美國 美國
美國證券交易委員會
華盛頓特區,20549
表格
(標記 一)
或
截至本財政年度止
或
的過渡期 至 .
或
需要該空殼公司報告的事件日期
佣金
文件編號:
(註冊人的確切姓名載於其章程)
+49
(3641) 508 180
(主要執行機構地址)
首席財務官
電話:
(公司聯繫人姓名、電話、電子郵件和/或傳真號碼和地址)
將 拷貝到:
索菲亞·哈德森
Kirkland&Ellis LLP
列剋星敦大道601號
紐約,NY 10022
電話:+1(212)446—4750
根據該法第12(B)條登記或將登記的證券:
| 每個班級的標題 | 交易代碼 | 註冊的每個交易所的名稱 | ||
根據該法第12(G)條登記或將登記的證券:
無
根據該法第15(D)條負有報告義務的證券:
無
説明截至年度報告所述期間結束時發行人所屬各類資本或普通股的流通股數量 。
截至2023年12月31日的流通普通股數量為
如果註冊人是證券法規則405中定義的知名經驗豐富的發行人,請用複選標記表示 。
是的
☐和。
如果此報告是年度報告或過渡報告,請勾選標記以確定註冊人是否不需要根據1934年《證券交易法》第 13或15(D)節提交報告。
是的
☐和。
用複選標記表示註冊人(1)是否在過去12個月內(或註冊人被要求提交此類報告的較短期限內)提交了1934年《證券交易法》第13或15(D)節要求提交的所有報告,以及(2) 在過去90天內是否符合此類提交要求。
在過去12個月內(或在註冊人 被要求提交此類文件的較短時間內),請勾選 是否以電子方式提交了根據法規S—T(本章第232.405節)第405條要求提交的每個交互式數據文件(如有)。
通過勾選標記確定註冊人是大型加速備案人、加速備案人、非加速備案人還是新興增長型公司 。參見《交易法》規則12b—2中"大型加速申報人"、"加速申報人"和"新興增長公司"的定義。
| 大型加速文件服務器☐ | 非加速文件管理器 | ||
| 新興成長型公司 |
如果 一家根據美國公認會計原則編制財務報表的新興成長型公司,用勾號表示註冊人 是否已選擇不使用延長的過渡期來遵守†根據交易法第13(A)節提供的任何新的或修訂的財務會計準則 。☐
| † | 新的或修訂的財務會計準則是指財務會計準則委員會在2012年4月5日之後發佈的對其會計準則編纂的任何更新。 |
用複選標記表示註冊人是否提交了一份報告,並證明其管理層根據《薩班斯-奧克斯利法案》(《美國法典》第15編第7262(B)節)第404(B)條對其財務報告內部控制的有效性進行了評估
編制或發佈其審計報告的註冊會計師事務所。
如果證券是根據該法第12(B)條登記的,請用複選標記表示備案文件中包括的註冊人的財務報表是否反映了對以前發佈的財務報表的錯誤更正。
用複選標記表示這些錯誤更正中是否有任何重述需要根據§240.10D-1(B)對註冊人的任何高管在相關恢復期間收到的基於激勵的薪酬進行恢復分析。☐
用複選標記表示註冊人在編制本文件所包含的財務報表時使用了哪種會計基礎:
美國公認會計準則
☒
其他
如果在回答上一個問題時勾選了“其他”,請用複選標記表示註冊人選擇遵循哪個財務報表項目。
刪除 項目17 項目18
如果這是年度報告,請用複選標記表示註冊人是否是空殼公司(如《交易所規則》第12b-2條所述)。
是
沒有
INFLARx 新澤西州
目錄表
| 頁面 | ||
| 前瞻性陳述 | 四. | |
| 判決的強制執行 | VI | |
| 第一部分 | 1 | |
| 項目1.董事、高級管理人員和顧問的身份 | 1 | |
| A. | 董事和高級管理人員 | 1 |
| B. | 顧問 | 1 |
| C. | 審計師 | 1 |
| 項目2.報價統計數據和預期時間表 | 1 | |
| A. | 報價統計 | 1 |
| B. | 方法和預期時間表 | 1 |
| 項目3.關鍵信息 | 1 | |
| A. | 資本化和負債化 | 1 |
| B. | 提供和使用收益的原因 | 1 |
| C. | 風險因素 | 2 |
| A. | 公司的歷史與發展 | 54 |
| B. | 業務概述 | 55 |
| C. | 組織結構 | 98 |
| D. | 財產、廠房和設備 | 98 |
| 項目4A。未解決的員工意見 | 99 | |
| 項目5.業務和財務審查及展望 | 99 | |
| A. | 經營業績 | 99 |
| B. | 財務運營概述 | 101 |
| C. | 流動資金和資本資源 | 108 |
| D. | 研發、專利和許可證等。 | 111 |
| E. | 趨勢信息 | 111 |
| F. | 關鍵會計估計 | 111 |
| 項目6.董事、高級管理人員和僱員 | 112 | |
| A. | 董事和高級管理人員 | 112 |
| B. | 補償 | 115 |
| C. | 董事會慣例 | 118 |
| D. | 員工 | 121 |
| E. | 股份所有權 | 121 |
| F. | 披露登記人追討錯誤判給的補償的行動 | 121 |
| 項目7.大股東和關聯方交易 | 122 | |
| A. | 大股東 | 122 |
| B. | 關聯方交易 | 125 |
| 項目8.財務信息 | 126 | |
| A. | 合併報表和其他財務信息 | 126 |
| B. | 重大變化 | 126 |
i
| 項目9.報價和清單 | 127 | |
| A. | 發售和上市詳情 | 127 |
| B. | 配送計劃 | 127 |
| C. | 市場 | 127 |
| D. | 出售股東 | 127 |
| E. | 稀釋 | 127 |
| F. | 發行債券的開支 | 127 |
| 項目10.補充信息 | 128 | |
| A. | 股本 | 128 |
| B. | 組織章程大綱及章程細則 | 128 |
| C. | 材料合同 | 128 |
| D. | 外匯管制 | 128 |
| E. | 税收 | 128 |
| 項目11.關於市場風險的定量和定性披露 | 151 | |
| 第12項.股權證券以外的證券的説明 | 151 | |
| A. | 債務證券 | 151 |
| B. | 認股權證及權利 | 151 |
| C. | 其他證券 | 151 |
| D. | 美國存托股份 | 151 |
| 第II部 | 152 | |
| 項目13.拖欠股息和拖欠股息 | 152 | |
| A. | 缺省值 | 152 |
| B. | 拖欠和拖欠 | 152 |
| 項目14.對擔保持有人的權利和收益的使用作出實質性修改 | 152 | |
| A. | 儀器的材料改裝 | 152 |
| B. | 對權利的實質性修改 | 152 |
| C. | 資產的撤回或替代 | 152 |
| D. | 受託人或付款代理人的更換 | 152 |
| E. | 收益的使用 | 152 |
| 項目15.控制和程序 | 153 | |
| A. | 披露控制和程序 | 153 |
| B. | 管理層關於財務報告內部控制的年度報告 | 153 |
| C. | 註冊會計師事務所認證報告 | 153 |
| D. | 財務報告內部控制的變化 | 153 |
II
| 項目16.保留 | 153 | |
| 項目16A。審計委員會財務專家 | 153 | |
| 項目16B。道德準則 | 153 | |
| 項目16C。首席會計師費用及服務 | 154 | |
| A. | 審計費 | 154 |
| B. | 審計相關費用 | 154 |
| C. | 税費 | 154 |
| D. | 所有其他費用 | 154 |
| E. | 審計委員會的審批前政策和程序 | 154 |
| F. | 審計工作由主要會計師以外的其他人執行,如果超過50% | 154 |
| 項目16D。豁免審計委員會遵守上市標準 | 154 | |
| 項目16E。發行人及關聯購買人購買股權證券 | 155 | |
| 項目16F。更改註冊人的認證會計師 | 155 | |
| 項目16G。公司治理 | 155 | |
| 第16H項。煤礦安全信息披露 | 155 | |
| 項目16I。關於妨礙檢查的外國司法管轄區的披露 | 155 | |
| 項目16J。內幕交易政策 | 155 | |
| 項目16K。網絡安全風險管理和戰略 | 155 | |
| 第三部分 | 157 | |
| 項目17.財務報表 | 157 | |
| 項目18.財務報表 | 157 | |
| 項目19.展品 | 157 | |
| 合併財務報表索引 | F-1 | |
三、
除非 另有説明或上下文另有要求,本年度報告表格20—F或本年度報告中所有提及的"InflaRx N.V.",“InflaRx”、“公司”、“我們的”、“我們的”或類似術語指InflaRx N.V.及其子公司。
財務報表列報
我們 根據國際會計準則理事會 或IASB頒佈的國際財務報告準則或IFRS以歐元進行報告。我們對本年報所載的部分數字作出四捨五入調整。因此, 在某些表中顯示為總計的數字可能不是之前數字的算術彙總。
在 本年度報告中,除非另有説明,否則與 2023年12月31日或之前支付的款項有關的美元換算為歐元(反之亦然),按相關付款時的有效匯率進行。
術語"美元"或"美元"指美元,術語"歐元"或"歐元"指 根據經修訂的建立 歐洲共同體條約在歐洲經濟和貨幣聯盟第三階段開始時引入的貨幣。
行業 和其他數據
我們 從我們自己的內部估計和研究以及 行業和一般出版物以及第三方進行的研究、調查和研究中獲得本年度報告中的行業、統計和市場數據。本年度 報告中使用的所有市場數據都涉及許多假設和限制。雖然我們相信這些行業出版物、調查 和研究中的信息是可靠的,但由於各種重要 因素,包括標題為"項目3"的章節中所述的因素,我們所經營的行業存在高度的不確定性和風險。關鍵信息—C.風險因素”。這些和其他 因素可能導致結果與獨立各方和我們所作估計中所表達的結果有重大差異。
商標
InflaRx®, GOHIBIC®關於Vilwaysi®是我們的商標。本 年度報告中出現的商標、商品名稱和服務標記均為各自所有者的財產。僅為方便起見, 本年度報告中提及的商標和商品名稱不含符號 ® 和™,但此類引用不應被解釋為任何表明其 各自的所有人不會在適用法律的最大範圍內主張其權利。
前瞻性陳述
本 年度報告包含涉及重大風險和不確定性的前瞻性陳述。在某些情況下,您可以通過諸如"可能"、"將"、"應該"、"預期"、"計劃"、"預期"、"可能"、"意圖"、"目標"、"項目"、"估計"、"相信"、"預測"、"潛在"或"繼續"等術語來識別前瞻性 陳述,或這些術語的否定或旨在識別未來陳述的其他類似表述 。這些陳述僅限於本年度報告日期,涉及已知 和未知的風險、不確定性和其他重要因素,這些因素可能導致我們的實際結果、業績或成就與前瞻性陳述中明示或暗示的任何未來結果、業績或成就有重大差異 。我們作出這些 前瞻性陳述主要基於我們對未來事件和財務趨勢的當前預期和預測, 我們認為這些事件和財務趨勢 可能會影響我們的業務、財務狀況和經營成果。這些前瞻性聲明包括但不限於 關於以下內容的聲明:
| ● | 我們有能力成功商業化GOHIBIC(vilobelimab)作為美國醫院治療COVID—19患者的治療藥物, 我們有能力積極影響醫療/保健機構、指南機構和其他第三方組織的治療建議 ; |
| ● | 我們的 對患者人羣規模、市場機會、覆蓋範圍的期望 和報銷、估計退貨和應計退貨以及臨牀效用 GOHIBIC(vilobelimab)在其批准或授權的適應症或vilobelimab和任何 根據緊急使用授權或EUA以及將來的其他候選產品 如果在美國或其他地方被批准用於商業用途; |
四.
| ● | 我們的 成功實施InflaRx承諾計劃的能力,我們的未來的成功 vilobelimab治療COVID—19和其他使人衰弱或危及生命的臨牀試驗 炎症適應症,包括壞疽性膿桿菌和任何其他候選產品, 包括INF904,以及此類臨牀結果是否反映了之前在 進行臨牀前研究和臨牀試驗; |
| ● | vilobelimab,INF904臨牀前研究和臨牀試驗的時間、進展和結果 以及任何其他候選產品,包括幾個項目的vilobelimab的開發 適應症,包括治療PG,以及關於啟動時間的聲明,以及 完成研究或審判及相關準備工作,期間 試驗的結果、這些試驗的費用和我們的研究, 發展計劃通常 |
| ● | 我們 與監管機構就臨牀試驗結果和潛在監管 批准或授權途徑的互動以及接受和批准,包括與我們的上市許可申請(MAA)、vilobelimab提交和 我們的GOHIBIC生物製品許可申請(BLA)相關(vilobelimab);vilobelimab、INF904或任何其他候選產品的監管批准或授權的任何討論或提交 的時間和結果,以及我們 獲得和維持針對任何適應症的vilobelimab或GOHIBIC(vilobelimab)的完全監管批准或EUA的時間和能力;我們 利用我們專有的抗C5a和抗C5aR技術發現和開發治療補體介導的自身免疫性疾病 和炎性疾病的療法的能力; |
| ● | 我們的 能夠保護、維護和實施我們對vilobelimab的知識產權保護, INF904和任何其他候選產品,以及此類保護的範圍; |
| ● | 如果 美國食品藥品監督管理局或FDA、歐洲藥品管理局或EMA, 或任何類似的外國監管機構將接受或同意數量、設計, 我們臨牀試驗的規模、實施或實施,包括任何擬議的主要或 此類試驗的次要終點; |
| ● | vilobelimab、INF904和任何其他候選產品的未來臨牀試驗取得成功 以及這些臨牀結果是否反映了之前進行的臨牀前研究中觀察到的結果 研究和臨牀試驗; |
| ● | 我們對Viloblimab、INF904或任何其他候選產品(如果獲得批准或授權用於商業用途)的患者數量、市場機會、醫療需求和臨牀效用的 預期; |
| ● | 我們的製造能力和戰略,包括我們製造方法和過程的可擴展性和成本,以及我們製造方法和過程的優化,以及 我們繼續依賴我們現有的第三方製造商的能力,以及我們 為我們計劃的未來臨牀試驗聘請更多第三方製造商的能力,以及 商業供應的Viloblimab和成品GOHIBIC(Viloblimab)的能力; |
| ● | 我們對費用、持續虧損、未來收入、資本需求和獲得額外融資的需求或能力的估計 ; |
| ● | 我們對任何批准的viloblimab適應症的範圍的期望; |
| ● | 我們 有能力對我們的候選產品在診所或任何商業銷售(如果獲得批准或授權)進行測試而產生的責任索賠進行辯護。 |
| ● | 如果我們的任何候選產品獲得監管批准或授權,則我們有能力 遵守和滿足持續的藥品監管義務和持續的監管概述; |
| ● | 在尋求市場批准或授權和商業化方面,我們 有能力遵守已制定和未來的法律; |
| ● | 我們未來的增長和競爭能力,取決於我們留住關鍵人員並招聘更多合格人員;以及 |
| ● | 我們的 競爭地位以及與競爭對手相關的發展和預測, C5a和C5aR抑制劑的開發以及正在開發的其他治療產品 在類似的醫療條件下,vilobelimab、INF904或我們的任何其他候選產品 正在開發或我們的行業。 |
v
Because forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified and some of which are beyond our control, you should not rely on these forward-looking statements as predictions of future events. The events and circumstances reflected in our forward-looking statements may not be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. You should refer to the “ITEM 3. KEY INFORMATION: - 3. Risk factors.” section of this Annual Report for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. Moreover, we operate in an evolving environment. New risk factors and uncertainties may emerge from time to time, and it is not possible for management to predict all risk factors and uncertainties. As a result of these factors, we cannot assure you that the forward-looking statements in this Annual Report will prove to be accurate. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein, whether as a result of any new information, future events, changed circumstances or otherwise. You should, however, review the factors and risks and other information we describe in the reports we will file from time to time with the Securities and Exchange Commission, or the SEC, after the date of this Annual Report.
