期間發生的辯護費用。公司提交了部分即決判決動議, 當事人現已充分聽取了簡要説明。該動議的聽證會於2024年1月11日舉行,但法官接受了提交的事項,尚未就該動議作出任何決定。 雙方已開始針對對方的書面證據程序, 預計也將發生證詞。公司打算繼續大力追究此事,以確定 公司有權獲得AmTrust和Freedom全額支付公司的主題預付費用。
雖然公司繼續 相信它對AmTrust和Freedom都有強有力的證據,並相信法院就上述事項 作出的對其有利的裁決對公司來説是一個重大的積極結果,但不能保證公司將在這一訴訟中獲勝。
我們從任何潛在產品中產生 收入的能力取決於我們獲得監管批准和滿足許多其他要求的能力 ,我們可能永遠無法成功產生收入或實現盈利。
我們的盈利能力和 取決於我們創造收入或執行其他業務發展安排的能力。我們不希望 產生大量收入(如果有的話),除非我們能夠獲得監管部門的批准,併成功將我們正在開發或可能開發的候選產品商業化 。成功的商業化,在一定程度上,將需要實現 許多關鍵里程碑,包括在臨牀試驗中證明安全性和有效性,獲得這些候選產品 的監管批准,製造、營銷和銷售,或簽訂其他協議以商業化,我們可能獲得監管批准的那些產品,滿足所有上市後要求,並從私人保險 或政府付款人那裏獲得產品報銷。由於與這些活動相關的不確定性和風險,我們無法準確和精確地預測 收入的時間和金額(如果有的話)、任何進一步損失的程度或我們何時可能實現盈利。我們可能永遠不會在這些活動中成功 ,即使我們成功了,我們也可能永遠不會產生足以讓我們實現盈利的收入。即使 我們確實實現了盈利能力,我們也可能無法維持或提高季度或年度盈利能力。
58
如果我們無法實現並 保持盈利,可能會壓低我們普通股的市場價格,並可能損害我們籌集資金、擴大業務、 多樣化產品供應或繼續運營的能力。
我們最近擴大了業務,未來需要增加組織的規模和複雜性,我們在管理增長和執行增長戰略方面可能會遇到困難 。
我們目前的管理、人員 和系統可能不足以支持我們的業務計劃和未來增長。我們將需要增加全職同等員工的 數量,以便對我們未來的產品進行1期、2期和3期臨牀試驗,並建立商業 組織和商業基礎設施。由於這些未來的活動,我們的業務運營的複雜性預計將大幅增加 。我們將需要開發和擴大我們的科學、製造、銷售和營銷、管理、合規、 運營、財務和其他資源,以支持我們計劃的研究、開發、製造和商業化活動。
為了有效管理 我們的運營、增長和各種項目,我們需要:
| ● | 繼續改進我們的業務、財務、管理和監管合規控制以及報告系統和程序; |
| ● | 吸引和留住足夠數量的優秀員工; |
| ● | 以經濟高效的方式有效地管理我們的商業化活動(目前,我們臨牀試驗的試驗和開發非常具有成本效益);以及 |
| ● | 有效管理我們的開發工作,同時履行我們對承包商和其他第三方的合同義務 。 |
我們已經並將繼續利用兼職外部顧問和承包商的服務為我們公司執行多項任務,包括與合規計劃、臨牀試驗管理、法規事務、配方開發和其他藥物開發職能相關的任務 。 我們的增長戰略可能需要擴大我們對顧問和承包商的使用,以實施這些和未來的其他任務。如果 我們不能通過招聘新員工和擴大顧問和承包商的使用來有效地擴大我們的組織, 我們可能無法成功執行有效執行我們計劃的研發、製造 和商業化活動所需的任務,因此可能無法實現我們的研究、開發和商業化目標。
我們將為之前重述的財務報表和/或導致此類重述的前管理層的某些行為承擔責任。
我們於2020年12月31日提交了當前的Form 8-K報告,並於2021年2月3日提交了另一份當前的Form 8-K報告,其中我們宣佈,由於我們 在業務合併之前發現的與KBL相關的事項,某些歷史財務報表不可靠。因此,我們重報了截至2020年6月30日的三個月和六個月的財務報表,以及截至2020年9月30日的三個月和九個月的財務報表,因為這些財務報表中存在錯誤,這些錯誤是在我們提交給美國證券交易委員會的原始季度報告中發現的 截至2020年6月30日和2020年9月30日的季度報告中發現的。雖然我們認為這些重述是KBL管理層行為的結果,也是KBL管理層的責任(他們都不再受僱於我們),但我們可能 因此類重述而受到股東訴訟、美國證券交易委員會訴訟、罰款和處罰、評級下調、負面宣傳以及難以吸引和 留住關鍵客户、員工和管理人員。此外,我們的證券的交易價格可能低於類似情況的公司,這些公司不必重新申報其財務報表。
我們未能對重大一次性交易導致的流程進行適當的調整,可能會導致財務報表中的錯誤陳述。
在我們的年度審計過程中但在備案之前,我們發現發生了一個錯誤,導致我們的公共認股權證的公允價值被誇大了 一個無關緊要的數額。在提交2022年財務報表之前,這一錯誤已得到更正。雖然我們相信權證在截至2022年12月31日止年度的財務報表中的公允價值 ,且由於該日期陳述正確, 類似的錯誤可能會對我們的財務狀況及經營業績產生重大不利影響,因此我們 可能需要我們重報前期或未來的財務報表。
59
未來一段時間內,運營結果可能會有很大變化。
我們的財務結果是 不可預測的,可能會因其他原因而波動,原因包括:我們未來候選產品的商業銷售;我們產品開發目標和里程碑的實現情況;臨牀試驗登記和費用;研發費用;以及合同製造和合同研究付款的時間和性質。我們很大一部分成本是按年預先確定的, 部分原因是我們的研發成本很高。因此,未來收入的小幅下降可能會對一個季度的財務業績產生不成比例的影響。
我們依賴於我們的關鍵人員以及吸引和留住員工的能力。
我們未來的增長和成功 取決於我們招聘、留住、管理和激勵員工的能力。我們高度依賴我們目前的管理和科學人員,包括我們的首席執行官James N.Woody博士、我們的聯席主席Marc Feldmann爵士博士和Lawrence Steinman醫學博士、我們的首席科學官Jonathan Rothbard博士和我們的科學家JagDeep Nanchahal。無法聘用或留住經驗豐富的管理人員可能會對我們執行業務計劃的能力產生不利影響,並損害我們的經營業績。由於我們的業務具有專門的科學和管理性質,我們在很大程度上依賴於我們吸引和留住合格的科學、技術和管理人員的能力。生物技術領域對合格人才的競爭非常激烈,我們可能無法繼續 吸引和留住業務發展所需的合格人員,或招聘合適的接班人。
我們的製造過程中出現的問題 我們未來的化學實體、不遵守制造法規或我們的製造成本意外增加 可能會損害我們的業務、運營結果和財務狀況。
我們負責製造和供應我們未來的CBD衍生品和α7nAChR計劃中的候選產品,用於商業用途和臨牀試驗。我們未來候選產品的製造需要遵守GMP和國際司法管轄區的其他法規要求 。我們成功製造未來候選產品的能力將包括在嚴格的 受控流程和程序下,製造成品以及標籤和包裝,包括產品信息、防篡改證據和防偽功能。此外,我們必須確保我們的批次之間的化學一致性,包括臨牀試驗批次和上市批次(如果獲得批准)。要證明這種一致性,可能需要典型的生產控制和臨牀數據。我們還必須確保我們的批次符合複雜的版本規格。如果我們無法按照法規規範生產我們未來的候選產品,或者如果我們的製造流程因損壞、 丟失或其他原因而中斷,或未能通過對我們製造設施的監管檢查,我們可能無法滿足需求或提供足夠的產品用於臨牀試驗,這還可能損害我們將未來候選產品及時商業化或具有成本競爭力的能力(如果有的話)。
我們可能無法及時開發和擴展我們的製造能力以滿足對我們候選產品的需求,FDA、MHRA或其他外國監管機構 可能不接受我們或我們合同製造商的設施適合生產我們的產品和候選產品 。我們製造過程中的任何問題都可能對我們的業務、運營結果和財務狀況產生重大不利影響。
我們與Celltrion Healthcare的諒解備忘錄 可能不會導致雙方達成最終協議。
2021年9月,我們與生物製藥公司Celltrion Healthcare簽署了一份不具約束力的諒解備忘錄,以供應我們正在進行的抗TNF產品開發中使用的抗TNF 生物類似藥。迄今為止,雙方尚未就此類關係達成最終協議,且此類最終協議可能不會按預期條款達成(如果有的話)。如果 我們無法與Celltrion Healthcare達成雙方同意的最終條款,公司已與表示有興趣繼續進行談判的抗TNF生物仿製藥替代供應商 。我們最終可能無法 找到替代供應商,或者此類替代供應商可能要求的條款比目前預期的要低。 上述任何情況都可能對我們的業務、經營業績和財務狀況造成重大不利影響。
60
我們預計將面臨來自比我們擁有更多資源和經驗的公司的激烈競爭;並可能面臨競爭對手的競爭,這些競爭對手試圖根據第505(B)(2)條的申請來營銷我們的產品。
製藥行業 競爭激烈,變化迅速。隨着越來越多的競爭對手 和潛在競爭對手進入市場,該行業將繼續擴大和發展。這些競爭對手和潛在競爭對手中的許多人擁有比我們公司更多的財務、技術、管理和研發資源和經驗。其中一些競爭對手和潛在的 競爭對手在藥品開發方面比我們公司擁有更多的經驗,包括驗證程序和 監管事項。此外,我們未來的候選產品(如果開發成功)將與大型 和成熟公司的產品競爭,這些公司擁有比我們公司或合作伙伴更豐富的營銷和銷售經驗和能力。特別是Insys Therapeutics,Inc.正在開發CBD在嬰兒痙攣("是"),以及潛在的 其他跡象。Zogenix公司在兩項低劑量芬氟拉明治療Dravet綜合徵的III期試驗中報告了陽性數據,並 開始使用該產品治療Lennox Gastaut綜合徵的III期試驗。Biocodex最近獲得FDA的監管批准,用於治療Dravet綜合徵的藥物Stiripentol(Diacomit)。其他比我們公司資源更大的公司 將來可能會宣佈類似的計劃。此外,醫用大麻行業的公司還提供了未經FDA批准的CBD製劑,這些公司可能會試圖與我們未來的候選產品競爭。
我們的許多競爭對手在以下方面擁有顯著更多的財務和技術資源、經驗和專業知識:
| ● | 研究和開發; |
| ● | 臨牀前試驗; |
| ● | 設計和實施臨牀試驗; |
| ● | 監管程序和審批; |
| ● | 生產和製造;以及 |
| ● | 銷售和營銷認可產品。 |
我們行業的主要競爭因素 包括:
| ● | 組織技術的質量和廣度; |
| ● | 組織的管理和組織戰略的執行; |
| ● | 組織員工的技能和經驗及其招聘能力 並留住技術熟練和經驗豐富的員工; |
| ● | 一個組織的知識產權組合; |
| ● | 從目標識別和驗證到藥物發現的各種能力 發展到製造和銷售;以及 |
| ● | 有大量的資本資源來資助發現、開發和 商業化活動。 |
Additionally, competitors may also seek to market versions of our drug products via a section 505(b)(2) application, which is a type of NDA provided in section 505(b)(2) of the FDC Act. A Section 505(b)(2) NDA is in contrast to an NDA under section 505(b)(1) of the FDC Act, which is commonly known as a Full NDA because it requires the applicant to undertake all of the nonclinical and clinical investigations necessary for the approval of the application. In contrast, a Section 505(b)(2) NDA application is one for which one or more of the investigations relied on by the applicant for approval were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted. Section 505(b)(2) applications may be submitted for drug products that represent a modification, such as a new indication or new dosage form, of a previously approved drug. Section 505(b)(2) applications may rely on the FDA’s previous findings for the safety and effectiveness of the previously approved drug in addition to information obtained by the 505(b)(2) applicant to support the modification of the previously approved drug. Preparing Section 505(b)(2) applications may be less costly and less time-consuming than preparing a Full NDA based entirely on new data and information. Section 505(b)(2) applications are subject to the same patent certification procedures as an ANDA.
61
如果我們無法成功競爭 ,我們的商業機會將減少,我們的業務、運營結果和財務狀況可能會受到實質性的損害 。
我們未來的候選產品 ,如果獲得批准,可能無法達到預期的市場接受度,從而限制我們從新產品中賺取收入的能力 。
Even when product development is successful and regulatory approval has been obtained, our ability to generate sufficient revenue depends on the acceptance of our products by physicians and patients. We cannot assure you that any of our future product candidates will achieve the expected level of market acceptance and revenue if and when they obtain the requisite regulatory approvals. The market acceptance of any product depends on a number of factors, including the indication statement, warnings required by regulatory authorities in the product label and new competing products. Market acceptance can also be influenced by continued demonstration of efficacy and safety in commercial use, physicians’ willingness to prescribe the product, reimbursement from third-party payors such as government health care programs and private third-party payors, the price of the product, the nature of any post-approval risk management activities mandated by regulatory authorities, competition, and marketing and distribution support. Further, our U.S. distribution depends on the adequate performance of a reimbursement support hub and contracted specialty pharmacies in a closed-distribution network. An ineffective or inefficient U.S. distribution model at launch may lead to inability to fulfill demand, and consequently a loss of revenue. The success and acceptance of a product in one country may be negatively affected by its activities in another. If we fail to adapt our approach to clinical trials in the U.S. market to meet the needs of EMA, MHRA or other European regulatory authorities, or to generate the health economics and outcomes research data needed to support pricing and reimbursement negotiations or decisions in Europe, we may have difficulties obtaining marketing authorization for our products from EMA/European Commission or the MHRA and may have difficulties obtaining pricing and reimbursement approval for our products at a national level. Any factors preventing or limiting the market acceptance of our products could have a material adverse effect on our business, results of operations and financial condition.
我們在抗腫瘤壞死因子和纖維化項目中的所有專利 都是使用方法專利,這可能導致未經我們許可使用生物仿製藥。
我們最先進的藥物開發平臺的成功取決於我們使用方法專利的可執行性,因為目前市場上有許多生物相似的抗腫瘤壞死因子 藥物。如果我們無法獲得組合物物質專利並強制執行這些專利,我們從抗腫瘤壞死因子平臺產生收入的能力可能會受到極大限制,競爭對手可能會利用我們的研究將競爭對手的藥物推向市場 ,這將減少我們的市場份額。
我們的大多數許可協議向許可方和/或交易對手提供使用和/或使用此類許可知識產權的權利。
我們的大多數許可協議 向許可方和/或交易對手提供了使用和/或利用此類許可知識產權的權利,在某些情況下,還向他們提供了此類知識產權、專有技術和研究成果的所有權。因此,我們可能會與與我們有許可協議的各方 競爭,他們可能不會擁有將我們的候選產品貨幣化、銷售或分銷的獨家權利,並且 可能會受到使用和銷售區域的限制。以上任何一項或全部內容可能會對我們的運營和現金流結果產生重大不利影響,並最終影響我們證券的價值。
62
隨着更多患者數據的出現,我們臨牀試驗中的中期數據、TOPLINE數據和初步數據可能會發生變化,並受到審計和驗證程序的影響,這可能會導致最終數據發生重大變化。
From time to time, we may publicly disclose preliminary, interim or topline data from our preclinical studies and clinical trials, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change as patient enrollment and treatment continues and more patient data become available. For example, any positive results from our preclinical testing, Phase 1 and Phase 2 clinical trials of our product candidate for any product candidate may not necessarily be predictive of the results from planned or future clinical trials for such product candidates. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical trials after achieving positive results in pre-clinical and early clinical development, and we cannot be certain that we will not face similar setbacks. These setbacks have been caused by, among other things, pre-clinical findings while clinical trials were underway or safety or efficacy observations in clinical trials, including adverse events. Adverse differences between previous preliminary or interim data and future interim or final data could significantly harm our business prospects. We may also announce topline data following the completion of a preclinical study or clinical trial, which may be subject to change following a more comprehensive review of the data related to the particular study or trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the interim, topline or preliminary results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data has been received and fully evaluated. Preliminary, interim, or topline data also remains subject to audit and verification procedures that may result in the final data being materially different from the data we previously published. As a result, preliminary, interim, and topline data should be viewed with caution until the final data is available.
此外,其他人(包括 監管機構)可能不接受或同意我們的假設、估計、計算、結論或分析,或可能以不同方式解釋 或權衡數據的重要性,這可能影響特定計劃的價值、特定候選產品或產品的批准性或商業化 以及我們公司的總體情況。此外,我們選擇公開披露 有關特定研究或臨牀試驗的信息是基於通常廣泛的信息,您或其他人可能不同意 我們確定哪些是重要的或其他適當的信息包括在我們的披露中。此外,我們對臨牀 數據的解釋或我們基於臨牀前體外和體內模型的結論可能被證明是不準確的,因為臨牀前和臨牀數據 可能容易受到不同的解釋和分析,許多認為其候選產品在臨牀前研究和臨牀試驗中表現令人滿意的公司 ,但卻未能獲得FDA批准或歐洲批准的上市許可 佣金.
如果我們在未來的臨牀試驗中未能產生積極的結果,則此類候選產品的開發時間表、監管批准和商業化前景,以及相應的我們的業務和財務前景,將受到實質性的不利影響。
我們的營銷經驗有限 ,即使未來的候選產品獲得批准,我們也可能無法成功地將它們商業化。
我們創造收入的能力 最終將取決於我們銷售經批准的產品和獲得足夠的第三方報銷的能力。我們目前沒有 營銷和銷售我們產品的經驗。我們未來產品的商業成功取決於 我們無法控制的許多因素,包括醫生向患者開我們未來產品的處方意願、支付方支付我們未來產品的意願和能力 、實現的定價水平、患者對我們未來產品的反應以及我們 未來營銷合作伙伴創造銷售額的能力。我們無法保證我們能夠建立或維持成功商業化我們未來產品或未來FDA、 MHRA或其他監管機構批准的任何候選產品所需的人員、系統、 安排和能力。如果我們未能建立或維持成功的營銷、銷售和報銷能力 ,或未能與第三方達成成功的營銷安排,我們的產品收入可能會受到影響。
63
如果我們未來批准的任何產品的價格下降,或者如果政府和其他第三方付款人沒有提供保險和足夠的報銷 水平,我們的收入和盈利前景將受到影響。
開了 藥物治療其病症的患者通常依賴第三方支付方報銷與其處方藥相關的全部或部分費用 。國際市場上的報銷制度因國家和地區而異,而且報銷 審批通常必須在每個國家的基礎上進行。政府醫療保健計劃(如Medicare和Medicaid)的覆蓋範圍和充分報銷以及商業支付方對新產品的接受至關重要。覆蓋範圍的決定可能取決於 臨牀和經濟標準,當已經 或隨後可用更成熟或更低成本的治療替代品時,這些標準不利於新藥產品。即使我們獲得了未來候選產品的承保範圍,最終的報銷 支付率可能需要患者認為高得無法接受的共同支付。如果未提供保險 或報銷不足以支付患者費用的很大一部分,患者可能不會使用我們未來的候選產品。
此外, 我們未來候選產品在美國的市場將在很大程度上取決於第三方付款人的處方藥,或第三方付款人提供承保和報銷的藥物清單 。納入此類處方集的行業競爭 通常會導致製藥公司面臨降價壓力。此外,第三方支付方可能會拒絕在其處方集中納入特定品牌 藥物,或在有成本較低的仿製藥或其他替代藥時限制患者獲得品牌藥物 。
第三方支付者,無論是外國還是國內,政府還是商業,都在開發越來越複雜的控制醫療成本的方法。 當前的環境給公司帶來了壓力,要求他們將產品定價低於他們認為合適的價格。如果我們以低於優化價格銷售未來候選產品,我們的未來收入 和整體成功可能會受到負面影響。另外,在美國,在第三方支付者之間沒有統一的藥品保險和報銷政策。因此,我們未來候選產品的承保範圍和 報銷可能因付款人而異。因此,覆蓋範圍確定 過程通常是一個耗時且成本高昂的過程,這將要求我們分別向每個付款人提供科學和臨牀支持,以使用我們的 未來候選產品,而不能保證將獲得覆蓋範圍。如果我們無法獲得未來候選產品的覆蓋範圍 和足夠的支付水平,醫生可能會限制他們將 開處方或管理這些產品的數量或在什麼情況下,患者可能會拒絕購買這些產品。這可能會影響我們成功將 候選產品商業化的能力,從而對我們的盈利能力、運營結果、財務狀況和未來的成功產生不利影響。
此外,如果我們已選擇與第三方就候選產品開發和商業化進行合作,我們的合作伙伴可能會選擇降低我們產品的 價格,以增加獲得報銷批准的可能性。在許多國家/地區,只有在報銷獲得批准之前,產品才能 投入商業使用,而且在某些國家/地區,談判過程可能會超過12個月。此外,某些國家/地區的定價和報銷決策可能會受到其他國家/地區決策的影響,這可能會導致其他一些國家/地區強制降價和/或額外的報銷限制,這可能會對銷售和盈利產生不利影響。 如果國家/地區實施的價格不足以讓我們或我們的合作伙伴盈利,我們的合作伙伴可能會 拒絕在這些國家/地區推出產品或將產品從市場上召回,這將對銷售和盈利能力產生不利影響。
業務中斷 可能會延誤我們開發未來候選產品的過程,並可能擾亂我們的產品銷售。
我們未來的製造設施、庫存或實驗室設施因火災、盜竊或其他原因而丟失,可能會對我們滿足未來候選產品的需求或繼續產品開發活動和開展業務的能力產生不利影響。未能 向合作伙伴供應商業產品可能會導致不良後果,包括合作伙伴有權承擔產品供應責任 。即使我們獲得保險以賠償此類業務中斷,此類保險也可能不足以完全補償因庫存的任何重大財產或意外損失而對我們的業務造成的損害。
64
如果針對我們的產品責任訴訟成功,我們將承擔大量責任,並可能被要求限制我們未來候選產品的商業化 。
儘管我們從未收到過任何針對我們的產品責任索賠或訴訟,但我們面臨着與在人體臨牀試驗中測試我們未來的候選產品有關的潛在產品責任風險,而且我們未來將在我們營銷和分銷的司法管轄區面臨索賠風險。當我們開始在美國和其他地方進行商業營銷和分銷我們的產品時,我們可能會面臨更多人提出的索賠。 將來,個人可能會向我們提出責任索賠,聲稱我們未來的候選產品之一造成了傷害。雖然我們計劃採取我們認為適當的預防措施,但如果對我們提起任何產品責任訴訟,我們可能無法避免重大責任。在集體訴訟或個人訴訟中,因藥物具有意想不到的副作用而做出的大額判決。雖然我們計劃購買保險來承保產品責任訴訟,但 如果我們不能針對產品責任索賠成功地為公司辯護,或者如果此類保險覆蓋範圍不足,我們將承擔大量責任。無論是非曲直或最終結果如何,責任索賠都可能導致對我們產品的需求減少、聲譽損害、臨牀試驗參與者退出、訴訟成本、產品召回成本、金錢賠償、責任保險成本增加、收入損失和業務中斷。
我們的員工之前或將來可能從事不當行為或其他不當活動,包括違反監管 標準和法律要求。
我們面臨員工欺詐或其他不當行為的風險。員工 不當行為還可能涉及不當使用信息,包括在臨牀試驗過程中獲得的信息,或非法 挪用藥品,這可能會導致政府調查並嚴重損害我們的聲譽。儘管我們採納了《道德準則》,但員工的不當行為並不總是能夠被識別和阻止。我們為檢測和防止這些被禁止的活動而採取的預防措施可能無法有效控制未知或未管理的風險或損失,或保護我們免受政府調查或因不遵守此類法律或法規而引起的其他行動或訴訟。如果對我們的公司採取任何此類行動,而我們未能成功捍衞我們的公司或維護我們的權利,這些行動可能會對我們的業務產生重大影響,包括施加鉅額罰款或其他制裁。
我們遵守 美國《反海外腐敗法》和其他反腐敗法律,以及出口管制法、海關法、制裁法和其他 管理我們業務的法律。如果我們未能遵守這些法律,我們可能會受到民事或刑事處罰、其他補救措施和法律費用,這可能會對我們的業務、經營業績和財務狀況產生不利影響。
我們的運營受到 反腐敗法律的約束,包括《美國反海外腐敗法》(“《反海外腐敗法》“),以及適用於我們開展業務的國家/地區的其他反腐敗法律。《反海外腐敗法》和這些其他法律一般禁止我們的公司、我們的員工和中介機構 向政府官員或其他人行賄、受賄或向其他人支付其他被禁止的款項,以獲得或保留業務 或獲得一些其他業務優勢。我們和我們的商業合作伙伴在許多具有潛在違反《反海外腐敗法》風險的司法管轄區開展業務,我們參與與第三方的合作和關係,這些第三方的行為可能使我們 承擔《反海外腐敗法》或當地反腐敗法律規定的責任。此外,我們無法預測未來監管要求的性質、範圍或效果,我們的國際業務可能受到這些要求的約束,也無法預測現有法律可能被管理或解釋的方式。
我們還受制於管理我們國際業務的其他 法律和法規,包括由美國、加拿大、以色列、英國政府和歐盟當局管理的法規,包括適用的出口管制法規、對國家和人員的經濟制裁、海關要求和貨幣兑換法規,統稱為貿易管制法律。
但是,不能保證 我們將完全有效地確保我們遵守所有適用的反腐敗法律,包括《反海外腐敗法》或其他法律要求,包括貿易控制法。如果我們不遵守《反海外腐敗法》和其他反腐敗法律或貿易管制法律,我們可能會受到刑事和民事處罰、返還和其他制裁和補救措施,以及法律費用, 這可能會對我們的業務、財務狀況、運營結果和流動性產生不利影響。同樣,美國或其他當局對任何潛在違反《反海外腐敗法》、其他反腐敗法律的調查也可能對我們的聲譽、業務、財務狀況和運營結果產生不利影響。
65
安全漏洞、數據丟失和其他中斷可能會危及與我們的業務相關的敏感信息,阻止我們訪問關鍵的 信息或使我們承擔責任,這可能會對我們的業務和我們的聲譽造成不利影響。
In the ordinary course of business, we expect to collect and store sensitive data, including legally protected patient health information, credit card information, personally identifiable information about our employees, intellectual property, and proprietary business information. The secure processing, storage, maintenance and transmission of this critical information is vital to our operations and business strategy, and we devote significant resources to protecting such information. Although we take measures to protect sensitive information from unauthorized access or disclosure, our information technology and infrastructure may be vulnerable to attacks by hackers, or viruses, breaches or interruptions due to employee error, malfeasance or other disruptions, or lapses in compliance with privacy and security mandates. Any such virus, breach or interruption could compromise our networks and the information stored there could be accessed by unauthorized parties, publicly disclosed, lost or stolen. We have measures in place that are designed to prevent, and if necessary, to detect and respond to such security incidents and breaches of privacy and security mandates. However, in the future, any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, such as HIPAA and European Union General Data Protection Regulation (“GDPR”)、政府執法行動和監管處罰。未經授權的 訪問、丟失或傳播也可能破壞我們的運營,包括我們處理樣本、提供測試結果、共享 和監控安全數據、賬單支付者或患者、提供客户支持服務、開展研發活動、處理 和準備公司財務信息、管理我們業務的各種一般和行政方面的能力,並可能損害我們的聲譽, 任何可能對我們的業務、財務狀況和經營業績產生不利影響的情況。
GDPR適用於所有 歐盟成員國,包括對違反數據保護規則的行為處以鉅額罰款,並可能要求我們建立額外的機制, 確保遵守新的和不斷變化的數據保護規則。GDPR是一項複雜的法律,監管指南仍在不斷髮展, 包括如何在臨牀試驗或我們可能訪問個人數據的其他交易中應用GDPR。GDPR增加了我們的合規成本,並導致更大的法律風險。
我們的研發計劃和候選產品正在開發中。因此,我們無法預測是否或何時會成功開發我們的候選產品或將其商業化。
我們的臨牀階段候選產品以及我們的其他候選藥物在 商業化之前將需要大量的進一步投資和監管批准。我們的每個候選產品都需要符合FDA、MHRA或其他類似外國監管機構的標準和要求的臨牀試驗設計,為我們的臨牀試驗選擇合適的終點和患者,以及額外的臨牀開發,管理臨牀、臨牀前和製造活動,獲得監管批准, 獲得製造供應,建立商業組織,大量投資和重大營銷工作,然後我們才能從產品銷售中獲得任何收入。在獲得FDA、MHRA或其他類似外國監管機構的監管批准之前,我們不允許營銷或推廣我們的任何候選產品,而且我們可能永遠不會對我們的任何候選產品獲得此類監管批准 。到那時,如果我們的候選產品可能獲得必要的監管批准, 與我們候選產品相關的疾病治療的護理標準可能會發生變化,因此有必要修改我們的全面批准計劃,並且我們產品的商業接受可能會受到護理標準變化的限制。
臨牀前測試和早期臨牀試驗的成功 不能確保以後的臨牀試驗將會成功,臨牀試驗過程可能無法證明我們的產品 候選產品對於其建議的用途是安全有效的。任何此類失敗都可能導致我們放棄任何一個或多個候選產品的進一步開發,並可能延遲其他候選產品的開發。臨牀試驗後期階段的候選產品可能無法顯示出所需的安全性和有效性特徵,儘管已通過臨牀前研究和初步臨牀試驗取得進展。
66
我們之前沒有向FDA或MAA或CMA提交過任何候選產品的NDA,或向MHRA提交過類似的藥品批准文件, 並且我們不能確定我們的任何候選產品是否會獲得監管部門的批准。此外,我們的候選產品即使在臨牀試驗中取得成功,也可能得不到監管部門的批准。如果我們的候選產品得不到監管部門的批准 ,我們可能無法繼續運營。即使我們成功獲得監管部門批准銷售我們的一個或多個候選產品 ,我們的收入在一定程度上也取決於我們或我們的合作者和未來的合作者是否有能力獲得監管部門的批准,以便在必要時將配套診斷用於我們的產品候選產品,以及我們獲得監管部門批准並擁有商業權的地區的市場規模。如果我們瞄準的患者亞羣市場沒有我們估計的那麼大,如果獲得批准,我們可能不會從此類產品的銷售中獲得可觀的收入。
此外,即使我們開發的任何候選產品 將獲得上市批准或商業化,以便與其他現有療法結合使用,我們 仍將繼續承擔FDA、MHRA或其他類似的外國監管機構可能撤銷與我們的候選產品組合使用的療法 的批准,或者這些現有療法可能出現安全性、有效性、製造或供應問題的風險。
我們候選產品的成功商業化 如果獲得批准,將取決於是否獲得市場認可,而我們可能無法獲得足夠的認可 以產生可觀的收入。
即使我們的候選產品 被成功開發並獲得監管部門批准,它們也可能無法獲得醫生、患者、醫療保健 付款人(如私人保險公司或政府以及其他資助方和醫療界)的市場認可。市場對我們任何產品的接受程度將取決於許多因素,包括:
| ● | 證明我們產品的臨牀療效和安全性; |
| ● | 任何不良副作用的流行率和嚴重程度; |
| ● | 產品批准標籤中包含的限制或警告; |
| ● | 成本效益和可接受價格的可用性; |
| ● | 競爭產品與替代治療方法的對比以及優效性 替代治療或療法; |
| ● | 營銷和分銷方法的有效性以及對產品的支持; 和 |
| ● | 保險範圍的可用性以及第三方付款人向 我們的產品獲得監管部門的批准。 |
疾病指徵可以是疾病的較小子集,當定義不同的疾病子集時,可以將其解析為越來越小的指徵。這種對疾病越來越精細的描述可能會產生負面後果;包括創建一個批准的適應症,它太小了,以至於我們沒有一個可行的市場。如果未來的技術允許以不同於用於大型關鍵研究的特徵的方式來描述疾病,則可能會使這些研究無效或降低其有效性,並且可能需要重複 全部或部分研究。未來的技術可能會提供更好的預後能力,這可能會減少預計需要新療法的患者比例。即使在監管部門批准後,一種產品隨後可能會被證明不安全或不具有其聲稱的效果,從而阻止其廣泛使用或要求從市場上撤出。
我們可能無法成功 建立開發和商業化合作,這可能會對我們開發候選產品的能力產生不利影響,並可能阻礙我們 的能力。
Developing pharmaceutical products, conducting clinical trials, obtaining regulatory approval, establishing manufacturing capabilities and marketing approved products is expensive, and therefore we have, and may in the future, seek to enter into collaborations with companies that have more resources and experience in order to continue to develop and commercialize our product candidates. We also may be required due to financial or scientific constraints to enter into additional collaboration agreements to research and/or to develop and commercialize our product candidates. The establishment and realization of such collaborations may not be possible or may be problematic. There can be no assurance that we will be able to establish such additional collaborations on favorable terms, if at all, or that our current or future collaborative arrangements will be successful or maintained for any specific product candidate or indication. If we are unable to reach successful agreements with suitable collaboration partners for the ongoing development and commercialization of our product candidates, we may face increased costs, we may be forced to limit the scope and number of our product candidates we can commercially develop or the territories in which we commercialize such product candidates, and we may be unable to commercialize products or programs for which a suitable collaboration partner cannot be found. If we fail to achieve successful collaborations, our operating results and financial condition will be materially and adversely affected.
67
此外, 任何合作協議的條款可能會限制我們在其他產品方面的活動,包括限制我們與其他第三方或在不同的適應症、疾病或地理位置授予許可證或開發產品的能力,或者可能 在我們的候選產品的開發或商業化方面對我們施加額外的義務。如果我們未能遵守 或違反協作協議的任何條款,協作者可能有權全部或部分終止此類協議 或尋求損害賠償。
我們的協作和許可 協議非常複雜,並且涉及各方之間共享或分割某些數據、專有技術和知識產權的所有權。因此,我們的協作者可能會與我們或我們的 其他協作者不同地解釋某些條款,這可能會導致與協作者發生意外或無意的糾紛。此外,這些協議可能會使 其他協作、合作或合併和收購變得困難。
無法保證 被第三方收購的合作者不會試圖更改可能對 合作產生負面影響的某些合同條款。收購公司也可能不接受我們合同的條款或轉讓,並可能尋求終止協議。 我們的任何一個合作者都可能違反契約、限制和/或分許可協議條款,導致我們陷入爭議和 潛在違反我們與其他合作伙伴的協議。
如果不在內部或通過合作伙伴建立銷售和營銷能力,我們將無法 成功地將我們的候選產品商業化。
我們可能無法為所有候選產品找到 合適的銷售和營銷人員以及合作者。市場營銷和銷售能力的發展 將需要大量支出、管理資源和時間。建立此類銷售團隊的成本可能超過任何潛在的 產品收入,或者我們的營銷和銷售努力可能會失敗。如果我們無法及時或根本無法開發內部營銷和銷售能力,或者如果我們無法以可接受的條款與第三方達成營銷和銷售安排,我們可能無法有效地營銷和銷售經批准的產品(如果有的話),這將妨礙我們能夠 產生收入和實現盈利。此外,如果我們無法 成功開發候選產品並尋求監管部門批准,我們可能無法開發內部營銷和銷售能力。
我們將依賴第三方承包商和服務提供商來執行我們開發計劃的某些方面。如果這些合作者未能在可接受的時間範圍內提供質量合適的服務,可能會導致我們的開發計劃延遲或失敗。
我們將某些功能、 測試和服務外包給第三方、合作伙伴、醫療機構和合作夥伴,並計劃將生產外包給合作伙伴 和/或合同製造商,我們將依靠第三方提供質量保證、臨牀監測、臨牀數據管理 和監管專業知識。特別是,我們依賴我們的合作伙伴來運行我們的臨牀試驗。無法保證此類個人 或組織將能夠按照約定或可接受的質量標準提供功能、檢測、藥物供應或服務, 並且我們可能會在產品或工藝的開發過程中遭受重大延誤。特別是,某些第三方服務提供商 可能無法履行其對我們的合同義務,原因是缺乏合格員工和顧問、 運營減少或人員減少或其他原因造成的中斷,在某些情況下,如果不遵守是由於服務提供商無法控制的因素導致 ,我們的追索權可能有限。
68
我們的業務高度 依賴於我們候選產品的成功。如果我們無法成功完成臨牀開發,無法獲得監管部門對我們的一個或多個候選產品的批准或將其商業化,或者如果我們在這方面遇到延誤,我們的業務將受到嚴重的 損害。
我們未來的成功和從我們的候選產品中產生可觀收入的能力 我們預計幾年內都不會發生,這取決於我們 成功開發、獲得監管機構批准並將我們的一個或多個候選產品商業化的能力,包括我們的 Dupuytren的Constrature候選產品,該產品之前已在英國成功完成了2b期臨牀試驗,這種情況 會影響手掌纖維結締組織的發育。我們所有其他候選產品都處於較早的 開發階段,需要在製造、臨牀前測試、臨牀開發以及一個或多個司法管轄區的監管審查和批准方面進行大量額外投資。如果我們的任何候選產品遇到安全或功效問題、 開發延遲或監管問題或其他問題,我們的開發計劃和業務將受到實質性損害。
我們可能沒有財力 繼續開發我們的候選產品。即使完成臨牀試驗,我們也可能遇到其他問題, 可能會推遲或阻止監管部門批准我們的候選產品,或阻止我們將其商業化,包括:
| ● | 無法向FDA、MHRA或其他類似的外國監管機構證明我們的候選產品是安全有效的; |
| ● | 我們的財政和其他資源不足,無法完成必要的臨牀試驗和臨牀前研究; |
| ● | 我們的臨牀試驗、臨牀前研究或與我們相似的候選產品的其他人的臨牀試驗 的陰性或非決定性結果,導致決定或要求進行額外的 臨牀試驗或臨牀前研究或放棄計劃; |
| ● | 在我們的臨牀試驗中受試者經歷的與產品相關的不良事件,包括意外的毒性結果,或者使用與我們的候選產品類似的藥物或治療性生物製劑的個人; |
| ● | 延遲提交研究用新藥申請或IND或類似的外國申請,或延遲或未能從監管機構獲得開始臨牀試驗所需的批准,或一旦臨牀試驗開始即暫停、終止或擱置; |
| ● | FDA、EMA、MHRA或其他類似的外國監管機構對我們臨牀試驗的範圍或設計施加的條件; |
| ● | 我們的候選產品在臨牀試驗期間效果不佳; |
| ● | 對照組的表現好於預期,如安慰劑組,這可能導致我們的臨牀試驗結果為陰性或不確定; |
| ● | 延遲招募受試者參加臨牀試驗; |
| ● | 受試者從臨牀試驗中的脱落率較高; |
| ● | 臨牀試驗所需的候選產品或其他材料的供應或質量不足 ; |
| ● | 高於預期的臨牀試驗或製造成本; |
| ● | 不利的FDA、EMA、MHRA或其他類似的監管機構 檢查和審查臨牀試驗場地; |
69
| ● | 我們的第三方承包商或調查人員未能及時或完全遵守監管要求或臨牀試驗方案或以其他方式履行其合同義務; |
| ● | 不利的FDA、EMA、MHRA或其他類似監管機構 對製造設施的檢查和審查,或這些設施無法保持FDA、EMA或類似監管機構可接受的合規狀態; |
| ● | 監管要求、政策和指南的延遲和變更, 包括對一般臨牀測試或針對我們的治療施加額外的監管監督 |
| ● | FDA、EMA、MHRA和其他類似的外國監管機構對數據的不同解釋。 |
在商業銷售之前,我們的候選產品將需要 額外耗時的開發工作,包括臨牀前研究、臨牀試驗和FDA、EMA、MHRA和/或其他適用的外國監管機構的批准 。所有候選產品都容易出現藥品開發固有的失敗風險 ,包括此類候選產品可能不會被證明足夠安全有效,無法獲得監管機構的批准。此外,我們不能向股東保證,獲得批准的任何此類產品 將以經濟的方式生產或生產,成功商業化或被市場廣泛接受,或者 比其他商業替代產品更有效。
由於開發我們的產品線需要大量的資源,並且根據我們獲得資金的能力,我們必須將某些候選產品的開發優先於其他候選產品。此外,我們可能無法將有限的資源用於可能更有利可圖或更有可能成功的候選產品或指示。
We have three separate programs for producing anti-inflammatory agents: (1) investigating new clinical opportunities for anti-TNF, (2) identifying orally available, small molecules that are agonists of α7 nicotinic acetylcholine receptor, and (3) identifying patentable analogs of CBD that initially will be used as pain medications, that are at various stages of preclinical development. Due to the significant resources required for the development of our product candidates, we must focus on specific diseases and disease pathways and decide which product candidates to pursue and advance and the amount of resources to allocate to each. Our decisions concerning the allocation of research, development, collaboration, management and financial resources toward particular product candidates or therapeutic areas may not lead to the development of any viable commercial products and may divert resources away from better opportunities. If we make incorrect determinations regarding the viability or market potential of any of our product candidates or misinterpret trends in the pharmaceutical industry, our business, financial condition, and results of operations could be materially adversely affected. As a result, we may (i) fail to capitalize on viable commercial products or profitable market opportunities, (ii) be required to forego or delay pursuit of opportunities with other product candidates or other diseases and disease pathways that may later prove to have greater commercial potential than those we choose to pursue, or (iii) relinquish valuable rights to such product candidates through collaboration, licensing, or other royalty arrangements in cases in which it would have been advantageous for us to invest additional resources to retain sole development and commercialization rights.
