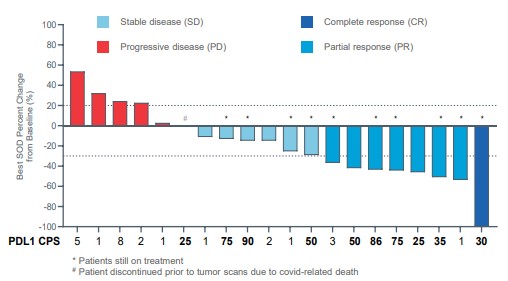
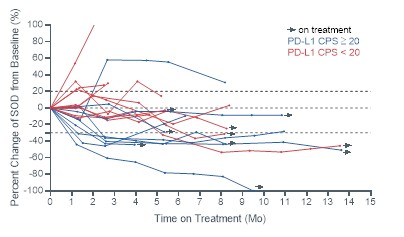
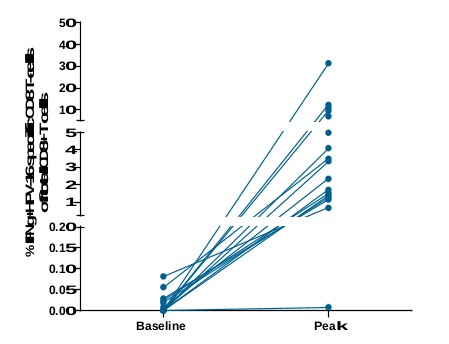
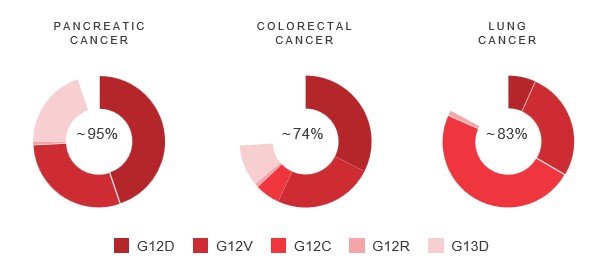
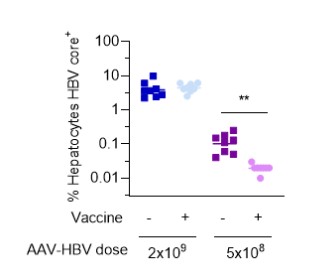
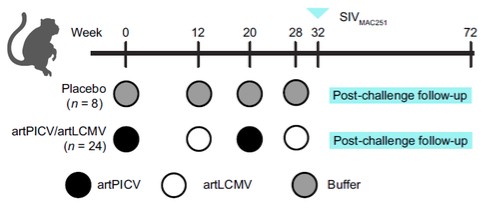
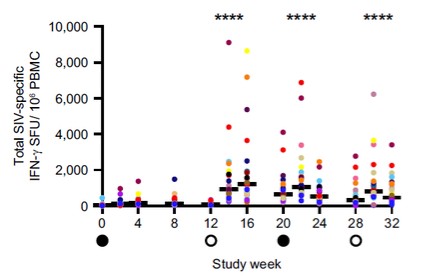
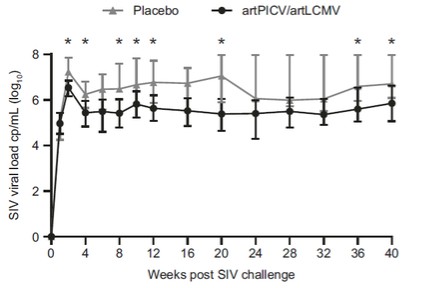
目錄表
現金等價物和限制現金,我們相信計劃中的削減將有助於節約資源,並更好地調整我們的組織,以直接支持後期臨牀開發工作。
領導力
我們由經驗豐富的管理人員、臨牀醫生和科學家組成的團隊領導,他們在腫瘤學、免疫學、疫苗學、臨牀開發和商業化領域擁有專注和轉化的專業知識。我們的首席執行官Joern Aldag曾是uniQure的首席執行官,該公司在他的領導下率先批准了第一個基因治療產品。我們沙粒病毒平臺的基本發現源於我們的聯合創始人,諾貝爾獎獲得者Rolf Zinkernagel,醫學博士,丹尼爾·平舍威爾醫學博士國際公認的沙粒病毒專家,擔任首席執行官的科學顧問。
我們的管道