判決的執行
我們 是一家有限責任公司(Naamloze Vennootschap)根據荷蘭法律註冊成立,我們的總部 位於德國。我們幾乎所有的資產都位於美國境外。我們的大多數執行官 和董事居住在美國境外。因此,投資者可能無法在美國境內 向這些人送達訴訟程序,或在美國法院對他們或我們執行強制執行,包括基於美國聯邦證券法民事責任條款的判決。
美國和荷蘭目前沒有一項條約規定在民事和商事事項中相互承認和執行判決(仲裁裁決除外)。因此,美國 法院作出的最終付款判決,無論是否僅基於美國證券法,都不會自動在荷蘭得到承認或執行。 為了獲得可在荷蘭執行的判決, 美國法院作出的最終和決定性判決的一方將被要求向荷蘭有管轄權的法院提交其索賠要求。
本 法院將有權酌情對相關美國法院作出的判決給予其認為適當的重視。根據當前 慣例,荷蘭法院可能會根據相關外國 法院的判決作出判決,但前提是,該判決(i)是最終判決,並且是由已根據國際公認的管轄權理由確立其對 相關荷蘭公司或荷蘭公司(視情況而定)的管轄權的法院作出的, (ii)沒有違反適當程序原則(理所當然地重演),(iii)不違反 荷蘭的公共政策,並且(iv)不與(a)荷蘭法院在同一當事方之間的爭議 中作出的先前判決,或(b)外國法院在同一當事方之間的爭議中作出的先前判決,涉及同一 標的事項並基於同一訴因,前提是這種事先判決在荷蘭是可承認的。荷蘭法院 可以拒絕承認和執行懲罰性賠償金或其他裁決。此外,荷蘭法院可以減少美國法院授予的損害賠償額 ,並僅在補償實際損失或損害所必需的範圍內承認損害賠償。執行 和承認美國法院在荷蘭的判決僅受《荷蘭民事訴訟法》的規定管轄。 如果未授予強制執行許可,申請人必須在荷蘭管轄法院再次提起訴訟。
荷蘭 民事訴訟程序在許多方面與美國民事訴訟程序有很大的不同。就證據的出示而言, 美國法律和基於普通法的其他幾個司法管轄區的法律規定了審前證據披露,這是一種程序,程序當事人 可以在審判前強迫對方或第三方出示文件並要求證人作證。 以這種方式獲得的證據可能對任何訴訟的結果具有決定性意義。根據荷蘭法律 不存在這樣的審前證據披露程序。
美國和德國目前沒有一項條約規定在民事 和商事事項上相互承認和執行判決。因此,美國法院作出的最終付款判決或宣告性判決,無論 是否完全基於美國證券法,都不會自動在德國得到承認或執行。如果德國法院認為美國法院不具備管轄權或判決違反德國公共政策原則,則德國法院可以拒絕承認和執行美國法院作出的判決。例如,在德國,判決懲罰性賠償通常不能執行 。德國法院可以減少美國法院授予的損害賠償金額,並僅在賠償實際損失或損害所必需的情況下才承認損害賠償。
此外,在德國法院針對我們、我們的董事、我們的高級管理層和本文中提到的專家提起的、以執行 基於美國聯邦證券法的責任的訴訟可能會受到某些限制。特別是,德國法院一般不會裁決懲罰性賠償。德國的訴訟也要遵守與美國規則不同的程序規則,包括 關於證據的獲取和可接受性、訴訟程序的進行和費用的分配。德國程序法沒有 規定審前文件披露,德國也不支持根據1970年《海牙證據公約》 進行審前文件披露。在德國的訴訟程序必須以德語進行,原則上,提交給法院的所有文件都必須翻譯成德語。由於這些原因,美國投資者可能難以根據美國聯邦證券法的民事責任條款,向德國法院起訴 針對我們、我們的董事、我們的高級 管理層和本年度報告中提到的專家。
VI
第 部分I
項目 1.董事、高級管理層和顧問的身份
| A. | 董事 及高級管理層 |
不適用 。
| B. | 顧問 |
不適用 。
| C. | 審計師 |
不適用 。
第 項2.優惠統計和預期時間表
| A. | 優惠 統計 |
不適用。
| B. | 方法 和預期時間表 |
不適用 。
第 項3.關鍵信息
| A. | 大寫 和負債 |
不適用 。
| B. | 原因 關於收益的要約和使用 |
不適用 。
-1-
| C. | 風險 因素 |
您 在投資我們的普通股之前,應 仔細考慮以下所述的風險和不確定性以及本年報中的其他信息。如果發生上述任何風險,我們的業務、財務狀況或經營業績可能受到重大不利影響 ,因此,我們普通股的市場價格可能下跌,您可能失去全部或部分投資 。本年報亦載有涉及風險及不確定性之前瞻性陳述。請參閲"前瞻性 聲明"。由於某些因素,我們的實際結果可能與這些前瞻性陳述中的預期結果存在重大不利差異 。
風險 因素摘要
以下是可能對我們的業務、運營和財務業績造成不利影響的主要風險的摘要。本摘要 並不涉及我們面臨的所有風險。有關我們業務面臨的重大風險的更完整討論,請參見下文 。
與我們的財務狀況和需要額外資本有關的風險
| ● | 風險 從未實現或維持盈利能力,投資者失去全部投資與獲得額外資金有關的風險 ,以及延遲、減少或取消產品發現和開發計劃或 商業化努力的風險 |
| ● | 風險 與有限的經營歷史和藥品商業化歷史有關風險 與德國聯邦政府資助的贈款有關 |
與我們候選產品的發現、開發、製造和商業化相關的風險
| ● | 風險 與我們候選產品的發現、開發和商業化有關, |
| ● | 風險 與我們對候選產品(包括主要產品)成功的依賴有關 候選人,vilobelimab |
| ● | 風險 與GOHIBIC(vilobelimab)的監管監督有關,我們為此獲得EUA, 這可能導致撤銷或撤銷授予的EUA, |
| ● | 風險 與臨牀開發的任何階段可能發生的臨牀失敗相關 |
| ● | 風險 未能遵守FDA要求和/或與FDA保持一致 反饋,可能會阻礙或延遲vilobelimab的開發、上市或生產 用於治療重症COVID—19患者,以及潛在的vilobelimab在 潰瘍性PG |
| ● | 風險 與持續監管義務相關的額外重大費用 以及繼續對GOHIBIC(vilobelimab)和任何其他候選產品進行監管審查 我們獲得批准或EUA |
| ● | 風險 導致額外成本或在完成時遇到延誤,或最終無法 完成候選產品的開發和商業化(如果是臨牀試驗) 我們的候選產品的安全性和有效性未能令人滿意地向FDA證明 和其他監管機構 |
| ● | 風險 如果我們的候選產品導致或被認為導致不良副作用,或 可能延遲或阻止其監管批准的其他屬性,限制商業 已批准標籤的特徵,或在上市後導致重大負面後果 批准,如果有的話 |
-2-
與我們依賴第三方有關的風險
| ● | 風險 依賴第三方進行我們的臨牀試驗 |
| ● | 風險 對第三方製造商和供應商的依賴,並維持關鍵製造 關係 |
| ● | 風險 與生產生物製品(如vilobelimab)的過程有關,這是非常 易受產品損失 |
| ● | 風險 由於產品風險和質量控制,風險 ,如果我們的第三方製造商無法擴大候選產品的生產規模並提高 產品產量,我們的製造成本可能會增加,產品商業化可能會延遲 |
| ● | 風險 如果我們無法在商業上合理的條款下建立合作,我們可以 必須改變我們的發展和商業化計劃, |
與我們知識產權相關的風險
| ● | 風險 與我們對獲取、維護、保護、捍衞和執行能力的依賴有關 專利、商業祕密和其他知識產權保護 |
| ● | 風險 與我們的專利有關,涵蓋我們專有的抗C5a和抗C5aR技術, 可能受到第三方質疑、縮小、規避和無效 |
| ● | 風險 與我們是第一個製造抗C5a和抗C5aR技術的不確定性有關 在我們的專利或專利申請中聲稱,或者我們是第一個申請專利的 保護 |
| ● | 風險 與專利申請過程和取得我們已經申請的專利有關 |
與員工事務和管理增長相關的風險
| ● | 風險 只有有限的員工來管理和運營我們的業務, |
| ● | 風險 嚴重依賴我們的某些執行官和董事 |
| ● | 風險 由於管理業務運營和數量的增長而導致業務中斷 人員 |
| ● | 風險 我們的員工和第三方承包商的不當活動對我們的業務承擔責任 |
風險 與我們的普通股和我們作為上市公司的地位有關
| ● | 風險 按照我們普通股的交易價格進行評級,這已經和將來可能是 高揮發性 |
| ● | 風險 與外國私人發行人相關,且不受美國代理規則約束, 遵循母國治理慣例,而不是納斯達克上市要求, | |
| 風險 與我們預計在可預見的將來不支付任何現金股息有關 |
一般 風險因素
| ● | 風險 金融市場、政治和監管變化對業務造成的影響 政策和經濟條件一般 |
| ● | 風險 為實現環境目標而採取的法律、監管或市場措施 |
| ● | 風險 通過籌集資本、限制和/或放棄的風險對股東造成的稀釋作用 技術和候選產品的權利 |
| ● | 風險 面對巨大的競爭 |
| ● | 風險 產品責任訴訟 |
| ● | 風險 網絡攻擊和電信故障對我們業務造成的損害和中斷 和信息技術設備 |
-3-
與我們的財務狀況和需要額外資本有關的風險
我們 有過重大經營虧損的歷史,預計在可預見的將來會發生重大且不斷增加的虧損;我們可能 永遠無法實現或維持盈利能力,投資者可能會損失全部投資
截至2023年12月31日、2022年和2021年12月31日止年度,我們 分別產生了4270萬歐元、2950萬歐元和4560萬歐元的淨虧損。此外,截至2023年12月31日,我們的累計赤字為2.861億歐元。
我們 預計,隨着我們將viloblimab和其他候選產品推向更多的臨牀試驗,以及更大規模和後期的臨牀試驗,我們的淨虧損將會增加。此外,我們預計,隨着我們根據歐盟協議在美國推進GOHIBIC(Viloblimab)商業化的實施,同時投資於初創企業商業化和營銷活動,我們的淨虧損將增加。截至 日期,除用於治療重症新冠肺炎的GOHIBIC(Viloblimab)外,我們尚未將其他任何產品商業化,也未從產品銷售中獲得任何有意義的收入,但GOHIBIC(Viloblimab)的初始銷售有限,如果 無法從產品銷售中實現足夠的收入,我們可能永遠無法實現盈利。我們將幾乎所有的財政資源和努力都投入到研究和開發上,包括臨牀前研究、臨牀試驗和製造開發。我們的淨虧損 可能會因季度和年度的不同而大幅波動。淨虧損和負現金流已經並將繼續對我們的股東權益和營運資本產生不利影響。
我們 預計我們的費用可能會增加,如果我們:
| ● | 繼續 開發和進行關於我們的主要候選產品viloblimab的臨牀試驗; |
| ● | 繼續 任何未來候選產品的研究、臨牀前和臨牀開發工作,包括IFX002和INF904; |
| ● | 積極 尋求確定更多研究項目和更多候選產品; |
| ● | 為成功完成 臨牀試驗的我們的候選產品尋求監管和營銷批准(如果有); |
| ● | 現在和將來建立 銷售、營銷、分銷和其他商業基礎設施,將我們可能獲得營銷授權或批准的各種產品商業化 ; |
| ● | 要求 擴大和驗證製造過程,並製造更大數量的候選產品,以用於臨牀開發,並有可能實現商業化; |
| ● | 與戰略合作伙伴協作,優化viloblimab、IFX002、INF904等流水線產品的製造工藝; |
| ● | 維護、擴大和保護我們的知識產權組合; |
| ● | 聘用並保留更多人員,如商業、營銷、臨牀、質量控制和科學人員;以及 |
| ● | 增加 運營、財務和管理信息系統和人員,包括人員 ,以支持我們的產品開發和商業化,並幫助我們履行作為上市公司的義務。 |
我們實現盈利並保持盈利的能力取決於我們的創收能力。我們預計不會產生可觀的收入 ,除非我們或任何未來的合作伙伴能夠獲得我們的一個或多個候選產品的營銷授權或批准,併成功將其商業化。成功的商業化將需要實現關鍵里程碑,包括完成對viloblimab和任何其他候選產品的臨牀試驗,獲得這些候選產品的營銷授權或批准, 製造、營銷和銷售我們或我們未來的任何合作伙伴可能獲得營銷授權或批准的產品,滿足任何上市後要求,並從私人保險或政府付款人那裏獲得我們產品的報銷 。由於與這些活動相關的不確定性和風險,我們無法準確預測收入的時間和金額,以及我們是否或何時可能實現盈利。我們和任何未來的合作者可能永遠不會在這些活動中成功,而且, 即使我們或任何未來的合作者成功,我們也可能永遠不會產生足夠大的收入來實現盈利。即使我們確實實現了盈利,我們也可能無法維持或提高季度或年度盈利能力。
-4-
我們 預計,由於各種因素,我們的財務狀況和經營業績將繼續在每個季度和每年波動,其中許多因素是我們無法控制的。為了取得成功,我們需要從一家專注於研發的公司轉型為一家能夠開展商業活動的公司。我們可能會遇到不可預見的費用、困難、複雜情況和延誤,在這樣的過渡中可能不會成功。
我們未能實現並保持盈利可能會壓低我們普通股的市場價格,並可能削弱我們籌集資金、支付股息、擴大業務、使我們的產品多樣化或繼續運營的能力。如果我們繼續像過去那樣蒙受損失,投資者可能得不到任何投資回報,可能會失去全部投資。
我們 將需要大量額外資金,如果我們無法在需要時籌集資金,我們可能被迫推遲、減少或停止 我們的產品發現和開發計劃或商業化努力
Developing pharmaceutical products, including conducting preclinical studies and clinical trials, is a very time-consuming, expensive and uncertain process that takes years to complete. For example, for the years ended December 31, 2023 and December 31, 2022, we used €37.8 million and €33.7 million, respectively, in net cash for our operating activities, most of which were related to research and development activities. We expect our expenses to increase in connection with our ongoing activities, particularly as we initiate new clinical trials of, initiate new research and preclinical development efforts for, establish robust manufacturing processes for, establish comprehensive commercialization and marketing processes and seek marketing authorization or approval for, our current product candidates or any future product candidates, including those that we may acquire. In particular, we will incur significant expenses as we conduct our planned clinical trial program and initiate new research and preclinical development efforts. In addition, after obtaining the EUA for GOHIBIC (vilobelimab) in the United States and if we obtain marketing approval for any of our product candidates, we may incur significant commercialization expenses related to product sales, marketing, manufacturing and distribution to the extent that such sales, marketing, manufacturing and distribution are not the responsibility of a future collaborator. Furthermore, we expect to incur significant additional costs associated with operating as a public company. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we may be forced to delay, reduce or eliminate our research and development programs or any future commercialization efforts.
我們 計劃將手頭現金主要用於資助我們計劃中的臨牀試驗項目、啟動新的研究和臨牀前開發 工作、建立商業規模的生產工藝以及用於營運資金和其他一般企業用途。我們將需要 花費大量資金,以便在臨牀開發後期推進vilobelimab的開發, 以及我們可能尋求開發的其他候選產品,包括IFX 002和INF904。我們還在評估vilobelimab的多個其他適應症 。我們的管線候選產品的任何未來開發活動將在很大程度上取決於vilobelimab在任何適應症中的臨牀 以及商業化和上市成功。
我們的 現有現金和現金等價物將不足以為我們計劃開展的所有工作提供資金,也不足以為完成 我們的任何候選產品的開發提供資金。因此,我們將需要通過公開或私人 股票發行、債務融資、基於特許權使用費的融資、合作和許可安排或其他來源獲得進一步的資金。目前,我們 沒有任何承諾的外部資金來源。我們可能無法以可接受的條件獲得足夠的額外融資,或 根本無法獲得。如果我們無法以我們可接受的條件籌集足夠數量的額外資金,我們可能不得不大幅 推遲、縮減或停止vilobelimab或我們任何其他候選產品的開發或商業化,或可能 完全停止運營。我們未能在需要時籌集資金,可能會對我們的財務狀況 和我們執行業務戰略的能力產生負面影響。
-5-
我們 相信,我們現有的現金、現金等價物和有價證券將使我們能夠至少在未來24個月內滿足當前業務計劃下的運營費用和資本支出需求。不斷變化的情況(其中一些情況可能 超出了我們的控制範圍)可能導致我們消耗資本的速度遠遠超過我們的預期,我們可能需要比計劃更快地尋求額外資金 。我們未來的短期和長期資金需求將取決於許多因素,包括:
| ● | 臨牀試驗的範圍、進展、時間、成本和結果,以及研究和臨牀前 我們當前和未來候選產品的開發努力,特別是vilobelimab; |
| ● | 未來候選產品的數量和我們追求的適應症及其開發 要求; |
| ● | 尋求監管批准的結果、時間和成本; |
| ● | 準備和實施商業化和商業化活動的費用 對於獲得上市許可批准的任何候選產品 此類成本不是任何未來合作者的責任,包括成本和 建立產品銷售、營銷、分銷和商業規模生產的時機 能力; |
| ● | 競爭的技術和市場發展的影響; |
| ● | 主題 收到上市授權或批准,從商業部門收到的收入(如有) 銷售我們當前和未來的候選產品; |
| ● | 我們 達成任何合作、許可或其他安排的能力以及條款和時間; |
| ● | 我們的 隨着我們擴大研究、開發、製造,員工人數增長和相關成本, 監管和商業活動; |
| ● | 準備、提交和起訴專利申請、維護和保護的費用 我們的知識產權,包括執行和保護知識產權 相關索賠;及 |
| ● | 作為一家上市公司的運營成本。 |
我們 的經營歷史和醫藥產品商業化歷史有限,這可能使我們難以評估 未來生存能力的前景
We commenced operations in 2008. Our operations to date have been limited to establishing our Company, raising capital, developing our proprietary anti-C5a and anti-C5aR technologies, identifying and testing potential product candidates and conducting clinical trials of our lead product candidate, vilobelimab and establishing a commercial scale manufacturing process for vilobelimab. In April 2023, we received the EUA for vilobelimab for the treatment of certain critically ill COVID-19 patients by the FDA. We have just started to arrange for third parties to manufacture and distribute our product at small commercial scale on our behalf. However, we have not yet demonstrated an ability to obtain full marketing approvals or conduct sales and marketing activities at large scale as necessary for successful product commercialization. Also, we are still in early stages of clinical development with our other product candidates. Accordingly, you should consider our prospects in light of the costs, uncertainties, delays and difficulties frequently encountered by companies in the early stages of development, especially clinical-stage biopharmaceutical companies such as ours. Any predictions you make about our future success or viability may not be as accurate as they could be if we had a longer operating history or a history of successfully developing and commercializing pharmaceutical products.
我們 在實現業務目標時,可能會遇到無法預見的費用、困難、併發症、延誤和其他已知或未知的因素。 我們最終將需要從一個以開發為主的公司轉變為一個能夠支持商業活動的公司 。我們在這樣的過渡中可能不會成功。
我們 預計我們的財務狀況和經營業績將繼續因各種因素而大幅波動,其中許多因素超出了我們的控制範圍。因此,您不應依賴任何季度 或年度的結果作為未來運營績效的指標。
我們 可能會面臨與德國聯邦政府收到的財政撥款有關的風險,這可能導致部分償還此類撥款
Vilobelimab在重症COVID—19患者中的 臨牀III期研究以及我們候選產品vilobelimab的某些生產開發相關活動 部分由德國聯邦政府通過2021年10月授予我們的贈款提供資金。 該補助金的結構是償還與vilobelimab臨牀開發和生產相關的某些預先指定費用的80%。授出期按計劃於二零二三年六月三十日結束。總的來説,在整個授予期內和 本協議之日,我們總共收到了3330萬歐元的資助。我們可能會接受德國聯邦政府或歐洲監管機構的財務監督機構 的未來審計,如果當局發現不遵守所有贈款條件,則未能通過這些審計,可能導致贈款授予的部分資金被追溯撤銷。
-6-
此外,在特殊公共利益的情況下,德國聯邦政府擁有使用作為資助工作一部分產生的知識產權的非排他性和可轉讓的權利。與第三方簽訂的與利用資助工作成果有關的合同 必須向代表德國聯邦政府管理贈款的機構披露,與歐盟以外各方簽訂的任何此類合同 在偏離 德國聯邦政府先前批准的商業開發計劃的情況下,都需要得到德國聯邦政府的事先同意。此外,我們可能需要授予第三方 許可,以便在某些條件下使用這些結果。在某些情況下,包括如果我們受到外國投資者的決定性影響 ,資助的成果未經德國 聯邦政府的事先同意而完全或主要用於德國境外,或者如果我們違反了我們在資助項下的義務,則資助資金(包括已經收到的資金) 可以被撤銷。
匯率 波動可能會對我們的經營業績和財務狀況產生重大影響
潛在的 未來費用和收入可能發生或產生於歐盟以外,特別是美國。因此,我們的 業務和股價可能會受到歐元和其他貨幣(特別是 美元)之間匯率波動的影響,這也可能對我們報告的經營業績和各期現金流量產生重大影響。 我們目前沒有任何匯率對衝安排。
由於GOHIBIC(vilobelimab)庫存過剩或過時,我們 可能面臨大量庫存減記的風險,因為在銷售前到期或不適合替代用途
過度購買原材料和生產產品或承諾購買或生產此類項目可能會導致高庫存 ,這在商業上可能是不可行的,並可能導致必須部分或全部減記這些項目。我們庫存的過剩和 過時風險可能源於保質期到期、積壓或供應鏈中斷、製造錯誤、可能導致錯誤、浪費或延誤的原材料缺陷等事件。這種過時的風險是否成為現實,最終取決於市場對我們產品的需求/滲透率和醫療需求,監管因素,如我們打算將產品商業化的地區的批准 或監管機構的批准撤回,我們運營所在市場的競爭格局,包括我們競爭對手開發類似產品的能力,以及我們掌握 具有競爭力的價格的能力,以及我們自己關於將庫存中的未完成或成品用於其他用途的計劃 ,例如用於其他適應症的臨牀試驗。此外,貨幣匯率的變化可能會影響進口商品的成本,並影響庫存估值。在評估我們的庫存時,我們會對這些因素做出假設,並在適用時更新這些假設。
1. 與我們候選產品的發現、開發、製造和商業化相關的風險
我們的開發工作正處於臨牀開發階段,我們針對C5a或C5aR抑制的方法是新穎的,我們可能 無法成功開發任何候選產品並將其商業化
Viloblimab是一種新的治療性抗體,其潛在的治療效果尚未得到證實,C5a或C5aR抑制治療補體介導的自身免疫性和炎症性疾病僅得到部分驗證。我們還沒有,也可能永遠不會成功地在關鍵的臨牀試驗中證明維洛貝利單抗的有效性和安全性,或者在此後獲得上市批准,用於重症新冠肺炎或任何其他適應症。上述情況仍然存在,儘管GOHIBIC(VILOBLIMAB)已獲得食品和藥物管理局的EUA批准,用於治療住院成人冠狀病毒病19,或新冠肺炎,在接受有創機械通氣或體外膜氧合後48小時內啟動。如果我們的開發努力不成功,我們 可能無法推進我們候選產品的開發,將產品商業化,籌集資金,擴大我們的業務,或繼續我們的業務 。
-7-
我們 依賴於我們的候選產品(包括我們的主要候選產品viloblimab)的成功,如果我們無法及時獲得對一個或多個適應症的候選產品的批准並將其商業化,我們的業務將受到實質性損害
我們的成功取決於我們是否有能力及時完成臨牀試驗,並獲得營銷授權或批准,然後成功地將我們的候選產品 商業化,包括我們的主要候選產品viloblimab,用於一個或多個適應症。我們的候選產品 將需要額外的臨牀開發、臨牀前和製造開發活動、政府監管機構的營銷批准、商業製造、大量投資和重大營銷努力,才能從產品銷售中獲得任何收入 。在獲得相關監管機構(包括FDA在美國市場營銷和EMA在歐洲市場營銷)的營銷授權或 批准之前,我們不允許在一個司法管轄區內營銷或推廣任何候選產品,而且我們可能永遠不會獲得超過FDA於2023年4月授予的GOHIBIC(Viloblimab)EUA之外的此類營銷批准或營銷授權。我們候選產品的成功將取決於許多因素,包括:
| ● | 籌集額外資金或進行合作,以完成我們候選產品的臨牀開發和商業化; |
| ● | 成功並及時完成我們正在進行的臨牀試驗; |
| ● | 啟動成功的患者登記並及時完成額外的臨牀試驗 ; |
| ● | 有效性、安全性和耐受性,符合FDA、EMA或任何類似的外國監管機構的上市審批要求; |
| ● | 及時 收到適用監管機構對我們的候選產品的營銷授權或批准 ; |
| ● | 向適用的監管機構作出任何必要的上市後審批承諾的程度; |
| ● | 與第三方藥品供應商和製造商 維護現有的或建立新的供應安排; |
| ● | 維護現有的或與第三方製造商建立新的規模化生產安排,以獲得經過適當包裝以供銷售的成品; |
| ● | 獲取 以及維護專利保護、商業祕密保護和監管排他性,在 美國和其他地方; |
| ● | 保護 我們在知識產權組合中的權利,包括我們的授權知識產權 財產; |
| ● | 成功 在任何上市授權或批准後啟動商業銷售; |
| ● | a 在任何上市許可或批准後,持續可接受的安全性特徵; |
| ● | 商業 患者、醫療界和第三方支付者的接受; |
| ● | 包含 醫療指南中已批准或(如GOHIBIC(vilobelimab))授權產品 機構,包括但不限於國家衞生研究所或NIH指南; |
| ● | 包含 醫院處方集上已批准或(如GOHIBIC(vilobelimab))授權的產品; 和 |
| ● | 我們的 與其他療法競爭的能力。 |
此外, 我們無法確定我們是否能夠及時獲得必要的監管授權或批准, 我們正在開發或製造的任何產品 也無法確定我們是否能夠對現有產品保持必要的監管授權或批准, 並且以下所有情況都可能對我們的業務產生重大不利影響:
| ● | 重要 延遲獲得或未能獲得所需授權批准; |
| ● | 損失 先前獲得的授權或批准的變更; |
| ● | 失敗 遵守現有或未來的監管要求; |
| ● | 更改 根據生產工藝或生產工藝標準,或 或改變對這些因素的解釋。 |
許多 此類因素仍不在我們的控制範圍內,如果我們無法實現上述一個或多個目標,我們的業務 將受到重大損害。
-8-
我們收到EUA的GOHIBIC (vilobelimab)受到持續監管監督,如果FDA認為不再提供EUA的基本要求或條件,則可能導致撤銷或 撤銷已授予的EUA。 如果我們的GOHIBIC(vilobelimab)EUA被 FDA撤銷或撤銷,我們可能會遭受重大損失,包括聲譽損失。
GOHIBIC (vilobelimab),根據《聯邦食品、藥品和化粧品法案》(《法案》)第 564節,我們獲得EUA,用於治療某些住院成人患者的COVID—19(21 U.S.C.§ 360bbb—3)受持續的監管監督,包括 確定和驗證基本要求,如公共衞生緊急情況或重大潛在公共衞生緊急情況,影響或有重大潛在影響國家安全或居住在海外的美國公民的健康和安全,並涉及導致COVID—19的病毒。授予的EUA將有效,直到 根據法案第564(b)(2)條終止 宣佈存在證明在COVID—19大流行期間授權緊急使用藥物和生物製品的情況,或根據法案第564(g)條撤銷EUA為止。如果授予EUA的條件或要求 將來不再明顯,或者如果報告的藥物安全性事件導致確定 維洛貝利單抗的總體藥物警戒出現新情況,FDA可以暫停、撤回或撤銷授予的EUA。這些事件中的任何一個都可能對我們的業務、財務狀況、經營業績和前景產生重大的 不利影響。
臨牀 失敗可能發生在臨牀開發的任何階段,並且我們的臨牀試驗結果可能不支持我們針對候選產品的擬定適應症
Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the results of later clinical trials will replicate the results of prior clinical trials and preclinical testing. Moreover, success in clinical trials in a particular indication does not ensure that a product candidate will be successful in other indications, even for the same underlying disease. A number of companies in the pharmaceutical industry, including biotechnology companies, have suffered significant setbacks in clinical trials, even after promising results in earlier preclinical studies or clinical trials or successful later-stage trials in other related indications, including in the context of controlling complement activation through C5 and C5a or C5aR inhibition. For example, while others in our industry have attempted to develop C5a-specific antibodies, there is no approved therapy inhibiting C5a. These setbacks have been caused by, among other things, preclinical findings made while clinical trials were underway and safety or efficacy observations made in clinical trials, including previously unreported adverse events as well as lack of efficacy and patient benefit as reported by clinical trial investigators. In particular, development of antibodies that target C5a rather than C5 to control complement activation is comparatively novel, and there is no approved therapy specifically targeting C5a. As a result, inhibition of C5a rather than C5, which blocks signaling to the two receptors C5aR and C5L2, may have unforeseen consequences or negative results that may lead to clinical failure or withdrawal in later stages of our product candidate development. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through preclinical and initial clinical trials for a variety of reasons, including differences in patient populations, changes in trial protocols and complexities of larger, multi-center trials among others. We may fail to complete clinical trials and/or to meet predetermined endpoints in the clinical trials, which may cause us to abandon a product candidate or an indication and may delay development of any other product candidates. Any delay in, or termination of, our clinical trials will delay the submission of the Biologics License Application, or BLA, or the EUA to the FDA, the MAA to the EMA or other similar applications with other relevant foreign regulatory authorities and, ultimately, our ability to commercialize any of our product candidates and generate revenue.