我們 臨牀試驗的時間表可能受到多種因素的影響,任何延遲都可能對我們執行當前業務戰略的能力產生不利影響。
臨牀測試費用高昂, 難以設計和實施,可能需要多年時間才能完成,而且結果不確定。在候選產品的開發和測試的任何階段,我們都可能會遇到臨牀試驗的延誤。我們計劃的臨牀試驗可能無法按時開始、 設計有效、招募足夠數量的受試者,或者無法如期完成(如果有的話)。
可能導致臨牀試驗延遲或不成功完成的事件包括:
| ● | 無法籌集啟動或繼續試驗所需的資金; |
| ● | 拖延獲得監管部門的批准才能開始審判; |
70
| ● | 延遲與FDA、MHRA或其他外國監管機構達成協議 最終試驗設計; |
| ● | 在檢查我們的臨牀試驗後實施臨牀試驗暫停 FDA、MHRA或其他監管機構的試驗操作或試驗中心; |
| ● | 與潛在CRO就可接受條款達成協議的延遲 臨牀試驗中心; |
| ● | 在 獲得所需機構審查委員會批准方面出現延誤 每個站點; |
| ● | 受試者完成試驗或返回的延遲 治療後隨訪; |
| ● | 受試者因副作用退出試驗導致的延遲 或以其他方式; |
| ● | 臨牀站點退出試驗,不利於入選; |
| ● | 增設新診所所需的時間;以及 |
| ● | 我們的合同製造商延遲生產和交付足夠的 臨牀試驗材料的供應。 |
如果我們的候選產品臨牀試驗的啟動或完成因上述任何原因或其他原因而延遲,我們的開發 成本可能會增加,我們的批准過程可能會延遲,商業上市後和專利保護到期前的任何時間可能會縮短 ,我們的競爭對手可能會有更多的時間在我們上市之前將產品推向市場。任何這些事件都可能損害 我們候選產品的商業潛力,並可能對我們的業務產生重大不利影響。
我們候選產品的臨牀試驗可能無法發現患者可能經歷的所有可能不良反應。
Clinical trials are conducted in representative samples of the potential patient population, which may have significant variability. Clinical trials are by design based on a limited number of subjects and of limited duration for exposure to the product used to determine whether, on a potentially statistically significant basis, the planned safety and efficacy of any product candidate can be achieved. As with the results of any statistical sampling, we cannot be sure that all side effects of our product candidates may be uncovered, and it may be the case that only with a significantly larger number of patients exposed to the product candidate for a longer duration, that a more complete safety profile is identified. Further, even larger clinical trials may not identify rare serious adverse effects or the duration of such studies may not be sufficient to identify when those events may occur. Patients treated with our products, if approved, may experience adverse reactions and it is possible that the FDA, MHRA or other regulatory authorities may ask for additional safety data as a condition of, or in connection with, our efforts to obtain approval of our product candidates. If safety problems occur or are identified after our product candidates reach the market, we may, or regulatory authorities may require us to amend the labeling of our products, recall our products or even withdraw approval for our products.
失敗可能發生在我們藥物開發工作的任何階段。
我們可能會在測試期間或測試結果中遇到許多不可預見的事件,這些事件可能會延遲或阻止我們獲得監管機構對我們候選藥物的批准或將其商業化 ,包括但不限於:
| ● | 監管機構或機構審查委員會(IRBs)不得授權我們在預期的試驗地點開始臨牀試驗或進行臨牀試驗; |
| ● | FDA和/或MHRA可能會就我們臨牀試驗的範圍或設計向我們強加條件,或者由於監管環境的變化,我們可能需要將我們的臨牀試驗方案重新提交給IRBs進行審查; |
| ● | 我們的臨牀試驗所需的受試者數量可能更多, 患者登記可能需要更長時間,或者患者退出臨牀試驗的比率可能比我們預期的更高; |
71
| ● | 如果我們、監管機構或IRBs確定參與者面臨不合理的健康風險,我們可能不得不暫停或終止我們的一項或多項臨牀試驗 ; |
| ● | 我們的第三方承包商、臨牀研究人員或合同合作者 可能無法遵守法規要求或未能及時履行其對我們的合同義務; |
| ● | FDA可能不接受在醫療標準可能與美國不同的國家進行的臨牀試驗的臨牀數據。 |
| ● | 我們的檢測可能會產生陰性或不確定的結果,我們可能會決定 或監管機構可能要求我們進行額外的檢測;以及 |
| ● | 我們的臨牀前和/或臨牀試驗的成本可能比我們預期的要高。 |
我們依賴第三方 進行臨牀前研究和臨牀研究和試驗,如果他們不履行對我們的義務,我們可能無法 獲得額外適應症的批准。
我們目前沒有能力獨立進行臨牀前研究或臨牀研究和試驗,到目前為止,我們必須依賴第三方,如第三方合同研究和政府組織和醫療機構來為我們進行研究和試驗。我們對第三方開發活動的依賴減少了我們對這些活動的控制。這些第三方可能無法按計劃完成 活動,或未按照法規 要求或我們的研究設計進行我們的臨牀前研究、臨牀研究和試驗。如果這些第三方不能成功履行合同職責或在預期的最後期限前完成,我們可能會受到不利影響,我們獲得監管部門批准並將適應症商業化的努力可能會被推遲。
如果我們與 其他各方一起進行研究,我們可能無法控制與該試驗相關的所有決定。如果我們在研究設計、研究時間等問題上與對方存在分歧,這可能會對我們的藥物開發計劃產生不利影響。
雖然我們也依賴第三方來管理我們研究和試驗的數據,但我們有責任確認我們的每項研究和試驗都是根據其總體調查計劃和方案進行的。此外,FDA和外國監管機構將要求我們遵守適用的法規和標準,包括良好實驗室規範(GLP)和良好臨牀實踐(GCP), 進行、記錄和報告此類研究和試驗的結果,以確保數據和結果可信和 準確,並確保人體研究和試驗參與者得到充分保護。我們對第三方的依賴並不解除我們的這些義務和要求,如果不滿足這些要求,我們可能無法獲得監管部門對任何其他適應症的批准 。
我們需要繼續 開發和維護分銷和生產能力或關係,才能取得成功。
我們可能無法成功地 製造任何產品,無論是獨立生產還是與第三方製造商達成製造安排(如果有)。此外, 如果任何製造商停止與我們的業務往來,或遇到延誤、供應短缺或產能需求過大的情況,我們可能無法及時或根本無法獲得足夠數量的產品。製造商及其供應商在某些情況下必須遵守FDA執行的現行保密協議承諾和現行良好製造規範(CGMP)及要求,以及其他國家/地區的類似要求。製造商未能遵守這些要求可能會影響 其向我們提供產品的能力。雖然我們打算依賴第三方合同製造商,但我們最終有責任確保我們的產品按照cGMP生產。此外,如果在對我們的第三方製造商的一個或多個工廠進行審批前檢查或其他檢查時,FDA確定該工廠不符合cGMP, 我們將該工廠列為製造商的任何營銷申請可能不會獲得批准,或者可能會推遲批准,直到 工廠符合cGMP並完成FDA的成功重新檢查。
任何製造問題、影響製造設施的自然災害或流行病或合同製造商的損失都可能中斷我們的 運營並導致銷售損失。此外,我們將依賴第三方提供生產我們的產品所需的原材料。對供應商的任何依賴都可能涉及幾個風險,包括可能無法獲得關鍵材料,以及減少對生產成本、交付時間表、可靠性和質量的控制。供應商問題對未來合同製造造成的任何意外中斷 都可能延誤產品發貨,增加我們銷售商品的成本,並導致銷售損失。如果我們的供應商 無法向我們供應充足的藥物,可能會對我們成功將候選藥物商業化的能力產生重大不利影響 。
72
如果我們無法發現、 開發其他候選產品並將其商業化,我們可能無法發展我們的業務,我們實現戰略目標的能力也將受到影響。此外,我們還可能尋求將某些可能不是我們專有的治療方法商業化。
雖然目前候選產品的開發 和商業化是我們最初的重點,但作為長期增長戰略的一部分,我們計劃開發 其他候選產品。雖然我們認為我們的計劃產品可能有潛在的適用性,但我們尚未對這些其他用途進行 任何臨牀試驗,我們可能無法成功開發用於其他用途的候選產品。此外,我們 打算投入資金和資源進行基礎研究,以發現和確定其他候選產品。這些研究項目 需要技術、財政和人力資源,無論最終是否確定任何候選產品。我們的研究項目 最初可能在識別潛在候選產品方面表現出希望,但由於多種原因,無法產生用於臨牀開發的候選產品 ,其中包括以下原因:
| ● | 使用的研究方法可能無法成功識別潛在產品 候選人; |
| ● | 競爭對手可能會開發替代產品,使我們的候選產品過時; |
| ● | 我們開發的候選產品可能仍受第三方的 專利或其他專有權保護; |
| ● | 在進一步的研究中,候選產品可能被證明具有有害的副作用或其他特徵,表明其不太可能有效或不符合適用的監管標準; |
| ● | 候選產品可能無法以可接受的成本進行商業批量生產,或者根本無法生產;以及 |
| ● | 候選產品可能不會被患者、醫療界或第三方付款人接受為安全有效。 |
如果我們不能在預期的時間範圍內實現預期的開發和商業化目標,我們的候選產品的開發和商業化可能會被推遲,我們的業務和運營結果可能會受到損害。
出於規劃目的,我們 尋求估計各種科學、臨牀、法規和其他產品開發目標的完成時間。 這些里程碑可能包括我們對開始或完成科學研究和臨牀試驗、提交法規備案文件或商業化目標的預期。我們可能會不時地公開宣佈其中一些里程碑的預期時間,例如完成正在進行的臨牀試驗、啟動其他臨牀計劃、收到 市場批准或產品的商業發佈。其中許多里程碑的潛在實現可能不在我們的控制範圍之內。 這些里程碑中的每一個都基於各種假設,如果不能按預期實現,可能會導致各自里程碑的潛在實現時間與我們的估計大不相同,包括:
| ● | 我們可用的資本資源或我們遇到的資本約束; |
| ● | 我們的臨牀試驗和研究以及開發活動的進度、成本和結果,包括與參與的臨牀醫生和合作者之間的日程衝突程度; |
| ● | 我們能夠識別和招募符合臨牀試驗資格標準的患者 ; |
| ● | 我們收到FDA、MHRA和其他監管機構的批准及其時間; |
73
| ● | 臨牀結果; |
| ● | 監管機構發佈的其他行動、決定或規則; |
| ● | 我們有能力獲得足夠、可靠和負擔得起的材料供應,用於生產我們的候選產品; |
| ● | 我們的合作者在我們的候選產品商業化方面所做的努力 ;以及 |
| ● | 產品製造 以及銷售和營銷活動的安全、相關成本和相關時間問題。 |
如果我們未能在預期的時間範圍內實現任何已宣佈的里程碑,我們候選產品的開發和商業化可能會被推遲,我們的業務和運營結果可能會受到損害,並可能對我們的股價表現產生負面影響。有關更多信息,請參閲本報告中題為“項目1.業務”的第 節。
如果我們依賴唯一的供應來源來生產我們的產品,我們可能會受到供應商生存能力的影響。
我們打算嘗試從多個供應商採購我們的產品。我們還打算與我們產品的任何供應商簽訂合同,使他們在合同中承擔滿足我們要求的義務。然而,如果我們依賴一家供應商,而該供應商不能或不會滿足我們的要求 (無論出於何種原因),我們的業務可能會受到不利影響。
我們可能無法 足夠規模地生產我們的候選藥物。
我們可能無法以足夠的方式成功地 增加我們的候選藥物的製造能力,無論是與第三方製造商合作還是我們自己,以及時或具有成本效益的方式或根本不能。如果合同製造商對我們的候選藥物的製造過程進行了改進 ,我們可能不擁有或可能必須共享這些改進的知識產權。
大幅擴大生產規模 可能需要額外的驗證研究,這些研究成本高昂,FDA必須審查和批准。此外,由於候選藥物本身或候選藥物與生產和包裝過程中添加的其他組分的組合 的固有性質,或在成品或 活性藥物成分的運輸和儲存過程中, 在這些規模擴大活動中可能會出現質量問題。如果我們無法以足夠的質量和數量成功擴大我們的任何候選藥物的生產 ,則該候選藥物的開發和監管批准或任何由此產生的藥物產品的商業上市 可能會被推遲,或者可能出現供應短缺,這可能會嚴重損害我們的業務。
我們和我們合作伙伴的 運營受到與正在進行的和未來潛在的全球衝突相關的風險,特別包括我們的研究合作伙伴Yissum的運營 。
2022年2月,俄羅斯和烏克蘭之間的武裝衝突升級。俄羅斯入侵烏克蘭後,美國和其他國家宣佈了對俄羅斯和白俄羅斯的制裁,包括限制在受影響地區或從受影響地區銷售或進口商品、服務或技術,以及旅行禁令和資產凍結,影響到俄羅斯和白俄羅斯的相關個人和政治、軍事、商業和金融組織。如果衝突進一步升級,美國和其他國家可能會實施更廣泛的制裁並採取其他行動。另外,2023年10月,以色列和某些伊朗支持的巴勒斯坦部隊在以色列、加沙地帶和周邊地區開始了武裝衝突,這場衝突有可能蔓延到包括黎巴嫩和伊朗在內的其他中東國家。
這些戰爭正越來越多地影響經濟和全球金融市場,並加劇持續的經濟挑戰,包括通脹上升和全球供應鏈中斷等問題。雖然我們認為這些衝突目前不會對我們的財務會計和報告產生實質性影響,但我們未來受到影響的程度在很大程度上取決於不確定和不可預測事件的性質和持續時間 ,我們的業務可能會受到影響。此外,我們目前與耶路撒冷希伯來大學有限公司的Yissm Research開發公司簽訂了協議並建立了關係(“亞瑟姆”),位於以色列,Yissum 的業務可能受到以色列正在進行的戰爭的重大影響,這可能會延遲、阻止或實質性增加Yissum需要向公司提供的 持續服務的成本。此外,未來的全球衝突或戰爭可能造成進一步的經濟挑戰,包括但不限於通脹加劇和全球供應鏈進一步中斷。因此,持續的 俄羅斯/烏克蘭哈馬斯/以色列衝突和/或其他未來的全球衝突可能導致運營費用增加和/或 未來收入減少,並可能進一步對我們的運營業績和現金流產生重大不利影響。
74
我們可能會在未來進行戰略交易,這可能會導致我們的業務發生實質性變化和/或控制權發生變化。
2023年12月,我們聘請了AG.P./Alliance Global Partners擔任財務顧問,以探索和評估提升股東價值的戰略選擇。作為這一過程的一部分,公司可能探索或評估的潛在戰略選擇包括但不限於收購、合併、反向合併、其他業務合併、出售資產、許可或涉及公司的其他戰略交易。 公司不打算在此過程中討論或披露進一步的發展,除非董事會已批准具體行動或以其他方式確定進一步披露是適當的。不能保證戰略審查流程將批准或完成任何特定交易或結果。
董事會和管理團隊致力於以公司、股東和利益相關者的最佳利益行事。沒有為完成戰略備選方案審查流程設定最後期限或最終時間表,也不能保證此流程 將導致公司尋求交易或任何其他戰略結果。
由於上述原因, 將來,我們可能會與尋求合併和/或收購我們和/或我們的業務的各方進行交易。雖然迄今為止,我們尚未 與任何此類方達成任何協議或諒解,但如果我們在未來確實達成此類交易或 交易,則可能會發行新的普通股或優先股,導致我們當時 的現有股東大幅攤薄和/或控制權的變更。因此,我們的新大股東可能會改變我們 董事會的組成,並可能取代我們目前的管理層。任何未來交易也可能導致我們的業務重點發生變化。截至本報告日期,我們尚未 就涉及本公司的任何戰略性交易訂立任何協議,今後也不會 訂立此類協議。任何涉及公司或其運營的未來戰略交易都可能對 我們的運營、現金流、運營結果、前景、運營計劃、我們在納斯達克的普通股上市、我們的管理人員、 董事和大股東以及我們的證券價值產生重大影響。
與我們未來候選產品的開發和監管審批相關的風險
臨牀試驗昂貴、耗時、不確定,而且容易發生變化、延遲或終止。臨牀試驗的結果可以有不同的解讀。
我們有三個不同的項目來生產抗炎藥:(1)研究抗腫瘤壞死因子的新臨牀機會,(2)確定口服用藥, 作為α7煙鹼型乙酰膽鹼受體激動劑的小分子,以及(3)確定最初將被用作止痛藥的CBD的專利類似物。然而,這些計劃,包括相關的臨牀試驗,都是昂貴的、耗時的 ,並且難以設計和實施。
監管機構可能不 接受我們提交的臨牀試驗設計,並且可能對臨牀試驗結果的分析或解釋與我們不同。即使 我們的臨牀試驗結果良好,我們的許多未來候選產品的臨牀試驗預計將持續 數年,並且可能需要更長的時間才能完成。此外,FDA、MHRA或其他監管機構, 包括州、地方和外國機構,或IRB,對於我們機構的試驗,可以隨時暫停、延遲或終止 我們的臨牀試驗,要求我們進行額外的臨牀試驗,要求特定臨牀試驗持續 比原計劃更長的時間,需要更改我們的開發計劃,以便我們以不同的順序對候選產品進行臨牀試驗 ,例如, 或者,由於各種原因(包括以下原因),其中任何一種可能會對我們的業務、財務狀況 和經營結果產生重大不利影響,而不是對同一候選產品並行進行兩次試驗, 或者可以暫停或終止我們採購和處理受控物質所需的註冊和配額分配:
| ● | 臨牀試驗期間任何候選產品缺乏有效性; |
75
| ● | 發現試驗中出現的嚴重或非預期毒性或副作用 受試者或其他安全性問題,如藥物相互作用,包括導致 水平混雜變化的問題 其他伴隨用藥; |
| ● | 受試者招募率和臨牀入組率低於預期 審判; |
| ● | 難以保留已啟動臨牀試驗但可能退出的受試者 任何時候,由於治療的不良副作用、療效不足、臨牀試驗過程疲勞或任何 其他原因; |
| ● | 延遲或無法制造或獲得足夠數量的材料 由於監管和生產限制,用於臨牀試驗; |
| ● | 我們的生產工藝或產品配方的不足或變更; |
| ● | 延遲獲得開始試驗的監管授權,包括在試驗開始之前或之後,延遲獲得監管機構(如FDA)暫停或終止試驗的“臨牀 暫停”; |
| ● | 與DEA相關的記錄保存、報告臨牀站點的安全或其他違規行為,導致DEA或州當局暫停或吊銷該站點的受控物質許可證,並導致計劃或正在進行的試驗的延遲 或終止; |
| ● | 適用的監管政策和法規的變化,包括對研究範圍、性質或時間的要求的變化; |
| ● | 延遲或未能就臨牀試驗合同或與預期臨牀試驗地點的協議中可接受的條款達成協議。 |
| ● | 關於適當劑量的不確定性; |
| ● | 延遲或未能提供符合 法規規範的臨牀試驗用產品; |
| ● | 正在進行的臨牀前研究和臨牀試驗的不利結果; |
| ● | 我們的CRO或其他第三方承包商未能遵守所有合同要求或未能以及時或可接受的方式履行其服務; |
| ● | 我們的公司、我們的員工、我們的CRO或他們的員工未能遵守與進行臨牀試驗有關的所有適用的FDA或其他法規要求; |
| ● | 與參與的臨牀醫生和臨牀機構的日程安排發生衝突; |
| ● | 未能設計適當的臨牀試驗方案; |
| ● | 監管機構對CBD衍生產品的普遍擔憂以及濫用的可能性,儘管只適用於非植物性、非精神活性產品; |
| ● | 數據不足,無法支持監管部門的批准; |
| ● | 醫療研究人員不能或不願意遵循我們的臨牀方案; 或 |
| ● | 難以在治療期間或治療後與患者保持聯繫,這可能會導致數據不完整。 |
76
我們可能會發現很難讓患者參加我們的臨牀試驗,這可能會推遲或阻止我們候選產品的臨牀試驗。
確定患者並使其有資格參與我們候選產品的臨牀試驗對我們的成功至關重要。我們臨牀試驗的時間取決於我們招募符合條件的患者參與測試我們候選產品的速度。我們在一些臨牀試驗中遇到了延遲,未來可能還會遇到類似的延遲。這些延遲可能會導致成本增加、產品開發延遲或臨牀試驗完全終止。
我們可能無法識別、招募和招募足夠數量的患者,或者那些具有在研究中實現多樣性所需或所需特徵的患者, 無法在預期時間內完成我們的臨牀試驗。患者登記可能受到多種因素的影響,包括但不限於:
| ● | 研究方案中要求的參與的設計和複雜性和/或承諾; |
| ● | 患者羣體的規模; |
| ● | 有關研究的資格標準; |
| ● | 臨牀用品供應情況; |
| ● | 參與研究中心識別、鑑定和後續激活的延遲 註冊; |
| ● | 所研究候選產品的感知風險和益處,包括 在類似或競爭療法中觀察到的不良反應的結果; |
| ● | 為潛在患者提供臨牀試驗地點的近似性和可用性; |
| ● | 競爭療法和臨牀試驗的可用性; |
| ● | 競爭 研究中心努力促進臨牀試驗的及時入組; |
| ● | 參與站點動機; |
| ● | 醫生的病人轉介做法; |
| ● | 患者權益倡導團體的活動; |
| ● | 在治療期間和治療後充分監測患者的能力;以及 |
| ● | 正在調查的疾病的嚴重程度。 |
特別是,我們計劃評估候選產品的每種 條件都是用於臨牀試驗的患者池有限的疾病。我們臨牀試驗的資格標準將進一步限制可用的研究參與者池。此外, 尋找和診斷患者的過程可能會證明成本高昂。我們臨牀試驗中的治療醫生也可以使用他們的醫療判斷力 建議我們臨牀試驗中的患者退出我們的研究,嘗試替代療法。此外, 大流行病或流行病可能會影響患者前往臨牀試驗中心的能力和意願,這可能會對我們臨牀試驗的入組產生負面影響,例如,由於COVID—19大流行,我們的試驗過去難以招募足夠數量的患者。
77
如果我們無法按照方案招募所需的合格患者參加FDA或EMA、MHRA或其他監管機構要求的臨牀試驗,我們可能無法啟動 或繼續臨牀試驗。我們在任何外國成功啟動、入組和完成臨牀試驗的能力 ,會面臨在外國開展業務所特有的許多風險,包括:
| ● | 難以建立或管理與CRO和醫生的關係; |
| ● | 進行臨牀試驗的不同標準; |
| ● | 我們無法找到合格的當地顧問、醫生和合作夥伴; |
| ● | 遵守各種外國法律、醫療標準的潛在負擔 和監管要求,包括對製藥和生物技術產品和治療的監管; |
| ● | 採購和交付執行所需的必要臨牀試驗材料的能力 研究;及 |
| ● | 在 時,無法遠程在參與研究中心實施充分的培訓 人的培訓無法完成。 |
如果我們難以招募足夠數量的患者來按計劃進行臨牀試驗,我們可能需要推遲、限制或終止正在進行或計劃中的臨牀試驗 ,其中任何一項都會對我們完成臨牀試驗的能力以及最終的手術結果產生不利影響。
如果我們 公司不遵守現有法規,可能會損害我們的聲譽和經營成果。
我們受到美國聯邦、州和外國政府的廣泛 監管,歐洲和加拿大市場,我們計劃 銷售我們的產品,或在我們的候選產品正在通過審批流程的市場。
We must adhere to all regulatory requirements including FDA’s GLP, GCP and GMP requirements, pharmacovigilance requirements, advertising and promotion restrictions, reporting and recordkeeping requirements, and their European equivalents. If we or our suppliers fail to comply with applicable regulations, including FDA pre-or post-approval requirements, then the FDA or other foreign regulatory authorities could sanction our company. Even if a drug is approved by the FDA or other competent authorities, regulatory authorities may impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-marketing trials. Any of our product candidates which may be approved in the U.S. will be subject to ongoing regulatory requirements for manufacturing, labeling, packaging, storage, distribution, import, export, advertising, promotion, sampling, recordkeeping and submission of safety and other post-market information, including both federal and state requirements. In addition, manufacturers and manufacturers’ facilities are required to comply with extensive FDA requirements, including ensuring that quality control and manufacturing procedures conform to GMP. As such, we and our contract manufacturers (in the event contract manufacturers are appointed in the future) are subject to continual review and periodic inspections to assess compliance with GMP. Accordingly, we and others with whom we work will have to spend time, money and effort in all areas of regulatory compliance, including manufacturing, production, quality control and quality assurance. We will also be required to report certain adverse reactions and production problems, if any, to the FDA, and to comply with requirements concerning advertising and promotion for our products. Promotional communications with respect to prescription drugs are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s approved label. Similar restrictions and requirements exist in the EU and other markets where we operate.
如果監管機構發現產品出現 以前未知的問題,例如嚴重度或頻率超出預期的不良事件,或產品生產所在地的問題 ,或不同意產品的推廣、營銷或標籤,則監管機構可能會對該產品或我們公司施加限制 ,包括要求產品退出市場。如果我們未能遵守適用的 監管要求,監管機構或執法機構可能會發出警告信、施加民事或刑事處罰、 暫停監管批准、暫停我們正在進行的任何臨牀試驗、拒絕批准我們提交的待審申請或已批准 申請的補充申請、限制我們的運營、扣押或扣留產品或要求產品召回。
78
此外,我們未來的產品可能會受到DEA的監管,根據《受控物質法》或其他地方的類似法律。DEA計劃 是一個單獨的過程,當藥物可能在NDA批准日期之後可供患者使用時,該過程可能會延遲,並且此類DEA過程的時間和結果 不確定。另請參閲“與受管制物質有關的風險“,見下文。
此外,任何政府 對涉嫌違法行為的調查都可能要求我們花費大量時間和資源進行迴應,並可能產生負面 的宣傳。任何不遵守現行法規要求的行為都可能嚴重影響我們商業化 的能力以及從未來候選產品中產生收入的能力。如果實施監管制裁或撤銷監管批准, 我們的業務價值和經營業績可能會受到不利影響。
任何針對我們違反這些法律的行為,即使我們成功地進行了辯護,也可能導致我們產生鉅額法律費用,轉移我們管理層對業務運營的 注意力,並損害我們的聲譽。我們預計將在合規工作上投入大量資源 ,而此類支出是不可預測的。不斷變化的法律、法規和標準也可能造成不確定性、更高的費用和增加保險成本 。因此,我們打算投入所有合理必要的資源來遵守不斷髮展的標準,這項投資 可能會導致管理和行政費用的增加,並將管理時間和注意力從創收活動轉移到合規活動上。
我們受聯邦、州和外國醫療法律法規的約束,此類醫療法律法規的實施或更改可能會對我們的業務和運營結果產生不利影響。
在美國和某些 外國司法管轄區,已經有許多立法和監管提案,以可能影響我們銷售未來候選產品的能力的方式改變醫療保健系統。如果我們被發現違反任何這些法律或任何其他 聯邦、州或外國法規,我們可能會受到行政、民事和/或刑事處罰、損害賠償、罰款、個人 監禁,我們從聯邦醫療保健計劃和我們的業務重組。其中任何一項都可能對我們的業務和財務業績產生重大不利影響 。由於這些法律中的許多尚未得到法院的充分解釋, 我們可能被發現違反其中一項或多項條款的風險增加。任何因違反這些法律而對我們提起的訴訟,即使 我們最終成功地進行了辯護,也將導致我們產生鉅額法律費用,並將我們管理層的注意力從我們的業務運營上轉移 。此外,在許多國外國家,特別是歐盟國家, 處方藥的定價受政府控制。
在一些外國國家,藥品的建議定價必須獲得批准,然後才能合法上市。各國對藥品定價的要求千差萬別。
例如,一些歐盟司法管轄區 實行正面清單和負面清單制度,只有在商定了報銷價格後,產品才能在市場上銷售。要獲得 報銷或定價審批,其中一些國家可能需要完成臨牀試驗,將特定候選產品的成本效益與當前可用的療法進行比較。其他成員國允許公司制定自己的藥品價格,但監測和控制公司利潤。各國定價機制的這種差異可能會造成歐盟成員國之間的價格差異。不能保證任何對藥品實行價格控制或報銷限制的國家/地區會 允許對我們的任何產品進行優惠的報銷和定價安排。從歷史上看,在英國和歐盟推出的產品並不遵循美國的價格結構。在英國和歐盟,醫療成本總體上的下行壓力變得很大,尤其是處方藥。因此,進入新產品的門檻越來越高,患者不太可能使用未得到政府報銷的藥物產品。我們可能會面臨來自國外低價產品的競爭,這些產品對藥品實施了價格管制。此外,進口外國產品可能會與我們未來可能銷售的任何產品競爭,這可能會對我們的盈利能力產生負面影響。
具體來説,在美國, 我們預計ACA以及未來可能採取的其他醫療改革措施可能會導致更嚴格的覆蓋範圍 標準,並對我們可能收到的任何批准的產品的價格造成額外的下行壓力。對ACA的某些方面提出了司法挑戰 ,並多次立法試圖全部或部分廢除和/或取代ACA,我們預計未來將對ACA提出更多挑戰和修訂。目前,ACA對我們未來業務的全面影響尚不清楚 。政府在美國醫療保健行業中作用的擴大可能會對處方藥產品的價格造成普遍的下行壓力,降低報銷或我們獲得監管批准的任何其他產品的價格, 降低產品利用率,並對我們的業務和運營結果產生不利影響。聯邦醫療保險或其他政府計劃的任何報銷減少都可能導致私人支付者支付的類似減少。實施成本控制措施或其他醫療改革可能會阻止我們創造收入、實現盈利或將我們可能獲得監管部門批准的任何未來候選產品商業化。
79
從擴大准入研究中獲得的信息可能無法可靠地預測我們未來的候選產品在公司贊助的臨牀試驗中的療效 ,並可能導致可能限制批准的不良事件。
我們目前支持的擴大准入研究 是不受控制的,由個別研究人員進行,通常不嚴格遵守GCP ,所有這些都可能導致與安慰劑對照試驗不同的治療效果。這些研究僅提供了監管審查有效性的軼事證據。這些研究不包含可供參考的對照或比較組,此 患者數據不旨在作為研究結果進行彙總或報告。此外,來自如此少數患者的數據可能具有很大的變數。從這些研究獲得的信息,包括我們和獨立調查人員選擇應用於數據的統計原理,可能無法可靠地預測通過系統評估公司贊助的臨牀試驗的療效收集的數據,或通過可能應用於這些試驗的其他統計原理進行評估的數據。依賴這些信息來設計我們的臨牀試驗可能會導致試驗不能充分設計以證明療效,並可能延遲或阻礙我們尋求批准我們未來的候選產品的能力 。
擴展訪問計劃 為監管審查提供支持性安全信息。進行這些研究的醫生可能會以與協議不一致的方式使用我們未來的候選產品 ,包括條件超出我們贊助的試驗所研究條件的兒童。 擴大准入計劃中的受試者遇到的任何不良事件或反應都可能歸因於我們未來的候選產品 ,並可能限制我們通過我們認為理想的標籤獲得監管批准的能力,或者根本不能。
候選藥物通過臨牀試驗的失敗率很高。
一般來説,通過臨牀試驗的候選藥物有很高的失敗率。我們的臨牀試驗可能會遭遇重大挫折,類似於製藥和生物技術行業的其他一些公司的經歷,即使在早期的試驗中獲得了令人振奮的結果 。此外,即使我們認為臨牀試驗的結果是積極的,FDA、MHRA或其他監管機構也可能不同意我們對數據的解釋。如果我們從候選產品的臨牀試驗中獲得否定結果,或者發生與潛在的化學、製造和控制問題或其他障礙相關的其他問題,而我們未來的候選產品 未獲批准,我們可能無法產生足夠的收入或獲得資金來繼續我們的運營,我們執行當前業務計劃的能力可能會受到實質性損害,我們在行業和投資界的聲譽可能會受到嚴重損害。此外,我們無法正確設計、啟動和完成臨牀試驗,可能會對我們臨牀試驗的時間和結果以及為我們的候選藥物尋求批准的能力產生負面影響。
如果我們被發現違反了聯邦或州“欺詐和濫用”法律或其他司法管轄區的類似法律,我們可能會被要求支付 罰款和/或被暫停參加聯邦或州醫療保健計劃,這可能會對我們的業務、財務狀況和運營結果產生不利影響。
在美國,我們 受到各種聯邦和州醫療保健“欺詐和濫用”法律的約束,包括反回扣法、虛假申報法和其他法律,旨在減少聯邦和州醫療保健計劃中的欺詐和濫用,這可能會影響我們的公司,尤其是當我們的產品在美國成功商業化 。1987年《聯邦醫療保險和醫療補助患者保護法》或聯邦反回扣法令 規定,任何人,包括處方藥製造商(或代表其行事的一方),在知情和故意的情況下索要、接收、提供或支付旨在引薦業務的任何報酬,包括購買、訂購或 根據聯邦醫療保健計劃(如Medicare或Medicaid)支付的特定藥物的處方。根據聯邦法律,一些被稱為安全港的安排被認為不違反聯邦反回扣法規。儘管我們尋求根據所有適用要求來構建我們的業務安排,但通常很難準確確定 法律在特定情況下將如何適用。因此,我們的做法可能會受到聯邦《反回扣法規》和《聯邦虛假申報法》的挑戰。違反欺詐和濫用法律的行為可能會受到刑事和/或民事制裁,包括罰款和/或聯邦和州醫療保健計劃(如Medicare和Medicaid)的排除或暫停,以及禁止 與美國政府簽訂合同。此外,個人有權根據聯邦《虛假申報法》以及幾個州的虛假申報法代表政府提起訴訟。
80
雖然我們相信我們 已組織我們的業務安排以遵守這些法律,但政府可能會指控我們違反或判定我們違反了這些法律。 如果我們被發現違反了這些法律中的一項,我們可能會被要求支付罰款,並可能被暫停或排除 參加聯邦或州醫療保健計劃,我們的業務、運營結果和財務狀況可能會受到不利的 影響。
歐盟成員國和其他國家也有反回扣法律,可以在侵權情況下施加懲罰,在一些司法管轄區,競爭對手也可以執行這一懲罰。
嚴重的不良事件 或其他安全風險可能要求我們放棄開發並阻止、推遲或限制對我們未來候選產品的批准, 限制任何已批准的標籤或市場接受度的範圍,或導致已經上市的產品召回或失去上市批准。
如果我們未來的任何產品 任何候選產品在任何商業銷售批准之前或之後,造成嚴重或意想不到的副作用,或與其他 安全風險相關,如誤用、濫用或轉移,可能會導致許多潛在的重大負面後果,包括:
| ● | 監管部門可以中斷、推遲或停止臨牀試驗; |
| ● | 監管機構可能會拒絕監管部門對我們未來的候選產品的批准; |
| ● | 監管機構可要求某些標籤説明,如警告或使用適應症的禁忌症或限制,和/或在批准或批准後以REMS的形式對分發施加限制。 |
| ● | 監管機構可能撤回他們的 批准,要求更繁瑣的標籤聲明,實施更嚴格的REMS,或要求我們召回任何已批准的產品; |
| ● | 我們可能被要求改變產品的給藥方式或進行額外的臨牀試驗; |
| ● | 我們與合作伙伴的關係可能會受到影響; |
| ● | 我們可能會被起訴,並對給患者造成的傷害承擔責任;或 |
| ● | 我們的聲譽可能會受損。對於我們正在為兒科適應症開發的未來候選產品 ,聲譽風險會增加。 |
如果我們在任何時候認為產品對參與者構成不可接受的風險,或者如果初步數據顯示我們未來的候選產品不太可能獲得監管部門的批准或不太可能成功商業化,我們可以自願暫停 或終止我們的臨牀試驗。 在收到產品商業銷售的批准後,如果 我們認為該產品的使用或接觸可能會導致不良健康後果或死亡,我們可能會自願從市場上撤回或召回該產品。到目前為止,我們 尚未撤回、召回或採取任何其他行動(自願或強制)將批准的產品從市場上移除, 尚未獲得任何產品的批准,也未銷售任何批准的產品。此外,如果監管機構、IRBs或數據安全監督委員會認為臨牀試驗未按照適用的法規要求進行,或對參與者構成不可接受的安全風險,則監管機構、IRBs或數據安全監督委員會可隨時建議暫時或永久停止我們的臨牀試驗,或要求我們停止在臨牀試驗中使用調查人員。儘管監管機構、IRB或數據安全監控委員會從未要求我們暫時或永久停止臨牀試驗,但如果我們選擇或被迫暫停或終止我們未來任何候選產品的臨牀試驗,該產品的商業前景將受到損害 ,我們從該產品獲得產品收入的能力可能會被推遲或取消。此外,這些事件中的任何一種都可能導致標記警告或禁忌症等聲明。此外,此類事件或標籤可能會阻止我們或我們的合作伙伴 獲得或保持對受影響產品的市場接受度,並可能大幅增加我們未來候選產品的商業化成本,並削弱我們通過我們公司或我們的協作合作伙伴將這些產品商業化而獲得收入的能力。
81
為我們未來候選產品開發 REMS可能會導致批准過程的延遲,並會增加額外的監管要求 ,這可能會影響我們在美國將未來候選產品商業化的能力,並降低其市場潛力。
即使FDA批准了 我們的未來候選產品的NDA,而沒有要求REMS作為NDA批准的條件,但FDA在批准後,如果發現新的安全性信息使得REMS需要 確保藥物的獲益大於潛在風險,則 可能要求我們的未來候選產品執行REMS。REMS元素可以包括用藥指南、 醫療保健專業人員的溝通計劃,以及確保安全使用的元素("ETASU”). ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring and the use of patient registries. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy. We may be required to adopt a REMS for our future product candidates to ensure that the benefits outweigh the risks of abuse, misuse, diversion and other potential safety concerns. There can be no assurance that the FDA will approve a manageable REMS for our future product candidates, which could create material and significant limits on our ability to successfully commercialize our future product candidates in the U.S. Delays in the REMS approval process could result in delays in the NDA approval process. In addition, as part of the REMS, the FDA could require significant restrictions, such as restrictions on the prescription, distribution and patient use of the product, which could significantly impact our ability to effectively commercialize our future product candidates, and dramatically reduce their market potential, thereby adversely impacting our business, financial condition and results of operations. Even if initial REMS are not highly restrictive, if, after launch, our future product candidates were to be subject to significant abuse/non-medical use or diversion from illicit channels, this could lead to negative regulatory consequences, including a more restrictive REMS.