我們正在利用我們的模塊化沙粒病毒平臺開發以下針對多種癌症和傳染病的候選產品:

站臺
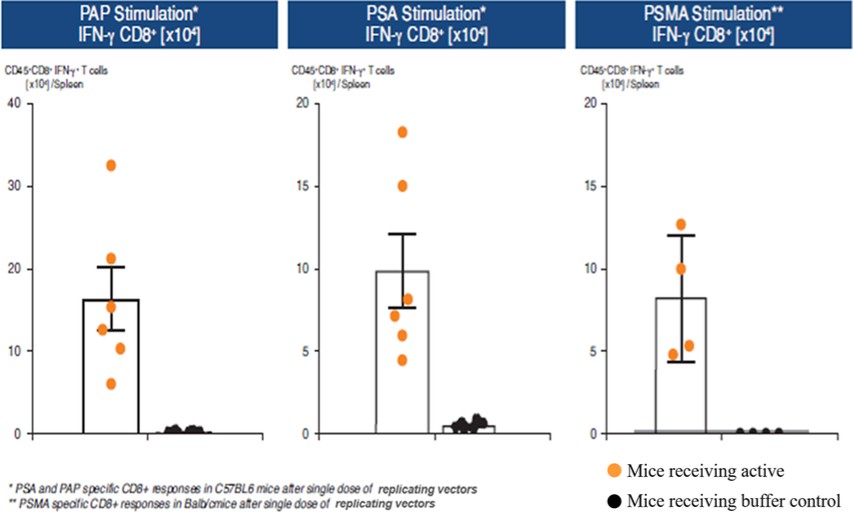
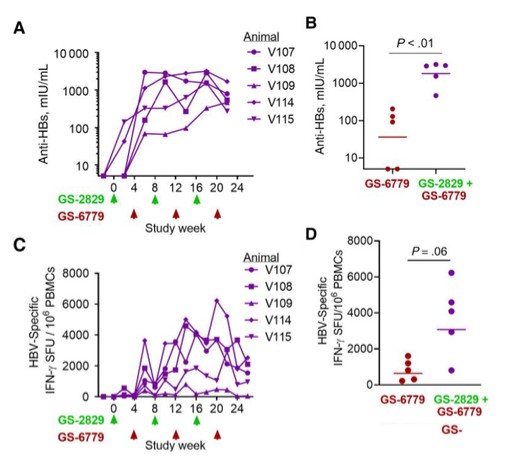
我們的差異化平臺基於工程沙粒病毒,旨在激活天然免疫防禦機制,以觸發強效和靶點特異性T細胞和B細胞免疫。幾十年來,沙粒病毒一直被用作研究CD8 + T細胞應答的臨牀前工具。我們的聯合創始人Rolf Zinkernagel因其基於Arenavirus的關於CD8 + T細胞如何識別病毒感染細胞的研究而獲得諾貝爾生理學或醫學獎。我們認為沙粒病毒具有幾個關鍵優勢,使它們具有最佳免疫療法的特徵,包括:
| ● | 通過直接靶向和激活抗原呈遞細胞("APC"),如樹突細胞誘導穩健的CD8 + T細胞應答的能力,所述抗原呈遞細胞是身體的最有效的抗原呈遞細胞; |
| ● | 誘導對疾病特異性靶抗原的穩健抗體應答的能力; |
| ● | 載體特異性抗體的中和減少,這有助於重複給藥的可能性; |
| ● | 不需要佐劑來刺激免疫系統;和 |
| ● | 在我們的臨牀前研究和臨牀試驗中觀察到耐受性良好。 |
我們相信我們是第一個為治療目的重組沙粒病毒的公司。我們的系統性和可重複性方法具有兩種技術,能夠提供疾病特異性抗原,用於疾病預防和治療。我們的非複製技術旨在誘導強烈的免疫反應,用於預防和治療傳染病。我們的複製技術被設計用來產生更強大的免疫力
8