未能 保持符合FDA要求和/或與FDA反饋保持一致可能會阻止或延遲用於治療重症COVID—19患者的vilobelimab的開發、上市 或生產,以及潛在的vilobelimab治療潰瘍性胰腺炎的潛在開發、上市或生產
我們的 生產和實驗室設施定期接受FDA和其他政府機構的檢查,以確保 滿足生產和質量要求。如果FDA或其他政府 機構認為此類檢查的結果令人不滿意,這些設施的操作可能會中斷或停止。此外,未能遵守FDA或其他有關vilobelimab開發、營銷、推廣、生產和分銷的監管要求可能導致罰款、意外合規支出 、召回或扣押我們的產品、全部或部分暫停生產或分銷、標籤和推廣限制、終止正在進行的研究、取消提交給監管機構的數據資格,執法行動, 禁令和刑事起訴。如果我們不符合適用的監管或質量標準,我們的產品可能會被召回, 在某些情況下,我們可能會被要求通知適用的監管機構。
-9-
GOHIBIC (vilobelimab)和我們獲得批准或EUA的任何其他候選產品均須接受持續監管監督, 我們將接受持續監管義務和持續監管審查,這可能導致大量額外 費用。倘我們未能遵守監管要求,我們可能會受到處罰。
GOHIBIC (vilobelimab)和我們已獲得或可能獲得批准的任何其他候選產品或EUA將受到持續的監管 監督,包括審查宣傳材料和其他安全性信息,且適用的監管機構可能 仍會對我們產品的指定用途或營銷施加重大限制,或對可能 成本高昂的事後批准研究或上市後監督。廣告和促銷材料必須符合FDA的規定,並且 接受FDA審查,以及其他可能適用的聯邦和州法律。在其他國家/地區,廣告和促銷材料 可能會受到類似規則的約束。如果在授權或批准我們的任何候選產品後,我們未能遵守適用的監管要求,監管機構可以:
| ● | 發出 警告信,聲稱我們違反了法律; |
| ● | 尋求 禁令或施加民事或刑事處罰或罰款; |
| ● | 暫停 或撤銷已授予的EUA或監管批准,或撤銷許可證; |
| ● | 暫停 任何正在進行的臨牀研究; |
| ● | 扣押 產品;或 |
| ● | 拒絕 允許我們簽訂供應合同,包括政府合同。 |
政府對涉嫌違法行為的任何調查都可能要求我們花費大量的時間和資源進行迴應,並可能 產生負面的宣傳。發生上述任何事件或處罰可能會抑制我們將任何授權 或批准的產品商業化併產生收入的能力。
這些事件中的任何 都可能妨礙我們實現或維持對我們可能識別和開發的任何產品的市場接受度,並可能 對我們的業務、財務狀況、經營業績和前景產生重大不利影響。
如果 我們候選產品的臨牀試驗未能令人滿意地向FDA和其他監管機構證明安全性和有效性,我們 或任何未來合作者可能會在完成或最終無法完成這些候選產品的開發和商業化過程中產生額外成本或經歷延遲
未經FDA的上市批准或EUA,我們 和任何未來合作者不得在美國商業化、營銷、推廣或銷售任何候選產品 。國外監管機構(如EMA)在其各自的市場上實施了類似的要求 。我們和任何未來的合作者必須完成廣泛的臨牀前開發和臨牀試驗,以證明我們候選產品在人體中的安全性和有效性,然後才能獲得這些批准。
我們候選產品的臨牀開發容易受到產品開發任何階段固有的失敗風險的影響。 即使我們的一個或多個候選產品具有有益效果,但由於一個或多個因素(包括我們臨牀試驗的規模、持續時間、設計、測量、實施或分析),在臨牀 評價期間也可能無法檢測到該效果。此外,我們的許多候選產品處於開發或臨牀測試的早期階段。因此, 我們的任何候選產品可能需要數年時間才能獲得監管部門的批准(如果有的話),並且其他臨牀試驗可能無法 證明我們的目標適應症的安全性、療效或耐受性。
-10-
任何 無法成功完成臨牀前和臨牀開發的情況都可能導致我們或任何未來合作者的額外成本 ,並削弱我們從產品銷售、監管和商業化里程碑以及版税中獲得收入的能力。此外,如果 我們或任何未來的合作者被要求對我們的候選產品進行額外的臨牀試驗或其他測試,超出 我們或他們預期的試驗和測試,如果我們或他們無法成功完成候選產品的臨牀試驗 或其他測試,或者這些試驗或測試的結果不利、不確定或只是適度有利,或存在不可接受的 與我們的候選產品相關的安全問題,我們或任何未來的合作者可以:
| ● | 招致 額外的計劃外費用,包括與額外的必要臨牀試驗相關的費用 或臨牀前測試; |
| ● | 是 延遲獲得vilobelimab或我們任何其他候選產品的上市批准; |
| ● | 不是 獲得FDA的上市批准或撤回或撤銷EUA; |
| ● | 獲得 批准的適應症或患者羣體不像預期或期望的那樣廣泛; |
| ● | 獲取 批准標籤,包括重大使用或分銷限制或重大 安全警告,包括盒裝警告; |
| ● | 是 需遵守額外的上市後檢測或其他要求;或 |
| ● | 是 在獲得上市批准後,或 EUA。 |
如果我們 未能成功完成候選產品的臨牀試驗,也未能證明獲得 監管部門批准以銷售候選產品所需的有效性和安全性,將嚴重損害我們的業務。
我們的 候選產品可能會導致或被認為會導致不良副作用,或具有其他可能延遲或阻止其 監管批准、限制已批准標籤的商業特性或導致上市後 批准(如果有的話)的重大負面後果的特性
我們的候選產品引起的不良副作用可能導致我們或監管機構中斷、延遲或停止臨牀試驗,並且 可能導致標籤限制性更強,或者FDA或類似的外國監管機構延遲、拒絕或撤回監管批准 。我們的臨牀試驗結果可能顯示出嚴重程度高且不可接受的副作用或非預期 特徵。此外,我們在vilobelimab臨牀試驗中招募的許多患者患有嚴重的既存 疾病。雖然此類疾病可能導致在試驗期間與 vilobelimab無關的嚴重不良事件或SAE,但此類事件可能產生負面安全性認知,並對任何 批准後的vilobelimab市場接受度產生不利影響。
如果我們的候選產品在開發過程中出現不可接受的副作用,我們、FDA或類似的外國監管機構、機構審查委員會或機構審查委員會或進行我們研究的機構或其他機構的獨立倫理委員會,或數據安全監測委員會或DSMB可以暫停或終止我們的臨牀試驗,FDA或類似的外國監管機構可以命令我們停止臨牀試驗或拒絕批准我們的任何或所有靶向適應症的候選產品。 副作用,無論是否與治療相關,還可能影響患者招募或登記患者完成試驗的能力或導致潛在的產品責任索賠。此外,治療醫務人員可能無法正確識別或處理這些副作用。我們希望對使用我們的候選產品的醫務人員進行培訓,以瞭解我們的臨牀試驗和任何候選產品商業化時的副作用。在識別或管理我們的候選產品的潛在副作用方面培訓不足可能會導致患者受傷或死亡。這些情況中的任何一種都可能對我們的業務、財務狀況和前景造成重大損害。
此外,我們候選產品的臨牀試驗是在精心定義的患者組中進行的,這些患者已同意進入臨牀 試驗。因此,我們的臨牀試驗或任何未來合作伙伴的臨牀試驗可能會顯示候選產品的明顯正面效果大於實際正面效果(如果有),或者無法識別不良的副作用 。如果在候選產品獲得批准後,我們或其他人發現該產品的有效性低於之前 認為的效果,或導致先前未確定的不良副作用,則可能會發生以下任何不良事件:
| ● | 法規 當局可撤銷其對產品的授權或批准或扣押產品; |
| ● | 我們, 或任何未來的合作者,可能需要召回產品,或被要求更改 產品的給藥方式或進行其他臨牀試驗; |
-11-
| ● | 附加 可能會對產品的銷售或製造過程施加限制 特定產品; |
| ● | 我們 可能受到罰款、禁令或民事或刑事處罰; |
| ● | 監管當局可能要求添加標籤説明,例如“黑匣子”警告或禁忌症; |
| ● | 我們, 或任何未來合作者,可能需要創建概述風險的藥物指南 以前未確定的副作用,以分發給病人; |
| ● | 我們, 或任何未來的合作者,可能需要實施強制分發的REMS 使用限制或進行上市後研究或臨牀試驗; |
| ● | 我們, 或任何未來的合作者,可能會被起訴並追究對患者造成的傷害的責任; |
| ● | 產品的競爭力可能會降低;以及 |
| ● | 我們的聲譽可能會受到影響。 |
這些事件中的任何事件都可能損害我們的業務和運營,並可能對我們的股價產生負面影響。
我們的 最先進的候選產品是嵌合或人源化抗體蛋白,它們可能會在患者中引起免疫應答, 導致產生針對這些治療性蛋白的有害抗體或中和抗體
In addition to the safety, efficacy, manufacturing and regulatory hurdles faced by our product candidates, the administration of proteins such as monoclonal antibodies that are chimeric or humanized, including our product candidates vilobelimab and IFX002, respectively, can cause an immune response, resulting in the creation of antibodies against the therapeutic protein. These anti-drug antibodies, or ADAs, can have no effect or can neutralize the effectiveness of the protein or require that higher doses be used to obtain a therapeutic effect. Whether ADAs will be created and how they react can often not be predicted from preclinical or even clinical studies, and their detection or appearance is often delayed. As a result, neutralizing antibodies may be detected at a later date or upon longer exposure of patients with our product candidates, such as following more chronic administration in longer lasting clinical trials. In some cases, detection of such neutralizing antibodies can even occur after pivotal clinical trials have been completed. Therefore, there can be no assurance that neutralizing antibodies will not be detected in future clinical trials or at a later date upon longer exposure (including after commercialization). If ADAs reduce or neutralize the effectiveness of our product candidates, the continued clinical development or receipt of marketing approval for any of our product candidates could be delayed or prevented and, even if any of our product candidates is approved, their commercial success could be limited, any of which would impair our ability to generate revenue and continue operations. Low levels of ADAs were detected in previously completed clinical studies.