FDA和類似外國機構的監管審批流程宂長、耗時且本質上不可預測,如果我們最終 無法為我們的候選產品獲得監管批准,我們的業務將受到嚴重損害。
未經FDA的監管批准,我們不得在美國商業化、營銷、推廣或銷售任何候選產品。外國監管機構,如EMA和MHRA,也實施了類似的要求。獲得FDA和類似外國當局的批准所需的時間本來是不可預測的 ,但通常需要在臨牀試驗開始後多年,並取決於許多因素,包括監管機構的相當大的自由裁量權。此外,在候選產品的臨牀開發過程中,審批政策、法規或獲得審批所需的臨牀數據的類型和數量可能會發生變化,並且可能因司法管轄區而異。到目前為止,我們還沒有向FDA提交保密協議或向可比的外國監管機構提交類似的藥品批准申請,以獲得我們用於Dupuytren‘s Constraint早期治療的最先進候選產品,或任何其他候選產品 。我們必須完成更多的臨牀前研究和臨牀試驗,以證明我們的產品在人體上的安全性和有效性 我們才能獲得這些批准。
臨牀測試費用昂貴,難以設計和實施,可能需要數年時間才能完成,而且結果本身也不確定。我們不能保證 我們提交的任何臨牀試驗設計將被FDA、MHRA或其他類似的外國監管機構接受,或者 臨牀試驗將按計劃進行或按計劃完成(如果有的話)。我們最初的和潛在的其他候選產品的臨牀開發容易受到任何開發階段固有的失敗風險的影響,包括未能在臨牀試驗中或在廣泛的患者羣體中證明 療效,發生嚴重的或醫學上 或商業上不可接受的不良事件,未能遵守協議或適用的監管要求,以及FDA、MHRA 或任何其他類似的外國監管機構確定候選產品可能無法繼續開發或不可批准。 即使我們的任何候選產品具有有益的效果,也可能由於一個或多個各種因素(包括我們臨牀試驗的規模、持續時間、設計、測量、進行或分析)而在臨牀評估過程中檢測不到該效果。相反,由於同樣的因素,我們的臨牀試驗可能表明該候選產品 的明顯積極效果大於實際積極效果(如果有的話)。同樣,在我們的臨牀試驗中,我們可能無法檢測到候選產品的毒性或耐受性 ,或者錯誤地認為我們的候選產品有毒或耐受性不佳,而事實並非如此。嚴重的不良事件或SAE或其他不良影響以及耐受性問題可能會阻礙或阻止 市場接受有爭議的候選產品。
82
我們當前和未來的候選產品 可能因多種原因而無法獲得監管批准,其中包括:
| ● | FDA、MHRA或其他類似的外國監管機構可能不同意我們臨牀試驗的設計或實施; |
| ● | 我們可能無法向FDA、MHRA或其他類似的外國監管機構證明候選產品對於我們建議的適應症是安全有效的; |
| ● | 臨牀試驗結果可能不符合FDA、MHRA或其他類似外國監管機構批准的統計意義水平 ; |
| ● | 我們可能無法證明候選產品的臨牀和其他 益處大於其安全風險; |
| ● | FDA、MHRA或其他類似的外國監管機構可能不同意我們對臨牀試驗或臨牀前研究數據的解釋; |
| ● | 從我們候選產品的臨牀試驗中收集的數據可能不足以 支持向FDA提交保密協議或其他提交或獲得美國、歐盟或其他地方的監管批准; |
| ● | FDA、MHRA或其他類似的外國監管機構可能會發現與我們簽訂臨牀和商業用品合同的第三方製造商的製造工藝存在缺陷。 |
| ● | FDA、MHRA或其他類似的外國監管機構的批准政策或法規可能會發生重大變化,導致我們的臨牀數據不足以獲得批准。 |
這一漫長的審批過程 以及臨牀試驗結果的不可預測性可能導致我們無法獲得監管部門的批准來銷售我們開發的任何候選產品 ,這將嚴重損害我們的業務、經營業績和前景。FDA、MHRA或其他類似的 外國監管機構在審批過程中有很大的自由裁量權,可以決定我們開發的任何候選產品何時或是否獲得監管批准 。即使我們認為從我們的候選產品的未來臨牀試驗中收集的數據 是有希望的,這些數據可能不足以支持FDA或任何其他監管機構的批准。
此外,即使我們 獲得批准,監管機構可能會批准我們的任何候選產品,適應症比我們要求的更少或更有限, 可能不會批准我們打算對產品收取的價格,可能會根據昂貴的上市後臨牀試驗的表現授予批准 ,或者可以批准帶有標籤的候選產品,該標籤不包括該候選產品的成功商業化所必需或期望的聲明。上述任何情況都可能嚴重損害我們候選產品的商業前景。
與我們對第三方的依賴相關的風險
我們與某些大學的現有合作 安排以及我們未來可能與其他合作伙伴達成的任何安排可能不會成功,這 可能會對我們開發未來候選產品並將其商業化的能力產生不利影響。
我們正在尋求與製藥或生物技術公司合作,以開發或商業化我們未來的候選產品。 對於我們未來的候選產品,我們可能會根據為自己保留 商業化權利的好處而選擇性地達成新的安排,而不是與美國和國際上領先的製藥 或生物技術公司就每個候選產品達成選擇性合作安排。如果我們決定簽訂 合作協議,我們在尋找合適的合作伙伴方面將面臨激烈的競爭,我們可能建立的任何合作條款或其他安排可能對我們不利。
83
我們進行的任何現有或將來的合作 可能不會成功。我們合作安排的成功將在很大程度上取決於我們合作者的努力和活動 。協作者通常在決定將 應用於這些協作的工作和資源方面有很大的酌處權。合作安排各方在開發、知識產權、監管 或商業化事宜上的分歧可能導致適用候選產品的開發過程或商業化 的延遲,在某些情況下,可能導致合作安排的終止。如果雙方 都沒有最終決策權,這些分歧就很難解決。任何此類終止或到期都可能損害我們的商業聲譽,並可能對其財務造成不利影響 。
我們希望 依靠有限數量的供應商提供材料和組件,以生產我們未來的候選產品。失去這些 供應商,或他們不能及時向我們供貨,可能會導致我們當前和未來的產能延遲,並對我們的業務產生不利影響。
We expect to depend on a limited number of suppliers for the materials and components required to manufacture our future product candidates. As a result, we may not be able to obtain sufficient quantities of critical materials and components in the future. A delay or interruption by our suppliers may also harm our business, results of operations and financial condition. In addition, the lead time needed to establish a relationship with a new supplier can be lengthy, and we may experience delays in meeting demand in the event we must switch to a new supplier. The time and effort to qualify for and, in some cases, obtain regulatory approval for a new supplier could result in additional costs, diversion of resources or reduced manufacturing yields, any of which would negatively impact our operating results. Our dependence on single-source suppliers exposes us to numerous risks, including the following: our suppliers may cease or reduce production or deliveries, they may be subject to government investigations and regulatory actions that limit or prevent production capabilities for an extended period of time, raise prices or renegotiate terms; our suppliers may become insolvent; we may be unable to locate a suitable replacement supplier on acceptable terms or on a timely basis, or at all; and delays caused by supply issues may harm our reputation, frustrate our customers and cause them to turn to our competitors for future needs.
與我們的知識產權有關的風險
我們可能無法 充分保護我們未來的候選產品或我們在市場上的專有技術。
我們的成功將部分取決於我們獲得專利、保護我們的商業祕密以及在不侵犯他人所有權的情況下運營的能力。 我們依靠專利、商業祕密保護(即,專有技術)和保密協議,以保護我們未來候選產品的知識產權。專利在製藥領域的優勢涉及複雜的法律和科學問題 ,並且可能是不確定的。在適當的情況下,我們為我們的產品和技術的某些方面尋求專利保護。在全球範圍內申請、起訴和捍衞專利的費用可能高得令人望而卻步。
我們的政策是在具有重大商業機會的司法管轄區尋找具有商業潛力的專利技術。但是,我們正在開發的某些產品或技術可能無法獲得專利保護 。如果我們必須花費大量的時間和金錢來保護、 捍衞或執行我們的專利,圍繞他人持有的專利進行設計或許可,可能會收取高額費用、專利或其他 他人持有的所有權,我們的業務、經營成果和財務狀況可能會受到損害。我們可能不會開發其他 專利產品。截至本報告日期,我們擁有廣泛的專利組合,包括許多已授權專利 和待批准的專利。
醫藥產品的專利地位是複雜和不確定的。我們未來候選產品的專利保護範圍和程度尤其不確定。 我們未來的候選產品將基於藥物化學而非大麻植物。雖然我們已經尋求專利保護, 在適當的情況下,針對其特定配方的物質組合物、其使用方法和 的製造方法,我們沒有並且將無法獲得對這些先前已知的CBD衍生物本身的物質組合物保護 。我們預計,我們未來開發的產品將基於我們可能發現的合成化合物。雖然我們 已經並將繼續在美國尋求專利保護,歐洲和其他國家的專利技術、未來的 候選產品、其使用方法和製造方法,其中任何或全部可能不受有效專利保護。 如果我們的任何產品獲得批准並銷售用於我們沒有已頒發專利的適應症,則我們使用 專利防止競爭對手針對該非專利適應症將我們商業產品的非品牌版本商業化的能力可能會受到嚴重損害,甚至被淘汰。
84
我們公司或其他人發佈與我們未來候選產品相關的信息 可能會阻止我們獲得或執行與這些 產品和候選產品相關的專利。此外,其他人可能會獨立開發類似產品,可能複製我們的產品,或可能圍繞我們的專利權進行設計 。此外,我們發佈的任何專利都可能遭到反對和/或宣佈無效或不可執行。如果我們未能 充分保護我們的知識產權,我們可能會面臨來自那些試圖創建仿製產品以與我們未來候選產品競爭的公司的競爭。我們還可能面臨來自那些開發與我們未來 候選產品基本相似的產品的公司的競爭,而這些產品不在我們的任何專利範圍之內。
許多公司在外國司法管轄區保護、捍衞和執行知識產權方面遇到了 重大問題。某些國家,特別是某些發展中國家的法律制度 不支持專利和其他知識產權 的執行,特別是與藥品有關的專利和其他知識產權,這可能使我們難以阻止侵犯我們的專利, 或銷售競爭產品,侵犯我們的所有權。在外國 司法管轄區執行我們的專利權的訴訟可能會導致大量成本,並轉移我們的精力和注意力,使我們的業務的其他方面。
如果第三方聲稱我們公司使用的知識產權侵犯了他們的知識產權,我們的運營利潤可能會受到不利影響。
在美國國內和國外都有大量的訴訟,涉及製藥行業的專利和其他知識產權。 我們可能會不時收到關於我們侵犯了第三方擁有的專利、商標、版權或其他知識產權的索賠的通知,我們不能保證其他公司將來不會向我們、我們的商業合作伙伴或我們許可的任何第三方專有技術提出此類侵權索賠。如果我們被發現侵犯了專利或其他知識產權,或者如果我們未能從第三方獲得或續簽專利或其他知識產權的許可,或者如果我們向其許可技術的第三方被發現侵犯了另一第三方的專利或其他知識產權,我們可能被要求支付損害賠償金,包括損害賠償金,最高可達發現或評估損害賠償金的三倍,如果侵權被發現是故意的,暫停某些產品的生產或重新設計 或重新命名我們的產品,如果可行,或者我們可能無法進入某些新產品市場。任何此類索賠都可能代價高昂 ,而且需要花費大量時間來辯護和轉移管理層的注意力和資源。因此,我們的競爭地位可能會受到影響。 此外,如果我們因任何原因拒絕或未能簽訂有效的保密或轉讓協議,我們可能不擁有該發明或我們的知識產權,我們的產品可能得不到足夠的保護。因此,我們不能保證我們未來的任何候選產品或其商業化不會也不會侵犯任何第三方的知識產權。
如果我們不能 充分防止商業祕密和其他專有信息泄露,我們的技術和產品的價值可能會 大幅縮水。
我們依靠商業祕密 來保護我們的專有技術,特別是在我們認為專利保護不合適或不可能獲得的情況下。然而,商業祕密很難保護。我們部分依賴與現任和前任員工、顧問、 外部科學合作者、贊助研究人員、合同製造商、供應商和其他顧問簽訂的保密協議來保護我們的商業祕密 和其他專有信息。這些協議可能無法有效阻止機密信息的泄露,也可能無法在未經授權泄露機密信息的情況下提供適當的補救措施。此外,我們不能保證我們已與可能或曾經訪問我們的商業祕密的每一方執行了這些協議。與我們或他們簽署此類協議的任何一方都可能違反該協議並泄露我們的專有信息,包括我們的商業祕密,我們可能 無法針對此類違規行為獲得足夠的補救措施。
強制執行一方非法披露或挪用商業祕密的指控是困難、昂貴和耗時的,結果是不可預測的。 此外,美國國內外的一些法院不太願意或不願意保護商業祕密。如果我們的任何商業祕密 由競爭對手合法獲取或獨立開發,我們將無權阻止他們或他們向其泄露此類商業祕密的人使用該技術或信息與我們競爭。如果我們的任何商業祕密被泄露給 ,或者由競爭對手或其他第三方獨立開發,我們的競爭地位將受到損害。
85
專利保護到期或喪失 可能會對我們未來的收入和運營收益產生不利影響。
我們在發現、開發、製造和銷售我們的候選產品時依賴於專利、商標、商業祕密和其他知識產權保護。 尤其是,專利保護在我們候選產品的開發和最終商業化過程中非常重要。涵蓋我們候選產品的專利通常提供市場排他性,這對於提高我們候選產品 能夠盈利的概率非常重要。
我們與纖維化和抗腫瘤壞死因子計劃相關的一項專利將於2033年到期;然而,大多數專利組合的壽命更長。雖然我們正在尋求可能保護這些專利背後的技術的額外專利覆蓋範圍,但不能保證 將授予此類額外專利保護,或者如果授予這些專利,則不會侵犯這些專利或以其他方式保持其可強制執行。 即使我們成功獲得專利,專利的壽命也是有限的。在美國,實用程序專利的自然失效時間通常是申請後20年。可以使用各種延期;但是,專利的有效期及其提供的保護是有限的。如果我們的候選產品沒有專利保護,我們可能會面臨來自此類方法和組合物的仿製版本的競爭。
如果我們沒有通過延長專利期而根據《哈奇-瓦克斯曼修正案》獲得保護,我們的業務可能會受到損害。
我們的商業成功將在很大程度上取決於我們在美國和其他國家/地區獲得和維護專利和其他知識產權的能力 對於我們的候選產品。考慮到新候選產品的開發、測試和監管審查所需的時間, 保護我們候選產品的專利可能會在候選產品開始商業化之前或之後不久到期。我們預計 將在美國尋求延長專利期限,如果有的話,還將在我們起訴專利的其他國家/地區尋求延長。
根據我們候選產品的時間、持續時間和FDA上市批准的具體情況,我們的一項或多項美國專利可能有資格根據1984年的《藥品價格競爭和專利期限恢復法》(簡稱《哈奇-瓦克斯曼修正案》)獲得有限的 專利期延長或PTE。Hatch-Waxman修正案允許專利恢復期限在專利正常到期後最多五年,作為對開發和FDA監管審查過程中丟失的專利期的補償,這僅限於專利涵蓋的經批准的適應症 (以及可能在延長期內批准的其他適應症)。此延期僅限於一項專利,該專利涵蓋經批准的產品、產品的經批准的用途或產品的製造方法。然而, 相關當局,包括FDA和USPTO在美國和其他國家/地區的任何同等監管機構, 可能不同意我們對此類延期是否可用的評估,並可能拒絕授予我們的專利延期,或者 可能批准比我們要求的更有限的延期。我們可能因為未能在適用的 截止日期內申請、未能在相關專利到期前申請或未能滿足適用要求等原因而無法獲得延期。此外, 適用的時間段或提供的專利保護範圍可能小於我們的要求。即使我們能夠獲得 延期,專利期仍可能在我們獲得FDA上市批准之前或之後不久到期。如果我們無法延長現有專利的 到期日或無法獲得更長到期日的新專利,我們的競爭對手可能能夠利用我們在開發和臨牀試驗方面的投資,參考我們的臨牀和臨牀前數據,在我們的專利到期後獲得競爭對手產品的批准,並比其他情況下更早推出他們的產品。
與受管制物質有關的風險
受控物質 各國立法不同,某些國家/地區的立法可能會限制或限制我們銷售未來產品的能力 候選產品。
大多數國家都是《1961年麻醉品單一公約》和《1971年精神藥物公約》的締約國,這兩項公約管理着包括大麻提取物在內的麻醉藥品的國際貿易和國內管制。各國可能會解釋和履行其條約義務 ,這可能會對我們未來產品在這些國家獲得營銷批准造成法律障礙。這些國家/地區 可能不願意或不能修改其法律法規以允許我們未來的產品上市,或者實現對法律法規的此類修改可能需要較長時間。在這種情況下,我們將無法在不久的將來或根本無法在這些國家/地區銷售我們未來的候選產品 。
86
我們正在開發的候選產品 可能受到美國受控物質法律法規的約束,如果不遵守這些法律法規或遵守這些法律法規的成本,可能會對我們的業務運營結果產生不利影響,無論是在臨牀 開發期間還是審批後,以及我們的財務狀況。
我們正在開發的候選產品可能包含1970年《美國聯邦受控物質法》和 CSA中定義的受控物質。作為藥品的受控物質受到CSA的高度監管,CSA除其他事項外,還規定了DEA管理的某些註冊、製造配額、安全、記錄、報告、進口、出口和其他要求 。DEA將受控物質分為五個附表:附表I、II、III、IV或V物質。附表 i根據定義,物質有很高的濫用可能性,目前沒有“公認的醫療用途"在美國,在醫療監督下使用缺乏 公認的安全性,且不得在美國開處方、銷售或銷售。在美國批准 使用的含有受控物質的藥品列於附表II、III、IV或V,其中,附表II物質被認為 存在最高的濫用或依賴可能性,而附表V物質在這些物質中濫用的相對風險最低。
雖然大麻是附表 I受控物質,但在美國批准用於醫療用途的含有大麻或大麻提取物的產品應列入 附表II—V,因為FDA的批准符合"公認的醫療用途"要求。如果我們的 未來候選產品獲得FDA批准,DEA將做出時間安排決定。如果FDA、DEA或任何外國監管機構 確定我們未來的候選產品可能有濫用的可能性,可能要求我們生成比我們目前預期的更多的臨牀或 其他數據,以確定該物質是否或在何種程度上具有濫用的可能性,這可能會增加 成本和/或延遲該產品的上市。
從事研究、 製造、分銷、進口或出口或分發受控物質的設施必須註冊(許可),以執行 這些活動,並具有DEA要求的安全、控制、記錄保存、報告和庫存機制,以防止藥物 損失和轉移。所有這些機構必須每年更新註冊,但配藥機構除外,其必須每 三年更新一次。DEA定期檢查某些經營受管制物質的註冊企業。獲得 必要的註冊可能會導致我們未來產品的進口、製造或分銷延遲。此外, 未能遵守CSA,特別是不遵守規定導致損失或轉移,可能導致監管行動 ,這可能對我們的業務、財務狀況和運營結果產生重大不利影響。DEA可能會尋求民事處罰, 拒絕更新必要的註冊,或啟動訴訟程序以限制、暫停或撤銷這些註冊。在某些情況下, 違反行為可能導致刑事訴訟。
個別州也 制定了受控物質法律和法規。雖然州控制的物質法律通常反映聯邦法律,但由於 州是獨立的司法管轄區,它們也可能單獨安排我們未來的候選產品。州計劃可能會延遲我們獲得聯邦監管部門批准的任何產品的商業銷售,而不利的計劃可能會對此類產品的商業吸引力產生重大不利影響。我們或我們的合作伙伴還必須獲得單獨的州註冊、許可證或許可證,以便 能夠獲得、處理和分銷受控物質用於臨牀試驗或商業銷售,如果不符合適用的 監管要求,除了來自DEA的要求外,還可能導致各州的執法和制裁,或者 根據聯邦法律產生的其他原因。
因為我們的產品在美國可能是受管制的物質,在美國進行臨牀試驗我們的每個研究中心都必須向DEA提交研究方案 ,並獲得和維護DEA研究員註冊,以便這些研究中心能夠處理和分配我們的產品,並 從我們的進口商處獲得產品。如果DEA延遲或拒絕向一個或多個研究中心授予研究註冊, 臨牀試驗可能會被顯著推遲,我們可能會失去臨牀試驗中心。臨牀試驗的進口商還必須 獲得進口商註冊和每次進口的進口許可。
87
歐盟的大麻立法 各成員國之間存在差異,因為這一領域尚未完全統一。例如,在德國,大麻被作為一種受管制物質加以管制(Bitäubungsmittel),它的處理需要特定的授權。
在美國和海外,醫療和娛樂用大麻的合法化和 使用可能會影響我們的業務。
在美國,醫療和娛樂大麻產品的使用發生了大量的變化。雖然未經FDA批准 的大麻產品是聯邦法律定義的附表一物質,並且根據聯邦 法律不允許擁有和使用它們(除非用於研究目的,在DEA註冊),但根據網站www.example.com,至少有38個州和 哥倫比亞特區已經頒佈了州法律,允許擁有和使用大麻用於醫療目的,worldpoplulationreview.com 2018年通過的《美國農業法案》(U.S. Farm Bill)禁止了從大麻植物中提取的某些材料,THC含量極低。雖然我們的業務與在線和藥房大麻公司的業務有很大的不同,但未來的立法授權銷售、分銷、使用和保險報銷未經FDA批准的大麻產品 可能會影響我們的業務。
會計風險
我們在過去、 和未來可能會損害長期資產和無形資產,包括商譽和收購的正在進行的研究和開發。
每當情況及情況發生變化以致 有跡象顯示賬面值可能無法收回時,我們便會審核長期資產 及若干可識別資產(包括無形資產)的減值準備。當長期使用的 或無形資產(包括商譽和收購中的研發)的賬面價值無法收回並超過其估計的 公允價值時,即存在減值。商譽是指在企業合併中收購的資產和負債的收購價與公允價值之間的差額。我們每年審查商譽,或在情況和情況發生變化時更頻繁地審查商譽,以確定報告單位的公允價值是否更有可能少於其賬面價值(包括商譽),以此作為確定是否需要進行量化分析的基礎。如確定報告單位的公允價值較有可能少於其賬面值,則會進行量化分析以確認商譽減值。
截至2021年12月31日,我們的上市股票 收於每股1,482.00美元;在2022年期間,我們單一報告單位的市值大幅下降。 截至2022年3月31日、2022年6月30日、2022年9月30日和2022年12月31日,我們上市股票的市值分別跌至每股984.20美元、322.24美元、252.70美元和64.41美元,因此,我們選擇對截至2022年9月30日和2022年12月31日的商譽進行量化分析 以評估減值。我們確定了我們的單一報告單位的公允市場價值,並將該價值與報告單位的賬面價值進行了比較,確定商譽在兩個計量日期均已減值。截至2022年9月30日和2022年12月31日,賬面價值分別比公平市場價值高出18,872,850美元和14,674,428美元 。為了確認商譽減值,我們在第三季度和第四季度末記錄了這些金額的損失,在截至2022年12月31日的年度損益表上顯示為商譽減值虧損33,547,278美元。
無形資產和正在進行的研發 (“知識產權研發“)資產是指分配給2019年7月16日收購的與重組相關的技術的公允價值,這些技術尚未達到技術可行性,未來也沒有其他用途 。知識產權研發資產被認為是無限期的--在相關研發項目完成或放棄之前 。在知識產權研發資產被視為無限期存續期間,如果我們發現知識產權研發資產的公允價值低於其賬面價值的任何事件或情況發生變化,則會按年度進行減值測試,或更頻繁地進行減值測試。如果開發完成(通常是在監管部門批准後完成),並且我們能夠將與知識產權研發資產相關的產品商業化,則這些資產將被視為已確定使用壽命,並根據其在該時間點的估計使用壽命進行攤銷。如果開發被終止或放棄,我們可能會記錄與知識產權研發資產相關的全部或部分減值費用,按知識產權研發資產的賬面價值超出其估計公允價值計算。
88
截至2022年12月31日,資產負債表上知識產權研發資產的賬面金額為12,405,084美元(包括分別與本公司的CBR Pharma子公司和180 LP子公司相關的賬面價值1,462,084美元和10,943,000美元)。根據截至年末從第三方獲得的估值,本公司知識產權研發資產的公平市值被確定為9,063,000美元(其中 包括與本公司的CBR Pharma子公司和180 LP子公司有關的公平市場價值分別為0美元和9,063,000美元)。 截至本計量日期,CBR Pharma和180 LP子公司的資產的賬面價值分別比其公平市場價值高出1,462,084美元和1,880,000美元。因此,管理層確定綜合知識產權研發資產減值3,342,084美元, 為確認減值,本公司於2022年第四季度記錄了這筆金額的虧損,在損益表中顯示為知識產權研發資產減值虧損。截至2022年12月31日,其CBR Pharma子公司 及其180個LP子公司的知識產權研發資產餘額分別降至零和9,063,000美元;減值後的綜合知識產權研發資產餘額 為9,063,000美元。
截至2023年9月30日,資產負債表上知識產權研發資產的賬面金額為9,063,000美元(包括與本公司180 LP子公司相關的餘額);本公司通常每年評估資產減值,除非觸發事件或其他事實或情況 表明應更早進行評估。於2023年第三季度末,本公司評估了一般經濟狀況、行業及市場因素、本公司的財務表現及所有可能顯示減值可能性的相關法律、法規、 及政治因素,並得出結論:綜合評估這些因素後,資產很可能已減值。本公司錄得虧損9,063,000美元,於截至2023年9月30日止三個月及九個月的損益表中顯示為知識產權研發資產減值虧損 。截至2023年12月31日,資產負債表上知識產權研發資產餘額 為0美元。
在一個期間確認的減值 可能不會在隨後的期間沖銷,即使我們的普通股未來價值增加。我們在過去和將來可能會產生額外的長期資產和/或無形資產減值,包括收購的正在進行的研發和商譽,這可能是實質性的。
我們在過去、並且未來可能會發現我們在披露控制程序和財務報告內部控制方面的重大弱點。 如果不加以補救,我們未能建立和維護有效的披露控制程序和財務報告內部控制 可能會導致我們的財務報表中出現重大錯報,並無法履行我們的報告和財務義務, 每一項都可能對我們的財務狀況和我們證券的交易價格產生重大不利影響。
重大缺陷是財務報告內部控制中的缺陷或缺陷組合,因此存在合理的可能性,無法及時防止或發現年度或中期財務報表的重大錯報。因此,重大 缺陷會增加我們報告的財務信息包含重大錯誤的風險。當控制的設計 或操作不允許管理層或員工在履行其指定職能的正常過程中及時防止或發現錯誤陳述時,則存在控制缺陷。
Maintaining effective disclosure controls and procedures and effective internal control over financial reporting are necessary for us to produce reliable financial statements and we are committed to remediating our material weaknesses in such controls as promptly as possible. However, there can be no assurance as to when these material weaknesses will be remediated or that additional material weaknesses will not arise in the future. Any failure to remediate the material weaknesses, or the development of new material weaknesses in our internal control over financial reporting, could result in material misstatements in our financial statements and cause us to fail to meet our reporting and financial obligations on a timely and accurate basis. If our financial statements are not accurate, investors may not have a complete understanding of our operations or may lose confidence in our reported financial information. Likewise, if our financial statements are not filed on a timely basis as required by the SEC and Nasdaq, we could face severe consequences from those authorities. Any of these cases could result in a material adverse effect on our business, on our financial condition or have a negative effect on the trading price of our common stock and warrants. Further, if we fail to remedy any future deficiencies or maintain the adequacy of our disclosure controls and procedures and our internal controls, we could be subject to regulatory scrutiny, civil or criminal penalties or stockholder litigation against us or our management.
89
我們不能保證 我們在未來採取的措施將糾正可能發現的任何其他重大弱點,或 我們的財務報表不會因未能實施和維持適當的內部控制 或規避這些控制而在未來出現重報 。
此外,在未來,如果 我們不能得出結論,我們有有效的內部控制對我們的財務報告,或如果我們的獨立註冊會計師事務所 無法提供無保留意見,我們的內部控制對財務報告的有效性( 在未來可能需要我們的情況下),投資者可能會對我們財務報表的可靠性失去信心,這可能 導致我們的股價下跌。未能遵守報告要求也可能使我們受到SEC或納斯達克(如適用)或其他監管機構的制裁和/或調查 。
此外,即使我們 成功地加強了我們的控制和程序,這些控制和程序可能不足以防止或識別 違規行為,也不足以促進我們的財務報表或我們提交給SEC的定期報告的公平列報。這可能需要 我們重述先前的財務報表。
由於採用新的會計準則或解釋,我們可能會對我們報告的經營業績產生不利的 影響。
我們執行和遵守會計規則的變更(包括新會計規則和解釋)可能會對我們報告的 財務狀況或經營業績造成不利影響,或導致我們報告的經營業績在未來期間出現意外波動。
與我們的普通股和認股權證相關的風險
我們普通股的市場價格一直非常不穩定,並且由於我們無法控制的許多情況,可能會繼續波動。
由於許多因素,其中一些可能超出我們的控制範圍,我們普通 股的市場價格已經波動,並可能繼續波動。這些因素 包括但不限於:
| ● | “空頭擠壓”; |
| ● | 證券分析師或其他第三方的評論,包括博客、文章, 留言板和社交及其他媒體; |
| ● | 大股東退出其在我們證券中的頭寸或增加或減少 在我們證券的短期利益; |
| ● | 財務和經營業績的實際或預期波動; |
| ● | 外幣匯率變動情況; |
| ● | 我們計劃或未來臨牀試驗的開始、入組或結果 我們的候選產品或競爭對手的候選產品; |
90
| ● | 競爭性藥物或療法的成功; |
| ● | 美國和其他國家的法規或法律發展; |
| ● | 有競爭力的產品或技術的成功; |
| ● | 關於專利申請、已頒發專利或其他的進展或爭議 所有權; |
| ● | 關鍵人員的招聘或離職; |
| ● | 與我們的候選產品或臨牀開發相關的費用水平 方案; |
| ● | 訴訟事項,包括根據 我們的高級管理人員和董事的保險政策、影響公司的監管行動及其結果; |
| ● | 我們努力發現、開發、獲取或授權其他產品的結果 候選產品; |
| ● | 關於財務結果、發展的估計的實際或預期變化 時間表或證券分析師的建議; |
| ● | 我們無法獲得或延遲獲得任何已批准的藥物供應 藥物或無法以可接受的價格這樣做; |
| ● | 與所有權(包括專利)有關的爭議或其他發展, 訴訟事項以及我們為我們的技術獲得專利保護的能力; |
| ● | 重大訴訟,包括專利或股東訴訟; |
| ● | 我們的財務業績或被認為是 與我們相似; |
| ● | 醫療保健支付體系結構的變化,包括覆蓋面和充分性 報銷任何獲批藥物; |
| ● | 製藥和生物技術部門的市場狀況; |
| ● | 總體經濟、政治和市場狀況以及總體波動 美國和海外的金融市場;以及 |
| ● | 投資者對我們和我們業務的總體看法。 |
整個股票市場,尤其是 我們的股票價格最近經歷了極端的價格和成交量波動,這些波動往往與這些公司和我們公司的經營業績無關或不成比例。例如,在2022年,我們普通股的收盤銷售價格範圍從每股1,482.04美元的分拆後調整後的高點到每股23.56美元的低點,在2023財年,我們普通股的收盤銷售價格範圍從每股100.70美元的高點到每股3.21美元的低點。在此期間,我們認為我們的財務狀況或經營業績沒有 發生任何重大變化,以解釋這種價格波動或交易量 ;然而,我們出售的股權對現有股東有攤薄作用。這些廣泛的市場波動可能會對我們證券的交易價格產生不利影響。此外,這些和其他外部因素已經導致並可能繼續導致市場價格 和對我們普通股的需求大幅波動,這可能限制或阻止我們的股東輕易出售他們持有的我們普通股股票 ,並可能對我們普通股的流動性產生負面影響。
91
由第三方發佈的公共媒體(包括博客、文章、留言板以及社交及其他媒體) 中的可用信息可能包含 不屬於我們的聲明,並且可能不可靠或不準確。
我們意識到第三方正在傳播大量與我們的業務相關的信息,包括在博客、留言板、社交媒體和其他媒體上傳播。第三方報告的此類信息可能不準確,可能導致我們的證券大幅波動,並可能最終導致我們的普通股或其他證券價值下降。
行使我們的未償還期權和認股權證,以及在行使後出售普通股,可能會對我們證券的交易價格產生不利影響。
截至2024年3月20日,我們有 (i)尚未行使的股票期權,以每股633.95美元的加權平均行使價購買總計17,788股普通股;及(ii)以加權平均行使價每股83.97美元購買983,473股普通股的尚未行使認股權證(不包括275,205份預先供資的認股權證)。在期權和權證的有效期內,持有人有機會 從我們普通股的市價上漲中獲利,而無需承擔所有權風險。 在行使已發行證券時發行股份也會稀釋我們現有股東的所有權權益。
這些 股票可供公開轉售,以及這些股票的任何實際轉售,都可能對我們普通股的交易價格產生不利影響。 我們無法預測根據未償還期權或認股權證的行使或其他證券的轉換而未來我們普通股的發行規模,或者未來我們普通股的股票發行和銷售可能對我們普通股的市場價格產生的影響(如果有的話)。出售或分發大量我們的普通股(包括與收購相關的股票),或認為可能發生此類出售,可能會導致我們普通股的市場價格下跌。
此外,在行使/轉換未行使可轉換證券時可發行的普通股 可能代表懸置,也可能對我們普通股的市場價格 產生不利影響。當市場上公司股票的供應大於對該股票的需求時,就會出現懸置 。當這種情況發生時,我們的股票價格將下降,股東試圖在市場上出售的任何額外股票 只會進一步降低股價。如果我們普通股的股份量不能吸收我們未發行可轉換證券持有人 出售的股份,那麼我們普通股的價值將可能下降。
我們的未償還公共認股權證 資金嚴重不足。
每份公開認股權證允許持有人以每1/760美元的行使價購買一股普通股的一千七百六十分之一TH:一股(每股4,370.00美元), 可作調整。於行使公眾認股權證時,概不會發行零碎股份。公開認股權證於首次公開募股結束後12個月內可行使,並於業務合併完成後五年(2025年11月6日)到期。公開 認股權證的金額嚴重不足,且由於在行使公開認股權證時不會發行零碎股份,所以 公開認股權證只能以760的倍數行使。因此,公共認股權證可能並無任何重大價值。此外, 未持有至少760份公開認股權證或持有可行使普通股零碎股份的公開認股權證的認股權證持有人,必須出售任何認股權證,以從零碎利息中獲得價值。因此,公開認股權證 的交易可能有限或零星,且該等公開認股權證可能沒有任何重大價值。任何持有少於 760份或不能被760整除的數量的公共認股權證持有人,在行使 公共認股權證時將不會收到任何普通股,因為行使公共認股權證時不會發行任何零碎普通股。
92
我們有相當數量的股票有資格出售,它們的出售或潛在出售可能會壓低我們普通股的市場價格,並對現有股東造成重大 稀釋。
Sales of a significant number of shares of our common stock in the public market could harm the market price of our common stock. Most of our common stock is available for immediate resale in the public market, including (a) options to purchase 17,788 shares of common stock with a weighted average exercise price of $633.95 per share; and (b) warrants to purchase 987,473 shares of common stock with a weighted average exercise price of $83.97 per share (such amount does not include 257,205 pre-funded warrants which were outstanding as of December 31, 2023, all of which have been exercised as of March 7, 2024). If a significant number of shares were sold, such sales would increase the supply of our common stock, thereby potentially causing a decrease in its price. The exercise of outstanding convertible securities will also cause significant dilution to existing stockholders and will likely cause the per-share value of our common stock to decline, possibly significantly. Some or all of our shares of common stock may be offered from time to time in the open market pursuant to effective registration statements and/or compliance with Rule 144, which sales could have a depressive effect on the market for our shares of common stock. Subject to certain restrictions, a person who has held restricted shares for a period of six months may generally sell common stock into the market. The sale of a significant portion of such shares when such shares are eligible for public sale may cause the value of our common stock to decline in value.