即使 我們完成了必要的vilobelimab和任何其他候選產品的臨牀前研究和臨牀試驗,上市 審批流程(包括EUA流程)是昂貴、耗時且不確定的,可能會妨礙我們或任何未來合作者 獲得部分或全部候選產品的商業化批准。因此,我們無法預測我們或任何未來的合作者何時或是否在哪些地區獲得營銷授權或批准,以將候選產品商業化
產品的 研究、測試、製造、貼標籤、批准、銷售、營銷、推廣和分銷均受FDA和類似外國監管機構的廣泛 監管。我們和任何未來的合作者不得在美國或其他國家銷售我們的 候選產品,直到我們或他們獲得FDA的BLA或EUA批准或 美國境外適用監管機構的上市批准。我們的候選產品處於不同的 開發階段,並面臨藥物開發固有的失敗風險。我們尚未在美國或任何其他司法管轄區提交申請或獲得 任何候選產品的上市批准。我們在進行 和管理獲得上市批准所需的臨牀試驗(包括FDA批准BLA或EUA)方面經驗有限。此外, 靶向C5a抑制的候選產品之前沒有監管部門批准的歷史。
-12-
The process of obtaining marketing authorizations or approvals, both in the United States and elsewhere is lengthy, expensive and uncertain. It may take many years, if approval is obtained at all, and can vary substantially based upon a variety of factors, including the type, complexity and novelty of the product candidates involved. Securing marketing approval, including the EUA, requires the submission of extensive preclinical and clinical data and supporting information to regulatory authorities for each therapeutic indication to establish the product candidate’s safety and efficacy. Securing marketing approval, including the EUA, also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, the regulatory authorities. The FDA or other regulatory authorities may determine that our product candidates are not safe and effective, only moderately effective or have undesirable or unintended side effects, toxicities or other characteristics that preclude our obtaining marketing approval or prevent or limit commercial use. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a drug candidate’s clinical development and may vary among jurisdictions. Any marketing approval, including the EUA, we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the approved product not commercially viable. For example, the EUA for the GOHIBIC (vilobelimab) has been granted for the treatment of COVID-19 in hospitalized adults when initiated within 48 hours of receiving IMV or ECMO. The FDA, EMA or any comparable foreign regulatory authorities may delay, limit or deny approval of vilobelimab for many reasons, including:
| ● | 我們 可能無法證明vilobelimab作為治療 我們的目標適應症符合FDA、EMA或類似國外法規的要求 機構; |
| ● | FDA、EMA或類似的外國監管機構可能需要進行額外的臨牀試驗 或vilobelimab的非臨牀研究, 無論是在批准前還是作為批准後的承諾,這將增加我們的成本, 延長vilobelimab的開發時間; |
| ● | 我們的臨牀試驗結果可能不符合統計學或臨牀顯著性水平 FDA、EMA或類似的外國監管機構要求獲得上市銷售 批准; |
| ● | FDA、EMA或類似的外國監管機構可能不同意數量、設計, 我們的臨牀試驗的規模、實施或實施,包括指定的臨牀終點; |
| ● | 臨牀計劃中研究的人羣可能不夠廣泛或具有代表性 以確保我們尋求批准的全部人羣的安全性; |
| ● | 我們聘請進行臨牀試驗的合同研究機構或CRO可能採取我們無法控制的 行動,對我們的臨牀試驗產生重大不利影響; |
| ● | FDA、EMA或類似的外國監管機構可能不會發現來自臨牀前研究和臨牀試驗的數據足以證明維洛利單抗和任何其他候選產品的臨牀和其他益處 大於其安全風險; |
| ● | FDA、EMA或類似的外國監管機構可能不同意我們對臨牀前研究和臨牀試驗數據的解釋。 |
| ● | FDA、EMA或類似的外國監管機構可能不接受臨牀試驗現場產生的數據,包括不符合CGCP; |
| ● | 如果我們的BLA在提交時被諮詢委員會審查,FDA可能難以及時安排諮詢委員會會議,或者諮詢委員會可能建議不批准我們的申請,或者可能建議FDA要求,作為批准的條件,額外的臨牀前研究或臨牀試驗,對批准的標籤或分銷的限制和使用限制; |
| ● | FDA、EMA或類似的外國監管機構可能要求制定風險評估和緩解戰略,或REMS,作為批准的條件; |
| ● | FDA、EMA或類似的外國監管機構可能會發現我們的第三方製造商的製造工藝或設施中存在缺陷,包括不符合當前的良好製造規範或cGMP;或 |
| ● | FDA、EMA或類似的外國監管機構可以改變各自的審批政策或採用新的法規。 |
-13-
在生物製藥行業大量正在開發的藥物中,只有一小部分藥物向FDA提交了BLA ,更少的藥物獲得了商業化批准。此外,即使我們確實獲得了上市viloblimab的監管批准, 任何此類批准都可能受到我們可能銷售該產品的指定用途或患者羣體的限制。因此, 即使我們能夠獲得必要的資金以繼續為我們的開發計劃提供資金,我們也不能向您保證viloblimab 和/或任何其他候選產品將成功開發或商業化。
此外,我們臨牀試驗的首席研究員可能會不時擔任我們的科學顧問或顧問,並獲得與此類服務相關的報酬 。在某些情況下,我們可能需要向FDA或其他監管機構報告其中一些關係。FDA或其他監管機構可能會得出結論,首席調查員可能會因為與我們的財務關係而包括 ,存在利益衝突,影響了研究的解釋。因此,FDA或其他監管機構可能會質疑在適用的臨牀試驗地點生成的數據的完整性,臨牀試驗本身的效用可能會受到威脅。這可能會導致FDA或其他監管機構延遲批准或拒絕我們的營銷申請 ,並可能最終導致拒絕我們的一個或多個候選產品的營銷授權或批准 。
任何延遲獲得或未能獲得所需批准的 都可能對我們或任何未來合作伙伴從特定候選產品獲得收入的能力產生負面影響,這可能會對我們的財務狀況造成重大損害,並對我們的股價產生不利影響。
對於我們的候選產品,我們 取決於臨牀研究中的患者登記人數。如果我們在臨牀試驗中招募患者時遇到困難 ,我們的臨牀開發活動可能會被推遲或受到其他不利影響
我們 還將被要求識別和招募足夠數量的PG患者,並可能有其他適應症,用於我們計劃的 或正在進行的這些適應症中的Viloblimab的臨牀試驗。其中一些是罕見的疾病適應症或適應症,患者人數相對較少。在未來的試驗中,試驗參與者的登記可能會受到限制,因為許多潛在的參與者可能不符合條件,因為他們已經在接受批准的藥物治療或正在參與其他臨牀試驗。
患者 登記受其他因素影響,包括:
| ● | 嚴重度 正在調查的疾病; |
| ● | 設計 臨牀試驗方案; |
| ● | 尺寸 患者羣體的性質; |
| ● | 資格 有關審判的標準; |
| ● | 感知 試驗候選產品的風險和受益; |
| ● | 感知 候選產品的安全性和耐受性; |
| ● | 對潛在患者的臨牀試驗地點的近似性和可用性; |
| ● | 競爭療法和臨牀試驗的可獲得性; |
| ● | 臨牀醫生 以及患者對所研究藥物潛在優勢的看法 與其他可用療法(包括標準治療和任何新藥, 可能被批准用於我們正在調查的適應症; |
| ● | 努力 促進臨牀試驗的及時入組; |
| ● | 患者 醫生的轉介做法;及 |
| ● | 我們的 能夠在治療期間和治療後充分監測患者。 |
此外, 治療這些疾病患者的專科醫生數量有限,主要臨牀中心集中 在少數幾個地理區域。我們在識別和入組此類患者時也可能遇到困難,其疾病階段適合我們正在進行的或未來的臨牀試驗 。此外,尋找和診斷患者的過程可能會證明是昂貴的。如果我們無法 為任何臨牀試驗招募足夠數量的患者(如有),則會導致重大延誤或可能需要我們 放棄一項或多項臨牀試驗。
我們 在重度COVID—19、PG和cSCC的臨牀試驗中,由於 在臨牀開發中針對相同患者人羣的其他化合物、疾病患病率低、診斷困難或 鑑於COVID—19大流行,臨牀試驗中心的限制, 招募速度低於預期。任何臨牀試驗的完成進一步延遲將 增加我們的成本,減慢我們候選產品的開發,並延遲或潛在地危及我們開始營銷 和產生收入的能力。此外,如果我們無法找到和招募足夠數量的合格患者參與這些臨牀試驗,那麼我們可能無法啟動或繼續FDA、EMA或其他外國 監管機構要求的vilobelimab或我們正在進行的任何其他候選產品的臨牀試驗。
-14-
即使 我們的一個候選產品獲得了市場批准(包括EUA),它也可能無法達到醫生、患者、第三方支付方和醫療界其他人獲得商業成功所必需的市場接受程度,在這種情況下,我們可能 無法產生可觀的收入或盈利
Even if our other product candidates are approved by the appropriate regulatory authorities for marketing and sale or receives the EUA, they may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors and others in the medical community. As a general proposition, physicians are often reluctant to switch their patients from existing therapies, such as for the treatment of cSCC, even when new and potentially more effective or convenient treatments enter the market. Further, patients often acclimate to existing therapies and do not want to switch therapies unless their physicians recommend doing so or they are required to do so due to lack of reimbursement for existing therapies. Further, we may face a lack of acceptance by the physician community of the efficacy of targeting C5a to inhibit terminal complement activation compared to targeting C5, which is well established in clinical practice (such as eculizumab). In addition, vilobelimab may not be accepted by physicians or patients if we cannot demonstrate, or if vilobelimab is perceived as not having, strong duration of effect, including compared to existing treatments. The duration of effect of vilobelimab has only been studied prospectively for durations less than the expected duration of any pivotal Phase III clinical trials. It is possible that the effects seen in shorter term clinical trials will not be replicated at later time points or in larger clinical trials. Further, even if we are able to demonstrate our product candidates’ safety and efficacy to the FDA and other regulators, safety concerns in the medical community may hinder market acceptance.
教育醫療界和第三方支付方瞭解我們候選產品的好處可能需要大量資源, 包括管理時間和財務資源,而且可能不會成功。如果我們的任何候選產品獲得批准,但沒有 達到足夠的市場接受度,我們可能不會產生顯著收入,我們可能無法盈利。如果我們的候選產品獲得商業銷售批准, 的市場接受程度將取決於多個因素,包括:
| ● | 產品的有效性和安全性; |
| ● | 與競爭療法相比,產品的潛在優勢,儘管取得了成功 達到或超過臨牀試驗終點; |
| ● | 任何副作用的流行率和嚴重程度; |
| ● | 如果 根據醫生治療指南,產品被指定為一線、二線或三線 治療; |
| ● | 我們的 能力或任何未來合作者的能力,以競爭力提供產品銷售 價格; |
| ● | 與替代治療相比,產品的方便性和易於給藥; |
| ● | 目標患者人羣嘗試該產品的意願,以及醫生開出處方的意願; |
| ● | 限制 或警告,包括產品批准的標籤中包含的分發或使用限制; |
| ● | 銷售、營銷和分銷支持力量; |
| ● | 更改產品的目標適應症的護理標準;以及 |
| ● | 可用性 以及政府支付者、管理式醫療計劃和其他第三方支付者的承保和報銷金額 。 |
如果我們的任何候選產品未能獲得市場認可(如果獲得授權或批准),將損害我們的業務,可能需要 我們尋求額外的融資。
-15-
即使 如果我們或任何未來的合作伙伴能夠將我們或他們開發的任何候選產品商業化,該產品可能會 受到不利的定價法規或第三方付款人保險和報銷政策的約束,其中任何一項都可能損害我們的業務
為其病情提供醫療服務的患者 通常依靠第三方付款人來報銷與其治療相關的全部或部分費用。因此,我們以及未來任何合作伙伴將我們的任何候選產品商業化的能力 將在一定程度上取決於這些產品和相關治療的承保範圍和報銷範圍 第三方付款人包括政府衞生行政部門和公共或私人健康保險公司。第三方 付款人決定他們將承保哪些藥物並建立報銷級別。我們不能確定是否可以為viloblimab或我們的任何候選產品提供報銷。此外,我們不能確定,如果獲得批准,不那麼繁瑣的報銷政策不會減少對我們產品的需求或我們可以收取的價格。新批准的孤兒疾病產品的保險覆蓋範圍和報銷狀態尤其不確定,如果不能為viloblimab 或任何其他候選產品獲得或保持足夠的保險覆蓋和報銷,可能會限制我們的創收能力。
如果 無法獲得保險和報銷,或者報銷僅限於有限級別,我們或任何未來的合作伙伴 可能無法成功將我們的候選產品商業化。即使提供了保險,批准的報銷金額 也可能不足以讓我們或任何未來的合作伙伴建立或維持足以實現我們或他們的投資的充分回報的定價。在美國,第三方付款人之間沒有統一的產品承保範圍和報銷政策,產品的承保範圍和報銷範圍因付款人而異。因此,承保範圍確定過程通常是一個耗時且昂貴的過程,需要我們為使用我們的產品分別向每個付款人提供科學和臨牀支持,但不能保證承保範圍和足夠的報銷將始終如一地應用或首先獲得 。
與第三方付款人覆蓋範圍和新藥報銷相關的重大不確定性。新藥產品的上市審批、定價和報銷因國家/地區而異。有些國家/地區要求藥品的銷售價格獲得批准後才能上市。在許多國家/地區,定價審查期在獲得營銷或產品許可批准後開始 。在一些國外市場,處方藥定價即使在獲得初步批准後仍受到政府的持續控制。因此,我們或任何未來的合作伙伴可能會獲得產品在特定國家/地區的營銷批准,但隨後會受到價格法規的約束,這些法規會推遲產品的商業發佈,可能會推遲很長時間,這可能會對我們在該國家/地區銷售產品所產生的收入產生負面 影響。不利的定價限制可能會阻礙我們的 能力或任何未來合作伙伴收回我們或他們在一個或多個候選產品上的投資的能力,即使我們的產品 候選產品獲得了市場批准。
醫療保健行業非常關注成本控制,無論是在美國還是在其他地方。政府當局和其他第三方付款人試圖通過限制特定藥物的承保範圍和報銷金額來控制成本,這 可能會影響我們或任何未來合作伙伴銷售我們的候選產品的盈利能力。這些付款人可能不認為我們的產品(如果有)具有成本效益,並且我們的客户或任何未來合作伙伴的產品可能無法獲得保險和報銷, 或者可能不足以使我們的產品(如果有)在競爭的基礎上進行營銷。政府當局的成本控制舉措或其他政策 措施可能會導致我們或任何未來的合作伙伴降低我們或他們可能為 產品制定的價格,這可能會導致產品收入低於預期。如果我們產品的價格(如果有的話)下降,或者如果政府 和其他第三方付款人沒有提供保險或足夠的補償,我們的收入和盈利前景將受到影響。
對於新批准的藥物,在獲得承保和報銷方面也可能存在延遲,並且承保範圍可能比FDA或類似的外國監管機構授權或批准的藥物適應症 更有限。此外,獲得報銷的資格 並不意味着任何藥物在所有情況下都會得到支付,或者支付的費率可以覆蓋我們的成本,包括研究、開發、製造、銷售和分銷。報銷率可能會有所不同,例如,根據產品的使用和使用的臨牀環境 。報銷費率也可以基於已為低成本藥品設置的報銷級別,也可以納入其他服務的現有付款中。
此外,第三方付款人對新技術的益處和臨牀結果的證據要求越來越高 ,並對所收取的價格提出質疑。我們不能確保 我們或任何未來的合作伙伴商業化的任何候選產品是否可以獲得報銷,如果有的話,報銷率是否足夠。此外,如果目前限制從藥品以低於美國的價格銷售的國家/地區進口藥品的法律發生變化,則藥品的淨報銷 可能會進一步減少。對於我們或任何未來的合作伙伴獲得營銷授權或批准的任何候選產品,如果不能迅速從政府資助和私人付款人那裏獲得承保範圍和足夠的付款費率 ,可能會嚴重損害我們的經營業績、我們籌集產品商業化所需資金的能力以及我們的整體財務狀況。
-16-
如果 我們不能單獨或通過與營銷合作伙伴的合作來發展我們的銷售、營銷和分銷能力, 我們將無法成功地將我們的候選產品商業化
我們 在組織內的營銷、銷售或分銷能力和經驗有限。如果我們的任何候選產品獲得了 授權或批准,我們要麼必須建立一個具有技術專業知識和支持分銷能力的銷售和營銷組織,將任何此類候選產品商業化,要麼將這一職能外包給第三方。這些活動是在2023年根據FDA對美國GOHIBIC(Viloblimab)的EUA發起的。這兩個選項都將非常昂貴且耗時 。部分或全部這些成本可能會在我們的候選產品(包括我們的主要候選產品)獲得批准之前發生。此外,我們可能無法在美國、歐洲或其他目標市場招聘足夠規模或在我們打算瞄準的醫療市場擁有足夠專業知識的銷售人員。這些風險可能會特別明顯,因為 我們關注的是嚴重的新冠肺炎,以及對PG的額外關注,這些疾病都是患者人數相對較少的疾病區域。 我們或第三方的內部銷售、營銷和分銷能力開發的任何失敗或延遲都將 對viloblimab和其他未來候選產品的商業化產生不利影響。
對於我們現有和未來的候選產品,我們可以選擇與擁有直接銷售隊伍和建立分銷系統的第三方合作,以增強或替代我們自己的銷售隊伍和分銷系統。 我們的產品收入可能低於我們直接營銷或銷售任何批准的產品。此外,我們獲得的任何收入將 全部或部分取決於這些第三方的努力,這些努力可能不會成功,而且通常不在我們的控制範圍內。 如果我們無法以可接受的條款或根本不能達成這些安排,我們可能無法成功地將任何授權或批准的產品商業化。如果我們沒有成功地將任何授權或批准的產品商業化,我們未來的產品收入將受到影響,我們可能會遭受重大的額外損失。
我們 可能面臨與GOHIBIC(Viloblimab)藥品的保質期到期有關的風險,包括但不限於資產負債表中的大量沖銷
根據美國EUA的許可,我們用於商業目的的GOHIBIC(Viloblimab)成品藥物 有有限的保質期。如果我們未能在保質期到期前將該成品用於預期用途, 該藥品將不得不相應銷燬,這可能會導致額外的大量成本和費用,並且 可能會導致銷燬藥品的大量註銷。
我們 可能會花費有限的資源來追求特定的候選產品或指示,而無法利用可能更有利可圖或成功可能性更大的候選產品或指示
我們 的財務和管理資源有限,因此我們打算專注於為我們確定為最有可能成功的特定適應症 開發產品候選,無論是在市場授權、批准和商業化方面。 因此,我們可能會放棄或推遲尋求其他候選產品或可能被證明具有更大商業潛力的其他適應症的機會。
我們的 資源分配決策可能會導致我們未能利用可行的商業產品或有利可圖的市場機會。我們 當前和未來的研發項目和特定適應症候選產品的支出可能不會產生任何商業上可行的 候選產品。如果我們沒有準確評估特定候選產品的商業潛力或目標市場, 我們可以通過合作、許可或其他版税安排放棄對該候選產品的有價值權利,如果 中保留對該候選產品的獨家開發和商業化權利對我們更有利。
-17-
臨牀 開發涉及一個漫長而昂貴的過程,其結果不確定。我們在完成或最終無法完成vilobelimab或我們可能開發的任何其他候選產品的開發和商業化過程中可能會產生額外成本或經歷延遲
vilobelimab和我們可能開發的任何其他候選產品(如INF904)的失敗風險很高。無法預測Vilobelimab何時 或是否將證明其在人體中有效和安全,或將獲得完全監管批准,用於治療嚴重 COVID—19或PG適應症或其他適應症。此外,在監管機構授予 vilobelimab的上市批准或EUA之前,我們將需要完成 正在進行的廣泛臨牀試驗,以證明在人體中的安全性和有效性。臨牀試驗費用昂貴,難以設計和實施,可能需要多年時間才能完成,而且結果本身就不確定。此外,臨牀前和臨牀數據往往 容易受到不同解釋和分析的影響,許多認為其候選產品在臨牀前研究和臨牀試驗中表現令人滿意的公司 ,但他們的藥物未能獲得上市批准。
我們 可能在監管授權和/或批准過程中或因此發生許多不可預見的事件,這些事件可能會延遲 或阻止我們從監管機構獲得上市批准或EUA,或使vilobelimab或任何未來候選產品商業化, 包括:
| ● | 調節器 或機構審查委員會不得授權我們或我們的研究者開始臨牀 在預期的試驗中心進行試驗或進行臨牀試驗; |
| ● | 臨牀 我們候選產品的試用可能會產生負面或不確定的結果,包括 未能證明統計顯著性,我們可能會決定,或者監管機構可能會要求 美國進行額外的臨牀試驗或放棄藥物開發計劃; |
| ● | 我們的 候選產品可能具有不良副作用或其他意外特性, 導致我們或我們的調查人員、監管機構、倫理委員會或機構審查委員會 暫停或終止審判; |
| ● | 我們的 第三方承包商可能無法遵守法規要求或履行合同 及時或根本對我們承擔義務;以及 |
| ● | 監管機構, 倫理委員會或機構審查委員會可能要求我們或我們的研究人員 因各種原因暫停或終止臨牀開發,包括不符合 監管要求或發現參與者暴露於不可接受的環境 健康風險。 |
We could also encounter delays if a clinical trial is suspended or terminated by us, by an overseeing ethics committee, by the institutional review boards of the institutions in which such trials are being conducted, by the data safety monitoring board for such trial or by the FDA or other regulatory authorities. Such authorities may impose such a suspension or termination due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. If we experience delays in the completion of, or termination of, any clinical trial of our product candidates, the commercial prospects of our product candidates will be harmed, and our ability to generate drug revenues from any of these product candidates will be delayed. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence drug sales and generate revenues. Any of these occurrences may harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval.
-18-
如果我們在測試或上市審批方面遇到延誤, 產品開發成本將進一步增加。臨牀試驗 的重大延誤還可能縮短我們可能擁有將候選產品商業化的獨家權利的任何時間,或允許 競爭對手在我們之前將藥物推向市場,並削弱我們成功將候選產品商業化的能力。我們正在 評估孤兒藥或vilobelimab在各種適應症中突破性治療指定的申請,但即使 獲得指定,我們可能無法 獲得任何此類指定或維持與孤兒藥狀態相關的益處,包括市場獨佔權
我們 正在評估一些適應症中的維羅布利單抗的孤兒藥物或突破性治療指定,我們可能會為我們正在流水線中或可能開發的其他臨牀前候選產品尋求孤兒藥物指定。在美國和其他 國家/地區,指定孤兒藥物使當事人有權獲得財政獎勵,例如為臨牀試驗費用提供贈款資金的機會、税收優惠和用户費用減免。在FDA或其他外國監管機構批准孤兒藥物指定後,FDA將公開披露藥物的仿製藥身份及其潛在的孤兒用途。孤兒藥物指定不會在FDA的審查和批准過程中傳遞任何優勢,也不會縮短持續時間。突破性治療指定是一個旨在加快用於治療嚴重疾病的藥物的開發和審查的過程,初步臨牀證據表明,在臨牀上具有重要意義的終點,該藥物可能比現有的治療方法有實質性的改善。突破性治療 指定可能使我們有資格獲得FDA對高效藥物開發計劃的密集指導,以及涉及FDA高級管理人員等的組織承諾 。雖然我們正在評估一些適應症的孤兒藥物或突破性治療指定 的申請,但不能保證我們會獲得這樣的指定。此外,獲得一個適應症的孤兒藥物或突破性治療指定並不意味着我們將能夠獲得另一個適應症的此類指定。
如果從FDA獲得孤兒藥物指定的產品隨後獲得FDA對其具有此類指定的疾病的特定有效成分的第一次批准,則該產品有權獲得孤兒藥物排他性,這意味着FDA在七年內不得批准任何其他申請,包括BLA,以銷售相同適應症的同一藥物。除非在有限的情況下 ,例如FDA發現孤兒藥物排他性持有者沒有證明它可以保證獲得足夠數量的孤兒藥物,以滿足指定藥物所針對的疾病或狀況的患者的需求。同樣,如果FDA得出結論 後一種藥物在臨牀上更好,意味着後一種藥物更安全、更有效,或者對患者護理做出了重大貢獻,則FDA隨後可以在排他期內批准具有相同活性部分的藥物用於相同的疾病。 即使我們從FDA獲得了Viloblimab的孤兒藥物指定,由於與開發藥品相關的不確定性,我們可能不是第一個獲得任何特定孤兒適應症的上市批准 ,因此,如果另一家公司獲得相同藥物的批准和孤兒藥物獨家經營權,並且我們面前的情況相同 ,則 viloblimab的批准可能會被阻止七年。如果我們確實在美國獲得了獨家營銷權,如果我們尋求批准比孤立指定適應症更廣泛的 適應症,則獨家營銷權可能會受到限制,如果FDA後來確定指定請求存在重大缺陷,或者如果我們無法保證足夠數量的產品來滿足相關患者的需求,則可能會失去獨家營銷權。 此外,獨家經營權可能無法有效地保護產品免受競爭,因為具有不同活性部分的不同藥物可以被批准用於相同的情況,相同的藥物可以被批准用於不同的適應症,然後可能在標籤外使用 我們批准的適應症,治療相同疾病的不同藥物可能已經獲得批准並投入商業使用。
即使我們獲得FDA對viloblimab或任何其他候選產品的授權或批准,我們也可能永遠無法獲得授權或批准或將我們的產品在美國以外的地方商業化
為了在美國境外銷售任何經批准的產品,我們必須建立並遵守其他國家/地區關於臨牀試驗設計、安全性和有效性的眾多且變化的法規要求。如果獲得相關政府當局的批准, 我們預計將在歐洲和美國以外的司法管轄區銷售用於治療新冠肺炎和其他適應症的維羅貝利單抗, 部分原因是與美國相比,歐洲的患者人數相對較多。在一個國家/地區進行的臨牀試驗 可能不會被其他國家/地區的監管機構接受,在一個國家/地區獲得監管批准並不意味着將在任何其他國家/地區獲得監管批准。審批程序因國家/地區而異,可能涉及額外的 產品測試和驗證以及額外的行政審查期。尋求外國監管機構的批准可能會導致我們的重大延誤、困難和成本,並可能需要額外的臨牀前研究或臨牀試驗,這將是昂貴和耗時的 ,並可能推遲或阻止在這些國家/地區推出viloblimab或我們的任何其他候選產品。
-19-
此外,如果我們獲得必要的批准,我們預計將面臨與在其他國家/地區運營相關的各種風險, 包括:
| ● | 美國以外國家的不同監管要求; |
| ● | 所謂平行進口的潛力(即當地銷售商面對當地價格較高或較高時,選擇從外國市場(以較低或較低的價格)進口貨物,而不是在當地購買); |
| ● | 關税、貿易壁壘、價格和外匯管制以及其他監管要求的意外變化; |
| ● | 經濟疲軟,包括通貨膨脹,或特別是外國經濟體和市場的政治不穩定; |
| ● | 國外的報銷、定價和保險制度; |
| ● | 對於居住或旅行在美國境外的員工,遵守税收、就業、移民和勞動法 ; |
| ● | 外國税,包括預扣工資税; |
| ● | 外國 貨幣波動,這可能導致運營費用增加和收入減少, 以及在另一個國家開展業務所附帶的其他義務; |
| ● | 困難 人員配備和管理海外業務; |
| ● | 勞動力 勞工騷亂比美國更普遍的國家的不確定性; |
| ● | 潛力 根據1977年《反海外腐敗法》或類似的外國法規承擔責任; |
| ● | 挑戰 執行我們的合同和知識產權,特別是在 不像美國那樣保護知識產權; |
| ● | 生產 任何影響原材料供應或製造能力的事件導致的短缺 以及美國以外的供應鏈中斷; |
| ● | 業務 中斷,包括由於地緣政治不確定性和不穩定(包括 與俄羅斯—烏克蘭衝突有關)。 |
如果 我們或我們的合作伙伴未能遵守監管要求或獲得並維持所需的批准,我們的目標市場將減少 ,包括我們無法在歐洲或其他地方銷售vilobelimab,我們實現候選產品全部市場潛力的能力將受到損害 。
我們 受到廣泛的政府法規的約束,不遵守這些法規可能會對 我們的運營和業務產生重大不利影響
在任何產品獲得批准之前和之後,我們和我們的供應商、合同製造商和臨牀研究者都要接受美國和其他國家政府機構的廣泛 監管,其中包括測試、製造、 質量控制、臨牀試驗、上市後研究、標籤、廣告、促銷、分銷、進出口、政府 定價,價格報告和回扣要求。不遵守適用要求可能導致以下一項或多項 行動:警告信;意外支出;延遲批准或拒絕批准候選產品;產品召回或 扣押;生產或臨牀試驗中斷;經營或銷售限制;禁令;刑事起訴和 民事或刑事處罰,包括罰款和其他罰款;負面宣傳;以及對我們業務的幹擾此外, 政府對潛在違反這些法律的調查將要求我們花費大量資源,並面臨負面 宣傳和我們業務的潛在中斷,即使我們最終被發現沒有違反。
獲得 FDA、EMA或其他監管機構對我們候選產品的授權或批准需要大量的時間、精力和 資源,可能會受到預期和不可預見的延遲,無法保證 我們的任何候選產品將及時獲得批准(如果有的話)。FDA、EMA或其他監管機構可能會決定我們的數據 不足以授權或批准我們的候選產品,並需要額外的臨牀前、臨牀或其他研究或 與化學、生產和控制或CMC相關的額外工作。如果我們需要對候選產品進行額外試驗或 其他測試,超出我們預期的監管批准範圍,如果我們無法成功完成 我們的臨牀試驗或其他測試,或者如果這些和其他試驗或測試的結果未能證明療效或引起安全性問題 ,我們可能會面臨大量額外費用,延遲獲得我們候選產品的市場批准,或可能永遠 無法獲得市場批准。
-20-
在產品獲得上市許可 或批准後,我們 還需要遵守廣泛的政府監管要求。監管機構可能要求進行可能對產品商業可行性產生負面影響的上市後研究 。一旦上市,產品可能與之前未檢測到的不良反應相關,並且/或者可能出現製造問題 。由於任何這些或其他問題,產品的監管批准可能會被撤銷,這可能會損害 我們的業務和經營成果。
我們 當前和未來與美國和其他地方的第三方付款人、醫療保健專業人員和客户的關係 可能直接或間接地受到適用的反回扣、欺詐和濫用、虛假索賠、醫生支付透明度、醫療保健 信息隱私和安全以及其他醫療保健法律法規的約束,這可能會使我們面臨重大處罰
美國和其他地方的醫療保健服務提供者、醫生和第三方支付者將在我們獲得上市批准的任何候選產品的推薦和處方方面發揮主要作用。我們當前和未來與醫療保健專業人員、 第三方付款人和客户的安排可能會使我們面臨廣泛適用的欺詐和濫用以及其他醫療保健法律法規,包括 聯邦反回扣法規和聯邦民事虛假索賠法案,這些法律法規可能會限制我們進行臨牀研究、銷售、銷售和分銷我們獲得上市許可的任何藥物。 此外,我們可能會遵守美國和 我們開展業務的其他司法管轄區的透明度法律和患者隱私法規。可能影響我們運營能力的適用聯邦、州和外國醫療保健法律和法規 包括以下內容:
| ● | the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under federal and state healthcare programs, such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation. Further, several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the Anti-Kickback Statute has been violated. Moreover, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act; |
| ● | 聯邦 民事和刑事虛假索賠法,包括聯邦民事虛假索賠法(即 可以通過民事舉報人或qui tam行動執行),以及民事罰款 法律,該法律對故意的個人或實體實施刑事和民事處罰 向聯邦政府(包括醫療保險)提交或促使提交。 以及醫療補助計劃、虛假或欺詐的付款索賠或作出虛假的 避免、減少或隱瞞向聯邦政府支付款項的義務的聲明; |
| ● | 1996年《聯邦健康保險攜帶和責任法案》,簡稱HIPAA,該法案規定 執行欺詐醫療保健計劃的刑事和民事責任(除其他外) 福利計劃或作出與醫療保健有關的虛假陳述。類似於 聯邦反回扣法規,個人或實體無需實際知情 (a)違反法規或違反法規的具體意圖; |
| ● | HIPAA, 經2009年《衞生信息技術經濟和臨牀健康法案》修訂, 或HITECH及其各自的實施條例,這些條例規定了所涵蓋的義務 醫療保健提供者、醫療計劃和醫療保健信息交換所及其業務 創建、接收、維護或傳輸個人可識別健康信息的員工 代表或代表受保護實體,保護隱私、安全 和傳送個人可識別的健康資料; |
-21-
| ● | 醫生支付陽光法案,根據《患者保護和平價醫療法案》6002節創建,經《醫療保健和教育協調法案》修訂,或統稱為《平價醫療法案》及其實施條例,它要求在Medicare、Medicaid或兒童健康保險計劃下可以付款的藥品、設備、生物製品和醫療用品的指定製造商 每年向Medicare和Medicaid服務中心(CMS)報告 。與支付給醫生的款項或其他“價值轉移”有關的信息,包括醫生、牙醫、驗光師、足科醫生和脊椎按摩師,和教學醫院 和適用的製造商每年在每個日曆年的第90天之前向CMS報告醫生及其直系親屬持有的所有權和投資權益。 所有此類報告的信息都是公開的;和 |
| ● | 類似的國家和外國法律法規,如國家反回扣和虛假索賠法律, 可能適用於銷售或營銷安排和索賠,涉及醫療保健項目 或由包括私人保險公司在內的非政府第三方付款人報銷的服務; 州和外國法律要求製藥公司遵守制藥行業的自願合規指南和聯邦政府頒佈的相關合規指南,或以其他方式限制可能向醫療保健提供者支付的費用; 國家和外國法律要求藥品製造商報告與向醫生和其他醫療保健提供者或營銷支出進行付款和其他價值轉移有關的信息; 以及在某些情況下管理健康信息隱私和安全的州和外國法律,其中許多法律在很大程度上彼此不同,並且通常不會被HIPAA 搶先,從而使合規工作複雜化。 |
確保我們與第三方的業務安排符合適用的醫療法律法規的努力 可能涉及鉅額 成本。政府當局可能會得出結論,我們的商業行為,包括我們與醫生和其他醫療保健提供者的關係,如果獲得批准,其中一些人可能會建議、購買或開出viloblimab,可能不符合當前或未來涉及適用欺詐和濫用或其他醫療保健法律和法規的現行或未來法規、法規或判例法。
如果 我們的運營被發現違反了任何這些法律或任何其他可能適用於我們的政府法規,我們可能會受到重大的民事、刑事和行政處罰,包括損害賠償、罰款、返還、個人監禁、 被排除在參與政府醫療保健計劃(如Medicare和Medicaid)之外、額外的報告要求和 監管(如果我們受到公司誠信協議或類似協議的約束,以解決有關違反這些法律的指控,以及削減或重組我們的業務,這可能對我們的業務產生重大不利影響)。如果我們預計與之開展業務的任何 醫生或其他醫療保健提供者或實體被發現不遵守適用法律,他們可能會受到刑事、民事或行政制裁,包括被排除在參與政府醫療保健計劃之外,這也可能對我們的業務產生重大影響。
頒佈和未來的立法可能會增加我們獲得上市授權或批准和商業化的難度和成本,並影響我們可能獲得的價格
在 美國和一些外國司法管轄區,有關醫療保健系統的多項立法和法規變更以及擬議的變更 可能會阻止或推遲viloblimab的上市審批,限制或規範審批後活動 ,並影響我們以盈利方式銷售我們獲得營銷授權或批准的任何候選產品的能力。
在美國和其他地方的 政策制定者和付款人中,有很大的興趣推動醫療保健系統的變革,其既定目標是控制醫療保健成本、提高質量和/或擴大准入。在美國,製藥 行業一直是這些努力的重點,並受到重大立法舉措的重大影響,如2010年的《平價醫療法案》(Affordable Care Act),該法案旨在擴大醫療保險的可及性,減少或限制醫療支出的增長,加強針對欺詐和濫用的補救措施,為醫療保健和醫療保險行業增加新的透明度要求, 對醫療行業徵收新的税費,並實施額外的醫療政策改革。在接下來的幾年裏,可能會對政府健康計劃進行更多的立法和監管改革,這可能會對製藥公司和我們候選藥物的成功 產生重大影響。
此外,自《平價醫療法案》頒佈以來,還提出並通過了其他立法修訂。這些變化包括: 從2013年4月1日起,每個財年向提供者支付的聯邦醫療保險總金額減少2%,由於隨後對法規進行的立法修訂,除非國會採取額外行動,否則這些變化將一直有效到2025年。2012年的《美國納税人救濟法》,除其他事項外,進一步減少了向幾家醫療服務提供者支付的醫療保險費用,並將政府向提供者追回多付款項的訴訟時效期限 從三年延長至五年。這些法律可能會導致聯邦醫療保險和其他醫療保健資金的進一步減少,如果獲得批准,這可能會對我們藥品的客户以及我們的財務運營產生實質性的不利影響。
-22-
醫療保險或其他政府計劃報銷的任何減少都可能導致私人支付者支付的類似減少。 成本控制措施或其他醫療改革的實施可能會妨礙我們創造收入、實現 盈利能力或將我們的藥物商業化。
已提出立法 和監管建議,以擴大藥品的批准後要求,並限制銷售和促銷活動。 我們無法確定是否會頒佈額外的立法變更,或者FDA法規、指南或解釋是否會變更 ,或者這些變更對vilobelimab(如有)的上市批准可能產生什麼影響。此外,美國國會對FDA批准過程的 審查力度加大,可能會大大推遲或阻止上市批准,並使 我們面臨更嚴格的藥物標籤和上市後檢測以及其他要求。
即使 我們或任何未來的合作者獲得了候選產品的市場批准或EUA,批准條款和對我們產品的持續監管可能會限制我們生產和銷售產品的方式,這可能會削弱我們的創收能力
一旦 上市批准或EUA獲得批准,已批准的產品及其製造商和營銷商將接受持續審查和 廣泛的監管。因此,對於我們或他們獲得上市許可的任何候選產品,我們和任何未來的合作者都必須遵守有關廣告和促銷的要求 。有關處方藥 的促銷通信受各種法律和法規限制的約束,並且必須與產品 批准標籤中的信息一致。因此,我們和任何未來的合作者將無法推廣我們為適應症或用途開發的產品 ,這些產品未經批准。
此外,授權和批准產品的製造商以及這些製造商的工廠必須遵守廣泛的 FDA要求,包括確保質量控制和生產程序符合cGMP,其中包括 與質量控制和質量保證有關的要求以及相應的記錄和文件維護和報告要求。 我們、我們的合同製造商、任何未來的合作者及其合同製造商都可能接受FDA的定期突擊檢查 ,以監測和確保其符合cGMP。
因此, 假設我們或任何未來合作者獲得了一個或多個候選產品的上市批准,我們和任何未來合作者、 以及我們和他們的合同製造商將繼續在所有監管合規領域(包括 製造、生產、產品監督和質量控制)上花費時間、金錢和精力。
GOHIBIC(vilobelimab)的 生產和分銷面臨許多可能損害我們聲譽、業務、財務 狀況和經營成果的風險。
GOHIBIC(vilobelimab)的 生產工藝複雜。我們可能會遇到製造困難,包括 與產品儲存和有效期有關的困難。此類困難可能是由於大規模生產產品批次的複雜性、設備故障、輔料和其他產品組分/成分的可用性(包括與原材料的選擇和質量相關的 )、分析檢測技術和產品不穩定性。具體而言,GOHIBIC(vilobelimab)或其組分的產品穩定性或有效期 不足可能嚴重限制或延遲我們或我們的合作者以當前價格或根本不銷售GOHIBIC(vilobelimab)的能力。此外,如果GOHIBIC(vilobelimab)因製造錯誤、設計/標籤缺陷或其他缺陷而受到產品召回, 我們的聲譽將受到不利影響。
GOHIBIC (vilobelimab)是一種"冷鏈產品",必須在低温下運輸和儲存。由於分銷困難,包括一般相關的供應鏈管理(例如,有效期到期), 特別涉及在低温下運輸和儲存GOHBIC(vilobelimab)。如果是這樣的話,我們可能會產生額外的製造成本 ,以便根據EUA向美國醫院供應所需的數量。
任何 此類製造和分銷困難都可能損害我們的聲譽、業務、財務狀況和經營成果。
-23-
政府,包括美國以外的政府,傾向於實施嚴格的價格控制,這可能會對我們的收入產生不利影響,如果有的話
在 許多國家,例如歐盟國家,處方藥的定價受到不同的價格控制 機制的制約,通常是國家衞生系統的一部分。其他國家允許公司自行定價醫療產品,但 監控和控制公司利潤。 在收到產品的上市批准後,與政府機構進行定價談判可能需要相當長的時間。為了在某些國家獲得報銷或定價批准,我們或任何未來的合作者 可能需要進行臨牀試驗,比較我們產品與其他可用療法的成本效益。如果 我們的產品無法報銷,或在範圍或金額上受到限制,或者定價水平不令人滿意,我們的業務可能會受到損害。 額外的價格控制或定價法規的其他變化可能會限制我們為候選產品收取的費用 ,我們認為歐盟日益重視成本控制措施已經並將繼續對候選產品的定價和使用造成 壓力。因此,鑑於嚴重COVID—19和PG的目標市場相對較小,對此類候選產品的任何減少償還可能不足以使我們產生商業上合理的收入 和利潤,並將對我們的財務狀況和經營業績造成不利影響。
我們或任何未來合作者獲得上市批准或EUA的任何 候選產品都可能受到 上市後限制或退出市場,如果我們或他們未能遵守監管要求,或者如果我們或他們,在 批准或EUA之後,我們的產品遇到了意外問題
我們或任何未來合作者獲得上市批准或EUA的任何 候選產品,以及此類產品的製造 工藝、批准後研究和措施、標籤、廣告和促銷活動等, 將遵守FDA、EMA和其他監管機構的持續要求和審查。這些要求包括 安全性和其他上市後信息和報告的提交、註冊和上市要求、 與生產、質量控制、質量保證和相應的記錄和文件維護有關的要求、 樣本分發給醫生和記錄保存有關的要求。即使獲得了候選產品的上市批准或EUA, 批准也可能受限於產品上市的指定用途或批准條件, 包括實施REMS的要求。
FDA、EMA和其他監管機構也可能會要求進行昂貴的上市後研究或臨牀試驗和監督 ,以監測產品的安全性或有效性。FDA和其他機構(包括司法部)密切監管和 監控產品的批准後營銷和推廣,以確保產品的生產、營銷和分銷僅用於 授權或批准的適應症,並符合批准標籤的規定。FDA對製造商關於標籤外使用的通信施加了嚴格的限制 ,如果我們或任何未來的合作者不銷售我們的任何候選產品 ,而我們或他們僅獲得其批准適應症的上市許可,我們或他們可能會受到警告 或針對標籤外營銷採取強制行動。違反FDCA和其他與推廣和廣告處方藥有關的法規可能導致調查或指控違反聯邦和州醫療保健欺詐和濫用法律以及州消費者保護法律,包括《虛假索賠法》。
此外,後來發現我們的產品或其製造商或製造過程中先前未知的不良事件或其他問題,或未能遵守法規要求,可能會產生各種結果,包括:
| ● | 限制 有關該等產品的製造; |
| ● | 限制 關於此類產品的標籤或營銷; |
| ● | 限制 產品分銷或使用; |
| ● | 要求 進行上市後研究或臨牀試驗; |
| ● | 警告 信件或無標題信件; |
-24-
| ● | 退出 市場上的產品; |
| ● | 拒絕 批准我們提交的待決申請或對已批准申請的補充; |
| ● | 召回 產品; |
| ● | 限制 第三方支付者的保險; |
| ● | 罰款, 歸還或沒收利潤或收入; |
| ● | 暫停 或撤銷上市許可,包括EUA; |
| ● | 拒絕 準許產品的進口或出口; |
| ● | 產品 扣押;或 |
| ● | 禁令或施加民事或刑事處罰。 |
我們 成功商業化GOHIBIC(vilobelimab)並從銷售中獲得收入的能力受到可能 損害我們業務、財務狀況和經營成果的若干風險的影響。
我們成功將GOHIBIC(Viloblimab)商業化的能力受到一些風險的影響,這些風險可能會影響我們的業務、財務狀況和經營業績。具體地説,我們從GOHIBIC(Viloblimab)的銷售中獲得收入的能力是不確定的,包括 由於市場機會以及對GOHIBIC(Viloblimab)的興趣和看法。特別是,考慮到因新冠肺炎感染而出現嚴重症狀的患者數量的波動,可尋址患者羣體的規模以及GOHIBIC(維洛利單抗)的市場機會 都是不確定的,可能會隨着時間的推移而縮小。此外,由於GOHIBIC(Viloblimab)獲得EUA,但未獲得FDA批准,因此GOHIBIC(Viloblimab)的銷售取決於美國醫院的醫療保健提供者是否有興趣和接受GOHIBIC(Viloblimab)作為新冠肺炎的治療方法。具體地説,如果GOHIBIC(Viloblimab)不包括在醫療機構和其他第三方醫療/保健組織(如NIH)發佈的治療指南中,或者如果這些機構和組織不推薦GOHIBIC(Viloblimab),醫院可能不願意提供GOHIBIC(Viloblimab)用於治療 患者。例如,美國國立衞生研究院的指南規定,沒有足夠的證據建議或反對使用GOHIBIC(維洛利單抗)治療危重新冠肺炎患者。NIH的這一中性指南對GOHIBIC(Viloblimab)的商業採用產生了負面影響,因為許多醫療保健提供者,尤其是醫院,在 決定將處方藥包括在其處方中允許醫院工作人員下產品訂單時,依賴NIH治療指南。最終, 如果我們不能成功地將GOHIBIC(Viloblimab)的銷售商業化併產生收入,我們的業務、財務狀況和經營業績可能會受到不利影響。
2. 與我們依賴第三方相關的風險
我們 依賴第三方進行臨牀試驗。如果他們的表現不令人滿意,我們的業務可能會受到損害
我們 不會獨立進行任何候選產品的臨牀試驗。我們依賴第三方(如CRO、臨牀數據 管理組織、第三方顧問、醫療機構和臨牀研究者)進行這些臨牀試驗 ,並希望依賴這些第三方對我們開發的任何其他候選產品進行臨牀試驗。在某些情況下,這些第三方 中的任何一方都可以終止與我們的合同。我們可能無法達成替代安排 或以商業上合理的條款進行。此外,當一個新的CRO開始工作時,有一個自然的過渡期。因此, 可能會出現延誤,這可能會對我們滿足預期臨牀開發時間表的能力產生負面影響,並損害我們的 業務、財務狀況和前景。
-25-