我們的證券市場上可能沒有足夠的流動性 以供投資者出售其股票。我們普通股的市場價格可能會繼續波動。
我們的普通 股票的市場價格可能會繼續高度波動。某些可能對我們普通股市場價格產生重大影響的因素 超出了我們的控制範圍,例如我們經營所在行業的條件或趨勢或我們普通股的銷售。這種情況 可歸因於多個因素,包括我們是一家小公司,對股票分析師、 股票經紀人、機構投資者和投資界其他產生或影響銷售量的人來説相對陌生,即使 我們引起了這些人的注意,他們傾向於規避風險,不願追隨我們這樣未經證實的公司,或購買或推薦購買我們的股票,直到我們變得更成熟和可行。
因此,與成熟的發行人相比,我們的股票可能有幾天或更長時間的交易活動很少或根本不存在,而成熟的發行人 擁有大量穩定的交易量,通常將支持持續銷售,而不會對股價產生不利影響。 我們普通股的更廣泛或更活躍的公開交易市場可能不會發展或維持,或者交易 水平將不會繼續。這些因素可能會對我們普通股的市場價格產生實質性的不利影響,無論我們的表現如何。 此外,公開股票市場經歷了極端的價格和交易量波動。這種波動嚴重影響了許多公司的證券市場價格,原因往往與特定公司的經營業績無關。這些廣泛的市場波動可能會對我們普通股的市場價格產生不利影響。
如果我們之前為登記在我們之前 發行中出售的某些證券而提交的登記聲明隨後被暫停或終止,我們將面臨嚴重的 處罰和損失。
根據先前的某些 證券私募發行,我們簽訂了登記權協議,要求我們提交某些登記聲明 ,以登記私募出售股份和某些證券在行使/轉換時的轉售,並在一定時間內維持 此類登記聲明的有效性。迄今為止,所有此類要求的註冊聲明 已由SEC宣佈生效。然而,如果註冊聲明隨後被暫停或終止,或者我們 未能滿足註冊權協議中規定的某些要求,我們可能被要求支付嚴重的 罰款,這可能會對我們的現金流造成不利影響,並導致我們的證券價值下跌。
某些未清償認股權證的條款可能會阻止第三方收購我們。
某些未執行 認股權證的條款可能會使第三方收購我們更困難或成本更高。某些尚未行使的認股權證禁止我們 參與構成“基本交易”的某些交易,除非(其中包括)尚存實體承擔 我們在就2022年7月發售、2022年12月發售、 2023年4月發售和2023年8月發售而發行的各項未行使認股權證下的義務(分別定義及╱或討論),以及尚未行使的二零二三年十二月預籌資金權證及二零二三年十二月普通權證。此外,該等尚未行使的認股權證規定,在某些交易 構成“基本交易”的情況下,除某些例外情況外,該等認股權證持有人將有權 按適用認股權證所述的價格(基於該等認股權證的布萊克—斯科爾斯價值)回購該等認股權證。 認股權證的這些和其他條款可以防止或阻止第三方收購我們,即使收購可能對股東有利 。
93
未來出售和發行我們的普通股或購買普通股的權利,可能會對我們的股東造成額外的稀釋,並可能導致我們普通股的價格 下跌。
我們未來可能會發行額外的普通股、可轉換證券或其他股權。我們還根據股權激勵計劃向員工、董事和其他服務提供商發行普通股。此類發行可能會稀釋投資者的權益,並可能導致我們普通股的價格下跌。此類發行的新投資者還可以獲得優先於現有股東的權利。
在公開市場上轉售我們的普通股可能會導致我們普通股的市場價格下跌。
大量 我們普通股股票的銷售可能在任何時候發生。發行新的普通股可能導致我們的 普通股被我們的現有股東轉售,擔心他們持有的股票的潛在所有權稀釋。反過來,這些轉售可能 有抑制我們普通股市場價格的效果。
未來出售我們的普通股可能會導致我們的股價下跌。
如果我們的股東在公開市場上出售 大量我們的普通股,我們的普通股的市場價格可能會大幅下跌。在公開市場上 我們的股東可能會出售我們的普通股股票的看法也會壓低我們的 普通股的市場價格。本公司已在一個"證券總價值高達125,000,000美元的"擱板“我們於2022年6月3日向委員會提交的表格S—3註冊聲明,並於2022年6月24日宣佈生效。然而, 截至本報告之日,我們的公眾持股量不足7500萬美元,根據SEC的規定,只要我們的公眾持股量 保持低於7500萬美元,我們在任何12個月期間使用 我們在表格S—3上的貨架登記聲明通過首次公開發行證券籌集的資金總額不得超過我們公眾持股量的三分之一。當我們的公眾 浮存量再次超過7500萬美元時,我們可以根據表格S—3登記聲明出售的證券數量將不再受此類規則的限制 。此外,如果我們的現有股東在公開市場上出售或表示有意出售大量我們的普通股 ,我們的普通股的交易價格可能會大幅下跌。當我們的普通股股票在公開市場上出售時, 的股票的市場價格可能會大幅下跌。我們普通股股價的下跌可能 阻礙我們通過發行額外的普通股或其他股本證券來籌集資金的能力。
與我們的管理文件和特拉華州法律相關的風險
我們的 公司註冊證書規定了對高級管理人員和董事的賠償,費用由我們承擔,並限制了他們的責任,因為公司資源可能會被用於高級管理人員或 董事的利益,這可能會給我們造成 重大成本並損害我們股東的利益。
我們的公司註冊證書規定的賠償如下:“在適用法律允許的最大範圍內,公司有權通過章程條款、與此類代理人或其他人的協議、股東投票或無利害關係的董事投票或其他方式,向公司的此類代理人(以及特拉華州法律允許公司向其提供賠償的任何其他人)提供賠償和墊付費用,超過《特拉華州公司法》第145條所允許的賠償和墊付費用,僅受特拉華州適用法律(法定或非法定)的限制,關於違反對本公司、其股東和其他人的義務的行為。
我們被告知 ,SEC認為,根據聯邦證券法產生的責任賠償違反了 《證券法》中所述的公共政策,因此無法執行。如果與我們的活動有關的董事、高級管理人員或 控制人員就聯邦證券法下產生的責任(除我們支付董事、高級管理人員或 控制人員為成功辯護任何訴訟、訴訟或程序而發生或支付的費用外)提出賠償要求,我們將(除非我們的律師認為,該事項已通過控制 先例解決)向具有適當管轄權的法院提交問題,我們的賠償是否違反《證券法》中所述的公共政策 ,並將受該問題的最終裁決的管轄。與此 事件有關的法律程序如果發生可能會非常昂貴,並可能導致我們收到負面宣傳,其中任何一個因素都可能 大幅降低我們股票的市場和價格。
94
我們的 公司註冊證書包含一項特定條款,限制了董事對我們和股東造成的金錢損失的責任 ,並要求我們在某些情況下賠償高級管理人員、董事和員工。
根據特拉華州法律,我們的董事、高級管理人員和員工的金錢責任受到限制,並且存在對他們的賠償權利,這可能會導致我們的鉅額支出,並可能阻礙針對我們的董事、高級管理人員和員工的訴訟。
我們的公司註冊證書 包含一項特定條款,該條款限制了我們的董事對我們和我們的股東造成的金錢損害的責任。根據我們與高管和董事簽訂的僱傭和聘用協議,我們還負有 合同賠償義務。 上述賠償義務可能導致我們產生鉅額支出,以支付我們董事和高級管理人員的和解或損害 賠償的費用,而我們可能無法收回這些費用。這些條款和由此產生的成本也可能會阻止我們因違反受託責任而對我們的董事和高級管理人員提起訴訟,並可能同樣阻止我們的股東對我們的董事和高級管理人員提起衍生品訴訟,即使此類訴訟如果成功,可能會 以其他方式使我們和我們的股東受益。
我們的董事有權 授權發行優先股股份和額外普通股股份。
我們的董事,在公司註冊證書中包含的 限制和限制範圍內,且無需股東採取進一步行動,有權 不時以一個或多個系列發行優先股,並確定股份數量和相關權利、 轉換權、投票權和贖回條款、清算優先權和任何其他優先權,任何此類系列的特殊權利和資格 。任何優先股的發行都可能對我們普通股持有人的權利造成不利影響。如果 我們稍後再發行普通股,那麼每個投資者在我們股票中的所有權權益將按比例減少 。
我們的第二份經修訂的公司註冊證書和我們的第二份經修訂和重新修訂的章程中的反收購條款 以及特拉華州法律的條款 可能會阻止、推遲或阻止我們公司控制權的變更或我們管理層的變更,從而壓低我們普通股的交易價格。
我們的第二次修訂和重新修訂的公司註冊證書,以及我們第二次修訂和重新修訂的章程和特拉華州法律包含的條款可能會阻止、推遲或阻止股東可能認為有利的合併、收購或其他控制權變化,包括您在 中的交易,否則您可能會從您的普通股或認股權證中獲得溢價。這些規定還可能阻止或推遲我們的股東更換或撤換我們管理層的嘗試。我們的公司治理文件包括以下規定:
| ● | 一個分類的董事會,因此我們的董事會被劃分 分為兩個班,每班交替任職兩年; |
| ● | 只有在有理由的情況下才能罷免董事; |
| ● | 要求提前通知股東的業務建議, 在股東會議上進行,並提名候選人蔘加董事會選舉; |
| ● | 禁止股東通過書面同意採取行動 行動; |
| ● | 規定股東特別會議只能召開 董事會主席、首席執行官或董事會根據董事會過半數成員通過的決議作出的決定; |
| ● | 授權空白支票優先股,可與 一起發行 投票權、清算權、股息和其他優於我們普通股的權利;以及 |
| ● | 限制董事的責任並向董事提供賠償 和軍官。 |
95
作為一家特拉華州公司, 我們還受特拉華州法律條款的約束,包括《特拉華州普通公司法》第203條,該條款限制了 持有代表我們已發行有表決權股票15%以上投票權的股份的股東與我們進行某些 業務合併的能力。我們的第二次修訂和重述的公司註冊證書(經修訂)或我們的第二次修訂和重述的章程或特拉華州法律中的任何條款,如果有延遲或阻止控制權變更的效果,可能會限制我們的股東 為其普通股股份獲得溢價的機會,並且可能會影響某些投資者 願意為我們普通股支付的價格。
上述條款和反收購措施的存在可能會限制投資者未來可能願意為我們的普通股或認股權證股票支付的價格。它們還可以阻止潛在的收購者收購我們公司,從而降低您在收購中獲得普通股或認股權證溢價的可能性。
我們的第二次修訂的 和重述的公司註冊證書(經修訂)包含排他性的訴訟條款,這些條款可能會阻止對我們和 我們的董事和管理人員提起訴訟。
我們的第二次修訂和重述的 公司註冊證書(經修訂)規定,除非公司書面同意選擇替代 法院,特拉華州高等法院將是(i)代表我們提起的任何衍生訴訟或程序 的唯一和專屬法院,(ii)就本公司任何現任或前任董事、高級職員、僱員或股東違反對本公司或本公司股東的受託責任提出索賠的任何訴訟,(iii)根據《特拉華州普通公司法》、我們的第二次修訂和重述的公司註冊證書或章程的任何條款提出索賠的任何訴訟,或(iv)主張受內部事務原則管轄的索賠的任何訴訟。
在我們的第二次修訂和重述的公司註冊證書中選擇法院條款 並不免除我們遵守聯邦 證券法及其規定的規則和法規的義務。此外,該規定不適用於為執行《交易法》或《證券法》規定的義務或責任而提起的訴訟。《交易法》第27條為執行《交易法》或其規定的規則和條例所規定的任何義務或責任而提起的所有訴訟創建了專屬聯邦管轄權,《證券法》第22條為聯邦和州法院創建了與執行《證券法》或其規定的規則和條例所規定的義務或責任而提起的訴訟相關的共同管轄權。因此,州法院和聯邦法院都有管轄權 根據《證券法》受理索賠。
這些排他性論壇條款 可能限制我們的股東在司法論壇上提出此類股東認為有利於與我們或我們的董事或高級管理人員發生糾紛的索賠,這可能會阻止針對我們及其董事和高級管理人員的此類訴訟。或者,如果法院 發現這些排他性法院條款中的一項或多項不適用於上述一種或多種指定的 訴訟或訴訟程序,我們可能會產生與在其他司法管轄區或法院解決此類問題相關的額外費用 ,這可能會對我們的業務、財務狀況或運營結果產生重大和不利的影響。
我們的第二次修訂的 和重述的公司註冊證書(經修訂)包含條款,據此,我們放棄向任何董事或高級管理人員提供的任何公司機會 中的任何權益,但某些例外情況除外。
我們的條款修訂和重申 公司註冊證書(經修訂)規定,在法律允許的範圍內,公司機會原則或任何其他 類似原則不適用於我們或我們的任何高級管理人員或董事,或他們各自的任何關聯公司,並且 我們放棄任何期望,即我們的任何董事或高級管理人員將提供他或她 可能意識到我們,但公司機會原則僅適用於我們的任何董事或高級管理人員 在以下情況下適用:(i)僅以我們的董事或高級管理人員的身份提供給該等人員的公司機會, (ii)我們在法律上和合同上被允許承擔並且我們可以合理追求的公司機會,及 (iii)在董事或高級人員獲準在不違反任何法律義務的情況下將該機會轉介予本行的範圍內。
此外,我們的每一位 高級管理人員和董事目前和將來可能對其他 實體負有額外的信託或合同義務,根據這些義務,這些高級管理人員或董事可能被要求向該實體提供商業機會,但須遵守其 或她在適用法律下的信託義務。因此,在是否向我們公司提供潛在業務合併機會方面,可能會產生利益衝突。這些衝突可能不會以有利於我們的方式解決。我們放棄公司機會 可能會對我們未來的經營業績產生重大不利影響,和/或產生利益衝突或感知利益衝突 ,這可能會對我們證券的價值產生重大不利影響。
96
我們的董事將他們的時間分配給其他業務,從而導致他們在決定將多少時間投入到我們的事務中存在利益衝突。
我們的董事不需要 ,也不需要將他們的全部時間投入我們的事務,我們的某些董事在 生命科學行業的其他公司擔任職務,包括其他董事職位,這可能導致在我們的運營和他們提供服務的其他公司之間分配他們的時間時產生利益衝突。如果董事的其他業務事務要求他們在這些事務上投入大量時間 超過其當前承諾水平,這可能會限制他們投入時間處理我們事務的能力,這可能會對我們的運營產生負面影響。此外,此類人員在 各種業務活動中分配時間時可能存在利益衝突。這些衝突可能不會以有利於我們的方式解決。此外,我們的董事,由於我們的公司 機會豁免,如上所述,可能選擇或被要求提供公司機會給他們 所屬的其他公司。實際或感知的利益衝突可能對我們的經營業績造成重大不利影響, 可能對我們證券的價值造成重大不利影響。
合規、報告和上市風險
為了確保符合美國和納斯達克的報告和公司治理要求,我們會產生巨大的 成本。
我們產生了與上市公司報告要求以及適用的美國和納斯達克公司治理要求相關的大量成本,包括 2002年《薩班斯—奧克斯利法案》和SEC和納斯達克實施的其他規則。納斯達克的規則包括要求 我們保留獨立董事、遵守其他公司治理要求以及支付年度上市和股票發行費用。 所有此類SEC和納斯達克義務都需要額外資源的承諾,包括但不限於額外費用, 並可能導致我們高級管理層的時間和注意力從我們的日常運營中轉移。我們預計所有 這些適用的規則和法規將顯著增加我們的法律和財務合規成本,並使某些活動 更加耗時和成本更高。我們還預計,這些適用的規則和法規可能會使我們更難獲得董事和高級管理人員責任保險, 可能會要求我們接受更低的保單限額和承保範圍,或者 為獲得相同或類似的承保範圍而產生更高的費用。因此,我們可能更難吸引和留住 合格的個人進入我們的董事會或擔任執行官。
由於作為一家報告公司,我們的 成本增加,並且鑑於我們的資本資源有限,此類額外成本可能會對我們的盈利能力產生不利影響 。
我們是一家美國證券交易委員會報告公司。 《交易法》下的規章制度要求報告公司提供包含交互式數據文件的定期報告, 這要求我們聘請法律、會計和審計專業人員,以及可擴展商業報告語言(XBRL)和服務提供商的 EDGAR(電子數據收集、分析和檢索)。提供此類服務的成本可能很高, 我們可能會繼續蒙受更多損失,這可能會對我們繼續經營下去的能力產生不利影響。此外,2002年的薩班斯-奧克斯利法案以及美國證券交易委員會實施的各種相關規則要求改變公司治理做法, 普遍提高了對上市公司的披露要求。例如,作為一家報告公司,我們被要求 向美國證券交易委員會提交定期和當前報告以及其他信息,我們已經採取了關於披露控制和程序的政策,並定期評估這些控制和程序。
作為一家報告公司,我們繼續 產生的額外成本(預計每年數十萬美元)將繼續 進一步耗盡我們有限的資本資源。由於我們的資源有限,我們必須將資源分配給其他生產性 用途,以繼續履行我們作為美國證券交易委員會報告公司的義務。此外,不能保證我們將有足夠的資源來繼續履行我們向美國證券交易委員會提交的到期報告和備案義務。
97
我們不符合納斯達克的持續上市標準,未來可能無法符合納斯達克的持續上市標準,因此我們的普通股和認股權證可能會從納斯達克退市。
我們的普通股和公共認股權證在納斯達克上交易,代碼為“ATNF”和“ATNFW,“這兩個字分別是。儘管有這樣的上市, 不能保證任何經紀商都有興趣交易我們的證券。因此,可能很難公開出售我們的證券 。也不能保證通過永久滿足納斯達克的持續上市要求,我們能夠在任何時間內保持納斯達克上的上市。
2023年9月7日, 公司收到納斯達克股票市場有限責任公司(“納斯達克”)上市資格部門的書面通知,通知 公司不符合納斯達克上市規則5550(a)(2)中規定的 繼續在納斯達克資本市場上市的最低出價要求。納斯達克上市規則5550(a)(2)要求上市證券維持每股1.00美元的最低 買入價,而上市規則5810(c)(3)(A)規定,如果不足持續連續三十(30)個營業日,則存在 未能滿足最低買入價要求。根據2023年7月26日至2023年9月6日連續三十(30)個工作日的 普通股收盤價,公司不再符合 最低投標價要求。
通知函指出,公司有180個歷日或至2024年3月5日,以重新遵守納斯達克上市規則第5550(A)(2)條。要重新獲得合規, 公司普通股的出價必須在至少連續10個工作日內至少達到每股1.00美元的收盤出價。如果公司在2024年3月5日之前仍未恢復合規,則只要公司滿足納斯達克資本市場初始上市標準(投標價格要求除外),並在必要時通過反向股票拆分的方式在第二合規期內書面通知納斯達克,就可以額外給予180天來恢復合規。如果本公司不符合第二個合規期的要求或未能在第二個180天的合規期內恢復合規,則本公司的普通股將被摘牌,屆時本公司將有機會向聽證小組就退市決定提出上訴。
The Company intends to monitor the closing bid price of its common stock and may, if appropriate, consider implementing available options to regain compliance with the minimum bid price requirement under the Nasdaq Listing Rules, including affecting a reverse stock split. The Company held a special stockholders’ meeting on February 16, 2024, to seek approval, for among other things, an amendment to our Second Amended and Restated Certificate of Incorporation, as amended, to effect a reverse stock split of our issued and outstanding shares of our common stock, by a ratio of between one-for-four to one-for-forty, inclusive, with the exact ratio to be set at a whole number to be determined by our Board of Directors or a duly authorized committee thereof in its discretion, at any time after approval of the amendment and prior to February 16, 2025. On February 16, 2024, the Company’s Board of Directors authorized a reverse stock split of our issued and outstanding shares of common stock in the amount of one-for-nineteen, which was effective on February 28, 2024. On March 13, 2024, the Company received a letter from Nasdaq notifying the Company that it has regained full compliance with the minimum bid price for continued listing on Nasdaq, pursuant to Nasdaq Listing Rule 5550(a)(2), because Nasdaq has determined that for 10 consecutive business days, the closing bid price of the Company’s common stock was at or above $1.00 per share.
於2023年10月11日,本公司收到納斯達克的書面通知,通知本公司,本公司不符合納斯達克上市規則第5635(D)條所載的股東批准要求 公開招股以外的交易涉及 以低於適用最低價格(定義見上市規則5635(D)(1)(A))的價格發行20%或以上的交易前已發行股份。
員工根據《上市規則》第5635(d)條作出的決定涉及公司發行和發行下列各項的合計:(i)35,102股公司普通股,價格為每股12.35美元,(ii)購買最多207,814股普通股的預融資認股權證,以每份預存資金認股權證12.3481美元的價格和(iii)購買最多242,915股普通股的認股權證。每股及相關普通認股權證 的發售價為12.35美元,而每份預存資金認股權證及相關普通認股權證的發售價為12.3481美元。
工作人員確定, 本次發行不是納斯達克股東批准規則的“公開發行”,原因是 發行的類型、根據配售代理協議進行的盡力而為發行,以及一名投資者購買了本次發行的98%。 因此,由於發行量佔已發行普通股的20%以上,且定價低於最低價格, 員工決定公司須根據上市規則第5635(d)條事先獲得股東批准。
98
2023年10月11日的信函 要求公司在45天內提交重新獲得合規的計劃。隨後,公司 於2023年11月9日向納斯達克提交了合規計劃,並於2023年11月14日,納斯達克批准公司將合規計劃中規定的某些交易延期至2023年12月15日,以糾正其先前違反納斯達克規則的行為,如納斯達克2023年10月11日的信函中所述。
本公司於2023年11月及12月進行了多項交易,包括修訂上述認股權證的條款,直至 本公司股東根據納斯達克上市規則批准該項發行,以重新遵守 上市規則第5635(D)(1)(A)條)。
由於這些交易,納斯達克於2023年12月14日向本公司發出書面通知,表示本公司已遵守先前延期的條款; 本公司遵守上市規則第5635(D)(1)(A)條);以及此事現已結束。
2023年11月15日, 公司收到納斯達克的函,通知公司不符合在納斯達克資本市場繼續上市的最低股東權益要求 。納斯達克上市規則第5550(B)(1)條(“規則“)要求 在納斯達克資本市場上市的公司保持至少250萬美元的股東權益。在公司截至2023年9月30日的季度報告10-Q表中,公司報告股東虧損(149,327美元),低於根據規則繼續上市所需的最低股東權益。此外,本公司並不符合納斯達克上市規則下的另類納斯達克持續上市標準。
納斯達克要求公司在2024年1月2日之前向納斯達克提交重新獲得合規的計劃。我們及時提交了恢復合規的計劃, 並且在2024年1月11日,納斯達克通知公司,它已決定批准公司延期,以重新遵守規則 。
The terms of the extension are as follows: on or before May 13, 2024, the Company must complete certain transactions described in greater detail in the compliance plan, contemplated to result in the Company increasing its stockholders’ equity to more than $2.5 million, and opt for one of the two following alternatives to evidence compliance with the Rule: Alternative 1: The Company must furnish to the SEC and Nasdaq a publicly available report (e.g., a Form 8-K) including: 1. A disclosure of the Staff’s deficiency letter and the specific deficiency(ies) cited; 2. A description of the completed transaction or event that enabled the Company to satisfy the stockholders’ equity requirement for continued listing; and 3. An affirmative statement that, as of the date of the report, the Company believes it has regained compliance with the stockholders’ equity requirement based upon the specific transaction or event referenced in Step 2; or Alternative 2: The Company must furnish to the SEC and Nasdaq a publicly available report including: 1. Steps 1 & 2 set forth above; 2. A balance sheet no older than 60 days with pro forma adjustments for any significant transactions or event occurring on or before the report date; and 3. That the Company believes it satisfies the stockholders’ equity requirement as of the report date. The pro forma balance sheet must evidence compliance with the stockholders’ equity requirement.
此外,在任何一種情況下,公司都必須披露納斯達克將繼續監督公司對股東權益要求的持續遵守情況,如果在下次定期報告時公司沒有證明遵守要求,公司可能會被摘牌 。
無論本公司選擇哪種方式 ,如果本公司在合規期結束後向美國證券交易委員會提交下一份定期報告(即截至2024年6月30日的季度報告)時未能證明其遵守情況,則本公司可能被摘牌。 如果本公司不滿足這些條款,納斯達克將以書面通知其證券將被摘牌。 屆時,本公司可就納斯達克的裁決向聽證會小組提出上訴。
公司目前正在 評估各種行動方案,以恢復合規性,並希望在合規期內恢復對納斯達克最低股東權益標準的遵守。然而,無法保證公司將能夠完成合規計劃中預期的交易 ,公司預計這些交易將使其重新遵守規則,或此類交易 將導致公司在納斯達克授予的合規期內重新遵守規則。
99
在繼續在納斯達克資本市場上市所需的 條件中,納斯達克要求我們在前兩年或前三年中的兩年中保持至少250萬美元的股東權益 或50萬美元的淨收入。截至2023年12月31日和2023年9月30日,我們的 股東權益低於250萬美元,我們沒有達到上述淨收入要求,因此, 我們目前不符合納斯達克關於最低股東權益的持續上市標準。如果 我們未能及時糾正我們遵守此類適用要求的情況,我們的普通股和公共認股權證可能會被摘牌。
由於上述原因或任何其他原因,我們未能滿足納斯達克的 持續上市要求,可能會導致我們的證券從納斯達克退市。
繼續在納斯達克上市所需的其他條件包括 要求我們擁有多數獨立董事、兩人薪酬委員會和三人審計委員會(每個 由所有獨立董事組成)。因此, 我們的普通股和公開認股權證可能會從納斯達克退市。
Even if we demonstrate compliance with the requirements of Nasdaq, we will have to continue to meet other objective and subjective listing requirements to continue to be listed on The Nasdaq Capital Market. Delisting from The Nasdaq Capital Market could make trading our common stock and Public Warrants more difficult for investors, potentially leading to declines in our share price and liquidity. Without a Nasdaq Capital Market listing, stockholders may have a difficult time getting a quote for the sale or purchase of our common stock and Public Warrants, the sale or purchase of our common stock and Public Warrants would likely be made more difficult, and the trading volume and liquidity of our common stock and Public Warrants could decline. Delisting from The Nasdaq Capital Market could also result in negative publicity and could also make it more difficult for us to raise additional capital. The absence of such a listing may adversely affect the acceptance of our common stock as currency or the value accorded by other parties. Further, if we are delisted, we would also incur additional costs under state blue sky laws in connection with any sales of our securities. These requirements could severely limit the market liquidity of our common stock and Public Warrants and the ability of our stockholders and warrant holders to sell our common stock and Public Warrants in the secondary market. If our common stock and Public Warrants are delisted by Nasdaq, our common stock and Public Warrants may be eligible to trade on an over-the-counter quotation system, such as the OTCQB Market or the OTC Pink market, where an investor may find it more difficult to sell our common stock and Public Warrants or obtain accurate quotations as to the market value of our common stock and Public Warrants. In the event our common stock and Public Warrants is delisted from The Nasdaq Capital Market, we may not be able to list our common stock on another national securities exchange or obtain quotation on an over-the counter quotation system.
一般風險因素
我們的公司註冊證書 和特拉華州法律中的條款可能會禁止收購我們,這可能會限制投資者 未來可能願意為我們普通股支付的價格,並可能會鞏固管理層。
我們的公司註冊證書 包含可能會阻止股東認為符合其最佳利益的主動收購建議的條款。 這些規定包括交錯的董事會,以及我們的董事會指定條款和發行新的 優先股系列的能力,這可能會使管理層的免職更加困難,並可能會阻礙交易,否則可能 涉及支付超過我們證券現行市場價格的溢價。我們還受到特拉華州法律下的反收購條款的約束,這可能會延遲或阻止公司控制權的變更。這些條款加在一起,可能會使管理層的免職變得更加困難,並且可能會阻礙交易,否則這些交易可能涉及支付超出我們證券現行市價的溢價 。
100
我們的專有信息或我們客户、供應商和業務合作伙伴的專有信息可能會丟失,或者我們可能會遭遇安全漏洞。
In the ordinary course of our business, we expect to collect and store sensitive data, including valuable and commercially sensitive intellectual property, clinical trial data, our proprietary business information and that of our future customers, suppliers and business partners, and personally identifiable information of our customers, clinical trial subjects and employees, patients, in our data centers and on our networks. The secure processing, maintenance and transmission of this information is critical to our operations. Despite our security measures, our information technology and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, regulatory penalties, disrupt our operations, damage our reputation, and cause a loss of confidence in our products and our ability to conduct clinical trials, which could adversely affect our business and reputation and lead to delays in gaining regulatory approvals for our future product candidates. Although we maintain business interruption insurance coverage, our insurance might not cover all losses from any future breaches of our systems.
我們的信息 技術系統故障,包括網絡安全攻擊或其他數據安全事件,可能會嚴重破壞我們的 業務運營。
Our business increasingly depends on the use of information technologies, which means that certain key areas such as research and development, production and sales are to a large extent dependent on our information systems or those of third-party providers. Our ability to execute our business plan and to comply with regulators’ requirements with respect to data control and data integrity, depends, in part, on the continued and uninterrupted performance of our information technology systems, or IT systems and the IT systems supplied by third-party service providers. As information systems and the use of software and related applications by our company, our business partners, suppliers, and customers become more cloud-based, there has been an increase in global cybersecurity vulnerabilities and threats, including more sophisticated and targeted cyber-related attacks that pose a risk to the security of our information systems and networks and the confidentiality, availability and integrity of data and information. In addition, our IT systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and backup measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we and our third-party service providers have taken to prevent unanticipated problems that could affect our IT systems, a successful cybersecurity attack or other data security incident could result in the misappropriation and/or loss of confidential or personal information, create system interruptions, or deploy malicious software that attacks our systems. It is also possible that a cybersecurity attack might not be noticed for some period of time. In addition, sustained or repeated system failures or problems arising during the upgrade of any of our IT systems that interrupt our ability to generate and maintain data, and in particular to operate our proprietary technology platform, could adversely affect our ability to operate our business. The occurrence of a cybersecurity attack or incident could result in business interruptions from the disruption of our IT systems, or negative publicity resulting in reputational damage with our stockholders and other stakeholders and/or increased costs to prevent, respond to or mitigate cybersecurity events. In addition, the unauthorized dissemination of sensitive personal information or proprietary or confidential information could expose us or other third–parties to regulatory fines or penalties, litigation and potential liability, or otherwise harm our business.
我們可能會收購其他 公司,這可能會轉移我們管理層的注意力,導致我們的股東進一步稀釋,並以其他方式擾亂 我們的運營並損害我們的經營業績。
我們將來可能會尋求 收購我們認為可以補充或擴展我們的產品供應、增強我們的技術 能力或以其他方式提供增長機會的業務、產品或技術。追求潛在收購可能會轉移管理層的注意力, 導致我們在識別、調查和追求合適的收購(無論這些收購是否完成)方面產生各種費用。 如果我們收購了其他業務,我們可能無法成功整合所收購的人員、運營和技術, 在收購後無法有效管理合並後的業務,或實現預期的成本節約或協同效應。我們也可能無法 從所收購業務中獲得預期利益,原因包括:
| ● | 產生與購置有關的費用; |
101
| ● | 將管理層的注意力從其他業務上轉移; |
| ● | 與收購相關的意外成本或負債; |
| ● | 收購對我們與協作合作伙伴的現有業務關係造成的損害 ; |
| ● | 損害我們的品牌和聲譽; |
| ● | 關鍵員工的潛在流失; |
| ● | 使用我們業務其他部分所需的資源;以及 |
| ● | 使用我們的大部分可用現金來完成收購 。 |
未來,如果我們的收購 不能產生預期回報,我們可能需要對減值評估過程產生的經營結果計入費用。 收購還可能導致股權證券的稀釋發行或債務的產生,這可能會對我們的經營業績產生不利影響 。此外,如果被收購的業務未能達到我們的預期,我們的業務、運營結果和財務狀況可能會受到不利影響 。
如果我們進行任何收購, 它們可能會擾亂我們的業務或對我們的業務產生負面影響。
如果我們在未來進行收購,在資金允許的情況下,我們可能難以將被收購公司的資產、人員和運營與我們自己的資產、人員和運營整合在一起。我們預計,我們未來可能進行的任何收購或合併都不會導致公司控制權的變更。此外,被收購業務的關鍵人員可能不願意為我們工作。我們無法預測擴張可能對我們的核心業務產生的影響。無論我們是否成功進行收購,談判都可能擾亂我們正在進行的業務,分散我們管理層和員工的注意力,並增加我們的 費用。除了上述風險外,收購還伴隨着一些固有風險,包括但不限於以下風險:
| ● | 整合收購的產品、服務或業務的困難; |
| ● | 潛在地擾亂正在進行的業務,分散我們的管理層和被收購公司管理層的注意力; |
| ● | 難以維持統一的標準、控制、程序和政策; |
| ● | 由於任何新管理人員的整合而可能損害與員工和客户的關係 ; |
| ● | 可能無法或無法通過向新客户和現有客户交叉營銷產品來實現更多銷售並增強我們的客户基礎。 |
| ● | 與收購的業務有關的任何政府法規的影響 ; |
| ● | 與被收購企業或產品線相關的潛在未知債務,或需要花費大量資金對被收購產品或運營的營銷和銷售進行重組、重新定位或修改,或對被收購公司在收購前的 行為導致的任何訴訟進行辯護,無論勝訴與否;以及 |
| ● | 根據不同司法管轄區的勞工、環境和其他法律規定的潛在費用。 |
如果我們無法成功解決與收購相關的任何這些風險或其他問題,我們的業務可能會受到嚴重的 損害,其中許多問題目前無法確定。這些風險和問題可能會擾亂我們正在進行的業務,分散我們的管理層和員工的注意力,增加我們的費用,並對我們的運營結果產生不利影響。
102
我們可能會將營運資本和未來資金用於最終不會改善我們的經營業績或增加我們證券價值的用途。
總體而言,我們對營運資本和未來可能獲得的任何新投資資本的使用擁有完全的 決定權。由於可能決定我們使用資金的數量和 各種因素,我們最終的資金支出(及其用途)可能與我們當前針對此類資金的預期運營計劃有很大差異 。
我們打算將現有的 營運資金和未來的資金用於研發和一般企業用途,包括 與完成反向合併相關的潛在費用和法律費用。我們亦會將資本用作一般營運資金用途。然而,我們沒有 關於我們資本的使用和支出更具體的計劃。我們的管理層有廣泛的自由裁量權使用我們的任何或全部可用的 資本儲備。我們的資本可能用於不改善我們的經營業績或以其他方式增加 股東投資價值的方式。
我們從未為普通股支付 或宣佈任何股息。
我們從未支付或宣佈我們的普通股或優先股的任何股息。同樣,我們預計在不久的將來不會對我們的普通股支付股息或分派 。未來普通股的任何股息將由我們的董事會酌情宣佈,並將取決於我們的收益、我們對未來運營和增長的財務需求以及我們認為適當的其他事實。 由於我們預計不會為我們的普通股支付現金股息,您的投資回報(如果有)將完全取決於我們普通股市值的增加。
通過我們通過發行額外的普通股來獲得融資和履行義務的努力,股東可能會被大幅稀釋。
在可能的情況下,董事會 將嘗試使用非現金對價來履行債務。在許多情況下,我們認為非現金對價將 包括我們普通股的限制性股份,或將向我們的高級職員、董事和適用顧問發行的股份。 我們的董事會擁有權力,無需股東採取行動或投票,但須遵守納斯達克規則和法規( 一般要求股東批准任何交易,如果交易導致發行超過我們當時流通的 普通股的20%,或代表我們當時流通的股票的20%以上的投票權,但某些例外情況除外), 發行全部或部分經授權但未發行的普通股股份。此外,我們可能會嘗試通過出售普通股股票 來籌集資金,可能會以低於市價的價格。這些行為將導致現有股東的所有權權益被稀釋, 這可能會進一步稀釋普通股賬面價值,並且這種稀釋可能是重大的。此類發行還可能有助於增強現有 管理層維持對公司控制的能力,因為這些股份可能會發行給承諾支持 現有管理層的各方或實體。
我們的增長部分取決於我們與第三方的戰略關係的成功 。
為了發展我們的業務, 我們預計我們將需要繼續依賴我們與第三方的關係,包括我們的技術提供商。確定 個合作伙伴,並與他們談判和記錄關係,需要大量的時間和資源。我們的競爭對手可能會有效地 激勵第三方偏愛他們的產品或服務,或使用我們的產品和服務。此外,我們的競爭對手收購我們的合作伙伴可能會導致我們現有和潛在客户的數量減少。如果我們 未能成功建立或維護我們與第三方的關係,我們在市場上競爭或 增長收入的能力可能會受到損害,我們的運營結果可能會受到影響。即使我們成功了,我們也不能向您保證這些 關係會增加客户對我們產品的使用或增加收入。
103
索賠、訴訟、政府調查和其他程序可能會對我們的業務和運營結果產生不利影響。
We are currently subject to, and expect to continue to be regularly subject to, actual and threatened claims, litigation, reviews, investigations, and other proceedings. In addition, we have filed lawsuits against certain parties for matters we discovered which related to KBL, prior to the Business Combination. Any of these types of proceedings may have an adverse effect on us because of legal costs, disruption of our operations, diversion of management resources, negative publicity, and other factors. Our current legal proceedings are described under, and incorporated by reference in, “Item 3. Legal Proceedings”. The outcomes of these matters are inherently unpredictable and subject to significant uncertainties. Determining legal reserves and possible losses from such matters involves judgment and may not reflect the full range of uncertainties and unpredictable outcomes. Until the final resolution of such matters, we may be exposed to losses in excess of the amount recorded, and such amounts could be material. Should any of our estimates and assumptions change or prove to have been incorrect, it could have a material effect on our business, consolidated financial position, results of operations, or cash flows. In addition, it is possible that a resolution of one or more such proceedings, including as a result of a settlement, could require us to make substantial future payments, prevent us from offering certain products or services, require us to change our business practices in a manner materially adverse to our business, requiring development of non-infringing or otherwise altered products or technologies, damaging our reputation, or otherwise having a material effect on our operations.
法律或 法規的變更,或未能遵守任何法律和法規,可能會對我們的業務、投資和經營業績造成不利影響。
我們受國家、地區和地方政府制定的法律、法規 和規則的約束。特別是,我們需要遵守某些SEC、Nasdaq和 其他法律或監管要求。遵守和監控適用的法律、法規和規則可能很困難、 耗時且成本高昂。這些法律、法規和規則及其解釋和應用也可能不時發生變化 ,這些變化可能對我們的業務、投資和經營業績造成重大不利影響。此外, 未能遵守適用的法律、法規和規則(如解釋和適用),可能會對我們的業務 和經營結果造成重大不利影響。
我們的某些高管 管理人員和董事現在隸屬於從事與我們開展的業務活動類似的業務活動的實體,因此在確定應向哪個實體提供特定業務機會 時可能存在利益衝突。
我們的高級管理人員和 董事現在或將來可能隸屬於從事與我們開展的業務活動類似的業務活動的實體。我們的高級管理人員和董事也可能意識到可能適合向我們和他們負有某些受託責任或合同義務的其他實體介紹的商機。因此,他們在確定特定業務機會應該呈現給我們公司還是其他實體時可能會有利益衝突 。這些衝突可能不會以對我們有利的方式解決,潛在機會可能會在呈現給我們之前呈現給另一個實體。我們的公司註冊證書 規定,我們放棄在向任何董事或高級管理人員提供的任何公司機會中的利益,除非 該機會僅以董事或我們公司高級管理人員的身份明確提供給該人,並且該機會是 我們合法和合同允許進行的,否則我們進行該機會將是合理的。
我們的高管、董事、證券持有人及其附屬公司可能存在與我們的利益衝突的競爭性金錢利益。
我們沒有采納 明確禁止我們的董事、執行官、證券持有人或關聯公司在我們將收購或出售的任何投資中,或在我們作為一方或擁有 利益的任何交易中,擁有直接或間接的金錢或財務利益。事實上,我們可能會與與我們的董事或執行官有關聯的目標企業達成戰略性交易。我們也沒有明確禁止任何此類人員為自己的利益從事我們開展的商業活動 的政策。因此,這些人或實體的利益可能與我們的利益發生衝突。我們的某些管理人員和董事在可能是我們競爭對手的公司任職。另請參見我們的高級管理人員和 董事的簡歷,以引用的方式併入本文下面的"第10項。董事、行政人員及企業管治”。
104
我們的業務已經 並可能繼續受到COVID—19疫情的不利影響。
2019年12月,據報道,一種新型冠狀病毒(COVID—19)在中國武漢浮出水面。2020年1月,COVID—19傳播到世界其他地區,包括美國和歐洲,遏制其傳播的努力已經加強,並取得了不同程度的成功。 因此,企業關閉,旅行和日常活動受到限制。COVID—19可能影響 我們業務的程度將取決於未來的發展,這些發展具有高度不確定性,無法自信地預測,例如疫情持續時間 、美國和其他國家的旅行限制和社交距離、企業關閉或業務中斷,以及美國和其他國家為遏制和治療該疾病而採取的行動的有效性。如果COVID—19大流行繼續 ,我們的計劃可能會被推遲或中斷。COVID—19的蔓延還造成了全球經濟的不確定性,這可能導致 合作伙伴、供應商和潛在客户密切監控其成本,並減少支出預算。上述情況可能對臨牀試驗、供應鏈、財務狀況和本公司的財務業績造成重大不利影響。
患者入組 我們的臨牀試驗、讓患者繼續參與我們正在進行的臨牀試驗、對招募的患者進行隨訪訪視以及收集 數據的工作已經並且可能繼續被推遲或受到限制,因為我們的某些臨牀試驗中心限制了他們的現場工作人員,或者由於COVID—19疫情和持續的政府限制而暫時 關閉。此外,由於聯邦或州政府施加或建議 旅行和物理距離的限制,或患者不願在大流行期間訪問臨牀試驗中心,患者可能無法或不願意訪問 臨牀試驗中心進行給藥或數據收集。COVID—19大流行導致的這些因素 可能會延遲或阻止我們臨牀試驗的預期讀數,最終可能會延遲或 阻止我們產生收入的能力,並可能對我們的經營業績產生重大不利影響。上述情況可能對臨牀試驗、供應鏈、財務狀況和本公司的財務業績造成重大不利影響。
此外,我們的冷凍 肩關節試模也受到了不利影響。2022年5月底,由於新冠肺炎和隨之而來的工作人員空缺,美國國立衞生研究所(NIHR)系統出現積壓,導致獲得批准的時間延遲 ,該試驗於2022年5月底開始接受招募。招募了9名受試者 參加試驗,直至2023年2月中旬。英國在COVID—19大流行之後,研究系統在支持服務和臨牀護理提供方面都面臨着前所未有的挑戰 。這導致 NIHR啟動了恢復和重置計劃,以識別並結束面臨挑戰的試驗。我們的凍結肩關節試驗 被認為是此類試驗之一,因為我們在開放招募中心和招募足夠的 受試者方面面臨相當大的挑戰。因此,NIHR要求首席調查員結束試驗,以便進一步招募。此關閉或未來 關閉或與未來研究招募參與者有關的困難,可能對 完成研究的能力、未來藥物的時間軸以及我們產生收入和支持運營的能力產生重大不利影響。
我們可能會受到氣候變化或法律、監管或市場對此類變化的反應的不利影響。
氣候變化的長期影響很難預測;然而,這種影響可能是廣泛的。氣候變化的影響可能包括物理風險 (例如海平面上升或極端天氣條件的頻率和嚴重程度—這可能會影響我們目前的運營,原因之一是我們的大部分運營基地位於加州,這容易受到惡劣天氣的影響)、社會和 人類影響(如人口失調或對健康和福祉的損害)、合規成本和過渡風險(如監管 或技術變化)以及其他不利影響。氣候變化的影響可能會增加某些產品、商品 和能源(包括公用事業)的成本,進而可能影響我們採購業務運營所需商品或服務的能力。氣候變化還可能導致成本增加,原因是我們設施的物理損壞或破壞、庫存損失 以及可能歸因於氣候變化的天氣事件導致業務中斷。這些事件和影響可能 對我們的業務運營、財務狀況或經營業績造成重大不利影響。
環境、社會和治理問題可能會影響我們的業務和聲譽。
Governmental authorities, non-governmental organizations, customers, investors, external stakeholders and employees are increasingly sensitive to environmental, social and governance, or ESG, concerns, such as diversity and inclusion, climate change, water use, recyclability or recoverability of packaging, and plastic waste. This focus on ESG concerns may lead to new requirements that could result in increased costs associated with developing, manufacturing and distributing our products. Our ability to compete could also be affected by changing customer preferences and requirements, such as growing demand for more environmentally friendly products, packaging or supplier practices, or by failure to meet such customer expectations or demand. We risk negative stockholder reaction, including from proxy advisory services, as well as damage to our brand and reputation, if we do not act responsibly, or if we are perceived to not be acting responsibly in key ESG areas, including equitable access to medicines, product quality and safety, diversity and inclusion, environmental stewardship, support for local communities, corporate governance and transparency, and addressing human capital factors in our operations. If we do not meet the ESG expectations of our investors, customers and other stakeholders, we could experience reduced demand for our products, loss of customers, and other negative impacts on our business and results of operations.
105
英國第我們退出歐盟 可能會導致監管和法律複雜性增加,這可能會使我們在 英國開展業務更加困難。和/或歐洲,並在確保我們的候選產品在英國的監管批准方面提出了額外的挑戰。或歐洲。
英國第自2020年1月31日起退出歐盟,過渡期至2020年12月31日,通常稱為“脱歐”, 導致了政治和經濟不確定性,包括適用於我們在英國的業務和候選產品的監管框架。和歐盟,這種不確定性可能會持續多年。英國脱歐可能會破壞英國之間貨物、服務和人員的自由流動。和歐盟,並導致法律和監管複雜性增加,以及 在歐洲開展業務的潛在成本上升。作為英國退歐的後果之一,EMA已經從英國搬遷。到門一條龍服務海運到荷蘭 這導致EMA員工大幅減少,導致並可能進一步導致其行政程序的重大中斷和 延誤,例如授予臨牀試驗許可或上市許可意見,中斷 活性物質和新藥製劑其他組分的進出口,以及臨牀試驗產品和最終授權制劑的供應鏈中斷。
監管框架中斷的累積影響可能會大大增加產品在歐盟和/或英國上市許可和商業化的開發週期 。監管複雜性可能會增加,這可能會擾亂我們臨牀試驗和監管批准的時間 。此外,國家和國際法律法規的變化和法律不確定性可能給我們的臨牀和監管策略帶來困難。由於英國退歐或其他原因,在獲得或無法獲得任何上市批准方面的任何延遲,都會阻止我們在英國將候選產品商業化。 和/或歐盟,並限制我們創造收入以及實現和維持盈利能力的能力。
此外,由於英國退歐,其他歐洲國家可能會尋求就其繼續加入歐盟進行全民公決。考慮到這些 可能性和我們可能無法預料的其他可能性,以及缺乏可比先例,目前尚不清楚英國退出會產生什麼財務、監管和法律影響。從歐盟將有哪些影響,以及我們的業務可能受到的負面影響。
越來越多的社交媒體平臺使用 給我們的業務帶來了新的風險和挑戰。
Social media is increasingly being used to communicate about pharmaceutical companies’ research, product candidates, and the diseases such product candidates are being developed to prevent. Social media practices in the pharmaceutical industry continue to evolve and regulations relating to such use are not always clear. This evolution creates uncertainty and risk of noncompliance with regulations applicable to our business, resulting in potential regulatory actions against us. For example, subjects may use social media channels to comment on their experience in an ongoing blinded clinical trial or to report an alleged adverse event. When such events occur, there is a risk that we fail to monitor and comply with applicable adverse event reporting obligations, or we may not be able to defend our business or the public’s legitimate interests in the face of the political and market pressures generated by social media due to restrictions on what we may say about our investigational product candidates. There is also a risk of inappropriate disclosure of sensitive information or negative or inaccurate posts or comments about us on any social media or networking website. Certain data protection regulations, such as the GDPR, apply to personal data contained on social media. If any of these events were to occur or we otherwise fail to comply with applicable regulations, we could incur liability, face regulatory actions or incur harm to our business, including damage to our reputation.