Further, although our reliance on these third parties for clinical development activities limits our control over these activities, we remain responsible for ensuring that each of our trials is conducted in accordance with the applicable protocol, legal and regulatory requirements and scientific standards. For example, notwithstanding the obligations of a CRO for a trial of one of our product candidates, we remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA, the EMA and potentially other regulatory agencies of different countries require us to comply with requirements, commonly referred to as current Good Clinical Practices, or cGCP, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. The FDA and regulatory agencies inside the European Union and other regulatory agencies enforce these cGCP regulations through periodic inspections of trial sponsors, principal investigators, clinical trial sites and IRBs. If we or our third-party contractors fail to comply with applicable cGCP regulations, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or other regulatory agencies may require us to perform additional clinical trials before approving our product candidates, which would delay the marketing approval process. We cannot be certain that, upon inspection, the FDA or other regulatory agencies will determine that any of our clinical trials comply with cGCP. We are also required to register clinical trials and post the results of completed clinical trials on a government-sponsored database, such as ClinicalTrials.gov in the United States, within certain timeframes. The same requirement applies to clinical trials outside the United States, such as EudraCT.ema.europa.eu in Europe. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
此外, 代表我們進行臨牀試驗的第三方不是我們的員工,除了根據我們與這些承包商的協議提供的補救措施 ,我們無法控制他們是否為我們的持續開發項目投入了足夠的時間、技能和資源。 這些承包商還可能與其他商業實體(包括我們的競爭對手)有關係,他們也可能為這些實體進行 臨牀試驗或其他藥物開發活動,這可能會妨礙他們將適當時間用於我們的臨牀項目的能力。 如果這些第三方(包括臨牀研究者)未能成功履行其合同職責,未能在預期期限內完成 或根據監管要求或我們的聲明方案進行臨牀試驗,我們可能無法獲得候選產品的上市批准,或可能 延遲獲得上市批准。如果出現這種情況,我們將無法成功地將候選產品商業化,或可能會推遲 我們的努力。在這種情況下,我們尋求開發的任何候選產品的財務業績和商業前景 都可能受到損害,我們的成本可能會增加,我們的創收能力可能會 被延遲、削弱或喪失抵押品贖回權。
我們對外國第三方製造商和供應商的依賴增加了我們獲得充足、及時和具有成本效益的候選產品和產品的風險
外國 製造業面臨許多風險,包括政治和經濟動盪、關税、配額和其他 進出口管制的實施、政府政策的變化以及地緣政治的不確定性和不穩定,這些都造成了市場波動。 尤其是,我們依賴中國和其他地方的第三方製造商供應vilobelimab。我們將候選產品和產品的所有制造 外包給第三方,同時在內部進行某些質量控制測試。中國的供應鏈和 製造業也可能由於當前的全球流行病以及全球政治局勢而對我們的運營造成嚴重影響 。
我們 聘請位於中國的第三方生產商為vilobelimab最終制劑的臨牀供應。 無法保證我們能夠以令人滿意的條件及時獲得所需的替代供應安排,或者如果需要的話,根本無法保證。 我們對製造商的依賴以及我們未能根據需要獲得替代供應安排,可能會對我們完成候選產品開發或商業化(如果獲得批准)的能力產生重大不利影響 。 在擴大到商業數量或優化工藝和處方方面可能存在困難, 生產成本可能過高。
即使 我們能夠與其他第三方製造商建立並維持協議,依賴第三方製造商通常 也會帶來超出我們控制範圍的額外風險,包括:
| ● | 信賴 為第三方提供製造工藝開發、法規遵從性和質量 保證; |
| ● | 信賴 第三方用於儲存和保護幾乎所有庫存品; |
| ● | 成本 以及進一步擴大或優化所需的新設備和設施的驗證 過程; |
| ● | 失敗 符合cGMP和類似的國外標準; |
| ● | 限制 第三方的能力和時間安排限制造成的供應; |
| ● | 缺少 合格的備份供應商,用於從單一或單一採購的組件 來源供應商; |
| ● | 閉包 以及對公共衞生危機造成的關鍵設施的限制; |
-26-
| ● | 能夠自由進口臨牀試驗材料和生產的潛在上市材料 我們在中國的第三方製造商進入臨牀試驗所在的國家 正在進行或可能出售的產品; |
| ● | 第三方可能違反生產協議,原因超出我們的 控制;及 |
| ● | 第三方可能終止或不續簽生產協議,地址為 一個對我們來説是昂貴或不便的時間,以及我們獲得替代供應的能力。 |
如果 我們不維持關鍵的製造關係,我們可能無法找到替代製造商或開發我們自己的製造能力 ,這可能會延遲或削弱我們為產品獲得監管部門批准的能力。如果我們確實找到了替代製造商, 我們可能無法就對我們有利的條款和條件與他們達成協議,並且在新設施獲得資格並在FDA和其他外國監管機構註冊之前,可能會有很大的延遲 。此外,生產設施的變更 包含固有風險,通常被視為生產工藝的重大變更,因此必須進行 可比性研究,以確保之前確立的生產工藝與新確立的 生產工藝之間的可比性,這可能導致製劑供應延遲,或者如果生產的製劑不具有可比性,保證對此類非可比製劑進行進一步的臨牀前和/或臨牀研究,這些研究也可能 由任何監管機構在審查可比性數據時強制執行。上述任何情況都可能對我們的業務、財務狀況、經營業績和前景產生重大不利影響。
生產生物製品(如vilobelimab)的過程極易發生產品損失
我們產品的生產流程複雜、監管嚴格,並受到其他幾個風險的影響。由於污染、設備故障或設備安裝或操作不當、供應商或操作員錯誤、產量不一致、產品特性的變化以及生產過程的困難,生物製劑的生產過程(如viloblimab)極易受到產品損失的影響。即使與正常製造流程的微小偏差也可能導致生產良率降低、產品缺陷 和其他供應中斷。如果在我們的候選產品中或在製造我們候選產品的製造設施中發現微生物、病毒或其他污染,則此類製造設施可能需要關閉較長時間 以調查和補救污染。此外,我們已生產並存儲以供以後使用的候選產品可能會 降級、受到污染或出現其他質量缺陷,這可能會導致受影響的候選產品不再適合 用於臨牀試驗或其他開發活動。如果不能 及時更換有缺陷的候選產品,我們的開發計劃可能會出現重大延誤,這可能會對候選產品的價值產生不利影響 ,從而對我們的業務、財務狀況、運營結果和前景產生不利影響。
我們的製造過程受到質量控制風險和相關法規要求的影響
我們 在自己的實驗室內通過關鍵的質量控制測試參與制造過程,我們持有製造商 許可證,因此我們負責監督整個製造過程,我們有責任確保我們的這部分業務 也根據cGMP標準運行。此外,我們還持有進口許可證。因此,我們在製造流程中聘用關鍵人員,如質量保證主管、製造主管和合格人員。
因此,我們的實驗室和我們的質量控制系統以及相關文件和人員也要接受政府的頻繁檢查,以確保遵守cGMP指南,並保持我們的製造和進口許可證。與這些活動相關的風險 可能會對我們滿足預期臨牀開發時間表的能力產生負面影響,並損害我們的業務、財務狀況和前景,包括:
| ● | 生產活動中關鍵人員的流失可能會導致我們候選藥物的製造和釋放測試的重大延遲 ,更換這些人員可能非常耗時,並與我們的額外成本相關; |
| ● | 釋放測試中的錯誤或不當行為可能導致錯誤的結果,這可能導致 錯誤地拒絕生產的藥物產品發佈或錯誤地接受功能失調的藥物產品,導致取得的數據和試驗結果為虛假或可能被錯誤解釋的藥物產品;和 |
| ● | 如果cGMP合規性不足,可能會因監管機構的檢查而導致製造或進口許可證暫時或永久丟失。 |
-27-
我們的 第三方製造商或我們可能無法遵守適用於viloblimab和生物製品的cGMP法規要求, 包括FDA藥品cGMP法規的適用條款、質量體系法規或QSR中體現的設備cGMP要求,或美國以外的類似法規要求。我們或我們的第三方製造商未能遵守適用法規可能導致對我們實施制裁,包括臨牀封存、罰款、禁令、民事處罰、延遲、暫停或撤回審批、扣押或自願召回候選產品、運營限制 和刑事起訴,其中任何一項都可能嚴重影響我們候選產品的供應。此外,我們的第三方製造商 和供應商以及我們都不定期接受FDA和其他地方監管機構的檢查。我們的第三方製造商和供應商或我們未能通過此類檢查並以其他方式圓滿完成FDA對我們的候選產品的批准方案,可能會導致監管行動,如發佈FDA Form 483觀察通知、警告信或禁令或吊銷運營許可證。
此外,我們和我們的第三方製造商和供應商須遵守多項環境、健康和安全法律法規, 包括管理廢物處理、使用、儲存、處理和處置的法律法規,如果不遵守這些法律法規 和法規,可能會導致與這些第三方的民事或刑事罰款和處罰相關的鉅額成本。 根據監管行動的嚴重程度,我們的臨牀或商業供應藥物和包裝及其他服務可能會中斷 或受到限制,這可能會對我們的業務產生重大不利影響,包括我們的臨牀研究活動和我們 開發候選產品和在獲得批准後銷售產品的能力(如有)。
如果 我們候選產品的任何第三方製造商無法增加我們候選產品的生產規模,和/或 提高其製造的產品產量,那麼我們的產品製造成本可能會增加,並且可能 延遲商業化
為了 生產足夠數量的產品以滿足臨牀試驗的需求,如果獲得批准,也可以滿足隨後的商業化的維洛貝利單抗 或我們正在開發的任何其他候選產品,我們的第三方生產商將被要求增加 其產量並優化其生產工藝,同時保持產品質量。向更大規模 生產的過渡可能很困難或成本高昂。此外,由於製造、供應和灌裝過程的放大或優化 ,我們的製造過程中的任何索賠都可能導致需要獲得監管部門的批准。如果我們的第三方製造商 無法優化製造工藝以提高我們候選產品的產品產量,或者無法在保持產品質量的同時增加 候選產品的數量,那麼我們可能無法滿足臨牀 試驗或市場需求的需求,這可能會降低我們產生利潤的能力。難以實現商業規模擴大生產 或生產優化,或因此需要額外的監管批准,可能會對我們的業務 和運營結果產生重大不利影響。
我們 希望尋求建立合作關係,如果我們無法以商業上合理的條款建立合作關係,我們可能不得不 改變我們的開發和商業化計劃
我們 希望尋求一個或多個合作者來開發和商業化我們的一個或多個候選產品。可能的合作者 可能包括大型和中型製藥公司、區域和國家制藥公司以及生物技術公司。 此外,如果我們從國外監管機構獲得了候選產品的上市許可或批准,我們可能會 與國際生物技術或製藥公司建立戰略合作關係,以便在美國境外將候選產品商業化 。
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the potential differentiation of our product candidate from competing product candidates, design or results of clinical trials, the likelihood of approval by the FDA, the EMA or comparable foreign regulatory authorities and the regulatory pathway for any such approval, the potential market for the product candidate, the costs and complexities of manufacturing and delivering the product to patients and the potential of competing products. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available for collaboration and whether such a collaboration could be more attractive than the one with us for our product candidate. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our product candidates or bring them to market and generate product revenue.
-28-
協作 是複雜的,而且談判和記錄耗時。此外,大型製藥公司最近進行了大量業務合併 ,這可能導致未來潛在合作者數量減少。我們將來簽訂的任何合作 協議可能包含對我們進行潛在合作或以其他方式 開發特定候選產品的能力的限制。我們可能無法及時、以可接受的條件或根本無法協商合作。
如果 我們無法做到這一點,我們可能不得不縮減我們尋求合作的候選產品的開發,減少 或推遲其開發計劃或我們的一個或多個其他開發計劃,推遲其潛在的商業化或減少 任何銷售或營銷活動的範圍,或增加開支並自行承擔 開發或商業化活動。
如果 我們與第三方合作開發和商業化我們的候選產品, 這些候選產品的前景將在很大程度上取決於這些合作的成功
我們 希望維持現有合作關係,並在某些地區為某些 候選產品的開發和商業化進行其他合作。我們可能對合作者 將用於我們候選產品的開發或商業化的資源數量和時間控制有限。我們從這些安排中產生收入的能力 將取決於任何未來合作者成功履行這些安排中分配給他們的職能的能力。 此外,任何未來的合作者都有權在商定的條款到期之前或到期之時放棄研發項目並終止適用的協議, 包括資助義務。
涉及我們候選產品的協作 會帶來許多風險,包括以下內容:
| ● | 協作者 在確定他們將應用於這些協作的工作和資源方面有很大的自由裁量權 ; |
| ● | 合作者 可能無法按預期履行其義務; |
| ● | 合作者 可能不追求我們的候選產品的開發和商業化,或可能選擇不 根據臨牀試驗結果,繼續或更新開發或商業化項目, 合作者戰略重點或可用資金或外部因素的變化, 例如轉移資源或產生競爭性優先事項的收購; |
| ● | 合作者 可以推遲臨牀試驗,為臨牀試驗計劃提供資金不足,停止臨牀試驗或放棄候選產品,重複或進行新的臨牀試驗,或者 要求臨牀試驗候選產品的新配方; |
| ● | 合作者 可以獨立開發或與第三方開發直接競爭的產品 或間接與我們的候選產品; |
| ● | a 對一個或多個產品擁有營銷和分銷權的合作者不得提交 有足夠的資源用於銷售和分銷該等產品; |
| ● | 分歧 與合作者的溝通,包括對所有權(包括商業祕密)的分歧 以及其他知識產權、合同解釋或首選研究課程 和開發可能導致研究、開發或商業化的延遲或終止 可能導致我們在產品方面承擔額外責任 候選人,或可能導致訴訟或仲裁,其中任何一種都將耗時 昂貴的; |
| ● | 合作者 可能無法正確起訴、維護、捍衞或執行我們的知識產權 或可能以以下方式使用我們的專有信息或其他知識產權: 引發可能危及我們知識產權或使其無效或暴露 潛在訴訟; |
| ● | 合作者 可能侵犯、盜用或以其他方式侵犯第三方的知識產權 當事人,這可能使我們面臨訴訟和潛在責任; |
| ● | 合作 可能被終止,如果終止,可能會導致需要額外的資本來進行投資 適用候選產品的進一步開發或商業化;以及 |
| ● | 協作 協議可能不會導致 中候選產品的開發或商業化 最有效的方式或所有。如果我們未來的任何合作者參與了某項業務 合併,它可以決定推遲、減少或終止開發或商業化 我們授權給它的任何候選產品。 |
-29-
由於資金短缺或全球健康問題,FDA和其他政府機構的資金變動 或中斷 可能會阻礙其履行我們業務運營所依賴的正常業務職能,這可能會對我們的業務產生負面影響
FDA審查和批准新候選產品和產品的能力可能受到各種因素的影響, 包括:
| ● | 政府 預算和資金水平,以及法律、法規和政策的變化; |
| ● | FDA僱用和保留關鍵人員並接受用户費用支付的能力; 和 |
| ● | 聯邦 政府關門和其他可能影響FDA能力的事件 執行例行功能。 |
因此,該機構的平均 審核時間近年來波動。
此外, 如果出現長期政府關閉,或者如果全球健康問題繼續阻止或推遲FDA或其他監管機構 進行定期檢查、審查或其他監管活動, FDA或其他監管機構及時審查和處理我們監管提交的能力可能會受到嚴重影響, 這可能對我們的業務產生重大不利影響。
3. 與我們的知識產權相關的風險
我們的 成功取決於我們獲得、維護、保護、捍衞和執行專利、商業祕密和其他知識產權保護的能力
我們 的成功取決於我們在美國和其他國家獲得、維護、保護、捍衞和執行專利、商業祕密和其他知識產權 保護的能力。如果我們不 充分保護、維護、捍衞和執行我們的知識產權,競爭對手可能會侵蝕、否定或搶佔 我們可能擁有的任何競爭優勢,這可能會對我們的業務和實現盈利能力產生不利影響。為了保護 我們的專利地位,我們在美國和某些其他國家提交了專利申請,涉及我們的新型候選產品 及其在不同醫療適應症中的潛在用途,這些適應症對我們的業務非常重要。專利申請和批准 過程既昂貴又耗時,我們可能無法以合理成本或及時的方式提交和起訴所有必要或理想的專利申請, 可能無法獲得和維護已發佈的專利。
如果 我們獲得的專利保護範圍不夠廣泛,我們可能無法阻止他人開發和商業化 與我們相似或相同的技術和產品。我們在市場上成功競爭所需的專利保護程度 在某些情況下可能無法獲得或受到嚴重限制,並且可能無法充分保護我們的權利或允許我們獲得或保持任何競爭優勢 。雖然我們與能夠訪問我們研發成果的機密或可獲得專利的 方面的各方(例如我們的員工、承包商、潛在的業務合作者、臨牀研究人員 和其他第三方)簽訂了保密協議和保密協議,但這些各方中的任何一方都可能違反協議並在提交專利申請之前披露此類成果, 這可能會危及我們尋求和獲得專利保護的能力。此外,科學文獻中發現的發表往往滯後於實際發現,美國和其他司法管轄區的專利申請通常在申請後18個月才發表,或者在某些情況下根本不發表。因此,我們不能確定我們是第一個製造我們專利或待審專利申請中要求保護的發明的人,或者我們是第一個申請此類發明的專利保護的人。
-30-
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions, and has been the subject of much litigation in recent years. As a result, the issuance, scope, validity, enforceability, and commercial value of our patent rights may be uncertain. Our pending and future patent applications may not result in patents being issued which protect our technology or product candidates or which effectively prevent others from commercializing competitive technologies and product candidates. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and its scope can be reinterpreted after issuance. Even if our patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors or other third parties from competing with us, or otherwise provide us with any competitive advantage. For example, there can be no assurance that our issued patents contain and pending patent applications will contain, when granted, claims of sufficient breadth to cover all antibodies alleged to be a biosimilar of our product candidates. Furthermore, there can be no assurance that our issued patents will not be challenged at the United States Patent and Trademark Office, or USPTO, or foreign patent offices or in court proceedings, and if any such challenge were successful, the scope of our issued patent claims could be limited so as to not cover antibodies alleged to be a biosimilar of our product candidates. In addition, changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection. In addition, the laws of other countries may not protect our rights to the same extent or in the same manner as the laws of the United States. For example, patent laws in various jurisdictions, including significant commercial markets, such as Europe, restrict the patentability of methods of treatment of the human body more than patent laws in the United States.
我們未來的一些專利和專利申請以及其他知識產權可能與第三方共同擁有。如果我們無法 獲得任何此類第三方共有人在此類專利或專利申請或其他知識產權中的權益的獨家許可,此類共有人可能會將其權利授權給其他第三方,包括我們的競爭對手,而我們的競爭對手可能會 銷售競爭產品和技術。此外,我們還需要我們專利的任何此類共有人的合作,以便 針對第三方執行此類專利,而此類合作可能不會提供給我們。此外,我們或任何未來的合作伙伴、 合作者或被許可人,可能無法識別在開發和商業化活動過程中所做的發明的可專利方面 在獲得專利保護之前為時已晚。因此,我們可能會錯過加強 專利地位的潛在機會。上述任何情況都可能對我們的業務、財務狀況、經營業績 和前景產生重大不利影響。
我們的 專利涵蓋我們的專利抗C5a和抗C5aR技術可能會受到第三方的質疑、縮小、規避和無效
我們的任何 專利都可能受到第三方的質疑、限制、規避或無效。專利的頒發並不是關於其發明人、範圍、有效性或可轉讓性的決定性 ,我們的專利可能會在 美國和其他地方的法院或專利局受到質疑。我們可能會受到第三方向美國專利商標局提交的現有技術或參與 反對、派生、撤銷、複審、授予後, 各方間審查或幹擾訴訟,挑戰我們的專利權或其他人的專利權。任何此類提交、訴訟或訴訟中的不利裁決可能會縮小我們專利權的範圍、可執行性或使其無效,允許第三方將我們的技術或產品商業化並在不向我們付款的情況下與我們直接競爭,或者導致我們無法在不侵犯第三方專利權的情況下製造或商業化產品 。此外,我們可能不得不參與美國專利商標局宣佈的幹擾程序,以確定發明的優先權 或授權後的挑戰程序,如外國專利局的異議,挑戰發明優先權或其他可專利性特徵。此類挑戰可能會導致專利權的喪失、排他性的喪失或專利主張的縮小、無效或無法執行,這可能會限制我們阻止他人使用類似或相同的技術和產品或將其商業化的能力,或者限制我們的技術和候選產品的專利保護期限。這樣的程序還可能導致大量成本,並需要我們的科學家和管理層花費大量時間,即使最終結果對我們有利。
此外,我們的競爭對手和其他第三方還可以通過以非侵權方式開發類似或替代技術或產品來規避我們的專利。例如,第三方可能開發一種競爭性療法,該療法提供與 viloblimab或其他候選產品類似的益處,但使用的技術不在我們的專利保護範圍內。我們的競爭對手 也可以尋求批准銷售任何已批准產品的仿製版本,在尋求此類批准的同時,可能會聲稱我們的專利無效、不可強制執行或未被侵犯。在這種情況下,我們可能需要捍衞或維護我們的專利,或者兩者兼而有之。 包括通過提起訴訟指控專利侵權。在上述任何類型的訴訟中,擁有管轄權的法院或其他機構可能會發現我們的專利無效或不可強制執行,或者我們的競爭對手正在以非侵權方式競爭。因此,即使我們擁有 有效且可強制執行的專利,這些專利仍可能無法針對競爭產品或流程提供足以實現我們的業務目標的保護。如果我們對候選產品 持有或申請的專利和申請提供的專利保護不夠廣泛,不足以阻礙此類競爭,我們成功將候選產品商業化的能力可能會受到負面影響,這可能會對我們的業務、財務狀況、運營結果和前景產生實質性的不利影響 。
-31-
我們 不能確定我們是第一個在我們的專利或專利申請中聲稱擁有抗C5a和抗C5aR技術的人,或者 我們是第一個申請專利保護的人
假設 滿足其他可專利性要求,目前,第一個提交專利申請的人通常享有專利。 然而,在2013年3月16日之前,在美國,第一個發明者享有專利。科學文獻中發現的發佈往往落後於實際發現,美國和其他司法管轄區的專利申請在提交後18個月才發佈,或者在某些情況下根本不發佈。因此,我們不能確定我們是第一個在我們的專利或未決專利申請中提出要求的發明,或者我們是第一個為此類發明申請專利保護的 。同樣,我們不能確定我們可以向其許可或購買專利權的各方是第一個做出相關權利要求的發明的人,或者是第一個為這些發明申請專利保護的人。如果第三方已在2013年3月15日或之前就我們的專利或申請中要求的發明提交了專利申請,則此類第三方可以在美國啟動幹預程序,以確定誰最先發明瞭涵蓋我們的專利申請的標的。如果第三方在2013年3月15日之後提交了此類申請,則此類第三方可以在美國啟動派生程序,以確定我們的發明是否源自他們的發明。
專利申請過程中存在許多風險,不能保證我們一定能成功獲得我們所申請的專利
待處理的專利申請不能針對實踐此類申請中所要求的技術的第三方強制執行,除非且直到 此類專利頒發。專利申請過程中存在許多風險和不確定性,不能保證我們或我們的任何未來開發合作伙伴都能成功地通過獲得和保護專利來保護我們的候選產品。 這些風險和不確定性包括:
| ● | 美國專利商標局和各種外國政府專利代理機構要求遵守多個 專利過程中的程序、文件、費用支付等規定。這裏 是指不符合規定可能導致專利或專利權的放棄或失效的情況 專利權的部分或全部喪失。 在這種情況下,競爭對手可能會比其他情況下更早進入市場 都是這樣; |
| ● | 在專利申請中所要求的覆蓋範圍可能會在專利被 發佈,發佈後可以重新解釋其範圍; |
| ● | 專利申請不得導致任何專利被授予; |
| ● | 專利 可能被質疑、無效、修改、撤銷、規避, 範圍縮小、無法執行或可能無法提供任何競爭優勢; |
| ● | 我們的 競爭對手,其中許多人擁有更多的資源,其中許多人已經 在競爭技術方面的重大投資,可能尋求或可能已經獲得 限制、幹擾或消除我們製造、使用和銷售我們的產品的能力的專利 潛在的候選產品; |
| ● | 美國政府和國際政府機構可能面臨巨大壓力,要求限制被證明成功的疾病治療方法在美國國內外的專利保護範圍。作為涉及全球衞生問題的公共政策問題 ;和 |
| ● | 國家 除美國以外,其他國家的專利法對專利權人的優惠可能比那些國家的專利法要低 得到美國法院的支持,使外國競爭者有更好的機會來創造、發展 和市場競爭的候選產品。 |
上述任何事件可能對我們的業務、財務狀況、經營業績和前景造成重大不利影響。
-32-
保護我們的知識產權和我們專有的抗C5a和抗C5aR技術是困難和昂貴的,我們可能無法 確保它們的保護
我們的商業成功將在一定程度上取決於獲得並保持對我們候選產品的成分、用途和結構、製造方法、相關治療靶點和相關治療方法的專利保護和商業祕密保護,以及成功保護這些專利免受潛在的第三方挑戰。我們保護我們的 候選產品不被第三方未經授權制造、使用、銷售、提供銷售或進口的能力取決於我們根據涵蓋這些活動的有效和可強制執行的專利所擁有的權利的程度。
美國專利商標局或美國法院或其他事實審查員,或任何相應的外國專利局或法院或其他審查員對權利要求是否符合所有可專利性要求的最終裁決無法得到保證。儘管我們的C5a和C5aR抑制劑產品組合包括我們擁有的針對C5a和C5aR抑制劑及其相關使用方法的六個專利和專利申請系列 ,但我們無法預測在我們的專利或專利申請、我們未來的許可專利或專利申請或第三方專利中可能允許或執行的權利要求的廣度。
我們 不能保證我們的任何專利申請將被發現是可申請專利的,包括我們自己的現有技術專利、 出版物或其他披露。此外,鑑於美國、歐洲和其他國家/地區專利法的不同, 例如,提交專利申請的寬限期以及可以被視為現有技術的內容,我們不能對我們在美國或其他司法管轄區的未決和未來專利申請可能提出的任何索賠的範圍作出 任何保證。同樣,我們不能保證在第三方對我們的專利和專利申請在美國或其他司法管轄區的專利和申請的可專利性、有效性或可執行性提出質疑的訴訟中,任何索賠的範圍仍然有效。任何此類挑戰如果成功,都可能限制對我們候選產品的專利保護,和/或對我們的業務造成實質性損害。
未來對我們所有權的保護程度不確定,因為法律手段只能提供有限的保護,可能無法充分 保護我們的權利或允許我們獲得或保持我們的競爭優勢。例如:
| ● | 我們 可能無法生成足夠的數據來支持保護我們一個或多個計劃的整個開發範圍的專利申請; |
| ● | 我們的一個或多個未決專利申請可能不會成為已發佈的專利,或者如果發佈,該專利(S)將不足以保護我們的技術或 產品,為我們提供商業上可行的產品基礎,或為我們提供任何競爭優勢; |
| ● | 如果我們的待決專利申請作為專利頒發,可能會被第三方質疑為未被侵犯、無效或根據美國或外國法律不可執行;或者 |
| ● | 如果 發佈,我們擁有權利的專利可能無效或不可強制執行。 |
此外,如果我們無法獲得並維護我們的某個候選產品的專利保護,或者在此類專利保護到期的情況下,通過進一步開發 後續適應症的產品或候選產品來擴展我們的產品組合可能不再具有成本效益。上述任何一項都可能對我們的業務、財務狀況、運營結果和前景產生重大不利影響。
獲得和維持我們的抗C5a和抗C5aR技術的專利保護取決於遵守政府專利機構提出的各種程序、文件 提交、費用支付和其他要求,如果不符合這些要求,我們的專利保護可能會減少或取消
定期 專利和申請的維護費、續期費、年費和各種其他政府費用需要在專利和申請的有效期內分幾個階段向USPTO和美國以外的各種政府專利機構支付 。美國專利商標局和各種非政府專利機構要求在專利申請過程中和專利頒發後遵守一些程序、文件、費用支付和其他類似條款。在 情況下,不遵守規定可能會導致專利或專利申請被放棄或失效,從而導致相關司法管轄區的專利權部分或全部喪失。我們可能會簽訂某些許可協議,在這些許可協議中,我們將無法維護或起訴產品組合中的專利,因此必須依賴第三方採取此類行動並遵守某些要求。 如果我們或我們的未來或任何現有許可方未能維持對我們的專利組合的保護,可能會對我們的業務、財務狀況、運營結果和前景產生重大不利影響。
-33-
此外,我們的專利或專利申請的準備或提交過程中可能存在形式上的缺陷,或者將來可能會出現 ,例如在適當的優先權要求、發明人、權利要求範圍或專利期限調整請求方面。 如果我們未能建立、維護或保護此類專利和其他知識產權,則此類權利可能會減少、取消、 無效和/或無法執行。如果我們目前或未來的任何合作伙伴、合作者、被許可方或許可方不完全合作 或不同意我們對任何專利權的起訴、維護或執行,則這些專利權可能會受到損害。 如果我們的專利或專利申請的形式、準備、審查或執行存在重大缺陷,則此類專利 可能無效和/或不可執行,且此類申請可能永遠不會產生有效、可執行的專利。任何這些結果都可能 損害我們防止來自第三方競爭的能力,這可能會對我們的業務、財務狀況、 經營業績和前景產生重大不利影響。
專利 條款可能不足以在足夠的時間內保護我們在候選產品上的競爭地位,如果我們 沒有 獲得1984年《藥品價格競爭和專利條款恢復法案》或Hatch—Waxman修正案以及類似的非美國法律的保護。 延長涵蓋我們每種候選產品的專利期限的立法,我們的業務可能會受到重大損害。
專利 的使用期限有限。在美國,專利的自然期滿通常是在申請後的20年。可以使用各種 擴展,但是,專利的壽命及其提供的保護是有限的。考慮到新候選產品的開發、測試和監管審查所需的時間 ,保護此類候選產品的專利可能在此類候選產品商業化之前 或之後不久到期。因此,我們的專利組合可能無法為我們提供充分和持續的 專利保護,足以將其他人排除在將與我們候選產品相似的產品商業化之外。
Depending upon the timing, duration and conditions of the FDA marketing approval of our product candidates, one or more of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Amendments and similar legislation in the EU and other jurisdictions. The Hatch-Waxman Amendments permit a patent term extension of up to five years for a patent covering an approved product as compensation for effective patent term lost during product development and the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent may be extended and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. In Europe, a maximum of five and a half years of supplementary protection can be achieved for an active ingredient or combinations of active ingredients of a medicinal product protected by a basic patent, if a valid marketing authorization exists (which must be the first authorization to place the product on the market as a medicinal product) and if the product has not already been the subject of supplementary protection. However, we may not receive an extension if we fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. Moreover, the length of the extension could be less than we request. If we are unable to obtain patent term extension or the term of any such extension is less than we request, the period during which we can enforce our patent rights for that product will be shortened and our competitors may obtain approval to market competing products sooner. As a result, our revenue from applicable products could be reduced and could have a material adverse effect on our business, financial condition, results of operations and prospects.
其他人 可能聲稱擁有我們的知識產權和專有的反C5a和反C5aR技術的所有權權益,這可能使我們 面臨訴訟,並對我們的前景產生重大不利影響
A third party may claim an ownership interest in one or more of our, or our future or any existing licensors’, patents or other proprietary or other intellectual property rights. A third party could bring legal actions against us and seek monetary damages and/or enjoin clinical testing, manufacturing and marketing of the affected product or products. While we are presently unaware of any material claims or assertions by third parties with respect to our patents or other intellectual property, we cannot guarantee that a third party will not assert a claim or an interest in any of such patents or other intellectual property. If we become involved in any litigation, it could consume a substantial portion of our resources, and could cause a significant diversion of effort by our technical and management personnel. If any of these actions are successful, in addition to any potential liability for damages, we could be required to obtain a license to continue to manufacture or market the affected product, in which case we may be required, for example, to pay substantial royalties or grant cross-licenses to our patents. We cannot, however, assure you that any such license will be available on acceptable terms, if at all. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations as a result of claims of patent infringement or other violations of other intellectual property rights. Further, the outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance, including the demeanor and credibility of witnesses and the identity of any adverse party. This is especially true in intellectual property cases that may turn on the testimony of experts as to technical facts upon which experts may reasonably disagree. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects.