106
我們未來可能會產生債務 ,這可能會降低我們的財務靈活性,增加利息支出,並對我們的運營和成本產生不利影響。
我們將來可能會產生大量 債務。我們的負債水平可能會在多個方面影響我們的運營,包括以下方面:
| ● | 我們很大一部分現金流需要用於服務 我們的債務; |
| ● | 高水平的債務增加了我們對普遍不利影響的脆弱性 經濟和工業條件; |
| ● | 管理我們未償債務的協議中包含的契約 限制了我們借入額外資金和提供額外擔保權益、處置資產、支付股息和進行 某些投資; |
| ● | 與 我們的競爭對手的槓桿率較低,因此可能能夠利用我們的債務可能會出現的機會 阻止我們追求; |
| ● | 債務契約可能會影響我們規劃和反應的靈活性 經濟和行業的變化。 |
高負債水平 增加了我們可能拖欠債務的風險。我們可能無法產生足夠的現金流來支付債務本金或利息,未來的營運資金、借款或股權融資可能無法支付或再融資此類債務。 如果我們沒有足夠的資金,並且無法安排融資,我們可能不得不出售大量資產或取消一部分資產的抵押品贖回權,這可能會對我們的業務、財務狀況和運營結果產生重大不利影響。
我們可能會受到會計準則變化的不利影響。
我們的合併財務報表受美利堅合眾國普遍接受的會計原則的適用(“美國“),定期修訂或重新解釋。我們需要不時採用由公認的權威機構發佈的新的或修訂的會計準則,包括財務會計準則委員會(“FASB“)和 美國證券交易委員會。未來的會計準則可能要求我們更改合併財務報表中的會計處理方式,並可能要求我們對財務系統進行重大更改。此類變化可能會對我們的財務狀況或運營結果產生重大不利影響。
由於上述原因和本文闡述的其他原因,對我們證券的投資涉及高度風險。
項目1B。未解決的 員工意見。
沒有。
項目1C。網絡安全。
本公司瞭解預防、評估、識別和管理與網絡安全威脅相關的重大風險的重要性。聘請了專門從事 網絡安全風險的顧問,以獨立於公司的其他 風險評估流程來評估、識別和管理網絡安全威脅的風險。評估的網絡安全風險包括操作風險、欺詐、災難恢復以及違反數據隱私或安全法。評估包括與使用第三方服務提供商有關的網絡安全威脅。
由於進行了網絡安全 風險評估,公司已對網絡安全流程和控制措施實施了某些增強措施。這包括對資本設備的正式 監控、驗證數據加密和安全更新是否到位,以及增強數據共享和傳輸的安全性 。顧問還提出了其他建議,但由於預算限制,正在考慮 今後加以改進。
我們在 標題"我們的信息技術系統故障,包括網絡安全攻擊或其他數據安全事件,可能 嚴重擾亂我們的業務運作",作為我們風險因素披露的一部分,包含在本 表格10—K年度報告第1A項。
網絡安全是我們風險管理流程的重要 部分,也是我們董事會和管理層的重點領域。我們的首席財務官負責監督網絡安全威脅的風險 ,擁有管理業務風險(包括與網絡安全相關的風險)的經驗。董事會收到 有關我們網絡安全和信息安全框架、數據隱私 和風險管理的有效性的信息並定期更新。還將向董事會提供與信息系統安全和網絡安全相關的任何重大事件的最新情況 。雖然管理層和董事會將瞭解可能對公司產生重大影響的網絡安全事件,但 計劃在未來實施時對所有網絡安全事件進行正式監控。
截至 2023年12月31日止年度,未發生對公司業務策略、運營業績 或財務狀況造成重大影響的網絡安全事件。
107
項目2.財產
公司總部位於加利福尼亞州的帕洛阿爾託。本公司相信其現有租賃辦公室空間適合進行其業務。我們認為, 這種安排適合我們的業務開展,但如果 我們認為有必要,我們將為我們的業務提供一個實際地點。
項目3.法律 訴訟程序。
有時,我們可能會 成為我們正常業務過程中出現的訴訟的一方。
此類當前訴訟或 其他法律程序在本“第3項”中進行了描述,並通過引用併入其中。本年度 報告表格10—K的法律訴訟程序”,自本文自第F—1頁開始包含的綜合財務報表中“訴訟和其他損失 或有事項”標題下的“附註9—承諾和或有事項”。本公司認為,目前未決事項的決議案 不會個別或整體對我們的財務狀況或經營業績 產生重大不利影響。然而,由於發現 公司或法官、陪審團或其他事實發現者目前不瞭解的事實,因此對當前訴訟或其他法律索賠的評估可能發生變化,這些事實與管理層對此類訴訟或索賠的可能責任或結果的評估 不符。
此外,訴訟的結果 本質上是不確定的。如果在報告期內解決了一個或多個針對公司的法律問題,金額 超出管理層的預期,則該報告期內公司的財務狀況和經營成果 可能會受到重大不利影響。
項目4.礦山 安全披露。
不適用。
108
第II部
第5項登記人普通股、相關股東事項和發行人購買股權證券的市場
市場信息
我們的普通股和認股權證 分別在納斯達克資本市場上市,代碼為“ATNF”和“ATNFW”。
持有者
截至2024年3月19日,共有852,758股普通股已發行和發行,由71名記錄持有人持有,以及180,828股相關普通股 11,500,000股已發行認股權證,用於購買我們普通股的股份。每份公開認股權證使登記持有人有權以每1/760股5.75美元的價格購買1股普通股的1-760股這是一股或每股4,370美元,可在2020年12月6日(我們的首次業務合併後30天) 開始至2025年11月6日(我們的初始業務合併後5年)、紐約市時間下午5:00或更早的贖回或清算時 開始的任何時間進行調整。如果認股權證持有人持有760份認股權證,則該等認股權證將可行使本公司普通股中的一股。 行使認股權證時不會發行零碎股份,而認股權證必須只行使全部股份。
股利政策
我們從未就普通股支付或宣佈任何現金股息,在可預見的未來也不會支付現金股息。我們預計,我們將 保留所有未來收益,用於我們的業務運營和一般公司用途。未來是否派發股息將由我們的董事會自行決定。因此,投資者必須依賴於在價格上漲後出售他們的普通股,而這種情況可能永遠不會發生,這是實現投資未來收益的唯一途徑。
最近出售的未註冊證券
在截至2023年12月31日的三個月內以及截至本報告之日,本公司未出售任何 股權證券,但如之前在Form 10-Q季度報告或Form 8-K當前報告中所述,除非如下所述:
2024年3月12日,根據證券法第3(A)(9)條,K類特別投票權股份的持有人 在一項豁免登記的交易中將該等股份轉換為本公司14股普通股。作為這種轉換的結果,不再有任何K類特別投票權股票。
109
發行人購買股票證券
沒有。
特別表決權股份
我們指定了兩類優先股,分別命名為C類特別表決權股票和K類特別表決權股票(統稱為“特殊 投票權股份“),並具有以下指定的權利和首選項。
特別投票權股票 的面值為每股0.0001美元。每股特別投票權股票的權利和優先權包括以下內容:
| ● | 在我們的普通股有投票權的所有情況下都有投票權,普通股是一類; |
| ● | 特別投票權股份使持有人奧德賽信託公司(受託人)有權獲得相當於可向持有之前已發行的CannBioRex Puracheco ULC和/或KatExco Puracheco ULC(加拿大子公司)的普通股發行的普通股數量的總投票權。可交換的 股”); |
| ● | 特別表決權股份持有人(以及間接地,可交換股份持有人)在通知、報告、財務報表和出席所有股東會議方面享有與普通股持有人相同的權利。 |
| ● | 沒有分紅的權利; |
| ● | 特別表決權股份的持有人無權在公司結束、解散或清算時獲得任何相關分派的任何部分;以及 |
| ● | 本公司可於以下情況下取消特別投票權股份: 並無已發行可交換股份,且CannBioRex Puracheco ULC及KatExo Puracheco ULC並無認購權或其他承諾,而 該等購股權或其他承諾可能需要CannBioRex Puracheco ULC及KatExo Puracheco ULC發行更多可交換股份。 |
如上所述,可交換股份的持有人 通過適用的特別投票權股份擁有與普通股相對應的投票權和其他屬性。可交換股份為CBR Pharma或KatExco證券的某些前加拿大居民提供了一個機會,以獲得與重組相關的加拿大所得税方面的應納税資本利得的延期。
截至本報告日期, 沒有已發行的C類特別表決權股票或K類特別表決權股票。
第六項。[已保留]
110
項目7.管理層對財務狀況和經營業績的討論和 分析。
以下對180生命科學公司截至2023年和2022年12月31日的經營業績和財務狀況的討論和分析應與我們的綜合財務報表以及本年度報告中其他部分包括的綜合財務報表的附註一起閲讀。本管理層對財務狀況和經營業績的討論和分析包含前瞻性陳述。請參閲上面的“關於前瞻性信息的警告聲明” 。實際結果可能大不相同,因為本年度報告 其他部分的“風險因素”中討論的因素,以及我們可能不知道的其他因素。
截至2023年12月31日,我們的累計赤字為127,343,657美元,營運資金赤字為1,422,710美元,截至2023年12月31日的年度淨虧損為19,935,112美元,運營活動中使用的現金為10,922,223美元。隨附的綜合財務報表 是假設本公司將繼續作為持續經營的企業而編制的。由於我們沒有產生收入,我們需要籌集大量資本來償還債務和支付運營成本。雖然公司在2022年7月、2022年12月、2023年4月、2023年8月和2023年11月籌集了資本,但不能保證我們能夠籌集到額外的所需資本,也不能保證這些 資本將以優惠的條款獲得。
在一個競爭激烈的行業中,我們面臨着發展新企業所固有的所有重大風險。由於缺乏長期的運營歷史和我們所在市場的新興性質,我們預計將出現運營虧損,直到 我們能夠成功實施我們的業務戰略,其中包括所有相關的收入流。我們可能永遠無法實現盈利 運營或產生可觀的收入。
我們目前的最低 每月現金支出約為350,000美元。我們相信,總的來説,我們將需要大量額外資金來支持和擴大我們產品的研發和營銷,資助未來的臨牀試驗,償還債務, 提供額外設備和開發成本、付款義務、辦公空間和業務管理系統的資本支出,並支付其他運營成本,直到我們計劃的產品收入流完全實施並開始抵消我們的運營成本 。
自我們成立以來,我們一直用股權和債務融資的收益為我們的運營提供資金。我們經歷了流動性問題,原因之一是,我們以可接受的條件籌集足夠資本的能力有限。我們歷史上一直依賴發行可轉換為普通股的股票和期票來為我們的運營提供資金,並投入了大量努力來減少這一風險敞口。我們預計,在可預見的未來,我們將需要發行股票來為我們的運營提供資金,並償還我們的未償債務。 如果我們無法實現運營盈利,或者我們無法成功獲得其他形式的融資,我們將不得不 評估其他行動,以減少我們的運營費用和節省現金。我們目前的現金餘額預計僅足以為我們計劃的業務運營提供資金,直至2024年5月左右。如果沒有額外的資本可用,我們可能無法 繼續我們計劃的業務運營,可能被迫改變我們計劃的業務運營,或者可能採取其他可能對我們的股東造成不利影響的行動,包括尋求破產保護。
隨附的綜合財務報表是根據美國公認的持續經營會計原則編制的,以持續經營為基礎,考慮在正常業務過程中資產的變現和負債的清償。 因此,綜合財務報表不包括與資產可收回和負債分類有關的任何調整,如果公司無法作為持續經營企業繼續經營,可能需要進行調整。本招股説明書中包含的綜合財務報表 還包括持續經營腳註。
此外,如果可能, 我們的董事會將嘗試使用非現金對價來履行債務。在許多情況下,我們認為非現金 對價將包括我們普通股的限制性股份、優先股或購買我們普通股股份的認股權證。 我們的董事會擁有權力,無需股東採取行動或投票,但須遵守納斯達克規則和法規(通常 要求股東批准任何導致發行超過20%的本公司當時已發行普通股股票或超過20%的本公司當時已發行股票的投票權的交易),發行全部或部分經授權但未發行的普通股、優先股或購買該等普通股股份的認股權證。此外,我們可能會嘗試通過出售我們的普通股股份來籌集資本,可能在未來以低於市場價格。這些行為將導致 現有股東的所有權權益被稀釋,可能進一步稀釋普通股賬面價值,並且這種稀釋可能是重大的。此類 發行還可能有助於增強現有管理層保持對我們控制的能力,因為這些股份可能會發行給 承諾支持現有管理層的各方或實體。
111
MD&A組織
除了隨附的 合併財務報表和附註外,還提供了我們的管理層對財務狀況和經營成果的討論 和分析(“MD & A”),以幫助讀者瞭解我們的經營成果、財務狀況和現金流量 。MD & A組織如下:
| ● | 業務概況和近期事件。 公司業務和某些重要近期事件的摘要。 |
| ● | 財務報表的重要組成部分。 公司重要財務報表組成部分的摘要。 |
| ● | 行動的結果。我們的財務業績 比較截至2023年和2022年12月31日止年度的分析。 |
| ● | 流動性和資本資源。分析我們資產負債表和現金流量的變化,並討論我們的財務狀況。 |
| ● | 關鍵會計估計。我們認為對 瞭解我們報告的財務業績和預測中包含的假設和判斷。 |
業務概述和最新事件
本MD & A及截至2023年12月31日止年度的相關 財務報表主要涵蓋180的運營,這是一家總部位於加利福尼亞州帕洛阿爾託的臨牀階段生物技術 公司,專注於開發治療慢性疼痛、炎症、 纖維化和其他炎症性疾病中未滿足的醫療需求,其中抗TNF治療將為患者提供明顯的益處,通過採用創新的研究, 和適當的聯合治療。我們有三個產品開發平臺:
| ● | 纖維化和抗腫瘤壞死因子("腫瘤壞死因子”); |
| ● | 大麻二酚衍生物的藥物("中央商務區“); 和 |
| ● | α 7煙鹼乙酰膽鹼受體("α7nAChR”). |
由於 公司資源的限制,公司尚未在α 7nAChR平臺方面取得進展,同時暫停了進一步的研發活動 。
我們有幾個未來的候選產品 正在開發中,其中一個候選產品最近在英國成功完成了一項針對Dupuytren攣縮(一種影響手掌纖維結締組織發育的疾病)的2b期臨牀試驗。 180由生物技術和製藥領域的幾位世界領先的科學家創立。
我們打算投資資源 成功完成正在進行的臨牀項目,發現新的候選藥物,並開發新的分子,以建立我們現有的管道,解決未滿足的臨牀需求。候選產品是通過一個由定義的 單元操作和技術組成的平臺設計的。這項工作在研發環境中進行,該環境評估和評估工藝每個步驟的變異性 ,以定義最可靠的生產條件。
我們可能會依賴第三方 代工組織(“Cmos“)和其他第三方在未來製造和加工候選產品。我們相信,對第一批候選臨牀產品使用合同製造和測試 具有成本效益,並使我們能夠根據我們的開發計劃快速準備臨牀試驗。我們預計, 第三方製造商將能夠提供和加工足夠數量的這些候選產品,以滿足預期的 臨牀試驗需求。
112
重要財務報表組成部分
研究與開發
到目前為止,S 180‘年的研發費用主要與其三個產品平臺的發現工作以及臨牀前和臨牀開發有關:(1)纖維化和抗腫瘤壞死因子,(2) 作為CBD衍生物的藥物,以及(3)α7nAChR(目前暫停)。研發費用主要包括與這三個產品平臺相關的 成本,其中包括:
| ● | 與S合作伙伴和第三方合同組織、代表其進行研究和開發活動的調查性臨牀試驗場地和顧問 達成的協議產生的費用; |
| ● | 與臨牀材料生產有關的成本,包括支付給合同製造商的費用; |
| ● | 與執行臨牀前和臨牀試驗有關的實驗室和供應商費用; |
| ● | 與員工有關的費用,包括工資、福利、 和股票薪酬;以及 |
| ● | 設施和其他費用,包括設施租金和維護費用、折舊和攤銷費用以及其他用品費用。 |
我們在所有研究和開發成本發生的期間內對其進行支出。我們通過監控每個項目的狀態和從外部服務提供商收到的發票來累計提供服務所產生的成本。我們根據實際成本情況調整應計項目。 如果根據研發安排或許可協議欠第三方或有里程碑付款,則里程碑付款義務將在實現里程碑結果時支出。
研發活動是我們業務模式的核心。處於臨牀開發後期階段的候選產品通常比處於臨牀開發早期階段的候選產品具有更高的開發成本,這主要是由於後期臨牀試驗的規模和持續時間增加了 。我們預計,隨着臨牀項目的進展,以及我們尋求啟動更多候選產品的臨牀試驗,未來幾年的研發費用將會增加。此外,隨着有選擇地確定和開發更多候選產品,預計研發費用將會增加。然而,很難確定 當前或未來候選產品的臨牀前計劃和臨牀試驗的持續時間和完成成本。
臨牀試驗的持續時間、成本、時間和候選產品的開發將取決於各種因素,包括但不限於以下 :
| ● | 每名患者的試驗成本; |
| ● | 參與試驗的患者數量; |
| ● | 包括在試驗中的地點數目; |
| ● | 在哪些國家進行試驗; |
113
| ● | 登記符合條件的患者所需的時間長度; |
| ● | 患者接受的劑量; |
| ● | 患者的輟學率或中途停用率; |
| ● | 監管機構要求的潛在額外安全監測或其他研究 ; |
| ● | COVID—19對我們審判時間的影響; |
| ● | 病人的跟進時間為何;及 |
| ● | 候選產品的有效性和安全性。 |
此外,每個候選產品的成功概率 將取決於許多因素,包括競爭、製造能力和商業可行性 。我們將根據每種候選產品的科學和臨牀成功情況,決定實施哪些項目並資助哪些項目, ,並評估每種候選產品的商業潛力。
由於候選產品 仍處於臨牀和臨牀前開發階段,且這些努力的結果尚不確定,因此我們無法估計成功完成候選產品的開發和商業化所需的實際金額,也無法估計我們是否或何時可能實現盈利。 由於這些計劃處於早期階段,我們不會逐個項目跟蹤成本。隨着這些計劃變得更加先進, 我們打算跟蹤每個計劃的外部和內部成本。
一般和行政
一般和行政費用 主要包括工資和其他與員工相關的費用,包括已發行普通股股份的股票報酬 和授予創始人、董事和行政、商業、財務、會計、法律、投資者關係、設施、 業務開發和人力資源職能部門人員的期權(包括歸屬條件)。
其他重大的一般 和行政費用包括與設施和間接費用有關的費用、與公司和專利事務有關的法律費用、 訴訟、SEC文件、保險、投資者關係費用、會計和諮詢服務費用以及其他一般和行政費用 。一般和管理成本在發生時支銷,我們通過監控所提供服務的狀態,接收服務提供商的估計數,並根據實際成本的已知調整 應計費用, 第三方提供的與上述費用相關的服務應計費用。
預計一般 和管理費用將在未來幾年內增加,以支持我們的持續研發活動、製造 活動、候選產品的潛在商業化以及作為上市公司運營成本的增加。預計這些增加 將包括與僱用額外人員、開發商業基礎設施、向外部顧問、律師和會計師支付的費用有關的費用增加,以及與上市公司相關的費用增加,以及 與保持遵守納斯達克上市規則和SEC要求有關的服務有關的費用、保險和投資者關係成本。
114
其他收入
其他收入主要指 為其他公司(其中一些公司為關聯方)進行研發工作所賺取的費用。
利息支出
利息支出主要包括與債務工具相關的利息支出。
商譽損失 減值
商譽減值虧損 指資產賬面值超出其於報告期間 估計公平市價的差額,且不可收回。
知識產權研發損失 資產減值
知識產權研發資產減值損失 指資產賬面值超出其估計公平市價的數額,且無法收回。
投資關係研發資產公允價值變動
知識產權研發資產公允價值變動 係指報告期內該資產公允價值的非現金變動。
衍生負債的公允價值變動
衍生負債公允價值變動 指報告期內衍生負債公允價值的非現金變動。 截至2023年12月31日及2022年12月31日止年度衍生負債公允價值變動所產生的收益,是由於期內股價下跌導致相關負債的公允價值較低所致。
發行服務性普通股的收益
發行服務普通股的收益是指服務支付的每股價格高於股票在發行之日的公平市價。
115
綜合經營成果
綜合經營成果
截至2023年12月31日的年度與截至2022年12月31日的年度比較
| 截至12月31日止年度, | ||||||||
| 2023 | 2022 | |||||||
| 運營費用: | ||||||||
| 研發 | $ | 2,303,751 | $ | 2,191,834 | ||||
| 與研發相關的各方 | 480,777 | 240,731 | ||||||
| 一般和行政 | 10,646,417 | 15,459,788 | ||||||
| 一般和與行政有關的當事人 | 46,555 | 5,612 | ||||||
| 總運營費用 | 13,477,500 | 17,897,965 | ||||||
| 運營虧損 | (13,477,500 | ) | (17,897,965 | ) | ||||
| 其他(費用)收入: | ||||||||
| 其他收入 | 21,074 | - | ||||||
| 利息支出 | (44,828 | ) | (28,175 | ) | ||||
| 利息收入關聯方 | - | 1,508 | ||||||
| 商譽減值損失 | - | (33,547,278 | ) | |||||
| 知識產權研發資產減值損失 | (9,063,000 | ) | (3,342,084 | ) | ||||
| 衍生負債的公允價值變動 | 75,323 | 15,144,986 | ||||||
| 發行服務性普通股收益 | 204,405 | - | ||||||
| 其他費用合計(淨額) | (8,807,026 | ) | (21,771,043 | ) | ||||
| 所得税前虧損 | (22,284,526 | ) | (39,669,008 | ) | ||||
| 所得税優惠 | 2,349,414 | 942,749 | ||||||
| 淨虧損 | $ | (19,935,112 | ) | $ | (38,726,259 | ) | ||
研究與開發
在截至2023年12月31日的年度內,我們產生的研發費用為2,303,751美元,與截至2022年12月31日的年度的2,191,834美元相比,增加了111,917美元或5%。這一變化是由於研發税收抵免的減少,這使本季度的研發支出增加了約400,000美元;牛津大學的支出也淨增加了約37,000美元。這些增長被以下兩項所抵消:i)因裁員/解僱而減少的薪金和獎金約570 000美元,以及ii)支付給科學諮詢委員會的費用減少約90 000美元。
與研發相關的各方
在截至2023年12月31日的年度內,我們產生的與研發相關的費用為480,777美元,與截至2022年12月31日的年度的240,731美元相比,增加了240,046美元或100%。這一變化歸因於研發税收抵免的減少,這使研發費用增加了約125,000美元,以及諮詢費增加了約200,000美元 。這些增加被基於股票的薪酬減少約80000美元所抵消。
一般和行政
於截至2023年12月31日止年度內,本集團產生一般及行政開支10,646,417美元,較截至2022年12月31日止年度的15,459,788美元減少4,813,371美元或31%。這一變化是由於法律費用、保險費用、應計獎金和基於股票的薪酬費用分別減少了約280萬美元、80萬美元、70萬美元和50萬美元。
116
一般和行政相關的當事人
在截至2023年12月31日的年度內,我們產生了46,555美元的一般和行政費用相關各方,與截至2022年12月31日的年度的5,612美元相比,增加了40,943美元,增幅為730%。增加的主要原因是關聯方諮詢費增加了41,000美元。
其他費用,淨額
於截至2023年12月31日止年度內,本集團產生其他開支淨額8,807,026美元,較截至2022年12月31日止年度的21,771,043美元減少12,964,017美元或60%。費用減少的主要原因是:1)上一年度商譽減值33,547,278美元(見附註5--無形資產和長期資產減值本年度知識產權研發資產減值較上年減少5,720,916美元,及iii)衍生負債公允價值變動較上年減少15,069,663美元。
流動性與資本資源
截至2023年12月31日和2022年12月31日,我們的現金餘額分別為1,975,799美元和6,970,110美元,營運資本赤字分別為1,422,710美元和3,270,608美元。
截至2023年12月31日和2022年12月31日止年度,營運活動所用現金分別為10,922,223美元和12,127,585美元。我們在截至2023年12月31日的年度運營中使用的現金主要 歸因於我們的淨虧損19,935,112美元,經非現金支出調整後總金額為9,026,993美元,以及運營資產和負債水平變化中使用的14,101美元現金淨額。本年度非現金支出的很大一部分涉及與知識產權研發資產減值相關的910萬美元非經常性支出(見附註5-無形資產和長期資產減值(見本文件所列合併財務報表)。我們在截至2022年12月31日的年度中在運營中使用的現金主要是由於我們的淨虧損38,726,259美元,經非現金支出調整後的總金額為23,876,048美元,以及用於運營資產和負債水平變化的現金淨額2,722,626美元。 年內非現金支出的很大一部分與與商譽和知識產權研發資產減值相關的非經常性支出3,690萬美元有關(請參閲附註5--無形資產和長期資產減值,本年度衍生工具負債公允價值變動15,144,986美元抵銷。
截至 12月31日、2023年和2022年12月31日止年度,並無投資活動提供現金。
在截至2023年12月31日和2022年12月31日的年度中,通過資助活動提供的現金分別為5,907,887美元和10,873,606美元。在截至2023年12月31日的年度內,融資活動提供的現金主要包括:i)出售2023年4月和8月發售的普通股和普通股認股權證的收益 5,999,488美元;以及ii)2023年8月發售的預融資權證和普通權證重新定價的收益830,769美元, 部分被iii)與我們2023年4月和8月發售相關的總髮售成本以及 2023年8月發售的重新定價634,816美元,以及iv)貸款的收益和償還分別為969,322美元和1,257,389美元所抵消。於截至2022年12月31日止年度內,融資活動提供的現金主要包括:i)2022年7月出售普通股及普通股認股權證所得款項6,499,737美元,ii)出售2022年12月普通股及普通權證所得款項5,999,851美元,及iii)應付貸款所得款項1,060,890美元,由iv)與本公司於2022年7月及2022年12月發售所支付的發售成本分別529,982美元及484,991美元抵銷,以及v)償還應付貸款及應付貸款- 關聯方分別為1,591,035美元及81,277美元。
我們的候選產品可能永遠不會實現商業化,我們預計在可預見的未來我們將繼續虧損。 我們預計我們的研發費用、一般和管理費用以及資本支出將繼續增加 。因此,在此之前,如果我們能夠產生可觀的產品收入,我們預計將通過股權發行、債務融資或其他資本來源的組合來滿足我們的現金需求,包括潛在的合作、許可證 和其他類似的安排,這些安排可能無法以優惠條款提供(如果根本沒有)。出售額外的股權或債務證券, 如果完成,可能會稀釋我們當時的股東。我們資本的主要用途是,我們預計將繼續是 薪酬和相關費用、第三方臨牀研發服務、可能出現的許可證付款或里程碑義務 、實驗室和相關用品、臨牀成本、潛在製造成本、法律和其他監管費用以及 一般管理費用。
117
我們的重大現金需求 以及已知合同和其他義務中此類需求的時間段包括與牛津大學和Yissm的許可協議相關的里程碑和特許權使用費付款、與D&O保險相關的付款、向顧問支付的款項以及與外部諮詢公司(如法律顧問、審計師、會計師等)相關的付款。這些現金需求總額為2024年約6,000,000美元,2025至2028年約為38,000,000美元。
此外, 我們的運營計劃可能會改變,我們可能需要額外的資金來滿足臨牀試驗和其他研發活動的運營需求和資本要求。我們目前沒有信貸安排,也沒有承諾的資金來源。由於與我們候選產品的開發和商業化相關的眾多風險和不確定性,我們無法估計與我們當前和預期的產品開發計劃相關的資本支出和運營支出增加的 金額。
我們尚未實現盈利 ,預計將繼續從運營中產生現金外流。預計我們的研發以及一般和行政費用將繼續增加,因此,我們最終將需要籌集更多資金來支持我們的運營。如果我們 無法以合理的條款獲得足夠的資金,我們可能會被要求大幅縮減或停止運營,或通過以不具吸引力的條款簽訂融資協議來獲得資金。我們的運營需求包括運營業務的計劃成本, 包括為營運資本和資本支出提供資金所需的金額。截至2023年12月31日,上述條件表明,在財務報表發佈日期後的一年內,我們作為持續經營企業的能力受到了極大的懷疑 。然而,在2022年7月、2022年12月、2023年4月、2023年8月和2023年11月,公司分別籌集了約600萬美元、550萬美元、270萬美元、270萬美元和80萬美元的額外資本,截至2024年3月18日,手頭現金約為80萬美元,公司預計能夠作為一家持續經營的企業持續經營到2024年5月。
我們的合併財務報表是按照美國公認會計原則(“美國公認會計原則”)編制的,該原則考慮了公司作為持續經營企業的持續經營以及在正常業務過程中的資產變現和負債清償。合併財務報表中列報的資產和負債的賬面價值不一定代表可變現或結算價值。合併財務報表不包括這一不確定性結果可能導致的任何調整。
最近的融資
2022年7月提供
於2022年7月17日,本公司 與若干買方訂立證券購買協議,據此,本公司同意出售合共9,211股普通股,併購買合共最多6,927股普通股的預融資認股權證(“2022年7月預籌資金 認股權證”),以及購買合共最多16,138股普通股的普通股認股權證(“二零二二年七月普通認股權證”),以每股402.80美元的合併購買價及認股權證(“二零二二年七月發售”)。2022年7月發行的 總收益為6,499,737美元。扣除配售代理費和本公司應付的其他估計發行費用後,本次發行給本公司的淨收益約為600萬美元。配售代理費和發行費用約為530,000美元,計入額外支付資本的減少。2022年7月發行於2022年7月20日結束。
2022年12月提供
2022年12月20日, 公司與某些買方簽訂了一份證券購買協議,據此,公司同意出售 總數為11,316股普通股,購買總數為78,910股普通股的預融資權證(“2022年12月 預融資認股權證”),以及購買最多135,339股普通股的普通股認股權證(“2022年12月普通認股權證”),以每股66.50美元的合併購買價和認股權證(“2022年12月發售”)。 2022年12月發行的總收益為5,999,851美元。扣除 配售代理費和本公司應付的其他估計發行費用後,本次發行給本公司的淨收益約為550萬美元。配售 代理費和發行費用約為500,000美元,計入額外支付資本的減少。2022年12月 發行於2022年12月22日結束。
118
2023年4月提供
於2023年4月5日,本公司 與若干買方簽訂了證券購買協議,據此,本公司同意出售總計21,053股普通股,併購買總計最多61,615股普通股的預融資認股權證(“2023年4月預籌資金 認股權證”),以及購買合共最多82,668股普通股的普通股認股權證(“2023年4月普通 認股權證”),以每股36.29美元的合併購買價及認股權證(“2023年4月發售”)。2023年4月發行的 總收益為2,999,882美元。扣除配售代理費和本公司應付的其他估計發行費用後,本次發行給本公司的淨收益約為270萬美元。配售代理費和發行費用約為300,000美元,計入額外支付資本的減少。2023年4月發行於 2023年4月10日結束。
2023年8月提供
2023年8月9日,本公司 與認可投資者簽訂了一份證券購買協議,以及依賴於本公司於2023年7月25日向SEC提交的 註冊聲明的某些購買者,該協議於2023年8月9日生效,根據該協議,本公司同意 出售總計35,102股普通股,購買總計最多207,814股普通股的預融資權證 (“2023年8月預出資認股權證”),以及購買總計最多242,915股普通股的普通股認股權證(“2023年8月普通認股權證”),以每股12.35美元的合併購買價和認股權證(“2023年8月 發售”)。二零二三年八月發售所得款項總額為2,999,606元。扣除配售代理費和本公司應付的其他估計發行費用後,本次發行給本公司的淨收益約為270萬美元。 配售代理費用及發行費用約300,000美元已計入額外已付資本的減少。 2023年8月發售於2023年8月14日結束。
修訂2023年8月發行
2023年10月11日,該公司收到納斯達克的一封信函,稱 納斯達克已確定,該公司未能遵守上市規則5635(d)中規定的納斯達克股東批准要求,該規則要求除公開發行以外的交易事先獲得股東批准,涉及以低於最低價格發行20%或 以上的交易前已發行股份。納斯達克的信件提到了2023年8月的發行 ,並要求該公司不遲於2023年11月27日提交合規計劃。2023年11月9日,公司向納斯達克提交了 合規計劃,2023年11月14日,納斯達克向公司提供了延期函。
根據提交給納斯達克的合規計劃, 2023年11月28日,本公司對2023年8月的發行進行了修訂,(“2023年8月發售的修訂”),據此, (i)買方同意就8月股份和8月預融資權證的重新定價支付額外830,769美元 (“重新定價金額”),(ii)公司同意向買方發行額外的預融資認股權證,以購買最多 257,205股普通股,行使價為每股0.0019美元(“額外預融資認股權證”),及 認購最多477,058股普通股,行使價為每股3.23美元(“額外普通認股權證”, 統稱為“額外認股權證”),以及(iii)公司和買方同意簽訂認股權證修訂協議。扣除配售代理費和公司應付的其他估計發行費用 後,重新定價給公司帶來的淨收益約為80萬美元。配售代理費和發行費用約為60,000美元, 作為額外支付資本的減少。2023年8月發售的修訂已於2023年12月1日結束。
關鍵會計估計
公司的合併財務報表是根據 美國公認的會計原則編制的。編制該等綜合財務報表要求管理層作出 影響其資產、負債、收入及開支的呈報金額的估計及假設。公司已確定某些估計對於其業務運營以及瞭解其過去或當前與過程中研發("IP研發")相關的運營結果至關重要。這些估計被認為是關鍵的,因為它們對公司的合併財務報表有重大影響,或有可能 產生重大影響,並且因為它們要求管理層作出重大 判斷、假設或估計。本公司認為,在對下文所述項目進行會計處理時作出的估計、判斷和假設 是合理的,基於作出這些估計、判斷和假設時的可用信息。但是,實際結果可能與這些 估計值不同,這些差異可能是重大的。
119
正在進行的研究和開發
本公司有大量 知識產權研發資產,至少每年進行一次減值評估。這些資產的減值分析 因其對公司的重要性而被視為至關重要。因業務合併或收購而產生的無形資產(例如IP R & D資產)初步按估計公允價值入賬。知識產權研發資產在相關研發項目完成 或放棄之前被視為無限期生存。
公司必須 至少每年評估一次知識產權研發資產,如果發生事件或情況變化,表明 公司報告單位的公允價值更有可能低於其賬面值,則更頻繁地評估一次。在評估知識產權研發資產減值時, 公司可能首先評估定性因素,以確定其報告單位的公允價值是否更有可能低於其賬面價值。
知識產權研發資產減值
截至2022年12月31日, 資產負債表上知識產權研發資產的賬面值為12,405,084美元(其中包括與公司的CBR Pharma子公司和180 LP子公司相關的賬面值分別為1,462,084美元和10,943,000美元)。根據 截至年底從第三方獲得的估值,公司知識產權研發資產的公允市值確定為9,063,000美元(其中 包括與公司CBR Pharma子公司和180 LP子公司相關的公允市值分別為0美元和9,063,000美元)。 截至本計量日期,CBR Pharma和180 LP子公司資產的賬面值分別超出其公允市值1,462,084美元和1,880,000美元。因此,管理層確定綜合知識產權研發資產減值3,342,084美元,為確認減值,本公司於2022年第四季度記錄了該金額的虧損, 在損益表中顯示為知識產權研發資產減值虧損。截至2022年12月31日,其CBR Pharma子公司 及其180 LP子公司的知識產權研發資產餘額分別減少至零和9,063,000美元;減值後綜合知識產權研發資產餘額總額 為9,063,000美元。
120
2023年期間,公司評估了最近的商業化延遲 時間軸、總體經濟狀況、行業和市場考慮因素、公司的財務表現以及所有可能表明減值可能性的相關 法律、監管和政治因素,並得出結論,當這些因素被 共同評估時,資產更有可能出現減值。該公司錄得9,063,000美元的損失, 在損益表中作為知識產權研發資產減值損失出現。因此,截至2023年12月31日,資產負債表上知識產權研發 資產的賬面值變為0美元(無)。此外,由於資產負債表上的IP研發資產 和利潤表上的IP研發資產減值虧損,公司在利潤表上記錄了與IP研發資產減值有關的遞延 税項負債減少230萬美元,作為與IP研發資產減值有關的所得税優惠,金額相同(見 附註5—無形資產和長期資產減值, 本報告所載之綜合財務報表,以獲取進一步資料)。
近期發佈的會計公告
看見附註3--主要會計政策摘要 本年度報告所載的綜合財務報表,以獲取最近發佈和採納的會計公告的摘要。
項目7A.關於市場風險的定量 和定性披露。
根據 法規S—K(§ 229.305(e))第305項,公司無需提供本項要求的信息,因為它是"規模較小的 報告公司,"但是,根據規則229.10(f)(1)的定義,公司提供了以下與利率風險有關的信息。
利率風險
我們在其日常業務過程中面臨市場風險。 這些風險主要包括利率敏感性。截至2023年12月31日,我們擁有1,975,799美元的現金及現金等價物。我們 打算將現金存入計息貨幣市場賬户。我們面對的主要市場風險是利率敏感性, 受美國利率總體水平變動的影響。由於我們的現金等價物的期限較短,且其投資的風險較低,利率立即變動100個基點不會對我們的現金等價物的公平市價產生重大影響。
項目8.財務 報表和補充數據。
本項目所要求的信息 包含在本報告中,如"合併財務報表索引"所述,該索引出現在本年度報告表格10—K的F—1頁,本報告簽名頁之後,並通過引用併入本文。
項目9.會計和財務披露方面的變化 和與會計師的分歧。
沒有。
121
項目9A.控制 和程序。
信息披露控制和程序的評估
我們的管理層在我們的首席執行官和首席財務官的參與下,評估了我們的披露控制和程序的有效性, 根據證券交易法於2023年12月31日頒佈的規則13a—15(e)和15d—15(e)中的定義。基於此評估,首席執行官和首席財務官得出結論,公司的披露控制措施 和程序於2023年12月31日生效。
管理層關於財務報告的內部控制報告
Management of 180 Life Sciences Corp. is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Internal control over financial reporting is a process designed by, or under the supervision of, our principal executive officer and principal financial officer, or persons performing similar functions, and effected by our board of directors to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that: (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the Company’s assets that could have a material effect on the financial statements.
由於其固有的侷限性, 對財務報告的內部控制可能無法防止或發現錯誤陳述。因此,即使那些被確定為有效的系統 也只能為財務報表的編制和列報提供合理的保證, 對未來期間的任何有效性的評估的預測 都可能面臨控制措施可能因條件的變化而變得不充分或 對政策或程序的遵守程度可能惡化的風險。
重大缺陷是指控制缺陷或控制缺陷的組合,導致無法及時防止或發現年度 或中期財務報表重大錯報的可能性極低。作為一家會計資源有限的公司, 管理層的大量時間和注意力已經並將從我們的業務中轉移,以確保遵守 這些監管要求。
根據《證券法》S—K條例第10(f)(1)項的定義,我們是一家“規模較小的報告公司”。只要我們繼續是一個規模較小的報告 公司,我們就可以利用適用於 規模較小的其他上市公司的各種報告要求的豁免。
管理180生命科學公司,包括我們的首席財務官在內,對截至2023年12月31日的公司內部控制 財務報告的有效性進行了評估,使用了Treadway委員會 贊助組織委員會(“COSO”)在《內部控制—綜合框架》(2013年)中規定的標準。
管理層得出結論,根據這些標準, 公司對財務報告的內部控制於2023年12月31日生效。
122
糾正財務報告內部控制中的重大缺陷
截至2022年12月31日,本公司先前報告稱,其在財務報告內部控制方面發現了以下重大缺陷:
| ● | 本公司的審查和控制程序沒有在適當的精確度水平上運作,以檢測與一次性反向股票分割有關的認股權證公允價值和知識產權研發資產公允價值的錯誤。 |
在截至2023年12月31日的年度內,公司已採取糾正措施和/或實施控制措施,以解決上述重大缺陷, 包括對損益表項目實施詳細的波動/差異分析,以及為SEC報告增加一層審查 。我們於二零二三年已執行以下步驟:
| 1. | 每季度對損益表的波動和差異進行分析,從美元數額和百分比變化角度檢測賬户餘額的重大變動,並研究超出規定閾值的任何差異。 |
| 2. | 對SEC的報告過程實施了額外的審查,並確保整個財務報表和準備工作受到SEC報告團隊成員(而不是SEC報告經理)的一致審查。 |
根據 上述糾正措施以及截至2023年12月31日止年度完成的測試,管理層得出結論,截至2022年12月31日存在的上述重大 缺陷已得到糾正。
對控制和程序有效性的限制
在設計和評估 披露控制和程序時,管理層認識到,任何控制和程序,無論設計和操作如何良好, 都只能提供實現預期控制目標的合理保證。此外,披露控制和 程序的設計必須反映這樣一個事實,即存在資源限制,管理層在評估 可能的控制和程序相對於其成本的好處時需要運用其判斷。
財務內部控制的變化 報告
在截至2023年12月31日的季度,公司對財務報告的內部控制沒有 的變化,這對公司對財務報告的內部控制造成重大影響, 或合理可能對公司的財務報告的內部控制造成重大影響。
項目9 B.其他 信息。
規則10b5—1交易計劃。
在截至2023年12月31日的季度,公司的董事或高級職員(定義見規則16a—1(f))
項目9 C. 關於阻止檢查的外國司法管轄區的披露。
沒有。
123
第三部分
第三部分第10、11、12、13和14項所要求的信息在本年報中省略,並將在本年報涵蓋的財政年度結束後120天內以最終委託書或本年報的修訂案 的形式提交。
項目10.董事、 執行官和公司治理。
本項目所需的信息 包含在標題下"選舉董事”, “行政人員”, “公司治理 ”, “道德守則”, “董事會委員會成員、和拖欠債務的 第16(A)節報告"(在適用和保證的範圍內)將在2023年12月31日之後的120天內提交給 美國證券交易委員會(“SEC”)的公司2024年委託書中,與 公司2024年股東年會委託書的徵集有關,並以引用的方式併入本文。
項目11.行政 薪酬。
本項目所需的信息 包含在標題下"高管和董事薪酬”, “高管薪酬”, “董事薪酬”, “財政年度結束時的傑出股票獎勵”, “薪酬 委員會聯鎖和內部人士參與“和”薪酬委員會報告”(在需要的範圍內), 將在2023年12月31日之後的120天內提交給SEC的公司2024年委託書中,並通過引用併入本文 。
項目12.證券 某些實益擁有人和管理層的所有權以及相關股東事項。
本項目所需的信息 包含在標題"投票權與大股東“和”股權薪酬 計劃信息在2023年12月31日之後的120天內向SEC提交的公司2024年委託書中,並以引用方式併入本文。
項目13.某些 關係和相關交易,以及董事獨立性。
本項目要求的信息包含在標題 "下某些關係和相關交易“和”公司治理” - “董事 獨立在公司將於2023年12月31日後120天內提交給美國證券交易委員會的2024年委託書中,並通過引用併入本文。
項目14.負責人 會計費用和服務。
我們的獨立公共會計師事務所是Marcum LLP,San Francisco,CA,PCAOB審計師ID審計師事務所ID:688。
本項目所需的信息 包含在標題"批准核數師的委任”-“審計費" 將在2023年12月31日之後的120天內向SEC提交的公司2024年委託聲明中,並通過引用將其納入本文 。
124
第四部分
項目15.附件、 財務報表和 時間表
| (1) | (A)作為本年度報告一部分提交的文件: |
以下是本表格10—K中包含的或以引用方式併入本表格的財務報表、附表和附件的索引。
所有財務報表
| 合併財務報表索引 | |
| 獨立註冊會計師事務所報告 | F-2 |
| 合併資產負債表 | F-3 |
| 合併經營報表和全面虧損 | F-4 |
| 合併股東權益變動表 | F-5 |
| 合併現金流量表 | F-7 |
| 合併財務報表附註 | F-9 |
(2)合併財務報表附表
除上述規定外, 所有財務報表附表均已被省略,原因是所需信息不適用或數量不足以 要求提交附表,或所需信息包含在本表格10—K中包含的合併財務報表及其附註 中。
(3)展品
展品索引
| 不是的。 | 描述 | |
| 1.1 | 配售代理協議,日期為2023年4月5日,由180 Life Science Corp.和A.G.P./Alliance Global Partners簽訂(作為註冊人於2023年4月10日提交的8-K表格當前報告的附件1.1提交,並通過引用併入本文)。 | |
| 1.2 | 配售代理協議,日期為2023年8月9日,由180生命科學公司和AG.P./Alliance Global Partners簽署。(作為註冊人於2023年8月15日提交的8-K表格當前報告的附件1.1提交,並通過引用併入本文)。 | |
| 3.1 | 第二次修訂和重新註冊的公司證書(作為註冊人於2020年11月12日提交的8-K表格當前報告的附件3.1提交,並通過引用併入本文)。 | |
| 3.2 | 2022年12月15日提交給特拉華州州務卿的第二次修訂和重新註冊的公司證書(作為註冊人於2022年12月16日提交的8-K表格當前報告的附件3.1提交,並通過引用併入本文)。 | |
| 3.3 | 第二次修訂和重述的180生命科學公司章程,自2023年9月4日起生效(作為2023年9月7日提交的註冊人關於表格8—K的當前報告的附件3.1提交,並通過引用併入本文)。 |
125
| 4.1 | 普通股證書樣本(作為2017年4月26日提交的註冊人註冊聲明表S-1的附件4.2提交,並通過引用併入本文)。 | |
| 4.2 | 根據《1934年證券交易法》第12條註冊的註冊人證券的説明(經修訂)(作為註冊人截至2022年12月31日財政年度的10-K表格年度報告的附件4.6於2023年3月31日向委員會提交,並以引用方式併入本文)。 | |
| 4.3 | 7月普通認股權證的格式(作為2022年7月19日提交的註冊人表格8-K當前報告的附件4.2提交,並通過引用併入本文)。 | |
| 4.4 | 12月普通認股權證表格(作為2022年12月22日提交的註冊人表格8-K當前報告的附件4.2提交,並通過引用併入本文)。 | |
| 4.5 | 180 Life Sciences Corp.和銷售股東之間於2023年1月12日對2022年12月普通認股權證的第1號修正案(作為2023年1月12日提交的登記人表格8-K當前報告的附件4.1提交,並通過引用併入本文)。 | |
| 4.6 | 180 Life Sciences Corp.與出售股東於2023年4月5日簽署的認股權證第1號修正案。(作為2023年5月15日提交的註冊人關於表格10—Q的季度報告的附件10.11提交,並通過引用併入本文)。 | |
| 4.7 | 4月普通認股權證表格(作為2023年4月10日提交的註冊人表格8-K當前報告的附件4.2提交,並通過引用併入本文)。 | |
| 4.8 | 180 Life Sciences Corp.和Continental Stock Transfer & Trust Company之間於2023年8月14日簽署的預融資認股權證和普通認股權證的認股權證代理協議。(作為2023年8月15日提交的註冊人表格8-K當前報告的附件4.1提交,並通過引用併入本文)。 | |
| 4.9 | 普通認股權證格式(見附件C—2附件4.8)。(作為2023年4月15日提交的註冊人當前表格8—K報告的附件4.3提交,並通過引用併入本文)。 | |
| 4.10 | 本公司與Armistice Capital Master Fund Ltd.於2023年8月9日簽署的認股權證修訂協議(作為2023年8月15日提交的註冊人表格8-K當前報告的附件4.4提交,並通過引用併入本文)。 | |
| 4.11 | 2023年12月預付款認股權證和普通認股權證的認股權證代理協議格式(作為2023年11月29日提交的登記人表格8-K當前報告的附件4.1提交,並通過引用併入本文)。 | |
| 4.12 | 2023年12月預付款認股權證的表格(作為附件4.11的附件A)(作為附件4.2提交給2023年11月29日提交的註冊人的表格8-K當前報告,並通過引用併入本文)。 | |
| 4.13 | 2023年12月普通認股權證的表格(作為附件4.11的附件B)(作為2023年11月29日提交的註冊人表格8-K的當前報告的附件4.3提交,並通過引用併入本文)。 | |
| 4.14 | 認股權證修訂協議,日期為2023年11月28日,由公司和買方(作為2023年11月29日提交的註冊人表格8-K當前報告的附件4.4提交,並通過引用併入本文)。 | |
| 4.15 | 根據1934年《證券交易法》第12條(經修訂)註冊的註冊人證券的描述(作為2023年3月31日提交的註冊人年度報告的附件4.6提交,並通過引用併入本文)。 |
126
| 10.1 | 註冊人和某些證券持有人之間的註冊權協議(作為2017年6月7日提交的註冊人表格8-K當前報告的附件10.3提交,並通過引用併入本文)。 | |
| 10.2 | 賠償協議格式(作為2017年4月26日提交的註冊人註冊聲明表S-1的附件10.8提交,並通過引用併入本文)。 | |
| 10.3 | 擔保和承諾協議表格(作為註冊人於2019年7月26日提交的8-K表格當前報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.4# | 180生命科學公司2020綜合激勵計劃(作為註冊人於2020年11月12日提交的8-K表格當前報告的附件10.3提交,並通過引用併入本文)。 | |
| 10.5# | 股票期權協議表格(獨立董事2021年8月授予)(作為登記人截至2021年8月16日提交的截至2021年6月30日的財政季度表格10—Q季度報告的附件10.9提交,並通過引用納入本文)。 | |
| 10.6# | 股票期權協議表格180生命科學公司2020綜合激勵計劃(作為登記人於2021年9月30日提交的S-8表格的附件4.2提交,並通過引用併入本文)。 | |
| 10.7# | 限制性股票授予協議和股票期權協議表格180生命科學公司2020綜合激勵計劃(作為登記人於2021年9月30日提交的S-8表格的附件4.3提交,通過引用併入本文)。 | |
| 10.8 | 本票,日期為2019年3月15日,發行給KBLIV保薦人有限責任公司(作為登記人於2019年11月12日提交的登記説明書S-4表格的第10.13號附件提交,並通過引用併入本文)。 | |
| 10.9 | 註冊權協議,由本公司及其簽字方簽署,日期為2020年6月12日(作為註冊人於2020年7月2日提交的8-K表格當前報告的附件10.2提交,並通過引用併入本文)。 | |
| 10.10 | 註冊權協議,由本公司及其簽字方簽署,日期為2020年9月8日(作為註冊人於2020年9月14日提交的8-K表格當前報告的附件10.2提交,並通過引用併入本文)。 | |
| 10.11 | 經修訂及重訂的本票,日期為2020年9月8日,簽發予KBLIV保薦人有限責任公司(作為登記人於2020年10月19日提交的S-1表格登記聲明的第10.24號附件提交,並以引用方式併入本文)。 | |
| 10.12# | 180 Life Corp.(f/k/a 180 Life Sciences Corp.)James N.伍迪醫學博士博士(作為2020年11月12日提交的註冊人當前表格8—K報告的附件10.4提交,並通過引用併入本文)。 | |
| 10.13# | 180 Life Corp.(f/k/a180 Life Sciences Corp.)James N.伍迪醫學博士博士(作為2020年11月12日提交的註冊人當前表格8—K報告的附件10.5提交,並通過引用併入本文)。 | |
| 10.14 | 2020年11月25日的修訂協議(作為2020年11月27日提交的註冊人當前表格8—K報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.15 | 180 Life Sciences Corp.與簽署該協議的買方簽署日期為2021年2月23日的註冊權協議(作為2021年2月24日提交的註冊人當前報告的附件10.4提交,並通過引用併入本文)。 | |
| 10.16# | 180 Life Sciences Corp.與其董事和執行官之間的鎖定協議表格(作為登記人於2021年2月24日提交的表格8—K當前報告的附件10.5提交,並通過引用併入本文)。 |
127
| 10.17# | 180 Life Sciences Corp.和Jagdeep Nanchahal教授於2021年2月22日簽署的諮詢協議(作為註冊人於2021年3月3日提交的表格8—K當前報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.18# | 180 Life Sciences Corp.與James N. Woody(作為登記人於2021年3月3日提交的表格8—K當前報告的附件10.2提交,並通過引用併入本文)。 | |
| 10.19# | 詹姆斯·N.伍迪—股票期權協議,2021年2月26日生效(作為登記人於2021年3月3日提交的表格8—K當前報告的附件10.3提交,並通過引用併入本文)。 | |
| 10.20# | 180 Life Sciences Corp.和Ozan Pamir於2021年2月24日簽署並於2020年11月6日生效的僱傭協議及其修正案和更正,日期為2021年3月1日(作為註冊人於2021年3月3日提交的表格8—K當前報告的附件10.4提交,並通過引用併入本文)。 | |
| 10.21# | Ozan Pamir—股票期權協議,2021年2月26日生效(作為登記人於2021年3月3日提交的表格8—K當前報告的附件10.5提交,並通過引用併入本文)。 | |
| 10.22# | 180 Life Sciences Corp.和Jagdeep Nanchahal教授於2021年3月31日簽署的諮詢協議第一修正案(作為註冊人於2021年4月2日提交的表格8—K當前報告的附件10.2提交,並通過引用併入本文)。 | |
| 10.23 | 180生命科學公司和EarlyBirdCapital,Inc.之間於2021年5月4日簽署的和解和相互釋放協議(作為註冊人於2021年5月7日提交的8-K表格當前報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.24# | 《就業協議第二修正案》,日期為2021年5月27日,2020年11月6日生效,由180生命科學公司、Katexo製藥公司和Ozan Pamir公司簽訂(作為註冊人於2021年5月27日提交的8-K表格當前報告的10.2號附件提交,通過引用併入本文)。 | |
| 10.25# | 董事被提名人邀請函表格(作為註冊人當前報告的附件10.1於2021年5月27日提交的Form 8-K,通過引用併入本文)。 | |
| 10.26# | 註冊人與Jonathan Rothbard於2019年8月21日簽訂的僱傭協議(作為註冊人於2021年7月9日提交的截至2020年12月31日的Form 10-K年度報告的附件10.44提交,並通過引用併入本文)。 | |
| 10.27 | Alpha Capital Anstalt和註冊人之間於2021年7月31日簽署的相互釋放和和解協議(作為註冊人於2021年8月2日提交的8-K表格當前報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.28 | 180生命科學公司及其簽字人之間於2021年8月23日簽署的註冊權協議(作為註冊人於2021年8月24日提交的8-K表格當前報告的附件10.3提交,並通過引用併入本文)。 | |
| 10.29 | 180生命科學公司與其董事和高管之間的鎖定協議表(作為註冊人於2021年8月24日提交的8-K表格當前報告的附件10.4提交,並通過引用併入本文)。 | |
| 10.30£ | 180生命科學公司與Mintz,Levin,Cohn,Ferris,Glovsky和Popeo,P.C.之間於2021年9月17日簽署的和解和相互發布協議(作為註冊人於2021年9月20日提交的8-K表格當前報告的附件10.1提交,並通過引用併入本文)。 |
128
| 10.31 | 180生命科學公司與Lawrence Steinman博士和Marc Feldmann爵士之間於2021年9月30日簽署的債務轉換協議(作為註冊人於2021年10月5日提交的8-K表格當前報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.32# | 180生命科學公司和Lawrence Steinman,M.D.於2021年11月17日簽訂的諮詢協議(作為註冊人於2021年11月2日提交的8-K表格當前報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.33# | 180生命科學公司和James N.Woody,M.D.,Ph.D.於2022年4月27日修訂和重新簽署的僱傭協議的第一修正案(作為註冊人於2022年4月28日提交的8-K表格當前報告的證據10.1提交,並通過引用併入本文)。 | |
| 10.34# | 180生命科學公司和喬納森·羅斯巴德博士於2022年4月27日簽署的僱傭協議第一修正案(作為註冊人於2022年4月28日提交的8-K表格當前報告的附件10.3提交,並通過引用併入本文)。 | |
| 10.35# | Cannbiorex製藥有限公司和馬克·費爾德曼爵士博士於2022年4月27日簽署的僱傭協議第一修正案(作為註冊人於2022年4月28日提交的8-K表格當前報告的第10.4號附件提交,並通過引用併入本文)。 | |
| 10.36# | 180生命科學公司和Lawrence Steinman,M.D.於2022年4月27日簽署的諮詢協議第一修正案(作為註冊人於2022年4月28日提交的8-K表格當前報告的附件10.5提交,並通過引用併入本文)。 | |
| 10.37# | Cannbiorex Pharma Ltd.和JagDeep Nanchahal教授於2022年4月27日簽署的諮詢協議第二修正案(作為註冊人於2022年4月28日提交的8-K表格當前報告的附件10.6提交,並通過引用併入本文)。 | |
| 10.38# | 180生命科學公司和James N.Woody,M.D.,Ph.D.於2022年5月26日簽署並於2022年6月1日生效的僱傭協議第二修正案(作為註冊人於2022年5月26日提交的8-K表格當前報告的證據10.1提交,通過引用併入本文)。 | |
| 10.39# | 180生命科學公司和Jonathan Rothbard博士之間於2022年5月26日簽署並於2022年6月1日生效的僱傭協議第二修正案(作為註冊人於2022年5月26日提交的8-K表格當前報告的證據10.3提交,並通過引用併入本文)。 | |
| 10.40# | 180生命科學公司和Lawrence Steinman,M.D.於2022年5月26日簽署並於2022年6月1日生效的諮詢協議第二修正案(作為註冊人於2022年5月26日提交的8-K表格當前報告的附件10.4提交,並通過引用併入本文)。 | |
| 10.41# | 180生命科學公司2022綜合激勵計劃(作為註冊人於2022年6月14日提交的8-K表格當前報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.42£ | 180生命科學公司和買方之間於2022年7月17日簽署的證券購買協議(作為註冊人於2022年7月19日提交的8-K表格當前報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.43 | 2022年7月29日,180 Life Sciences Corp.和大陸股票轉讓和信託公司簽署的2022年7月普通權證的權證代理協議(作為2023年5月5日提交的註冊人在表格S—1上的註冊聲明的附件10.43提交,並通過引用併入本文)。 | |
| 10.44 | 鎖定協議的格式(2022年7月發售)(作為登記人於2022年7月19日提交的關於表格8—K的當前報告的附件10.2提交,並通過引用併入本文)。 | |
| 10.45£ | 2022年12月20日,180 Life Sciences Corp.與買方簽署的證券購買協議(作為登記人於2022年12月22日提交的表格8—K當前報告的附件10.1提交,並通過引用納入本文)。 |
129
| 10.46 | 180 Life Sciences Corp.和Continental Stock Transfer & Trust Company於2022年12月22日簽署的2022年12月普通認股權證的認股權證代理協議(作為登記人於2022年12月22日提交的表格8—K當前報告的附件10.3提交,並以引用方式併入本文)。 | |
| 10.47 | 鎖定協議的格式(2022年12月發售)(作為登記人於2022年12月22日提交的關於表格8—K的當前報告的附件10.4提交,並通過引用併入本文)。 | |
| 10.48# | 2022年12月28日,180 Life Sciences Corp.,Cannbirex Pharma Ltd.和Prof. Jagdeep Nanchahal(作為2022年12月29日提交的註冊人當前表格8—K報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.49# | 180 Life Sciences Corp.和Continental Stock Transfer & Trust Company於2023年1月13日對權證代理協議的修訂(作為登記人於2023年1月18日提交的表格8—K當前報告的附件10.1提交,並通過引用納入本文)。 | |
| 10.50# | 180 Life Sciences Corp.和Quan Vu於2023年1月18日簽署的分離和釋放協議(作為2023年1月20日提交的註冊人當前表格8—K報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.51# | 180 Life Sciences Corp.和Quan Vu於2023年3月29日簽署的《分離和釋放協議第一修正案》(作為附件10.59提交給委員會,截至2022年12月31日的財政年度註冊人表格10—K年度報告,並通過引用併入本文)。 | |
| 10.52£ | 2023年4月10日,180 Life Sciences Corp.與買方簽署的證券購買協議(作為登記人於2023年4月10日提交的表格8—K當前報告的附件10.1提交,並通過引用納入本文)。 | |
| 10.53 | 180 Life Sciences Corp.和Continental Stock Transfer & Trust Company於2023年4月10日簽署的2023年4月普通認股權證的認股權證代理協議(作為登記人於2023年4月10日提交的表格8—K當前報告的附件10.3提交,並以引用方式併入本文)。 | |
| 10.54 | 鎖定協議的格式(2023年4月發售)(作為2023年4月10日提交的註冊人當前報告的附件10.4提交,並通過引用併入本文)。 | |
| 10.55# | 180生命科學公司和James N.Woody,M.D.,Ph.D.於2023年4月27日簽署的就業協議第三修正案,於2023年1月1日生效(作為註冊人於2023年4月28日提交的8-K表格當前報告的證據10.1提交,通過引用併入本文)。 | |
| 10.56# | 180生命科學公司和Ozan Pamir於2023年4月27日簽署並於2023年1月1日生效的僱傭協議第三修正案(作為註冊人於2023年4月28日提交的8-K表格當前報告的附件10.2提交,並通過引用併入本文)。 | |
| 10.57# | 180生命科學公司和Jonathan Rothbard博士之間於2023年4月27日簽署並於2023年1月1日生效的僱傭協議第三修正案(作為註冊人於2023年4月28日提交的8-K表格當前報告的證據10.3提交,並通過引用併入本文)。 | |
| 10.58# | 修訂並更正了180 Life Sciences Corp.和Ozan Pamir之間日期為2023年5月9日的僱傭協議第三次修訂案(作為2023年5月15日提交的註冊人季度報告的附件10.12提交,並通過引用納入本文)。 | |
| 10.59# | 180Life Science Corp.2022綜合激勵計劃(作為註冊人於2023年7月10日提交的8-K表格當前報告的附件10.2提交,並通過引用併入本文)。 |
130
| 10.60£ | 2023年8月9日180生命科學公司和機構投資者之間的證券購買協議。(作為註冊人於2023年8月15日提交的8-K表格當前報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.61 | 鎖定協議表格(參考本公司於2023年6月16日提交的S-1表格(文件編號333-272749)註冊説明書附件10.56)。 | |
| 10.62# | 股票期權協議表格(首次修訂和重新修訂的2022年綜合激勵計劃)(作為註冊人於2023年9月7日提交的8-K表格當前報告的附件10.2提交,並通過引用併入本文)。 | |
| 10.63 | 終止函(牛津許可證)2023年9月22日(作為註冊人於2023年9月28日提交的8-K表格當前報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.64£ | 180生命科學公司和買方之間於2023年11月28日簽署的證券購買協議的第1號修正案(作為註冊人於2023年11月29日提交的8-K表格當前報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.65# | 180生命科學公司和James N.Woody,M.D.,Ph.D.於2024年1月10日簽署的就業協議第四修正案(作為註冊人於2024年1月17日提交的8-K表格當前報告的證據10.1提交,通過引用併入本文)。 | |
| 10.66# | 180生命科學公司和Jonathan Rothbard博士之間於2024年1月10日簽署並於2024年1月1日生效的就業協議第四修正案(作為註冊人於2024年1月17日提交的8-K表格當前報告的證據10.2提交,並通過引用併入本文)。 | |
| 10.67# | 180生命科學公司和Lawrence Steinman,M.D.於2024年1月10日簽署並於2024年1月1日生效的諮詢協議第三修正案(作為註冊人於2024年1月17日提交的8-K表格當前報告的附件10.3提交,並通過引用併入本文)。 | |
| 10.68# | Cannbiorex製藥有限公司和馬克·費爾德曼爵士於2024年1月10日簽署的《諮詢協議第二修正案》(作為註冊人於2024年1月17日提交的8-K表格當前報告的第10.4號附件提交,並通過引用併入本文)。 | |
| 10.69# | 180 Life Science Corp.和Blair Jordan(董事)之間的要約函,日期為2024年2月24日,生效日期為2024年2月28日(作為註冊人於2024年2月29日提交的表格8—K當前報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.70# | 180 Life Science Corp.和Omar Jimenez(董事)之間的要約函,日期為2024年3月4日,生效日期為2024年3月7日(作為註冊人於2024年3月11日提交的表格8—K當前報告的附件10.1提交,並通過引用併入本文)。 | |
| 10.71# | 180 Life Science Corp.和Ryan L. Smith(董事)日期:2024年3月5日,2024年3月7日生效 (作為2024年3月11日提交的註冊人當前表格8—K報告的附件10.2提交,並通過引用併入本文)。 | |
| 14.1# | 商業和道德準則(作為2017年4月26日提交的註冊人在表格S—1上的註冊聲明的附件14提交,並通過引用併入本文)(文件。第333—217475號) | |
| 21.1 | 子公司名單(作為註冊人截至2022年12月31日的財政年度Form 10-K年度報告的附件21.1提交給委員會,於2023年3月31日提交,並通過引用併入本文)。 | |
| 23.1* | 獨立註冊會計師事務所Marcum LLP的同意 | |
| 31.1* | 根據《薩班斯—奧克斯利法案》第302條認證首席執行官 | |
| 31.2* | 根據《薩班斯—奧克斯利法案》第302條認證首席會計官 | |
| 32.1** | 根據《薩班斯-奧克斯利法案》第906條對主要行政人員的認證 | |
| 32.2** | 根據《薩班斯—奧克斯利法案》第906條頒發的首席會計官證書 | |
| 97.1 | 180生命科學公司,關於收回錯誤獎勵獎勵的獎勵性補償的政策(作為附件10.4提交的表格10—Q季度報告由註冊人於2023年11月9日提交,並通過引用併入本文)。 | |
| 101.INS* | 內聯XBRL實例文檔—實例文檔未顯示在交互式數據文件中 因為它的XBRL標籤嵌入在內聯XBRL文檔中 * | |
| 101.Sch* | 內聯XBRL分類擴展架構文檔 | |
| 101.卡爾* | 內聯XBRL分類擴展計算鏈接庫文檔 | |
| 101.定義* | 內聯XBRL分類擴展定義Linkbase文檔 | |
| 101.實驗所* | 內聯XBRL分類擴展標籤Linkbase文檔 | |
| 101.前期* | 內聯XBRL分類擴展演示文稿Linkbase文檔 | |
| 104* | 內嵌XBRL,用於本年度報告的表格10—K封面頁,包含在附件101內嵌 XBRL文檔集 |
| * | 現提交本局。 |
| ** | 隨信提供。 |
| # | 管理合同或補償計劃或安排。 |
| £ | 根據法規S—K第601(a)(5)項,某些附表、附件和類似附件被 省略。任何遺漏的時間表或附件的副本將根據要求以書面方式提供給 證券交易委員會;但是,180生命科學公司可根據經修訂的1934年證券交易法第24b—2條要求對如此提供的任何時間表或附件進行保密處理。 |
項目16.表格 10—K摘要
沒有。
131
簽名
根據1934年《證券交易法》第13條或第15條(d)款的要求,註冊人已正式促使下列簽署人代表其簽署本報告,並經正式授權。
| 180生命科學公司。 | ||
| 日期:2024年3月22日 | /s/詹姆斯·N·伍迪 | |
| 發信人: | 詹姆斯·N. Woody,首席執行官 (首席執行官) | |
| 日期:2024年3月22日 | /s/Ozan Pamir | |
| 發信人: | Ozan Pamir,首席財務官 (首席財務和會計官) | |
根據1934年《證券交易法》的要求 ,本報告已由以下人員代表註冊人在下面簽署,並在指定的日期以註冊人的身份簽署。
| 簽名 | 標題 | 日期 | ||
| /s/詹姆斯·N·伍迪 | 董事首席執行官兼首席執行官 | 2024年3月22日 | ||
| 詹姆斯·N·伍迪 | (首席行政主任) | |||
| /s/Ozan Pamir | 首席財務官 | 2024年3月22日 | ||
| 奧贊·帕米爾 | (首席財務會計官) | |||
| /s/勞倫斯·斯坦曼 | 董事執行主席兼首席執行官 | 2024年3月22日 | ||
| 勞倫斯·斯坦曼 | ||||
| /s/布萊爾·喬丹 | 引領董事 | 2024年3月22日 | ||
| 布萊爾·喬丹 | ||||
| /s/Omar Jimenez | 董事 | 2024年3月22日 | ||
| 奧馬爾·希門尼斯 | ||||
| /s/Ryan L.史密斯 | 董事 | 2024年3月22日 | ||
| 瑞安湖史密斯 |
132
180生命科學公司。及附屬公司
截至2023年和2022年12月31日的合併財務報表
目錄
| 頁面 | |
| 獨立註冊會計師事務所報告(PCAOB ID 688) | F-2 |
| 合併財務報表: | |
| 截至2023年12月31日和2022年12月31日的合併資產負債表 | F-3 |
| 截至2023年及2022年12月31日止年度的綜合經營報表及全面虧損 | F-4 |
| 截至12月31日止年度股東(虧損)權益合併變動表, 2023年和2022年 | F-5 |
| 截至2023年及2022年12月31日止年度的綜合現金流量表 | F-7 |
| 合併財務報表附註 | F-9 |
F-1
獨立註冊會計師事務所報告
致本公司股東及董事會
180生命科學公司
對財務報表的幾點看法
我們審計了隨附的180生命科學公司的合併資產負債表,(“本公司”)截至2023年和2022年12月31日,截至2023年12月31日止兩年各年的相關合並經營及全面虧損表、股東權益變動(虧損)及現金流量表,及相關附註(統稱“財務報表”)。我們認為, 財務報表在所有重大方面公允列報了截至2023年12月31日的公司財務狀況,以及 截至2023年12月31日止兩年期間各年的經營成果和現金流量,符合美國普遍接受的會計 原則。
解釋性段落--持續關注
隨附財務報表 的編制假設本公司將持續經營。如附註2所述,本公司存在重大 營運資金不足,已產生重大虧損,需要籌集額外資金以履行其義務和維持其 運營。這些情況令人對公司持續經營的能力產生重大懷疑。管理層關於這些事項的 計劃也在附註2中描述。財務報表不包括 此不確定性結果可能導致的任何調整。
意見基礎
這些財務報表由公司管理層負責。我們的責任是根據我們的審計對公司的財務報表發表意見。我們 是一家在美國上市公司會計監督委員會(PCAOB)註冊的公共會計師事務所,根據美國聯邦證券法以及美國證券交易委員會和PCAOB的適用規則和法規,我們 必須與公司保持獨立。
我們按照PCAOB的 標準進行審計。這些標準要求我們計劃和執行審計,以獲得關於財務報表是否沒有重大錯報的合理保證,無論是由於錯誤還是欺詐。本公司不需要,也沒有聘請 對其財務報告的內部控制進行審計。
作為我們審計的一部分,我們需要了解財務報告的內部控制,但不是為了對公司財務報告的內部控制的有效性發表意見。因此,我們不表達這樣的意見。
我們的審計包括執行程序以評估財務報表重大錯報的風險(無論是由於錯誤還是欺詐),以及執行應對這些風險的程序。這些程序包括在測試基礎上審查與財務報表中的金額和披露有關的證據。 我們的審計還包括評估所使用的會計原則和管理層做出的重大估計,以及評估財務報表的整體列報。我們相信,我們的審計為我們的觀點提供了合理的基礎。
關鍵審計事項
關鍵審計事項是指因對已傳達或要求傳達給審計委員會的財務報表進行當期審計而產生的事項,以及 :(1)涉及對財務報表具有重大意義的賬目或披露,以及(2)涉及我們特別具有挑戰性的、 主觀或複雜的判斷。我們確定不存在關鍵的審計事項。
/s/Marcum有限責任公司
馬庫姆有限責任公司
我們自2019年以來一直擔任本公司的審計師
加州舊金山
2024年3月22日
PCAOB ID#688
F-2
180生命科學公司。及附屬公司
合併資產負債表
(以美元表示)
| 12月31日, | 12月31日, | |||||||
| 2023 | 2022 | |||||||
| 資產 | ||||||||
| 流動資產: | ||||||||
| 現金 | $ | $ | ||||||
| 預付費用和其他流動資產 | ||||||||
| 流動資產總額 | ||||||||
| 無形資產,淨額 | ||||||||
| 正在進行的研究和開發 | ||||||||
| 總資產 | $ | $ | ||||||
| 負債與股東權益 | ||||||||
| 流動負債: | ||||||||
| 應付帳款 | $ | $ | ||||||
| 應付帳款--關聯方 | ||||||||
| 應計費用 | ||||||||
| 應計費用--關聯方 | ||||||||
| 應付貸款--本期部分 | ||||||||
| 衍生負債 | ||||||||
| 流動負債總額 | ||||||||
| 應付貸款--非流動部分 | ||||||||
| 遞延税項負債 | ||||||||
| 總負債 | ||||||||
| 承付款和或有事項(附註9) | ||||||||
| 股東(赤字)權益: | ||||||||
| 優先股,$ | ||||||||
| C類優先股; | ||||||||
| K類優先股; | ||||||||
| 普通股,$ | ||||||||
| 額外實收資本 | ||||||||
| 累計其他綜合收益 | ( | ) | ( | ) | ||||
| 累計赤字 | ( | ) | ( | ) | ||||
| 股東(虧損)權益總額 | ( | ) | ||||||
| 總負債和股東權益 | $ | $ | ||||||
附註是這些合併財務報表的組成部分。
F-3
180生命科學公司。及附屬公司
合併經營報表和全面虧損
(以美元表示)
| 截至該年度為止 | ||||||||
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 運營費用: | ||||||||
| 研發 | $ | $ | ||||||
| 與研發相關的各方 | ||||||||
| 一般和行政 | ||||||||
| 一般和與行政有關的當事人 | ||||||||
| 總運營費用 | ||||||||
| 運營虧損 | ( | ) | ( | ) | ||||
| 其他收入(支出): | ||||||||
| 其他收入 | ||||||||
| 利息支出 | ( | ) | ( | ) | ||||
| 利息收入關聯方 | ||||||||
| 商譽減值損失 | ( | ) | ||||||
| 知識產權研發減值損失 | ( | ) | ( | ) | ||||
| 衍生負債的公允價值變動 | ||||||||
| 清償應計負債的收益 | ||||||||
| 其他費用合計(淨額) | ( | ) | ( | ) | ||||
| 所得税前淨虧損 | ( | ) | ( | ) | ||||
| 所得税優惠 | ||||||||
| 淨虧損 | $ | ( | ) | $ | ( | ) | ||
| 其他綜合損失: | ||||||||
| 外幣折算調整 | ( | ) | ( | ) | ||||
| 全面虧損總額 | $ | ( | ) | $ | ( | ) | ||
| $ | ( | ) | $ | ( | ) | |||
| 已發行普通股加權平均數: | ||||||||
附註是這些合併財務報表的組成部分。
F-4
180生命科學公司。及附屬公司
合併股東權益變動表(虧損)
(以美元表示)
| 截至2023年12月31日止的年度 | ||||||||||||||||||||||||
| 普通股 | 額外實收 | 累計 其他 全面 | 累計 | 總計 | ||||||||||||||||||||
| 股票 | 金額 | 資本 | 收入 | 赤字 | (赤字) | |||||||||||||||||||
| 餘額-2023年1月1日 | $ | $ | $ | ( | ) | $ | ( | ) | $ | |||||||||||||||
| 與反向股票分割有關的調整 | ( | ) | ||||||||||||||||||||||
| 為董事提供專業服務而發行的股份 | ||||||||||||||||||||||||
| 為專業服務而發行予執行主席的股份 | ||||||||||||||||||||||||
| 發行2023年4月預備及普通認股權證,淨額 (a) | - | |||||||||||||||||||||||
| 因行使二零二三年四月預撥備認股權證而發行的股份 (a) | ||||||||||||||||||||||||
| 與2023年4月發售有關的已發行股份 (a) | ||||||||||||||||||||||||
| 發行2023年8月預備及普通認股權證,淨額 (b) | - | |||||||||||||||||||||||
| 行使2023年8月預撥備認股權證而發行的股份 (b) | ||||||||||||||||||||||||
| 與2023年8月發售有關的已發行股份 (b) | ||||||||||||||||||||||||
| 從修訂案到 的預存資金和普通認股權證的發行 2023年8月SPA,淨值 | - | |||||||||||||||||||||||
| 基於股票的薪酬 | - | |||||||||||||||||||||||
| 綜合損失: | ||||||||||||||||||||||||
| 淨虧損 | - | ( | ) | ( | ) | |||||||||||||||||||
| 其他綜合損失 | - | ( | ) | ( | ) | |||||||||||||||||||
| 餘額-2023年12月31日 | $ | $ | $ | ( | ) | $ | ( | ) | $ | ( | ) | |||||||||||||
| (a) | |
| (b) |
附註是這些合併財務報表的組成部分。
F-5
180生命科學公司。及附屬公司
股東 (虧損)權益變動綜合報表,續
(以美元表示)
| 截至2022年12月31日止的年度 | ||||||||||||||||||||||||
| 普通股 | 額外實收 | 累計 其他 全面 | 累計 | 總計 股東的 | ||||||||||||||||||||
| 股票 | 金額 | 資本 | 收入 | 赤字 | 權益 | |||||||||||||||||||
| 餘額-2022年1月1日 | $ | $ | $ | $ | ( | ) | $ | |||||||||||||||||
| 與反向拆股相關的調整 | ||||||||||||||||||||||||
| 發行2022年7月預備權證 (c) | - | |||||||||||||||||||||||
| 因行使2022年7月預撥備認股權證而發行的股份 (c) | ||||||||||||||||||||||||
| 與2022年7月發行相關的股份(c) | ||||||||||||||||||||||||
| 發行2022年12月預融資權證 | - | |||||||||||||||||||||||
| 因行使2022年12月預融資權證而發行的股份 | ||||||||||||||||||||||||
| 與2022年12月發行相關的股份 | ||||||||||||||||||||||||
| 為董事提供專業服務而發行的股份 | ||||||||||||||||||||||||
| 基於股票的薪酬 | ||||||||||||||||||||||||
| 綜合損失: | ||||||||||||||||||||||||
| 淨虧損 | - | ( | ) | ( | ) | |||||||||||||||||||
| 其他綜合損失 | - | ( | ) | ( | ) | |||||||||||||||||||
| 餘額-2022年12月31日 | $ | $ | $ | ( | ) | $ | ( | ) | $ | |||||||||||||||
| (c) |
附註是這些合併財務報表的組成部分。
F-6
180生命科學公司。及附屬公司
現金流量簡明合併報表
(以美元表示)
| 在過去幾年裏 | ||||||||
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 經營活動的現金流 | ||||||||
| 淨虧損 | $ | ( | ) | $ | ( | ) | ||
| 對淨虧損與經營活動中使用的現金淨額進行的調整: | ||||||||
| 基於股票的薪酬 | ||||||||
| 為服務而發行的股票 | ||||||||
| 股票期權和限制性股票單位的攤銷 | ||||||||
| 商譽減值 | ||||||||
| 知識產權研發資產減值準備 | ||||||||
| 無形資產攤銷 | ||||||||
| 遞延税項負債 | ( | ) | ( | ) | ||||
| 衍生負債的公允價值變動 | ( | ) | ( | ) | ||||
| 經營性資產和負債變動情況: | ||||||||
| 預付費用和其他流動資產 | ||||||||
| 應付帳款 | ||||||||
| 應付帳款--關聯方 | ||||||||
| 應計費用 | ( | ) | ||||||
| 應計費用--關聯方 | ( | ) | ||||||
| 調整總額 | ||||||||
| 經營活動中使用的現金淨額 | ( | ) | ( | ) | ||||
| 融資活動產生的現金流 | ||||||||
| 與2022年7月出售普通股及普通股認股權證有關的發售費用 | ( | ) | ||||||
| 與2022年12月出售普通股及普通股認股權證有關的發售費用 | ( | ) | ||||||
| 與2023年4月出售普通股及普通股認股權證有關的發售費用 | ( | ) | ||||||
| 與2023年8月出售普通股及普通股認股權證有關的發售費用 | ( | ) | ||||||
| 與2023年8月發行的重新定價相關的發行成本 | ( | ) | ||||||
| 應付貸款收益(附註8) | ||||||||
| 償還應付貸款,扣除調整後的淨額(附註8) | ( | ) | ( | ) | ||||
| 償還應付貸款--關聯方 | ( | ) | ||||||
| 出售2022年7月普通股及普通股認股權證所得款項 | ||||||||
| 出售2022年12月普通股及普通股認股權證所得款項 | ||||||||
| 出售2023年4月普通股及普通股認股權證所得款項 | ||||||||
| 出售2023年8月普通股及普通股認股權證所得款項 | ||||||||
| 2022年7月行使預先出資認股權證的收益 | ||||||||
| 行使2022年12月預付資助權證的收益 | ||||||||
| 行使2023年4月預付資助權證的收益 | ||||||||
| 行使2023年8月預付資助權證所得款項 | ||||||||
| 2023年8月發行的普通股和預籌資權證的重新定價所得收益 | ||||||||
| 融資活動提供的現金淨額 | ||||||||
附註是這些合併財務報表的組成部分。
F-7
180生命科學公司。及附屬公司
現金流量簡明合併報表 ,續
(以美元表示)
| 在過去幾年裏 | ||||||||
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 匯率變動對現金的影響 | ( | ) | ||||||
| 現金淨減少 | ( | ) | ( | ) | ||||
| 現金--期初 | ||||||||
| 現金--期末 | $ | $ | ||||||
| 現金流量信息的補充披露: | ||||||||
| 在此期間支付的所得税現金 | $ | $ | ||||||
| 期內支付的利息現金 | $ | $ | ||||||
附註是這些合併財務報表的組成部分。
F-8
180生命科學公司。及附屬公司
合併財務報表附註
(以美元計算的金額,但股份金額除外)
注1 -業務組織和 運營性質
180生命科學公司,前身為KBL Merger Corp. IV(“180LS”,或與其子公司一起統稱為“公司”),是一家空白支票公司 於2016年9月7日根據特拉華州法律成立。本公司成立的目的是實現合併、股票交換、資產收購、股份購買、重組或與一個或多個業務的類似業務合併。
180 Life Corp.("180", f/k/a 180 Life Sciences Corp. and CannBioRx Life Sciences Corp.)是本公司的全資子公司,於2019年1月28日在特拉華州註冊成立。本公司位於美國(“美國”)。是一家醫療製藥 公司,通過180旗下的三家全資子公司180 Therapeutics L.P.(“180 LP”)、CannBioRex Pharmaceuticals Corp.(“CBR Pharma”)和Katexco Pharmaceuticals Corp.(“Katexco”),專注於炎症性疾病、纖維化和慢性疼痛領域未滿足的醫療需求。180 LP、CBR Pharma和Katexco合併為“180子公司”。Katexco於2018年3月7日根據不列顛哥倫比亞省《英國公司法》的條款註冊成立。此外,180的全資子公司Katexco Callco,ULC,Katexco Producaseco, ULC,CannBioRex Callco,ULC和CannBioRex Producaseco,ULC於2019年5月31日在加拿大不列顛哥倫比亞省成立,以 促進對Katexco,CBR Pharma和180 LP的收購。於二零二一年七月一日,加拿大公司(Katexco 及CBR Pharma)的資產及負債已轉移至其各自的附屬公司,即Katexco Pharmaceuticals Corp.(“Katexco U.S.”)。 和CannBioRex Pharma Limited(“CBR Pharma U.K.”)。
該公司是一家臨牀 階段的生物技術公司,專注於開發治療慢性疼痛、炎症、纖維化和其他炎症性疾病的未滿足醫療需求,其中抗TNF治療將通過採用創新研究和 (如果適當)聯合治療為患者提供明顯的益處。我們有三個產品開發平臺:
| ● | 肝纖維化和抗腫瘤壞死因子(“TNF”); |
| ● | 大麻二酚(“CBD”)衍生物類藥物;及 |
| ● | α7煙鹼型乙酰膽鹼受體(“α7nAChR”)。 |
由於 公司資源的限制,公司尚未在α 7nAChR平臺方面取得進展,並在此期間暫停了 進一步的研發活動。
風險和不確定性
管理層繼續評估 COVID—19疫情、俄羅斯—烏克蘭戰爭、以色列—哈馬斯戰爭以及利率波動對經濟和 資本市場的影響,並得出結論認為,儘管此類事件合理可能對公司 財務狀況產生負面影響,但截至該等綜合財務報表日期,具體影響尚不容易確定。綜合 財務報表不包括可能由這些不確定性的結果導致的任何調整。
當前充滿挑戰的經濟環境可能導致現金流、營運資金水平和/或債務餘額發生不利變化,這也可能對公司未來的經營業績和財務狀況產生直接影響。 目前尚不清楚政府幹預經濟的最終持續時間和規模以及政府幹預對公司的財務影響的效果。這種影響的程度將取決於未來的事態發展,這些事態發展具有高度的不確定性,不在本公司的控制之下。
2024年期間反向庫存拆分
2024年2月26日,
公司董事會批准了對公司發行在外普通股進行一股19股反向股票分割
,並批准了對我們的公司註冊證書進行修改,以影響此類反向股票分割,該修改證書於
2024年2月26日提交。反向股票拆分於2024年2月28日東部時間上午12:01生效,股票在2024年2月28日開市時開始交易
。與反向股票分割有關,截至生效時間,公司
已發行和流通的普通股每19股自動轉換為
關於反向
拆分,所有未行使的期權、認股權證和其他使其持有人有權購買或以其他方式接收普通股股票的證券都按照每種證券的條款進行了調整。根據本公司的
股權激勵計劃可授予的股份數量也進行了適當調整。反向拆分後,普通股的面值保持不變
為美元
19股反向股票分割的影響已追溯 在本報告中得到反映;有關更多詳細信息,請參見附註13—後續事項。
F-9
注2-持續經營和管理層的計劃
公司自成立以來沒有產生
任何收入,併產生了重大損失。截至2023年12月31日,公司累計虧損
美元
該等綜合財務報表 乃根據持續經營的假設編制,即假設本公司將能夠在正常業務過程中變現其資產 並清償其負債。公司繼續經營的能力取決於 為其正在進行的經營獲得新的融資。本公司未來可用的融資選擇包括股權融資和貸款 ,如果本公司無法及時或以優惠條件獲得此類額外融資,本公司可能不得不縮減其開發、 營銷和促銷活動,這將對其業務、財務狀況和經營業績產生重大不利影響, 它最終可能被迫停止運營並進行清算。這些事項對公司 在合理期限內持續經營的能力產生了重大疑問,該期限定義為綜合 財務報表發佈之日起一年內。本公司資產的變現可能與本綜合財務報表中呈列的賬面值存在重大差異,且隨附的綜合財務報表不包括如果本公司無法持續經營, 可能需要的任何調整。
注3-重要會計政策摘要
陳述的基礎
隨附的合併財務報表是根據美國公認會計原則(“美國公認會計原則”)編制的。
合併原則
合併財務報表包括公司及其全資子公司180 LP、CBR Pharma、Katexco和180 Life Corp.(“180 LC”)的賬目。 所有公司間結餘及公司間交易已於合併時對銷。合併財務報表 以美元列報。
預算的使用
編制符合美國公認會計準則的財務報表需要管理層作出估計、判斷和假設,這些估計、判斷和假設會影響資產、負債、收入和支出的報告金額,以及合併財務報表相關附註中披露的金額。本公司在這些財務報表中使用的重大 估計和假設包括但不限於金融工具、 認股權證、期權和衍生負債的公允價值;研發税收抵免和應計費用,以及與知識產權研發資產減值分析相關的估計和假設 。公司的某些估計可能受到外部條件的影響,包括公司特有的那些 和總體經濟條件。這些外部因素可能對公司的 估計產生影響,並可能導致實際結果與這些估計不同。
外幣折算
公司報告
貨幣為美元。
全面收益定義為:
除所有者投資或向所有者的分派以外,所有來源的實體權益變動,包括上文所述的外幣換算調整。截至2023年及2022年12月31日止年度,本公司錄得其他全面虧損
以外幣計值的交易(包括公司間交易)產生的外幣收益和
損失計入
經營結果。本公司確認美元
F-10
現金和現金等價物
本公司將原到期日為三個月或以下的所有
高流動性投資視為財務報表中的現金等價物。本公司
於2023年或2022年12月31日無現金等價物。截至2023年12月31日,該公司在美國和英國有銀行賬户;在其可用現金餘額中,
商譽
商譽指在業務合併中收購的資產和負債的購買價格與公允價值之間的差額 。本公司每年審查商譽 ,或當情況和情況發生變化,有跡象表明賬面值可能 無法收回時,更頻繁地審查商譽減值,方法是初步考慮定性因素,以確定報告單位的公允價值是否更有可能低於其賬面值,包括商譽,作為確定是否有必要 進行定量分析的基礎。如果確定報告單位的公允價值更有可能低於 其賬面值,則進行定量分析以識別商譽減值。如果確定報告單位的公允價值不太可能 低於其賬面值,則無需進行定量 分析。本公司可選擇繞過定性評估而直接進行定量分析。有關進一步信息,請參閲“附註5—無形資產和長期資產減值”。
無形資產和在製品研發("IP研發")
無形資產 包括Katexco持有的許可專利以及與重組有關獲得的技術許可。許可專利在專利的剩餘壽命內進行攤銷。技術許可證指為開發 和商業化某些許可證和知識而獲得的許可證的公允價值。技術許可在相關專利的 估計使用壽命內以直線法攤銷。有必要根據公司研發活動的結果,監控並可能調整許可專利和 技術許可證的使用壽命。
知識產權研發資產指
分配給2019年7月16日就重組收購的技術的公允價值,該等技術尚未達到
技術可行性且無替代未來用途。知識產權研發資產被視為具有無限壽命,直至相關研發項目完成
或放棄。在知識產權研發資產被視為無限期存續期間,
每年對其進行減值測試,或如果公司意識到任何事件或情況下發生的變化
表明知識產權研發資產的公允價值低於其賬面值,則進行更頻繁的減值測試。如果開發
完成(通常在監管部門批准後進行),並且公司能夠將與知識產權
研發資產相關的產品商業化,則這些資產將被視為具有固定壽命,並根據其當時的估計使用壽命進行攤銷。
如果開發被終止或放棄,本公司可記錄與知識產權研發資產相關的全部或部分減值費用,
計算方法為知識產權研發資產的賬面值超出其估計公允價值的差額。於二零二三年,本公司錄得
知識產權研發資產減值虧損金額為美元,
金融工具的公允價值
本公司根據《會計準則法典》("ASC")820 "公允價值計量"("ASC 820")的指導,計量金融資產和負債的公允價值,該準則定義了公允價值,建立了公允價值計量框架,並擴大了公允價值計量的 披露。
F-11
ASC 820將公允價值 定義為在計量日市場參與者之間有序交易中為資產或負債在本金或最有利的 市場上轉移負債而收取的交換價格(退出價格)。ASC 820還建立了公允價值等級,要求實體在計量公允價值時最大限度地使用可觀察到的投入,最大限度地減少使用不可觀察到的投入 。ASC 820描述了可用於計量公允價值的三個級別的投入:
| ● | 第1級—相同資產或負債在活躍市場的報價; | |
| ● | 第2級—活躍市場或可觀察輸入數據中類似資產和負債的報價;及 | |
| ● | 第三級—不可觀察的輸入(例如,基於假設的現金流量建模輸入)。 |
由於本公司某些 金融工具(主要包括應付貸款)的賬面值與這些 綜合財務報表中呈列的公允價值相若,原因是這些工具的短期性質。本公司的衍生負債採用第3級輸入值估值 (更多信息請參見附註7—衍生負債)。
基於股票的薪酬
本公司根據獎勵的公允價值計量為換取權益工具獎勵而獲得的服務成本 。獎勵的公允價值 於授出日期計量,並由管理層根據對普通股近期現金銷售價格的觀察進行估計。 公允價值金額隨後在需要提供服務以換取獎勵的期間內確認,通常為 歸屬期。在行使期權或認股權證時,公司將從其授權但 未發行的股票中發行新的普通股。
衍生負債和可轉換工具
公司評估其 債務和股權發行,以確定這些合同或這些合同的嵌入式組成部分是否符合需要在公司財務報表中單獨 確認的衍生工具的資格。實體必須考慮是否將可能以其自身股票(如認股權證)結算的合同分類為實體的股權或資產或負債。如果不屬於實體控制範圍的事件 可能需要現金淨額結算,則合同應分類為資產或負債,而不是權益。
這種會計處理的結果是嵌入衍生工具的公允價值在每個資產負債表日按市價計價,並記錄為負債 ,公允價值變動計入綜合經營報表中的其他(費用)收入淨額。在有多個需要分叉的嵌入式儀器的情況下,所有分叉的衍生工具將作為單個、複合衍生工具入賬。衍生工具的分類,包括該等工具是否應記作負債或權益,將於每個報告期結束時重新評估。最初被歸類為權益並須重新分類的權益工具,將按工具於重新分類日期的公允價值重新分類為負債。衍生工具負債在資產負債表內分類為流動或非流動,以衍生工具是否預期於資產負債表日起十二個月內以現金淨額結算為基礎。
F-12
如果嵌入式轉換 期權不需要分叉,則本公司隨後通過將本公司相關股票截至承諾日的公允 價值與工具的有效轉換價格(內在 價值)進行比較,評估是否存在有利轉換特徵。
這些 安排下的債務折價在相關債務的期限內攤銷至其所述的贖回日期,並在綜合經營報表中分類為利息支出 。只有當贖回成為可能時,優先股折扣才會增加到其贖回價值。
對可轉換票據的修訂進行評估,以確定其是否應被視為對原始票據的修改而不改變 會計,或者如果條款發生重大變化,則應被視為原始票據的終止和新票據的發行。
本公司已使用柏力克—舒爾斯期權定價模式計算已發行認股權證及期權的 公允價值。認股權證、可轉換 票據和可轉換優先股使用的預期期限是合同有效期,而發行期權使用的預期期限是所授出期權預期未行使的估計期限。本公司利用"簡化"方法來制定"普通香草"期權授予的預期期限的估計 。本公司使用的是一個基於歷史波動率審查的預期波動率數字,在一段時間內(相當於被估值工具的預期壽命), 在其行業內處於類似地位的上市公司。無風險利率是根據剩餘期限與估值工具預期期限一致的美國國債零息 的隱含收益率確定的。
每股普通股淨虧損
每股普通股 基本淨虧損的計算方法是,淨虧損除以該期間已發行普通股的加權平均數。每股普通股攤薄淨虧損 的計算方法是:淨虧損除以已發行普通股加權平均數,加上如果發行普通股等價物(使用庫存股票或 轉換方法計算),如果具有攤薄效應,則會發行的額外普通股數量。
| 截至12月31日止年度, | ||||||||
| 2023 | 2022 | |||||||
| 選項 | ||||||||
| 認股權證 | (1) | |||||||
| 潛在稀釋股份總數 | ||||||||
| (1) |
研究與開發
研發費用
在發生時計入運營部門。截至二零二三年及二零二二年十二月三十一日止年度,本公司產生美元
所得税
公司根據ASC主題740 "所得税"("ASC 740")的規定對所得税進行核算。
對於財務報表或納税申報表中已計入或未計入的項目的預期未來税務後果,本公司確認遞延的 納税資產和負債。遞延税項資產及負債乃根據資產及負債的課税基準與其各自按現行税率計算的財務報告金額(“暫時性差異”)之間的差額(“暫時性差異”)而釐定。
本公司利用確認門檻和計量程序確認財務報表,並對納税申報表中已採取或預期採取的納税狀況進行計量 。本公司的政策是將税務相關利息的評估(如有)歸類為利息支出,並將罰款 歸類為綜合經營報表和全面虧損中的一般和行政費用。
F-13
近期發佈的會計公告
2016年6月,FASB發佈了ASU 2016—13, 金融工具.信用損失(主題326):金融工具信用損失的測量. 該準則的主要目標是通過要求提前確認融資應收款 和範圍內其他金融資產的信貸損失,來改進財務報告。新指南代表了對信貸損失會計處理的重大變化,包括i)將在初始確認範圍內資產時確認全 全存續期預期信貸損失;ii)當達到可能閾值時確認虧損的當期已發生虧損減值 模型將被不設 確認閾值的預期信貸損失減值法取代;以及iii)預期信貸損失估計將基於歷史信息、當前條件和 合理且可支持的預測。ASU 2016—13對SEC申報人(不包括有資格成為較小報告 公司的申報人)有效期為2019年12月15日之後,對所有其他實體有效期為2022年12月15日之後。 公司已採納本準則,自2023年1月1日起生效,其採納對公司的綜合 財務報表及相關披露並無重大影響。
2023年11月27日,FASB發佈了2023—07會計準則更新(“ASU”), 分部報告(主題280). 該等修訂旨在改善可報告分部披露要求,主要通過加強有關重大 分部開支的披露。此外,該等修訂加強了中期披露規定,澄清實體可披露 多個分部損益計量的情況,為具有單一可報告分部的實體提供新的分部披露規定, 幷包含其他披露規定。該等修訂旨在使投資者能夠更好地瞭解實體的 整體表現並評估潛在的未來現金流量。ASU適用於所有需要根據ASC 280報告分部 信息的公共實體。由於本公司無需報告分部信息,本聲明 對本公司的合併財務報表和相關披露沒有重大影響。
2023年12月14日,FASB發佈ASU 2023—09, 所得税(專題740):所得税披露的改進. ASU側重於圍繞實際税率和支付的現金所得税的所得税披露。ASU 2023—09在很大程度上遵循了2023年早些時候發佈的擬議ASU,並進行了幾項重要的修改和澄清,下文將討論。ASU 2023—09在2024年12月15日(通常為2025年日曆年度)之後的年度期間對公共 商業實體有效,一年後對所有其他商業 實體有效。實體應在未來的基礎上採納本指引,但允許追溯應用。 公司目前正在評估該ASU的影響。
管理層認為,任何其他最近發佈但尚未生效的會計公告(如果目前採用)不會對公司的綜合財務報表產生重大影響。
附註4-預付費用和其他流動資產
| 12月31日, | ||||||||
| 2023 | 2022 | |||||||
| 保險 | $ | $ | ||||||
| 研發費用應收税額抵免 | ||||||||
| 專業費用 | ||||||||
| 增值税應收賬款 | ||||||||
| 税費 | ||||||||
| $ | $ | |||||||
F-14
注5—無形資產和 長期資產的損害
| 加權
平均值 剩餘 攤銷 期間 | 截至2023年12月31日 | 截至2022年12月31日 | ||||||||||||||||||||||||||
| 以年數為單位 12月31日, 2023 | 毛收入 資產 價值 | 累計 攤銷 | 網絡 攜帶 價值 | 毛收入 資產 價值 | 累計 攤銷 | 網絡 攜帶 價值 | ||||||||||||||||||||||
| 獲得許可的專利 | $ | $ | ( | ) | $ | $ | $ | ( | ) | $ | ||||||||||||||||||
| 技術許可證 | ( | ) | ( | ) | ||||||||||||||||||||||||
| $ | $ | ( | ) | $ | $ | $ | ( | ) | $ | |||||||||||||||||||
自獲得之日起,許可專利和技術許可的總資產 值的變化是外匯匯率變化的結果。
公司記錄了攤銷費用
$
| 截至12月31日止的年度, | ||||
| 2024 | $ | |||
| 2025 | ||||
| 2026 | ||||
| 2027 | ||||
| 2028 | ||||
| 此後 | ||||
| $ | ||||
商譽減值
該公司公開交易的股票收盤價為$
截至2023年12月31日和
2022年12月31日,商譽餘額為美元
F-15
| CBR製藥 商譽 | 180 LP 商譽 | 已整合 商譽 | ||||||||||
| 餘額,2022年1月1日 | $ | $ | $ | |||||||||
| 貨幣換算 | ( | ) | ( | ) | ||||||||
| 商譽減值 | ( | ) | ( | ) | ( | ) | ||||||
| 平衡,2022年12月31日 | $ | $ | $ | |||||||||
知識產權研發資產減值
截至2022年12月31日,資產負債表上知識產權研發資產的賬面價值為$
F-16
在2023年期間,本公司評估了其商業化時間表的最新延遲、一般經濟狀況、行業和市場考慮因素、公司的財務業績以及可能表明減值可能性的所有相關法律、法規和政治因素,並
得出結論,當綜合評估這些因素時,資產更有可能減值。公司記錄了一筆虧損,金額為#美元。
由於資產負債表中知識產權研發資產的註銷和損益表中知識產權研發資產的減值損失,本公司計入與知識產權研發資產減值相關的遞延税項負債減少
$
以下是本公司截至2023年12月31日及2022年12月31日止年度的知識產權研發活動摘要,其中包括上述知識產權研發資產的已記錄虧損。
| CBR 製藥IP 研發資產 | 180 LP IP 研發 資產 | 已整合 知識產權研發 資產 | ||||||||||
| 餘額,2022年1月1日 | $ | $ | $ | |||||||||
| 貨幣換算 | ( | ) | ( | ) | ||||||||
| 知識產權研發資產減值準備 | ( | ) | ( | ) | ( | ) | ||||||
| 平衡,2022年12月31日 | ||||||||||||
| 知識產權研發資產減值準備 | ( | ) | ( | ) | ||||||||
| 平衡,2023年12月31日 | $ | $ | $ | |||||||||
附註6--應計費用
| 12月31日, | ||||||||
| 2023 | 2022 | |||||||
| 諮詢費 | $ | $ | ||||||
| 專業費用 | ||||||||
| 訴訟應計費用(1) | ||||||||
| 僱員及董事薪酬 (2) | ||||||||
| 研發費 | ||||||||
| 利息 | ||||||||
| 其他 | ||||||||
| $ | $ | |||||||
| (1) | |
| (2) |
截至2023年12月31日和2022年12月31日,應計費用相關方為及$
F-17
附註7--衍生負債
| 截至2023年12月31日止的年度 | ||||||||||||||||||||||||
| 認股權證 | ||||||||||||||||||||||||
| 公眾 | 私 | Alpha | ||||||||||||||||||||||
| SPAC | SPAC | 管道 | AGP | 資本 | 總計 | |||||||||||||||||||
| 截至2023年1月1日餘額 | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
| 衍生負債的公允價值變動 | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||
| 截至2023年12月31日的餘額 | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
| 截至2022年12月31日止的年度 | ||||||||||||||||||||||||
| 認股權證 | ||||||||||||||||||||||||
| 公眾 | 私 | Alpha | ||||||||||||||||||||||
| SPAC | SPAC | 管道 | AGP | 資本 | 總計 | |||||||||||||||||||
| 截至2022年1月1日的餘額: | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
| 衍生負債的公允價值變動 | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| 截至2022年12月31日的餘額 | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
F-18
12月31日, 2023 | |||||
| 無風險利率 | % | ||||
| 預期期限(以年為單位) | |||||
| 預期波動率 | % | ||||
| 預期股息 | % | ||||
SPAC認股權證
公共SPAC搜查證
KBL首次公開募股的參與者總共獲得了
F-19
私人SPAC認股權證
KBL首次私募的參與者總共獲得了
喉管搜查證
2021年2月23日,
公司發行了為期5年的認股權證(“管道權證”),用於購買
以下假設 用於在發行時對管道認股權證進行估值:
| 2021年2月23日 | ||||
| 無風險利率 | % | |||
| 預期期限(以年為單位) | ||||
| 預期波動率 | % | |||
| 預期股息 | % | |||
其他手令
AGP授權
鑑於業務合併於2020年11月6日完成,本公司有義務承擔購買以下股份的五年認股權證
F-20
於2021年3月12日,本公司
向AGP(“AGP認股權證”)發出認股權證,購買合共
以下假設 用於在發行時對AGP認股權證進行估值:
| 2021年3月12日 | ||||
| 無風險利率 | % | |||
| 預期期限(以年為單位) | ||||
| 預期波動率 | % | |||
| 預期股息 | % | |||
Alpha Capital Anstalt(“Alpha”) 認股權證
關於2021年7月29日達成的阿爾法
和解協議(於2021年7月31日簽署),本公司發行了一份為期三年的購買權證,
以下假設 用於在發行時對Alpha認股權證進行估值:
| 7月29日, 2021 | ||||
| 無風險利率 | % | |||
| 預期期限(以年為單位) | ||||
| 預期波動率 | % | |||
| 預期股息 | % | |||
F-21
授權證活動
| 手令的數目 | 加權平均 行使價 | 加權平均 剩餘生命 年 | 固有的 值 | |||||||||||||
| 未償還,2022年1月1日 | $ | $ | - | |||||||||||||
| 已發佈 | ||||||||||||||||
| 已鍛鍊 | ( | ) | ||||||||||||||
| 未清償,2022年12月31日 | $ | |||||||||||||||
| 中國發布了一份聲明。 | ||||||||||||||||
| *被行使。 | ( | ) | ||||||||||||||
| 未清償,2023年12月31日 | $ | $ | ||||||||||||||
| 可行使,2023年12月31日 | $ | |||||||||||||||
| 未清償認股權證 | 可行使的認股權證 | |||||||||||||
| 加權 | ||||||||||||||
| 平均值 | ||||||||||||||
| 鍛鍊 | 數量 | 剩餘 | 數量 | |||||||||||
| 價格 | 股票 | 以年為單位的壽命 | 股票 | |||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | (1) | |||||||||||||
| $ | (2) | |||||||||||||
| (1) |
| (2) |
F-22
注8—可支付貸款
| 本金餘額為 1月1日, 2023 | 調整 | 本金 償還 現金 | 新股發行 | 的效果 外國 交易所 費率 | 本金 餘額為 12月31日, 2023 | |||||||||||||||||||
| 重振旗鼓貸款計劃 | $ | $ | $ | ( | ) | $ | - | $ | $ | |||||||||||||||
| 首次保障--2022年 | ( | ) | ||||||||||||||||||||||
| 第一次保險—2023 | ( | ) | ||||||||||||||||||||||
| 其他應付貸款 | ||||||||||||||||||||||||
| 應付貸款總額 | $ | $ | ( | ) | $ | $ | ||||||||||||||||||
| 減去:應付貸款--當期部分 | ||||||||||||||||||||||||
| 應付貸款--非流動部分 | $ | $ | ||||||||||||||||||||||
| 本金 餘額為 1月1日, 2022 | 調整 | 本金 償還 現金 | 新股發行 | 的效果 外國 交易所 費率 | 本金 餘額為 12月31日, 2022 | |||||||||||||||||||
| 工資保障計劃 | $ | $ | $ | ( | ) | $ | $ | $ | ||||||||||||||||
| 重振旗鼓貸款計劃 | ( | ) | ( | ) | ||||||||||||||||||||
| 首次保障--2021年 | )(1) | ( | ) | |||||||||||||||||||||
| 首次保障--2022年 | ||||||||||||||||||||||||
| 其他應付貸款 | (2) | |||||||||||||||||||||||
| 應付貸款總額 | $ | $ | ( | ) | $ | $ | ( | ) | ||||||||||||||||
| 減去:應付貸款--當期部分 | ||||||||||||||||||||||||
| 應付貸款--非流動部分 | $ | $ | ||||||||||||||||||||||
| (1) |
| (2) |
F-23
| 單利率 | 12月31日, 2023 | 12月31日, 2022 | ||||||||||
| 2019年9月18日發放的應付貸款 | % | $ | $ | |||||||||
| 2019年9月18日發放的應付貸款 | % | |||||||||||
| 2019年10月8日發放的應付貸款 | % | |||||||||||
| 2019年10月29日發放的應付貸款 | % | |||||||||||
| 2019年12月31日發放的應付貸款 | % | |||||||||||
| 2020年2月5日發放的應付貸款 | % | |||||||||||
| 2020年2月5日發放的應付貸款 | % | |||||||||||
| 2020年3月31日發放的應付貸款 | % | |||||||||||
| 2020年3月31日發放的應付貸款 | % | |||||||||||
| 2020年6月8日發放的應付貸款 | % | |||||||||||
| 2020年6月17日發放的應付貸款 | % | |||||||||||
| 2020年7月15日發放的應付貸款** | % | |||||||||||
| 2020年7月15日發放的應付貸款 | % | |||||||||||
| 2020年10月8日發放的應付貸款* | % | |||||||||||
| 2020年10月13日發放的應付貸款 | % | |||||||||||
| 2020年10月14日發放的應付貸款 | % | |||||||||||
| 當前部分Bounce Back Loans (1) (2) | % | |||||||||||
| 2023年12月和2022年12月發放的第一保險應付款(2) | % | |||||||||||
| $ | $ | |||||||||||
| * |
| (1) |
| (2) |
F-24
應付貸款,非流動部分
| 單利率 | 2023年12月31日 | 12月31日, 2022 | 成熟性 日期 | |||||||||||
| BBLS應付貸款於2020年6月10日發放 | % | |||||||||||||
| 小計 | ||||||||||||||
| 減:BLS貸款的流動部分(見上文) | ( | ) | ( | ) | ||||||||||
| 非流動部分 | $ | $ | ||||||||||||
於二零二零年六月十日,本公司收到英鎊英鎊。
2022年12月10日,
公司與
First Insurance Funding就董事和高級管理人員保險單(“D & O保險”)達成融資安排,以融資$
應付貸款的利息支出(收入)
截至2023年12月31日、2023年12月31日及2022年12月31日止年度,本公司確認與應付未償還貸款有關的利息開支相關人士為$
截至2023年12月31日、2023年12月31日及2022年12月31日止年度,本公司確認與應付未償還貸款有關的利息收入相關人士為$
截至2023年12月31日,
公司已累計利息和與未償還貸款相關的應計利息相關方為$
截至2022年12月31日,
公司已累計利息和與未償還貸款相關的應計利息相關方為$
F-25
附註9--承付款和或有事項
訴訟和其他或有損失
本公司記錄因索賠、評估、訴訟、罰款和其他來源引起的或有損失的負債 當很可能已發生負債 並且損失金額可以合理估計時。截至2023年12月31日,本公司沒有記錄或有損失的負債 。
潛在的法律問題
針對KBL前行政人員的行動
2021年9月1日,本公司在特拉華州衡平法院對公司前首席執行官兼董事首席執行官Marlene Krauss博士(“Krauss博士”)及其兩家關聯公司KBLIV贊助商有限責任公司和KBLHealthcare Management Inc.(統稱為“KBL關聯公司”)提起法律訴訟,罪名包括:從事未經授權的公司資產貨幣轉移、在公司合併財務報表內未披露金融負債、未經適當授權發行股票;以及不正當地允許股東贖回。公司的起訴書
指控Krauss博士和/或KBL關聯公司違反受託責任、越權行為、不當得利、疏忽和聲明救濟,並要求超過$的補償性損害賠償。
2021年10月5日,Krauss博士 和KBL關聯公司針對公司 和12名現任或曾經擔任公司董事和/或高級管理人員的個人(即,Marc Feldmann,Lawrence Steinman,James N.伍迪,特蕾莎·德盧卡,弗蘭克·克努特爾二世,帕梅拉·馬龍,勞倫斯·戈爾德,唐納德·A.麥戈文,羅素T. Richard W. Barker、Shoshana Shendelman和 Ozan Pamir(統稱為"第三方被告")。 2021年10月27日,本公司和Ozan Pamir提交了對克勞斯反訴的答覆,所有其他第三方被告就第三方投訴提交了駁回動議。
2022年1月28日,克勞斯博士和KBL關聯公司提交了一份動議,要求允許提交經修訂的反訴
和第三方投訴,並解僱先前提名的六名現任和前任董事,即,解僱特蕾莎·德盧卡,弗蘭克·克努特爾二世,帕梅拉·馬龍,羅素·T. Richard W.巴克和Shoshana Shendelman 該動議根據規定獲得批准,並於2022年2月24日,克勞斯博士提交了一份經修訂的答覆、反訴和第三方投訴(“經修訂的反訴”)。
實質上,經修訂的反訴聲稱:(a)公司和其餘第三方被告違反了對克勞斯博士的信託義務,在SEC的文件中對克勞斯博士作出了所謂的錯誤陳述,並且沒有將她的股份登記在公司中以便
這些股份可以交易,以及(b)公司違反了公司與克勞斯博士之間關於登記這些股份的合同,並且
沒有向克勞斯博士支付據稱根據本金額為美元的期票所欠的款項
2022年3月16日,唐納德·A。小麥戈文Lawrence Gold提交了駁回針對他們的修正反訴的動議,公司和其餘 第三方被告提交了對修正反訴的答辯書,否認了上述內容。 2022年4月19日,克勞斯博士規定駁回她對唐納德·A的所有反訴和指控。小麥戈文和勞倫斯黃金,從而動議駁回對他們的修正反訴。公司和第三方被告打算繼續為 所有經修訂的反訴進行有力辯護,但是,無法保證他們將成功為此類 經修訂的反訴進行法律辯護。2022年4月,唐納德·A.小麥戈文勞倫斯·戈爾德被駁回訴訟案件尚未開始 的發現。公司和第三方被告打算繼續大力抗辯所有 經修訂的反訴,但是,不能保證他們將在經修訂的反訴的法律抗辯中成功。
F-26
克勞斯博士對公司採取的行動
2021年8月19日,克勞斯博士在特拉華州衡平法院對公司提起訴訟。最初的申訴尋求快速救濟,並提出以下兩項主張:(1)指控公司有義務預付費用,包括律師費、向克勞斯博士預付向SEC辯護的費用和SEC向克勞斯博士發出的某些傳票;和(2)它聲稱,公司還被要求償還克勞斯博士對本公司提起訴訟的費用。2021年9月3日左右,克勞斯博士在這起訴訟中提交了經修訂的補充申訴(經修訂的申訴),其中 補充了另一項指控,即克勞斯博士據稱還有權獲得公司墊付她的費用,包括律師費,用於在下文提到的Tyche Capital LLC訴訟中針對第三方申訴進行辯護的費用。以及上述針對公司自己對克勞斯博士的申訴的辯護費用。2021年9月23日左右,公司提交了對修改後的申訴的答覆,其中公司否認了克勞斯博士的每一項指控,並進一步提出了許多積極的抗辯。
2021年11月15日,克勞斯博士就案件中的某些問題提交了簡易判決動議,遭到公司的反對。2021年12月7日就該動議舉行了聽證會,2022年3月7日,法院就此事做出了裁決,部分駁回了簡易裁決動議,並部分批准了該動議。法院隨後於2022年3月29日發佈了執行該決定的命令。目前,各方正在進行該實施令中規定的訴訟程序。法院批准了克勞斯博士關於提前支付克勞斯博士在其動議中要求的部分法律費用的請求,公司被要求支付這些費用的一部分,而公司反對爭議費用的剩餘部分。
2022年10月10日,克勞斯博士提交申請,要求公司支付克勞斯博士要求的2022年5月至7月的全部費用,並修改法院命令。公司對此提出反對意見。2023年1月18日,克勞斯博士提交了第二份申請,要求公司支付克勞斯博士要求的2022年8月至10月的全部費用,並修改法院命令。公司對此提出了
反對意見。2023年5月3日,法院發佈了一項命令,批准克勞斯博士的兩項申請,要求全額支付
所要求的律師費,共計美元。
針對Tyche Capital LLC的訴訟
本公司於2021年4月15日在紐約州最高法院開始並
對被告Tyche Capital LLC(“Tyche”)提起訴訟。 在其投訴書中,公司聲稱Tyche因其違反其書面
合同義務而對Tyche提出索賠,該合同義務載於日期為2019年7月25日的“擔保和承諾協議”以及日期為2019年4月10日的“KBL與CannBioRex業務合併的條款表”(統稱為“標的擔保”)。
公司在其投訴書中聲稱,儘管已要求Tyche履行其在標的
擔保項下的義務,Tyche未能履行並拒絕履行義務,並且目前欠公司的債務金額為$
2021年5月17日左右,Tyche對公司的投訴作出了迴應,對公司提出了答辯和反訴,聲稱違反標的擔保的是公司,而不是Tyche。Tyche還對六名第三方被告 提出了第三方申訴,其中包括公司管理層的三名成員Marc Feldmann爵士、James Woody博士和Ozan Pamir(統稱為公司被告個人),聲稱他們在標的擔保方面違反了對Tyche的受託責任。在這方面,2021年6月25日,每一名個別公司被告都提出動議,駁回泰奇對他們的第三方投訴。
2021年11月23日, 法院批准了公司的請求,對Tyche在託管中持有的所有公司股票 發出扣押令。 在此過程中,法院認定,基於公司投訴書中所指控的事實,公司證明瞭案件的是非曲直 有可能勝訴。
F-27
2022年2月18日,Tyche 提交了修改後的答辯書、反訴和第三方申訴。2022年3月22日,公司和每一名個別公司被告提出動議,駁回Tyche的所有索賠。該駁回動議的聽證會於2022年8月25日舉行,法院批准了動議,完全駁回了每一名公司被告以及針對公司的四項反訴中的三項,只剩下Tyche的聲明性救濟索賠。2022年9月9日,Tyche就法院的裁決提交了上訴通知,但尚未通報或裁決。2022年8月26日,Tyche提交了一項動議,要求騰出或修改公司現有的扣押令,以解除或修改Tyche以託管方式持有的公司股票。本公司已對此提出反對意見,法院於2023年1月3日未經審理即駁回了該動議。Tyche隨後就此拒絕提交了上訴通知,並於2023年1月30日提交了開庭陳述。本公司 於2023年3月2日提交了反對意見陳述,上訴法院提交了該案件。2023年5月4日,上訴法院作出裁決,一致確認下級法院做出的有利於本公司的裁決。
2023年1月30日,公司 提交了一份即決判決動議通知,並駁回了對Tyche的正面抗辯。泰奇對此提出了反對意見,最終於2023年9月11日和19日就該公司的動議舉行了聽證會。在其裁決中,法院批准了公司的動議,但將公司對Tyche的損害賠償金問題提交給了一名特別仲裁人。法院和各方目前正在任命特別裁判,以便就公司對Tyche的損害賠償金額做出決定。泰奇於2023年10月12日提交了上訴通知,作為法院簡易判決的裁決。目前還沒有關於上訴的簡報。 公司打算繼續積極地向Tyche提出索賠,公司和個別公司被告打算 如果他們被上訴,將繼續積極抗辯Tyche的所有索賠;然而,不能保證他們 將在此類努力中勝訴。
對羅納德·鮑爾和薩曼莎·鮑爾的訴訟
2022年2月25日,本公司及其兩家全資子公司KatExco PharmPharmticals Corp.和CannBioRex PharmPharmticals Corp.(統稱為公司原告)在不列顛哥倫比亞省最高法院對Ronald Bauer和Samantha Bauer以及他們的兩家公司Theseus Capital Ltd.和Astatine Capital
Ltd.(統稱為“Bauer被告”)提起訴訟。公司原告
要求Bauer被告賠償挪用資金和股票、未經授權的股票銷售和不當差旅費用,合計金額至少為$
Bauer被告於2022年5月6日提交了對公司民事索賠投訴的答覆,其中Bauer被告否認了公司的索賠 並闡述了他們對此事事實的自己版本。該案的證據開示尚未開始。不能保證 公司原告將在此法律訴訟中勝訴。
AmTrust International對該公司採取的聲明性救濟行動
2022年6月29日,KBL的合併前董事和高級職員保險單承保人AmTrust 國際保險人DAC(“AmTrust”)向美國加利福尼亞州北區地區法院(“宣告性救濟行動”)提交了針對公司的聲明性救濟訴訟,要求宣佈AmTrust根據董事和高級職員保險單承擔的義務。在宣佈救濟行動中,AmTrust聲稱由於合併 公司不再是保險標的下的被保險人。儘管本公司 尋求向AmTrust追回的費用涉及合併前發生的事項。
2022年9月20日,公司對AmTrust提出了答辯和反訴,指控AmTrust違反了AmTrust根據標的董事和高級管理人員保險單對
公司承擔的保險責任,並要求至少賠償$
F-28
2022年11月22日,公司提交了針對AmTrust和Freedom的簡易判決動議。*該動議得到了充分的簡報,並於2023年3月9日舉行了聽證會。 法院對簡易判決動議的勝訴標準很高,需要 法官發現沒有爭議的事實問題,以便他們能夠從法律上對這些問題做出裁決。在這種情況下, 法官發現三個主要問題可以作為法律問題做出有利於公司的裁決,其中一個問題,即控制權排除的變更, 需要進一步發現。
2023年4月21日,法院 發佈了一項命令,部分批准和部分拒絕公司的部分簡易判決動議。
具體而言,法院就下列問題作出了有利於公司的簡易判決:(A)公司事實上是美國信託公司和自由公司保單下的被保險人;(B)被告美國證券交易委員會傳票的某些相關費用,公司前首席執行官兼董事首席執行官馬琳·克勞斯博士和前董事會主席喬治·霍尼格都在美國信託公司和自由公司保險單的基本承保範圍之內;以及(C)AmTrust and Freedom所依賴的投保人與投保人之間的排除不適用于禁止任何此類保險。
法院還發現,保單中關於控制權變更排除的事實存在爭議,因此,根據法律規定,法院不能批准公司其餘的簡易判決請求。因此,法院此時駁回了本公司進一步的簡易判決請求,並認為控制權變更問題暫時待審理時確定,以認定保單:(I)承保本公司已預付的費用,並將預付給Marlene Krauss博士和George Hornig博士;(Ii)AmTrust違反政策;(Iii)AmTrust必須支付本公司的此類 費用;並且,一旦AmTrust保單用完,(Iv)Freedom將有義務根據其保單支付公司的此類費用。
2023年8月4日,法院批准了公司在此案中提出第二項部分簡易判決的動議,這一動議是關於是否應要求AmTrust向公司預付Marlene Krauss博士和George Hornig博士在案件懸而未決期間發生的辯護費用。當事人充分聽取了部分即決判決動議的簡報,並於2024年1月11日就該動議舉行了聽證會。案件提交審理後,於2024年2月12日,法院批准了公司對AmTrust和Freedom作出部分即決判決的動議,命令如下:(A)根據其與公司的保險單,AmTrust有義務向公司墊付超過公司已經墊付或將墊付給克勞斯博士和霍尼格先生的某些美國證券交易委員會傳票的免賠額之外的所有抗辯費用,以及(B)在AmTrust保險單用完後,自由有義務根據其與公司的超額責任保險單向公司墊付同樣的費用。本命令適用於整個案件的審理過程, 但不構成最終判決,公司和兩家保險公司均保留在定於2025年5月12日進行的庭審中對所有適用問題提出異議的權利。審判後的最終判決可能會確認保險公司的這些義務,或者,或者,推翻並要求公司償還全部或部分此類預付款。目前還不能保證最終判決可能會有什麼結果。
雙方已開始對對方進行書面證據開示程序,並預計也將進行證詞陳述。本公司打算繼續積極地 處理此事,以確定本公司有權獲得AmTrust和Freedom對本公司預支費用的全額和最終付款 。雖然公司仍然相信它對AmTrust和Freedom都有很強的理由,但 不能保證公司會在這場訴訟中獲勝。
斯坦福大學許可協議
2018年5月8日,Katexo與斯坦福大學(“Stanford”)簽訂了為期六個月的期權協議(“Stanford期權”),根據該協議,Stanford
授予公司為期六個月的期權,以獲得與用於治療自身免疫性疾病的生物物質相關的專利(“已授權專利”)的獨家許可。作為對斯坦福期權的對價,該公司向斯坦福支付了#美元
2018年7月25日,Katexco
行使了六個月的選擇權,並與斯坦福大學簽訂了獨家許可協議(“斯坦福大學許可協議”)。
根據斯坦福許可協議,自生效日期的第一週年起,以及此後的每個週年,
公司將提前向斯坦福支付每年的許可維護費,
F-29
此外,公司將 有義務支付以下里程碑付款:
| i) | $ |
| Ii) | $ |
| Iii) | $ |
斯坦福許可協議
可由公司在30天內予以取消。使用費,計算為
牛津大學協議
2020年9月18日,CBR
Pharma與Oxford簽訂了一份為期3年的研發協議(“3年牛津協議”),以研究和
研究纖維化的潛在機制,以換取總對價$
2020年9月21日,CBR
Pharma與牛津大學簽訂了一份為期兩年的研發協議(“2年牛津協議”),用於
用於治療炎症性疾病的大麻素藥物的臨牀開發,以換取總對價$
截至2023年12月31日和 2022年12月31日,公司沒有欠牛津兩年期協議的任何款項。
2021年5月24日,本公司
與牛津大學簽訂了研究協議,(“牛津”和“第五次牛津協議”),根據該協議,公司將贊助牛津大學的工作,開展多中心、隨機、雙盲、平行組,
抗腫瘤壞死因子注射治療疼痛主導期成人凍結肩的可行性研究。
| 應付金額 | ||||
| 里程碑 | (不含增值税) | |||
| £ | ||||
| £ | ||||
| £ | ||||
| £ | ||||
F-30
該公司支付了第一個
里程碑$
2021年11月2日,公司與牛津大學簽訂了一項為期20年的HMGB1分子許可技術協議,該技術與組織再生有關。根據協議,牛津大學同意將該技術許可給公司,用於研究、開發和使用許可專利。該公司同意向牛津大學支付過去的專利費用$
由於HMGB1研究計劃的持續成本,以及公司需要將其資源集中在公司使用抗腫瘤壞死因子(TNF)治療纖維化的主要平臺上,公司董事會於2023年9月22日決定終止公司與牛津大學的HMGB1許可協議,並於2023年9月22日,公司和牛津大學簽訂了終止
函,正式終止許可證,自2023年9月22日起生效。解約信還澄清了我們在許可證終止後欠我們的金額,包括大約$
由於最近的資金緊張,公司無法及時支付牛津公司的款項,牛津公司是公司大部分許可證和專利的許可方
和公司的研究合作伙伴。牛津大學聲稱,大約一GB的總和
肯尼迪許可協議
2019年9月27日,180 LP與美國、日本、英國和歐盟國家的肯尼迪風濕病研究信託基金(“Kennedy Trust for Rheumatology Research”) 就某些特許專利(“肯尼迪特許專利”)簽訂了許可協議(“肯尼迪許可協議”),包括授予再許可的權利,以及研究、開發、銷售或製造任何醫藥產品的權利(I)其研究、開發、製造、使用、如果沒有根據《肯尼迪許可協議》授予的許可,進口或銷售將侵犯肯尼迪許可專利,或(Ii)含有抗體片段或源自抗體的抗體,而該抗體的研究、開發、製造、使用、進口或銷售將侵犯沒有根據《肯尼迪許可協議》授予的許可的肯尼迪許可專利,用於所有人類用途,包括診斷、預防和治療疾病和狀況。
作為授予肯尼迪許可專利的對價,180 LP向肯尼迪支付了一筆英鎊的預付款GB
本公司支付給肯尼迪的專利費 的有效期將在(I)肯尼迪許可專利中包括的涵蓋或聲稱在適用國家/地區開發產品的專利的最後有效主張 ;(Ii)該產品在該國的監管排他性到期;或(Iii)該產品在該國首次商業銷售的10年內到期。肯尼迪許可協議 可以通過提供90天的通知而無故終止。
F-31
Petcanna分許可協議
2018年8月20日,CBR Pharma 與其全資子公司Petcanna Pharma Corp. (“Petcanna”)簽訂了一份分許可協議(“分許可協議”),該公司前首席財務官是董事的首席財務官。Petcanna是一傢俬人公司,與公司有一個共同的委託人。
根據
分許可協議的條款,公司已授予許可專利的分許可,以尋求
所有獸醫病症治療的開發和商業化。考慮到,Petcanna將(a)發佈
經營租約
2016年2月,FASB 發佈了對會計準則編纂主題842的更新, 租契("ASC 842")。新準則要求 大多數租賃在資產負債表上確認為使用權(“ROU”)資產和租賃負債。使用權 資產初步按預期於租期內支付的金額現值計量。這些 租賃成本在收益表中的確認被分解並確認為經營費用(用於攤銷使用權資產) 和利息費用(用於租賃付款中與利息相關的部分)。本公司於發佈時採納該準則。
根據ASC 842, 公司可以選擇(按資產類別)不在資產負債表上記錄租期為12個月或更短且不包括 承租人合理確定將行使的購買選擇權的租賃。如果選擇,租賃將按照 以前的公認會計原則作為經營租賃對待;付款將在租賃期內以直線法確認。在確定租賃是否符合 此選擇時,公司將僅在將其視為租賃期的一部分時包括續訂選擇權,即, 公司合理確定會行使的選擇權。如果租賃期延長至12個月以上,或者如果合理確定公司 將行使購買選擇權,則公司將不再能夠應用該實用權宜方法,並將應用ASC 842指南。
關於租賃( 目前沒有租賃),公司將對12個月或更短的短期經營租賃使用實際權宜方法。 此實際權宜方法已被選為一攬子方案,並將一致地應用於所有租賃。此外,如果公司的 租賃被視為經營性質,因此未反映在資產負債表中,公司將每月在利潤表中確認短期 租賃付款為租金/租賃費用。
截至2023年12月31日和2022年12月31日,公司擁有不是租賃以及截至該日期沒有租賃或租金支出。
諮詢協議
南查哈爾諮詢協議
2021年2月22日,公司與關聯方賈格迪普·南查哈爾教授(“顧問”)簽訂了一份諮詢協議(經修訂,“諮詢協議”)。該諮詢協議於2020年12月1日生效。
根據諮詢協議
,公司同意向顧問支付
| ● | £的總和 |
| ● | £的總和 |
| ● | £的總和 |
| ● | £的總和 |
F-32
The Consulting Agreement has an initial term of three years, and renews thereafter for additional three-year terms, until terminated as provided in the agreement. The Consulting Agreement can be terminated by either party with 12 months prior written notice (provided the Company’s right to terminate the agreement may only be exercised if the Consultant fails to perform his required duties under the Consulting Agreement), or by the Company immediately under certain conditions specified in the Consulting Agreement if (a) the Consultant fails or neglects efficiently and diligently to perform the services required thereunder or is guilty of any breach of its or his obligations under the agreement (including any consent granted under it); (b) the Consultant is guilty of any fraud or dishonesty or acts in a manner (whether in the performance of the services or otherwise) which, in the reasonable opinion of the Company, has brought or is likely to bring the Consultant, the Company or any of its affiliates into disrepute or is convicted of an arrestable offence (other than a road traffic offence for which a non-custodial penalty is imposed); or (c) the Consultant becomes bankrupt or makes any arrangement or composition with his creditors. If the Consulting Agreement is terminated by the Company for any reason other than cause, the Consultant is entitled to a lump sum payment of 12 months of his fee as of the date of termination.
自2021年3月30日起生效
,以清償欠顧問的
自2021年8月27日起
為清償欠顧問的剩餘獎金2,公司發行
2021年12月,Dupuytren的收縮臨牀試驗數據被提交給同行評議的期刊發表,並向顧問支付了獎金1。
2022年4月27日,公司
簽訂了諮詢協議修正案,在接受Dupuytren合同的2b期臨牀試驗數據公佈後,諮詢費將增加到GB
2022年12月28日,
公司簽訂了諮詢協議修正案,顧問月費將增加到GB
Feldmann諮詢協議
2018年6月1日,CannBioRex
Pharma Limited(“CannBioRex公司”)和Prof. Marc Feldmann博士,我們的執行聯合主席,簽訂了諮詢
協議;根據該協議,Feldmann教授擔任CannBioRex的主席、首席執行官和執行董事,或擔任與其身份相符的其他
職位。Feldmann爵士獲得的賠償
該協議包含一個習慣性的 非競爭條款,禁止Feldmann爵士在其受僱期間為任何競爭企業工作,或持有 其他企業的股權,除非他和他的家人的總實益權益不超過該類別證券的5%,他可以持有或實益擁有上市公司的證券。本協議沒有固定期限,任何一方 均可提前9個月發出書面通知終止本協議。CannBioRex也可隨時終止協議 ,並通過發出書面通知立即生效。如果CannBioRex在未提供9個月 書面通知的情況下終止Feldmann教授的僱傭關係,他將有權獲得相當於他在9個月 通知的情況下有權獲得的基本工資的報酬。
F-33
2021年11月17日,
董事會根據薪酬委員會的建議,將Feldmann教授的工資提高至$
自2022年4月27日起,CannBioRex和Prof. Feldmann爵士簽署了諮詢協議的修正案,根據該修正案,雙方同意自2022年3月1日起生效,Feldmann爵士的薪酬將減少$
2024年1月1日,公司 對本僱傭協議進行了另一項修訂;見附註13—後續事項, 補償協議的修正 瞭解更多信息。
拉森諮詢協議
2021年4月29日,本公司
與180 LP前首席執行官Glenn Larsen簽署了一份諮詢協議,以談判代表的身份
為四項專利的許可進行談判。作為所提供服務的對價,公司同意向Larsen先生賠償美元。
2023年2月22日,公司與Glenn Larsen簽訂了提供諮詢服務的第二份諮詢協議;作為對所提供服務的補償,公司同意賠償Larsen先生#美元。
斯坦曼諮詢協議
於2021年11月17日,自2021年11月1日起,本公司與本公司執行聯席主席勞倫斯·施泰因曼(醫學博士)簽訂了諮詢協議。諮詢協議“)。根據諮詢協議,Steinman博士同意向公司提供某些諮詢服務,包括但不限於,參與確定和制定公司的戰略目標;積極尋找收購和合並候選者;以及對公司的á7nAChR平臺(統稱為服務").協議的期限為一年( "初始項");條件是協議在初始 期限後自動延長一年(每個"自動續約期限以及初始條款和所有自動續訂條款(如果有), 術語“)如果任何一方在初始 期限或任何自動續期期限結束前至少30天向另一方發出不自動延長協議期限的書面通知,則在符合續約要求(如下所述)的情況下。如果(I)Steinman博士再次當選為董事會成員,則該任期只能延長為自動更新任期。衝浪板")在緊接 自動續訂期限開始之日之前的公司股東年會上;(ii)董事會確認其為適用的自動續訂聯席主席的任命 任期(或未能在適用的自動續訂期限之前任命其他人擔任聯席主席)以及(iii)Steinman博士繼續任職 在他的職責是負責公司的a'7nAChR平臺的科學開發("續訂 要求").諮詢協議也在下列日期(以較早者為準)立即到期:(i)Steinman博士不再擔任聯合主席,並且不再對我們的a'7nAChR平臺承擔主要科學責任的日期;及(ii)公司要求的任何較早 日期,(由董事會會議上多數成員(不包括Steinman博士)的投票證明),或(2)Steinman博士(由Steinman博士給董事會的書面通知證明)。此外,如果Steinman博士無法或拒絕履行服務,公司可立即終止 諮詢協議,且無需事先通知;如果另一方違反諮詢協議的任何重要條款 ,任何一方 均可立即終止諮詢協議,且無需事先通知。
F-34
公司同意向斯坦曼博士支付美元
自2022年4月27日起,公司和斯坦曼博士簽署了諮詢協議的修訂案,根據該修訂案,雙方同意
自2022年3月1日起生效,斯坦曼博士的工資將減少$
行政總裁聘用協議
2021年2月25日,
公司與公司首席執行官James Woody博士(“CEO”)簽訂了一份日期為2021年2月24日並於2020年11月6日生效的修訂協議(
“A & R協議”),該協議取代了CEO與公司的先前協議
。根據A & R協議,首席執行官同意擔任本公司高級管理人員,任期為三年,此後
可自動續期一年,除非任何一方向另一方提供至少90天的書面通知,説明其不續簽協議的意向。根據該協議,首席執行官的年基本工資最初為美元,
作為首席執行官同意簽署協議的額外考慮
,公司授予他購買
首席執行官還有資格獲得年度獎金,目標獎金相當於
A&R協議可由公司以正當理由(符合協議的補救條款)或無正當理由(提前60天書面通知首席執行官)、首席執行官有充分理由(如協議中所述,且符合協議的補救條款)、 或首席執行官在沒有充分理由的情況下隨時終止。如果任何一方提供如上所述的不續訂通知,本協議也將在初始期限或任何續訂期限結束時自動到期。
如果公司無故終止A & R協議 ,或首席執行官有充分理由終止,公司同意向他支付18個月工資 或協議剩餘期限中較低者,支付上一年度的任何應計獎金,在他將收到遣散費的同一時期,他在本年度的獎金和健康保險費中按比例分配(如上所述)。
A&R協議包含 標準和慣例的發明轉讓、賠償、保密和非徵集條款,這些條款在協議終止後24個月內仍然有效。
F-35
2022年4月27日,公司
簽訂了僱傭協議修正案,根據該修正案,公司將提供
首席財務官聘用協議
2021年2月25日,
公司與公司臨時首席財務官Ozan Pamir
簽訂了日期為2021年2月24日並於2020年11月6日生效的僱傭協議(下稱“CFO協議”)。根據該協議,首席財務官同意擔任本公司的臨時
首席財務官(“首席財務官”),起薪為美元
作為首席財務官同意簽署協議的額外考慮
,公司授予他購買
根據協議,CFO
有資格獲得年度獎金,目標金額為
此外,2024年1月1日,公司對本僱傭協議進行了另一項修訂;見附註13—後續事項, 補償協議的修正案,以獲取更多信息。
本協議 公司可隨時終止,無論是否有任何理由,並提前60天書面通知;首席財務官可隨時終止,並提前60天書面通知。如果本協議 在某些情況下因原因終止,本公司也可提前六天通知終止本協議。在公司無故終止CFO協議或 CFO有充分理由終止時,公司同意向其支付三個月的遣散費。
該協議包含標準 和習慣發明轉讓、賠償、保密和非招標條款,這些條款在協議終止後的24個月內仍然有效 。
首席運營官/首席業務官聘用協議
2021年10月29日,本公司
與Quan Vu訂立日期為2021年10月27日的僱傭協議(“首席運營官/首席運營官協議”),並於2021年11月1日(
“開始日期”)生效。根據該協議,武先生同意擔任本公司的首席運營官/首席業務官(“首席運營官/首席業務官”),初步薪酬為美元。
F-36
作為Vu先生同意簽訂協議的額外對價
,公司授予他購買
根據協議,Vu
有資格獲得年度獎金,目標金額為
本協議 可在任何時候由公司提前30天發出書面通知,無論是否有理由終止,Vu先生也可在任何時候由提前30天發出書面通知。如果協議 在某些情況下因原因終止,公司也可以提前十天通知終止協議。在公司無故終止武先生的協議或 武先生有充分理由終止後,公司同意向他支付十二個月的遣散費,除非高管在一年週年前離職 。
該協議包含標準 和習慣發明轉讓、賠償、保密和非招標條款,這些條款在他的協議終止後的24個月內仍然有效。
2022年4月27日,公司
簽署了《僱傭協議》的修訂案,據此,公司同意提供
自2023年1月15日起,公司和Vu共同同意終止Vu的僱傭關係。根據終止協議,雙方簽訂了
離職協議,據此,公司同意向武先生支付一筆協議的遣散費,包括累計的欠薪、約定的
醫療保險費用和累計的帶薪休假,總額為美元。
首席科學官僱傭協議書
2019年8月21日,公司與Jonathan Rothbard博士簽訂了僱傭協議。本協議的有效期為三年,此後自動延長
一年的有效期,除非任何一方在
下一次續訂日期前至少提前90天書面通知終止本協議,並規定工資為$
該協議規定,
Rothbard博士將獲得年度獎金,但須滿足董事會設定的某些目標,目標獎金金額
為
F-37
如果我們、羅斯巴德博士有正當理由(如僱傭協議中所述)終止僱用羅斯巴德博士,或者我們沒有續簽協議,則需要向他支付36個月的遣散費(如果終止發生在任期的第一年); 24個月的遣散費(如果終止發生在任期的第二年);和12個月的遣散費(如果這種終止發生在任期的第二年之後),以及任何累積的獎金金額和根據目標獎金按比例計算的年度獎金, 以及支付與他被要求支付遣散費的同期的健康保險費。
《僱傭協議》於2022年1月1日生效,以推翻自動年薪增長,
注10—股東(虧損)權益
2022年期間的反向股票拆分
2022年12月15日,在公司股東特別會議上,公司股東批准了對經修訂的公司第二份公司註冊證書的修正案,以實現我們普通股的已發行和已發行股票的反向股票拆分,面值為$。
由於《證書修正案》沒有減少普通股的授權股份數量,反向股票拆分的效果是相對於已發行和已發行股票的數量增加了可供發行的普通股數量。反向股票拆分不會改變普通股的面值,也不會修改普通股的任何投票權或其他條款。反向股票拆分後剩餘的任何零碎股份 將向上舍入到最接近的完整股份。
關於公司的 2020年綜合激勵計劃和2022年綜合激勵計劃,公司薪酬委員會和董事會認為,(i)根據激勵計劃將可發行的公司普通股股票數量下調20倍,符合公司及其股東的最佳利益(任何零碎股份向下舍入至最接近的整股);(ii)減少因購買公司普通股股份的每份未行使期權而可發行的普通股股份的數量 ,以及所有其他未行使 獎勵,20倍(任何零碎股份向下舍入至最接近的整股);及(iii)調整 購買先前根據激勵計劃授予的普通股股份的任何未行使購股權的行使價,(四捨五入 至最接近的整數美分),在每種情況下,公平地調整反向股票拆分的兑換比率,該等調整 在反向股票拆分生效後自動生效。20分之一反向股票分割的影響 已在整個財務報表和財務報表附註中追溯反映。
2024年反向股票拆分
2024年2月16日,在公司股東特別會議上,公司股東批准了對經修訂的公司第二份公司註冊證書的修正案,以實現我們普通股的已發行和已發行股票的反向股票拆分,面值為$。
此外,因行使購股權及其他股權獎勵而可發行的普通股股數(包括根據本公司股權補償計劃預留供發行的股份)由適用的管理人按比例調整,按1比19比例調整,並向下舍入至最接近的整體股份。該公司優先股的轉換率(其中 沒有流通股)也將按1比19的比例進行調整。在行使本公司的已發行認股權證以購買已發行普通股的股份時,可發行的股份數目亦將根據該等證券的條款進行公平調整 與19股換1股反向分拆有關。此外,每項已發行購股權及認股權證的行權價將與19份認股權證中1份認購權及認股權證的分拆比率成反比增加,以致於行權時,根據該等證券的條款,購股權持有人或認股權證持有人就受購股權或認股權證約束的股份向本公司支付的總行權價將保持與反向股票分拆前的總行權價大致相同。
F-38
此外,
根據本公司K類特別投票權股份(“有表決權的股票“),在生效時間
之後,有表決權股票可轉換為相當於
的數量的普通股,並投票數量等於
的有表決權股票
由於反向股票拆分,每個股東在公司的百分比所有權權益和比例投票權幾乎保持不變 ,但因將零碎股份四捨五入為完整股份而產生的微小變化和調整除外。 普通股股東的權利和特權將基本不受反向股票拆分的影響。
優先股
根據2020年11月6日提交的公司第二次修訂和重新註冊證書,公司已
特別投票權股份
的面值為$
| ● | 在我們的普通股有投票權的所有情況下都有投票權,普通股是一類; |
| ● | 特別表決權股份使持有人奧德賽信託公司(受託人)有權獲得相當於可向之前已發行的加拿大子公司CannBioRex Puracheco ULC和/或Katexo Puracheco ULC的先前已發行股票的持有者發行的普通股數量的總表決權。可交換股份”); |
| ● | 特別表決權股份持有人(以及間接的可交換股份持有人)在通知、報告、財務報表和出席所有股東大會方面與普通股持有人享有相同的權利; |
| ● | 沒有分紅的權利; |
| ● | 特別表決權股份的持有人無權在公司結束、解散或清算時獲得任何相關分派的任何部分;以及 |
| ● | 當沒有已發行的可交換股份,且CannBioRex Puracheco ULC和Katexo Puracheco ULC沒有認購權或其他承諾時,本公司可取消特別投票權股份,這可能需要CannBioRex Puracheco ULC和Katexo Puracheco ULC發行更多可交換股票。 |
如上所述,可交換股份的持有人 通過適用的特別投票權股份擁有與普通股相對應的投票權和其他屬性。可交換股份為CBR Pharma或KatExco證券的某些前加拿大居民提供了一個機會,以獲得與重組相關的加拿大所得税方面的應納税資本利得的延期。
F-39
普通股
本公司獲授權
發行
2022年7月提供
於2022年7月17日,本公司與若干買方訂立證券購買協議,據此,本公司同意出售合共
2022年7月的預融資權證
的行權價相當於1美元
2022年7月普通權證
的行權價相當於$
2023年,本公司
於4月、8月和11月對這些權證進行了一系列修訂;這些修訂將行權價格從
$
截至2023年12月31日,所有
F-40
2022年12月提供
於2022年12月20日,本公司與若干買方訂立證券購買協議,據此,本公司同意出售總額為
2022年12月預籌資金的認股權證的行權價相當於$
二零二二年十二月普通認股權證
的行使價等於$
於二零二三年,本公司於四月、八月及十一月對該等認股權證訂立
一系列修訂;該等修訂將行使價由
截至2023年12月31日,所有
2023年4月提供
於2023年4月5日,本公司
與若干買方訂立證券購買協議,據此,本公司同意出售總額為
F-41
2023年4月預先出資的認股權證的行權價相當於$
2023年4月的普通權證
的行使價相當於$
於二零二三年,本公司於八月及十一月對該等認股權證訂立
一系列修訂;該等修訂將行使價由
截至2023年12月31日,所有
2022年7月和12月普通權證修正案
於2023年4月5日,本公司
就2022年7月及2022年12月發行的共同認股權證協議訂立修訂,據此認股權證可購買
最多
為評估
相對公允價值的變化,本公司執行了布萊克—斯科爾斯期權模型計算,以量化截至修改日期的原始條款下的普通認股權證
的公允價值,使用以下假設:
F-42
2022年綜合激勵計劃第一修正案
在
2023年7月6日舉行的公司2023年股東年會上,公司股東批准了180生命科學公司2022年綜合激勵計劃(經第一修正案修訂
,簡稱"OIP")的第一修正案
(簡稱"第一修正案")。第一修正案最初於2023年5月5日由公司董事會批准,須經股東批准,第一修正案於股東批准時生效。《第一修正案》
提高了根據OIP可發行的最大股份數量,
2023年8月提供
2023年8月9日,除了依賴於本公司於2023年7月25日向美國證券交易委員會提交的
註冊聲明的某些購買者之外,本公司
與一名認可投資者簽訂了一份證券購買協議,該協議於2023年8月9日生效,根據該協議,本公司同意
出售總計
2023年8月預籌資金的認股權證的行權價相當於$
2023年8月的普通權證
的行權價相當於$
於二零二三年,本公司於八月及十一月對該等認股權證訂立
一系列修訂;該等修訂將行使價由
截至2023年12月31日,所有
2023年8月預融資認股權證已獲行使,價值為美元
F-43
2022年7月、2022年12月和2023年4月的共同認股權證協議第二次修訂
於2023年8月9日,本公司
就2022年7月、2022年12月及2023年4月發行的普通認股權證協議訂立修訂,據此,普通認股權證
可購買最多
為
評估相對公允價值的變動,本公司執行了布萊克—斯科爾斯期權模型計算,以量化截至修改日期的普通認股權證在其原始條款下的公允價值
,使用以下假設,就2022年7月、2022年12月和2023年4月普通認股權證:
修訂2023年8月發行
2023年11月28日,
公司對2023年8月發行進行了修訂(“2023年8月發行的修訂”),據此,(i)買方
同意支付額外的$
除行使價和可行使性外,額外認股權證與2023年8月的預籌資權證和2023年8月普通權證的條款和條件相同,因此被確定為股權類別,因為它們在控制權變更的情況下滿足有限的例外。
由於額外的權證屬於股權類別,配售代理費用和發售費用將計入額外實繳資本的減少額
。額外認股權證將不得行使,直至本公司就發行
根據對2023年8月發售的修訂,本公司訂立權證修訂協議,以修訂買方持有的以下未償還認股權證:(I)最多可購買
本公司將2023年8月發售的修訂計入認股權證修訂,據此修訂的影響以緊接修訂前及修訂後的相對公允價值的差額 衡量;任何對相對公允價值的增加 均確認為股權發行成本。
F-44
為評估
相對公允價值的變化,本公司採用布萊克·斯科爾斯期權模型計算普通權證截至修改日期的公允價值
,使用2022年7月、2022年12月、2023年4月和2023年8月普通權證的以下假設:股價為$
截至本報告提交時,
所有額外的
2023年和2022年期間為服務發行的普通股
截至2023年12月31日止年度,本公司共發行
在截至2022年12月31日的年度內,本公司共發行了
2023年及2022年發行的受限制股票
在截至2022年12月31日的年度內,本公司發行了
| 非既有限制 | 加權 平均值 格蘭特 日期 | |||||||
| 庫存 | FV價格 | |||||||
| 截至2022年1月1日未歸屬 | $ | |||||||
| 授與 | ||||||||
| 既得 | ( | ) | ||||||
| 被沒收 | ||||||||
| 截至2022年12月31日未歸屬 | ||||||||
| 授與 | ||||||||
| 既得 | ( | ) | ||||||
| 被沒收 | ( | ) | ||||||
| 截至2023年12月31日未歸屬 | ||||||||
特別表決權股份
特別表決權股份 是向CBR Pharma和KatExco的前股東發行的,與業務合併前的180重組有關。 特別表決權股份可由持有者交換為公司普通股的股份,並與公司的普通股股東作為一個單一的 類別一起投票。特別表決權股份無權獲得任何分派股息。
於截至二零二三年及二零二二年十二月三十一日止年度, 股票是在交換與特別表決權股份相關的普通股等價物時發行的,
餘額為
F-45
股票期權
| 選項數量 | 加權 平均值 鍛鍊 價格 | 加權 平均值 剩餘 術語 (按年計算) | 固有的 價值 | |||||||||||||
| 未償還,2022年1月1日 | $ | |||||||||||||||
| 授與 | - | |||||||||||||||
| 未清償,2022年12月31日 | - | |||||||||||||||
| 授與 | - | |||||||||||||||
| 被沒收 | ( | ) | - | |||||||||||||
| 未清償,2023年12月31日 | $ | $ | ||||||||||||||
| 可行使,2023年12月31日 | $ | $ | ||||||||||||||
| 未償還的股票期權 | 可行使的股票期權 | |||||||||||||
| 加權 | ||||||||||||||
| 平均值 | ||||||||||||||
| 鍛鍊 | 數量 | 剩餘 | 數量 | |||||||||||
| 價格 | 股票 | 以年為單位的壽命 | 股票 | |||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
下文 所列二零二三年及二零二二年的假設乃使用i)美聯儲於授出日期公佈的無風險利率,ii)所用的預期 年期為合約年期加上加權平均歸屬年期的平均值,iii)波動率是使用 適用季度其他金融工具的第三方估值報告得出的,以及iv)使用的預期股息率 取自適用的期權授予協議。
F-46
2022年5月19日,公司
授予了購買
2022年5月19日,公司
還授予了購買
| 無風險利率 | % | |||
| 預期期限(年) | ||||
| 預期波動率 | % | |||
| 預期股息 | % |
2023年9月4日,
公司授予了購買
2023年9月4日,
公司還授予了購買
2023年9月4日,該公司還授予了購買的十年期權
| 無風險利率 | % | |||
| 預期期限(年) | ||||
| 預期波動率 | % | |||
| 預期股息 | % |
公司確認了以股票為基礎的
薪酬支出$
F-47
納斯達克合規
不遵守納斯達克上市規則5550(a)(2)的通知
2023年9月7日,公司收到納斯達克上市資格部的書面通知(“納斯達克")通知
該公司不符合納斯達克上市規則5550(a)(2)中規定的
繼續在納斯達克資本市場上市的最低出價要求。納斯達克上市規則5550(a)(2)要求上市證券維持最低出價
價格為美元
通知函稱,公司有180個日曆日或截至2024年3月5日,重新遵守納斯達克上市規則5550(a)(2)。為了重新獲得合規性,
公司普通股的投標價必須具有至少$
本公司於2024年2月16日舉行了一次特別 股東大會,並獲得批准,除其他事項外,對我們的第二次修訂和 重述的公司註冊證書進行了修訂,以實現我們的已發行和流通股的反向股票分割,比例為一比四到一比四十(含),確切比率將設定為整數,由我們的董事會或其正式授權的委員會酌情決定,在修訂批准後和 之前的任何時間 。董事會隨後於2024年2月16日批准了發行在外普通股的19股反向拆分,反向拆分於2024年2月28日生效,下文將在附註13—後續 事件中詳細討論。
不遵守納斯達克上市規則5635(d)的通知
2023年10月11日,本公司
收到納斯達克上市資格部的書面通知,通知本公司不符合納斯達克上市規則5635(d)中規定的
股東批准要求,該規則要求事先獲得股東批准,
除公開發行外,涉及發行
員工根據上市規則第5635(d)條作出的決定
涉及本公司發行及發行下列各項的合計:(i)
2023年10月11日,納斯達克上市資格部門發出的書面通知為公司提供了45天的時間提交重新合規的計劃。該公司隨後於2023年11月9日向納斯達克提交了該合規計劃 ,2023年11月14日,納斯達克批准該公司 延期至2023年12月15日,以完成該合規計劃中規定的某些交易,以補救其先前 違反納斯達克規則的行為,如10月11日所述,2023年納斯達克的信。
如上文更詳細披露的 ,2023年11月29日,本公司進行了多項交易,包括修改普通認股權證的條款,使其 在本公司股東根據納斯達克上市規則批准該發行之前不得行使,以恢復 遵守上市規則5635(d)(1)(A)。
由於這些交易, 2023年12月14日,納斯達克向公司提供書面通知,稱公司已履行先前延期的條款; 遵守上市規則5635(d)(1)(A);該事項現已結束。
未能遵守納斯達克上市規則第5550(b)(1)條的通知
2023年11月15日,
公司收到納斯達克的信函,通知公司在截至2023年9月30日的
表格10—Q季度報告(“表格10—Q”)中報告的股東權益不符合繼續在納斯達克資本市場上市的最低股東權益要求
。納斯達克上市規則5550(b)(1)要求在納斯達克資本市場上市的公司保持股東權益至少為美元
F-48
本違規通知 對公司普通股在納斯達克資本市場的繼續上市或交易沒有立即影響,該公司普通股將繼續在納斯達克上市和交易,但須視公司遵守其他持續上市要求而定。 納斯達克已要求公司在2024年1月2日前向納斯達克提交恢復合規的計劃。我們及時提交了恢復合規性的計劃 ,2024年1月11日,納斯達克通知公司,它已決定批准公司延期,以恢復 遵守該規則。參見附註13—後續事件。
附註11--所得税
公司須繳納 美國、加拿大和英國的聯邦和州/省所得税,每個法人實體均以非合併 為基礎進行申報。180LP重組前淨經營虧損的利益已轉移至其擁有人。
| 在過去幾年裏 | ||||||||
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 國內 | $ | ( | ) | $ | ( | ) | ||
| 國際 | ( | ) | ( | ) | ||||
| $ | ( | ) | $ | ( | ) | |||
| 在過去幾年裏 | ||||||||
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 遞延税項優惠: | ||||||||
| 國內: | ||||||||
| 聯邦制 | $ | $ | ||||||
| 狀態 | ||||||||
| 國際 | ||||||||
| 更改估值免税額 | ( | ) | ( | ) | ||||
| 所得税淨額優惠 | $ | $ | ||||||
F-49
| 在過去幾年裏 | ||||||||
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 美國聯邦法定利率 | % | % | ||||||
| 國內和國外聯邦利率之間的差異 | ( | )% | ( | )% | ||||
| 扣除聯邦福利後的州税和省税 | % | % | ||||||
| 永久性差異: | ||||||||
| 商譽減值 | ( | )% | ||||||
| 衍生工具及應計可發行股本的公允價值變動 | % | % | ||||||
| 其他 | ( | )% | ||||||
| 更改估值免税額 | ( | )% | ( | )% | ||||
| 有效所得税率 | % | % | ||||||
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 遞延税項資產: | ||||||||
| 淨營業虧損結轉 | $ | $ | ||||||
| 攤銷 | ||||||||
| 應計薪酬目前不可扣除 | ||||||||
| 股票薪酬 | ||||||||
| 應計利息 | ||||||||
| 應收票據壞賬準備 | ||||||||
| 為税務目的遞延的組織成本 | ||||||||
| 遞延税項負債: | ||||||||
| 賬面和計税基礎之間的差異與以下方面有關: | ||||||||
| 技術許可證 | ( | ) | ( | ) | ||||
| 收購正在進行的研究和開發 | ( | ) | ||||||
| 其他 | ( | ) | ( | ) | ||||
| ( | ) | ( | ) | |||||
| 遞延税項資產和負債合計 | ||||||||
| 估值免税額 | ( | ) | ( | ) | ||||
| 遞延税項資產和負債合計,淨額 | $ | ( | ) | $ | ( | ) | ||
F-50
| 在過去幾年裏 | ||||||||
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 期初 | $ | ( | ) | $ | ( | ) | ||
| 根據税務規定作出的估值變動 | ( | ) | ( | ) | ||||
| 真實-截至上一年的納税申報單 | ( | ) | ||||||
| 期末 | $ | ( | ) | $ | ( | ) | ||
截至2023年12月31日, 公司在不同司法管轄區的淨營業虧損(NOL)結轉可用於抵消未來應納税所得額 如下:
| ● | 大約$ |
| ● | 大約
$ |
| ● | 大約
$ |
由於所有權變更,利用國內NOL抵銷未來應納税所得額可能受到《國税法》第382條和類似州法規的年度限制。
本公司已根據ASC 740的規定評估了遞延税項資產變現的可能性。ASC 740要求此類審查考慮所有可用的正面和負面證據,包括遞延税項資產的預定沖銷、預計的未來應納税所得額和税務籌劃策略。美國會計準則第740條要求,當“很有可能”全部或部分遞延税項資產無法變現時,應設立估值準備。在進行了如2023年12月31日及2022年12月31日等審核後,管理層認為其遞延税項資產的未來變現存在不確定性
,因此已設立全額估值撥備。公司計入的估值津貼增加了#美元。
管理層已評估並得出結論,截至2023年12月31日和2022年12月31日,公司合併財務報表中沒有需要確認的重大不確定税務狀況 。本公司預計其未確認的税務優惠在報告日期起計12個月內不會有任何重大變化 。
截至2023年、2023年及2022年12月31日止年度內,並無展開或進行税務審核,亦無於該等期間產生任何與税務有關的利息或罰款。公司自成立以來在美國、加拿大和英國提交的納税申報單仍需接受 審查。
附註12--關聯方
應付賬款—關聯方
應付賬款—相關
方為$
應計費用--關聯方
應計費用—相關
當事方為 及$
F-51
與研發費用相關的各方
研發費用
相關方$
與一般和行政費用相關的當事人
截至2023年及2022年12月31日止年度的一般及行政開支
—關聯方為美元
利息收入—關聯方
截至2023年12月31日及2022年12月31日止年度,本公司記錄 及$
附註13--後續活動
本公司已評估 2023年12月31日之後至財務報表發佈之日的事件和交易。除以下事項外, 沒有發現需要在財務報表中披露的後續事項。
補償協議的修正
2024年1月10日,自2024年1月1日起,公司與首席執行官 兼董事Woody博士簽訂了第四項修訂和重述的僱傭協議;(b)與 公司首席科學官(CSO)Rothbard博士簽訂了第四項修訂僱傭協議;(c)與本公司執行主席斯坦曼博士的諮詢協議第三次修訂;及(d)與本公司前任執行聯席主席Feldmann教授簽訂的諮詢協議的第二次修訂(統稱為“修訂”), 每項修訂當時與這些個人訂立的補償協議(見附註9—承諾和或有事項)。
根據修正案,伍迪博士和羅斯巴德博士自2024年1月1日起同意減少各自修訂後的僱傭協議中規定的基本工資,
此外,根據修正案,
博士和費爾德曼爵士,自2024年1月1日起生效,同意減少
各自諮詢協議中規定的基本工資,
批准延期以恢復納斯達克合規
如之前在附註10—股東(虧損)權益中披露的
,本公司收到納斯達克的信函,通知本公司
不符合繼續在納斯達克資本市場上市的最低股東權益要求。納斯達克
上市規則5550(b)(1)(以下簡稱"規則")要求在納斯達克資本市場上市的公司保持
股東權益至少為美元
延期的條款如下:2024年5月13日或
之前,公司必須完成合規計劃中更詳細描述的某些交易,預計
導致公司將其股東權益增加至超過美元,
F-52
應計獎金的退還
截至2023年12月31日止年度,公司餘額為美元
D & O保險訴訟
2024年2月12日, 公司的美國加利福尼亞州北區地區法院聖何塞分部的未決訴訟批准 對AmTrust International Underwriters DAC("AmTrust"),該公司是合併前 董事和高級管理人員的保險單承保人,公司的超額保險公司,自由 專業保險公司("自由”,命令如下:
| (a) | AmTrust有義務根據其與 公司簽訂的保險單,向公司預付超過公司已經預付或將預付給博士的免賠額的所有辯護費用。 公司前首席執行官兼董事Marlene Krauss和前董事會主席George Hornig,就美國證券交易委員會發出的某些傳票;以及 |
| (b) | AmTrust保險單用盡後,Freedom 有義務根據其與公司簽訂的超額責任保險單進行同樣的處理。 |
本命令適用於 案件最終處置,但不構成最終判決,公司和兩家保險公司保留 在定於2025年5月12日舉行的審判中對所有適用問題提出異議的權利。
目前尚不清楚 被告是否將採取措施對本命令提出上訴,任何此類上訴的結果,我們收到我們可能收到的任何資金的時間 ,根據與償還與證券及監察委員會傳票(如有)相關的款項有關的命令,或我們最終可能收到的此類款項 。
審判後的最終判決可能會確認保險公司的這些義務,或者推翻並要求公司償還全部或某些 部分預付款。目前還不能保證最終判決可能涉及什麼。
2022年綜合激勵計劃第二次修正案
2024年2月16日,
公司召開股東特別會議,截至2023年12月18日,批准通過180生命科學公司2022年綜合激勵計劃第二修正案。該修訂增加了根據本計劃可發行的最大股份數量,
特別股東大會/反向 股票分割
2024年2月16日,
公司於2023年12月18日召開股東特別會議,就三項提案進行表決:i)批准
第二次修訂和重述的公司註冊證書的修正案,以實現已發行和流通的普通股的反向股票分割,面值為美元
2024年2月16日, 公司董事會批准了一個反向拆分比例為19分之一,其中 反向拆分後剩餘的任何零碎股份將被四捨五入至最接近的整股。公司認為,拆分對於維持 公司普通股在納斯達克資本市場的上市以及糾正公司目前不遵守納斯達克資本市場上市規則的問題是必要的;董事會還認為,反向拆分還將提高普通股的市場流通性和流動性。
2024年2月26日, 公司向特拉華州務卿提交了經修訂的第二次修訂和重述的公司註冊證書的修訂證書(“修訂證書 ”),以實現反向股票分割。根據修訂證書 ,反向股票拆分於2024年2月28日東部時間上午12:01(“生效時間”)生效。 本公司普通股股票於2024年2月28日開始在納斯達克資本市場(“納斯達克”)交易 ,新的CISIP編號:68236V302。公司普通股或公開認股權證的交易代碼分別為"ATNF"和"ATNFW",與反向股票分割有關。
F-53
委任新董事會成員
2024年2月28日,
公司董事會任命Blair Jordan為董事會成員,2024年3月7日,
公司董事會任命Omar Jimenez和Ryan L。史密斯為董事會成員。與此同時,董事會將董事會成員人數定為5名。董事會認定Jordan先生、Jimenez先生和Smith先生均為獨立人士,且不參與與公司訂立的任何重大
計劃、合同或安排(無論是否書面)。關於他們的董事會任命,並於2024年2月24日,2024年3月4日和2024年3月5日,公司分別與喬丹先生,希門尼斯先生和史密斯先生簽署了一份協議
,根據協議,每人將支付美元。
董事會成員辭職
2024年3月7日,馬克·費爾德曼爵士博士向董事會發出辭去董事會成員職務的通知,並於同日生效。費爾德曼爵士的辭職並非由於與本公司就本公司的運營、政策或做法或其他方面存在任何分歧所致。在辭職前,費爾德曼爵士曾擔任本公司聯席執行主席,但並未在董事會任何委員會任職。費爾德曼爵士將繼續擔任該公司的一家子公司的員工。
預先出資的認股權證演練
在2024年2月和
3月期間,2023年8月發行修正案中剩餘的額外預籌資權證以
$的價格行使
納斯達克重新遵守上市規則第5550(A)(2)條
2023年9月7日,納斯達克員工通知該公司,其普通股未能將最低投標價格維持在$
K類特別表決權股份的轉換
2024年3月12日,K類特別投票權股份的一名持有人將此類股份轉換為
F-54