-34-
如果 我們因侵犯、盜用或以其他方式侵犯第三方知識產權而被起訴,此類訴訟可能 代價高昂且耗時,並可能阻止或延遲我們開發或商業化候選產品
我們的 商業成功部分取決於我們開發、製造、營銷和銷售我們的候選產品的能力,而不侵犯、 盜用或以其他方式侵犯第三方的所有權或任何其他知識產權。第三方可能 擁有與化合物、化合物製造方法和/或 用於治療我們正在開發的候選產品的疾病適應症的方法有關的美國和非美國已頒發的專利和待審專利申請,這些疾病適應症可能涵蓋我們的候選產品 候選產品或補體抑制方法。如果發現任何第三方專利或專利申請涵蓋了我們的候選產品 或其使用或生產方法,或我們的補體抑制方法,我們可能無法在未獲得許可的情況下按計劃自由生產或銷售我們的候選產品 ,這些許可可能無法以商業上合理的條款獲得,或根本無法獲得。
在生物技術和製藥行業有大量的知識產權訴訟,我們可能成為與我們的產品有關的知識產權訴訟或其他對抗性訴訟的一方或威脅方 ,包括在USPTO的幹擾和授權後訴訟。可能存在與我們候選產品的組成、使用或製造有關的材料、配方、製造方法或處理方法的第三方專利或專利申請 。由於專利申請可能需要數年時間才能發佈,因此可能存在待處理的專利申請,這些申請可能會導致我們的候選產品可能被指控侵權。此外,第三方未來可能會獲得專利 並聲稱使用我們的技術侵犯了這些專利。因此,第三方可以根據現在存在或將來出現的知識產權向我們提出侵權索賠。知識產權訴訟的結果 受到不確定因素的影響,這些不確定因素無法預先充分量化。製藥和生物技術行業產生了大量專利,包括我們在內的行業參與者可能並不總是清楚哪些專利涵蓋各種類型的產品或使用或製造方法。專利提供的保護範圍取決於法院的解釋,而且解釋並不總是一致的。如果我們因專利侵權而被起訴,我們需要證明我們的候選產品、 產品或方法沒有侵犯相關專利的專利主張,或者專利主張無效或不可執行, 我們可能無法做到這一點。證明無效性是困難的。例如,在美國,要證明專利的無效性,就需要提出明確而令人信服的證據,以推翻專利授權所享有的有效性推定。即使我們在這些訴訟中取得成功 ,我們也可能會產生鉅額成本,我們的管理層和科學人員的時間和注意力可能會被轉移到這些訴訟中,這可能會嚴重損害我們的業務和運營結果。此外,我們可能沒有足夠的 資源來成功完成這些操作。
如果我們被發現侵犯、挪用或以其他方式侵犯第三方的知識產權,我們可能會被迫停止開發、製造或商業化侵權候選產品或產品,包括法院命令。或者,我們可能需要從該第三方獲得許可,才能使用侵權技術並繼續開發、製造或商業化侵權候選產品或產品。但是,我們可能無法以商業上的合理條款或根本無法獲得任何所需的許可證。即使我們能夠獲得許可證,它也可能是非排他性的,從而使我們的競爭對手能夠訪問向我們許可的相同技術;或者,或者另外,它可能包括阻礙或破壞我們在商業市場上成功競爭的能力的條款。此外,如果我們被發現故意侵犯專利,我們可能被判承擔金錢損害賠償責任,包括三倍損害賠償 和律師費。侵權發現可能會阻止我們將候選產品商業化,或迫使我們停止一些業務運營,這可能會損害我們的業務。聲稱我們盜用了任何第三方的商業祕密或其他機密信息,這可能會對我們的業務產生類似的負面影響。 上述任何一項都可能對我們的業務、財務狀況、運營結果和前景產生重大不利影響。
-35-
我們 可能會受到第三方的索賠,聲稱我們的員工或我們挪用了他們的知識產權,或者要求 擁有我們自己的知識產權和專有的抗C5a和抗C5aR技術
我們的許多現任和前任員工以及我們許可方的現任和前任員工,包括我們的高級管理層,以前 受僱於大學或其他生物技術或製藥公司,包括一些可能是競爭對手或潛在競爭對手的 。儘管我們盡力確保我們的員工在工作中不使用他人的專有信息或專有技術 ,但我們或這些員工可能會被要求使用或披露任何此類第三方的知識產權,包括商業祕密 或其他專有信息。可能有必要提起訴訟來對此類索賠進行辯護。如果我們未能為任何此類索賠辯護,除了支付金錢損害賠償外,我們還可能失去寶貴的知識產權或人員或遭受損害。 此類知識產權可能會被授予第三方,我們可能需要從該第三方獲得許可證 才能將我們的技術或產品商業化。這樣的許可可能不會以商業上合理的條款或根本不存在。即使我們成功地對此類索賠進行了辯護,訴訟也可能導致鉅額成本,並分散管理層的注意力。
此外,雖然我們通常要求參與我們知識產權開發的員工、顧問和承包商在此類員工、顧問和承包商的僱傭或我們的其他僱傭範圍內執行 將此類知識產權轉讓給我們的協議,我們可能無法與實際上 開發我們視為自己的知識產權的每一方執行此類協議,或此類協議可能被違反或被指稱無效,這可能導致我們提出或針對我們的索賠,涉及此類知識產權的所有權。如果我們未能起訴或辯護任何此類索賠,除了支付金錢損失外,我們可能會失去寶貴的知識產權。即使我們成功起訴或辯護此類索賠,訴訟也可能導致大量費用,並分散我們的高級管理人員 和科學人員的注意力。上述任何情況都可能對我們的業務、財務狀況、 經營業績和前景產生重大不利影響。
我們 可能會失去德國聯邦政府對我們某些知識產權的獨佔權
我們 通過德國的全資子公司InflaRx GmbH持有我們所有的知識產權。如果發生全國性流行病 或大流行病,德國聯邦政府、聯邦衞生部和其他當局有權出於德意志聯邦共和國的公共福利或安全的利益下令使用 我們擁有的和正在授權的專利。德國 聯邦政府可能會針對我們擁有的和正在授權的專利發佈此類命令,我們可能會失去 此類專利所涵蓋的技術的排他性。
此外, 導致我們某些專利和技術的研究,包括與我們在重症COVID—19中的臨牀開發相關的專利和技術,部分由德國聯邦政府資助。此類政府資助的研究項目的結果必須在符合某些條件的情況下免費提供給德國的學術研究和教學,並且必須在每半年的中期報告中公佈,並在資助工作完成後發表一份最終報告。與所產生的知識產權、 商業期望、科學成功機會、下一步措施和某些附加信息有關的信息,必須根據書面保密協議向德國政府和學術研究和教學的第三方披露。在特殊公共利益的情況下,德國 聯邦政府還擁有使用作為資助工作一部分產生的知識產權的非排他性和可轉讓權利。
-36-
我們的某些 員工和董事受德國法律約束,包括與發明的所有權和補償有關的法律
我們的許多員工(包括我們的部分董事)在德國工作,可能受德國僱傭法的約束。 可能屬於專利或作為實用新型受保護的發明,以及可能不屬於專利或作為實用新型受保護的其他技術創新的技術改進建議 由這些僱員作出,受 德國僱員發明法(Gesetzüber Arbeitnehmererfindungen),它規範了僱員發明的所有權和報酬。我們面臨的風險是,我們與現任或前任員工之間可能發生爭議,涉及 我們支付的賠償金是否充足、根據《德國僱員發明法》分配發明權利或 據稱不遵守本法規定,其中任何一項爭議都可能花費高昂的成本,無論我們在此類爭議中獲勝還是失敗。此外,根據《德國僱員發明法》,某些僱員 保留他們在2009年10月1日之前發明或共同發明並向我們披露的專利權。雖然我們相信,我們所有 當前和過去的德國僱員發明者隨後都將其發明或 共同發明的專利和發明的權益轉讓給了我們,但我們不能保證所有此類轉讓都是完全有效的。即使我們合法擁有受《德國僱員發明法》約束的僱員 發明人的所有發明,根據德國法律,我們仍須就使用專利給予 此類僱員合理補償。
如果 我們的任何現任或過去的員工獲得或保留我們認為 我們擁有的任何發明或其他知識產權的所有權,我們可能會失去寶貴的知識產權,並可能被要求從這些員工那裏獲得和維護對 此類發明或知識產權的許可,這些許可可能無法以商業上合理的條款提供或根本無法提供,或者可能是非排他性的。 如果我們無法獲得並維持任何此類員工對此類發明或知識產權的興趣的許可, 我們可能需要停止我們可能開發的一個或多個候選產品的開發、製造和商業化。 此外,我們知識產權的任何專有權的喪失都可能限制我們阻止他人使用或商業化 類似或相同技術和產品的能力。上述任何事件都可能對我們的業務、財務狀況、經營業績和前景造成重大不利影響。
我們 可能無法在全球範圍內執行我們的知識產權
在世界各地所有國家對我們的候選產品進行申請、起訴、維護、執行和保護將是非常昂貴的 ,而且我們在美國以外的某些國家的知識產權可能比在美國 的知識產權更少。在某些國家,特別是在發展中國家,對專利性的要求可能有所不同;因此,即使在我們確實尋求專利保護的國家,也不能保證任何專利將涉及我們的候選產品的權利要求。
Moreover, our ability to protect and enforce our intellectual property rights may be adversely affected by unforeseen changes in the United States and foreign intellectual property laws. Additionally, laws of some countries outside of the United States and Europe do not afford intellectual property protection to the same extent as the laws of the United States and Europe. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, including India, China and other countries, do not favor the enforcement of patents and other intellectual property rights. This could make it difficult for us to stop the infringement of our patents or the misappropriation or other violations of our other intellectual property rights. For example, many countries outside the United States have compulsory licensing laws under which a patent owner must grant licenses to third parties. Consequently, we may not be able to prevent third parties from practicing our inventions in certain countries outside the United States and Europe. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop and market their own products and, further, may export otherwise infringing products to jurisdictions where we have patent protection, if our ability to enforce our patents to stop infringing activities is inadequate. These products may compete with our products, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
我們可能獲得任何專利權許可的協議 可能無法給予我們足夠的權利,允許我們在所有相關司法管轄區執行 我們的許可專利或為聲稱這些專利無效的任何索賠進行辯護(或控制執行或辯護),因為要求可能有所不同。
在美國或外國司法管轄區執行我們專利權的訴訟程序,無論是否成功,都可能導致大量成本 ,並將我們的精力和資源從我們業務的其他方面轉移出去。此外,此類程序可能使我們的專利面臨 失效或狹義解釋的風險,我們的專利申請面臨無法發佈的風險,並可能引發第三方對我們提出 索賠。我們可能不會在我們發起的任何訴訟中獲勝,並且所裁定的損害賠償或其他補救措施(如有)可能沒有 商業意義。此外,雖然我們打算在主要市場為我們的候選產品 尋求保護我們的知識產權,但我們無法確保我們能夠在我們可能希望 營銷我們的候選產品的所有司法管轄區發起或保持類似的努力。因此,我們在這些國家保護知識產權的努力可能不夠。 上述任何情況都可能對我們的業務、財務狀況、經營業績和前景產生重大不利影響。
-37-
我們 可能會捲入訴訟,以保護或執行我們的專利或其他知識產權,這可能是昂貴的、耗時的 和不成功的
我們的 競爭對手和其他人可能侵犯、盜用或以其他方式侵犯我們的專利或其他知識產權。為了對抗 侵權或未經授權的使用,我們可能需要提出侵權或其他索賠,這可能會花費昂貴且耗時 ,並會分散我們的管理人員和科學人員的時間和注意力。
除了聲稱我們的專利無效或不可執行,或兩者兼而有之的反訴之外, 我們對被認為侵權者提出的任何索賠都可能促使這些當事人對我們提出反索賠,聲稱我們侵犯了他們的專利。在任何專利侵權訴訟中,法院都有可能判定我們的專利全部或部分無效或不可強制執行,並且我們無權阻止對方使用相關發明。還有一種風險是,即使此類專利的有效性得到支持,法院也會狹隘地解釋該專利的權利要求,或者以我們的專利權利要求不包括該發明為理由,裁定我們無權阻止 另一方使用該發明。 涉及我們的一項或多項專利的訴訟或訴訟中的不利結果可能會限制我們針對這些當事人或其他競爭對手主張這些專利的能力,並可能限制或排除我們將第三方排除在開發、製造和銷售類似或 競爭產品的能力。即使我們認定侵權行為成立,法院也可能決定不對進一步的侵權行為頒發禁令,而只判給金錢損害賠償金,這可能是也可能不是足夠的補救措施。此外,由於與知識產權訴訟相關的發現需要大量的 ,因此我們的一些機密信息可能會在訴訟期間被泄露。還可能公佈聽證會、動議或其他臨時程序或事態發展的結果。如果證券分析師或投資者認為這些結果是負面的,可能會對我們普通股的價格產生不利影響。此外,不能保證我們將有足夠的財政或其他資源來提交和追查此類侵權索賠,這些索賠通常會持續數年才能結案。即使我們最終在這類索賠中獲勝,此類訴訟的金錢成本以及我們管理層和科學人員注意力的轉移也可能超過我們從訴訟中獲得的任何好處。任何此類訴訟都可能對我們的業務、財務狀況、運營結果和前景產生實質性的不利影響。
如果我們的商標和商號沒有得到充分的保護,那麼我們可能無法在我們感興趣的商標中建立品牌認知度 ,我們的業務可能會受到不利影響
我們的商標或商品名稱可能會受到質疑、侵犯、規避或宣佈為通用商標,或被確定為侵犯了其他商標。 我們可能無法保護我們對這些商標和商品名稱的權利,或者可能被迫停止使用這些名稱,我們需要這些名稱 才能在我們感興趣的市場中獲得潛在合作伙伴或客户的名稱認可。在商標註冊程序中,我們可能會 收到我們無法克服的拒絕,在美國專利商標局和許多外國司法管轄區的類似機構中,第三方 有機會反對未決的商標申請並尋求註銷註冊商標。可能會對我們的商標提起反對或取消訴訟,而我們的商標可能無法繼續使用。如果我們無法根據我們的商標和商號建立名稱 認可,我們可能無法有效競爭,我們的業務可能會受到不利影響。
如果 我們未能履行我們在任何未來或與第三方的其他知識產權許可下的義務,我們可能會失去對我們的業務非常重要的許可 權利
我們 可能依賴第三方授予的某些專利權和專有的抗C5a和抗C5aR技術以及其他知識產權 ,這些對我們的候選產品的開發以及我們產品的製造和其他商業化是重要或必要的 。這些許可證和其他許可證可能不會提供在所有相關使用領域以及我們希望在未來開發、製造或商業化我們的技術和產品的所有地區使用此類知識產權和技術的獨家權利。因此,我們可能無法阻止競爭對手在我們所有許可證所包括的地區開發、製造和商業化競爭產品 。我們的許可人可能再許可了由第三方擁有的專利和其他知識產權,或者依賴於第三方顧問或協作者或來自第三方的資金,這些第三方擁有或擁有此類許可內知識產權的所有權或其他權利、所有權或權益,因此我們的許可人不是我們許可的專利和其他知識產權的唯一和獨家所有者。這可能會對我們的競爭地位、業務、財務狀況、運營結果和前景產生重大不利影響。
-38-
此外,我們可能根據的協議許可專利權,可能無法讓我們控制專利申請的起訴或維護, 因此我們可能無法控制提出的索賠或論點,並且可能無法確保、維護或成功 執行和捍衞必要或理想的專利保護,使其免受這些專利權的侵害。我們不能確定我們的許可方的專利申請起訴 和維護活動將遵守適用的法律法規,或者將產生有效的 和可執行的專利。即使我們被允許進行此類強制執行或抗辯,我們仍將需要我們未來 或任何現有許可方的合作,並且不能保證我們將收到該協議以及以何種條件收到該協議。我們不能確定我們未來的許可人 將分配足夠的資源,或優先考慮他們或我們執行此類專利或對此類索賠進行辯護,以保護我們在任何許可專利中的利益 。如果我們無法獲得專利保護或對第三方強制執行現有或未來的專利, 可能會對我們的業務、財務狀況、經營業績和前景產生重大不利影響。
此外, 我們可以根據哪些協議向第三方或從第三方獲得技術或任何其他知識產權,這些協議中的某些 條款可能會受到多種解釋的影響。解決可能出現的任何合同解釋分歧 可能會縮小我們認為我們對相關技術或任何其他知識產權的權利範圍, 或增加我們認為我們在相關協議下的財務或其他義務。此外,如果關於我們可能許可的技術 或其他知識產權的爭議妨礙或損害我們以商業上可接受的條款 維持許可安排的能力,我們可能無法成功開發、製造和商業化受影響的候選產品,這可能 對我們的業務、財務狀況、經營業績和前景造成重大不利影響。
受許可協議約束的知識產權方面可能出現糾紛,包括:
| ● | 根據許可協議和其他與解釋相關的權利範圍 問題; |
| ● | 我們的技術和工藝侵犯知識產權的程度 不受許可協議約束的許可人; |
| ● | 在當前和未來的合作開發下對專利和其他權利進行分許可 關係; |
| ● | 我們的 任何許可協議下的勤勉義務,以及哪些活動能夠滿足此類義務; |
| ● | 發明和專有技術以及其他知識產權的發明和所有權, 我們的許可交易對手共同創建或使用知識產權,以及 我們和我們的合作伙伴; |
| ● | 專利技術發明的優先權。 |
儘管我們盡了最大努力,但我們的許可證交易對手可能會得出結論,認為我們實質上違反了我們的許可證協議,因此可能 終止許可證協議,這可能會使我們喪失開發、製造和商業化候選產品 和這些許可證協議涵蓋的技術的能力。如果任何內許可被終止,競爭對手可能能夠尋求監管部門的批准 ,並銷售與我們相同的產品。我們可能無法以合理的成本或合理的條款(如果有的話)獲得我們需要的任何額外許可證。在這種情況下,我們可能需要花費大量的時間和資源來 重新設計我們的候選產品、技術或其製造方法,或者開發或許可替代技術,所有這些 在技術或商業基礎上都可能不可行。如果我們無法做到這一點,我們可能無法開發、生產 或將受影響的候選產品商業化,這可能會損害我們的競爭地位、業務、財務狀況、運營結果 和前景。
-39-
如果 我們無法保護我們的商業祕密的機密性,我們的技術價值可能會受到負面影響,我們的業務 將受到損害
In addition to the protection afforded by patents, we also rely on trade secret protection for certain aspects of our intellectual property. However, trade secrets are difficult to protect. We seek to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, consultants, independent contractors, advisors, contract manufacturers, suppliers and other third parties. We also enter into confidentiality and invention or patent assignment agreements with employees and certain consultants and independent contractors. Any party with whom we have executed such an agreement may breach that agreement and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. Additionally, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating the trade secret. Further, if any of our trade secrets were to be lawfully obtained or independently developed by a competitor or other third party, we would have no right to prevent such third party, or those to whom they communicate such technology or information, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed or otherwise obtained by a competitor or other third party, it could have a material adverse effect on our business, financial condition, results of operations and prospects.
| 4. | 風險 與員工事務和管理成長有關 |
我們 只有有限的員工來管理和運營我們的業務
截至2023年12月31日,我們有66名全職或兼職員工。我們專注於vilobelimab的開發和商業化,要求 我們優化現金利用率,並以有限的人員管理和運營我們的業務。我們無法向您保證,我們將能夠 僱傭額外的員工和/或保留足夠的人員配置水平,以開發和商業化vilobelimab或運營我們的運營,或 實現我們本來會尋求實現的所有目標。
我們 嚴重依賴我們的執行官和董事,失去他們的服務將對我們的業務造成重大損害
我們 的成功取決於並可能繼續取決於我們是否有能力聘用和保留我們現有的執行官、 董事、首席顧問和其他人的服務。我們高度依賴首席執行官Niels Riedemann、首席科學官郭仁峯、首席財務官Thomas Taapken以及首席醫療官Camilla Chong(自2023年7月起)的管理、開發、臨牀、財務和業務開發專業知識。我們在生物技術 和製藥行業的競爭力取決於我們吸引和留住高素質的管理、科學和醫療人員的能力。
我們的 行業近年來管理人員的高流動率。我們的任何人員都可以隨意終止其僱傭 。如果我們失去了一名或多名執行官或其他關鍵員工,我們成功實施業務戰略的能力可能會受到嚴重損害 。此外,更換執行官或其他關鍵員工可能很困難,而且可能需要較長的時間 ,因為我們行業中具備開發、 成功獲得產品市場批准和商業化所需技能和經驗的人員數量有限。
從這個有限的人才庫中招聘的競爭 非常激烈,考慮到眾多製藥和生物技術公司對類似人員的競爭,我們可能無法以可接受的條件招聘、培訓、留住或激勵這些額外的關鍵員工。我們還經歷了 從大學和研究機構招聘科學和臨牀人員的競爭。
我們 依靠顧問和顧問,包括科學、戰略、監管和臨牀顧問,協助我們制定 研發和商業化戰略。我們的顧問和顧問可能受僱於其他實體,並且可能與這些實體簽訂諮詢或諮詢合同,從而限制了他們對我們的可用性。如果我們無法繼續 吸引和留住高素質人才,我們開發和商業化候選產品的能力將受到限制。
-40-
我們 希望擴大我們的組織,因此,我們可能會在管理增長方面遇到困難,這可能會擾亂我們的運營
We expect to expand scope of our operations, particularly in the areas of clinical development and regulatory affairs. To manage such growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Our management may need to devote a significant amount of its attention to managing these growth activities. Moreover, our expected growth could require us to relocate to a different geographic area of the country. Due to our limited financial resources and the limited experience of our management team in managing a company with such anticipated growth, we may not be able to effectively manage the expansion or relocation of our operations, retain key employees, or identify, recruit, attract and train retain human capital. Competition for qualified, motivated, and highly-skilled executives, professionals and other key personnel in biotechnology and pharmaceuticals industries is significant. Our inability to manage the expansion or relocation of our operations effectively may result in weaknesses in our infrastructure, give rise to operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. Our expected growth could also require significant capital expenditures and may divert financial resources from other projects, such as the development of additional product candidates. If we are unable to effectively manage our expected growth, our expenses may increase more than expected, our ability to generate revenues could be reduced and we may not be able to implement our business strategy, including the successful development and commercialization of our product candidates.
我們的 員工、獨立承包商、顧問、合作者和CRO可能會從事不當行為或其他不當活動,包括 不遵守監管標準和要求,這可能會給我們造成重大責任並損害我們的聲譽
我們 面臨員工、獨立承包商、顧問、合作者和CRO可能從事欺詐行為或其他非法活動的風險。這些當事人的不當行為可能包括故意、魯莽或疏忽的行為或向我們披露未經授權的活動,違反:(I)FDA法規或類似的非美國監管機構的類似法規,包括要求向此類機構報告真實、完整和準確信息的法律,(Ii)製造和臨牀試驗 行為標準,(Iii)聯邦和州醫療欺詐和濫用法律法規以及由類似的非美國監管機構制定並執行的類似法律法規,(4)要求準確報告財務信息或數據的法律 。受這些法律約束的活動還涉及在臨牀試驗過程中獲得的信息的不當使用, 這可能導致監管制裁和對我們的聲譽造成嚴重損害。並非總是能夠識別和阻止不當行為, 我們為檢測和防止此類活動而採取的預防措施可能無法有效控制未知或未管理的風險或損失 或保護我們免受因未能遵守此類法律、標準或法規而引起的政府調查或其他行動或訴訟。如果針對我們提起任何此類訴訟,而我們未能成功為自己辯護或維護我們的權利,這些訴訟可能會對我們的業務和運營結果產生重大影響,包括施加民事、刑事和行政處罰、損害賠償、罰款、可能被排除在聯邦醫療保險、醫療補助和其他聯邦醫療保健計劃之外、合同損害、聲譽損害、利潤和未來收益減少以及我們的業務縮減, 任何這些都可能對我們運營業務的能力和運營結果產生實質性的不利影響。
| 5. | 與我們的普通股和上市公司地位有關的風險 |
我們普通股的交易價格一直很不穩定,未來可能也很不穩定,這可能會給我們普通股的持有者造成重大損失,導致我們的股價下跌,並引發針對我們公司或我們管理層的證券訴訟
我們的股價一直是,未來也可能是高度波動的。股票市場,尤其是規模較小的製藥公司和生物技術公司的市場經歷了極端的波動,這種波動往往與特定公司的經營業績 無關。您應該認為投資我們的普通股是有風險的,只有在您能夠承受投資的重大損失和市值大幅波動的情況下才進行投資。我們普通股的市場價格可能受到許多 因素的影響,包括:
| ● | 維羅貝利單抗和任何其他候選產品的臨牀試驗的時間、登記和結果; |
| ● | 針對viloblimab、我們的其他候選產品或競爭對手的 產品和候選產品的監管 行動; |
| ● | GOHIBIC(Viloblimab)商業化的成功; |
-41-
| ● | 現有或新的有競爭力的產品或技術的成功; |
| ● | 我們對viloblimab或任何未來候選產品的開發或監管備案的任何 延遲,以及與適用的監管機構審查此類備案有關的任何不利發展或被認為不利的發展,包括FDA簽發了一封“拒絕提交”的信函或要求提供更多信息; |
| ● | 我們或我們的競爭對手宣佈重大收購、戰略夥伴關係、合資企業、合作或資本承諾。 |
| ● | 開始 或終止我們開發計劃的合作; |
| ● | 失敗 或我們的任何開發計劃中斷; |
| ● | 競爭對手候選產品的臨牀試驗結果; |
| ● | 美國和其他國家/地區的監管或法律發展; |
| ● | 與專利申請、已頒發專利或其他專有權利有關的發展情況或糾紛; |
| ● | 關鍵人員的招聘或離職; |
| ● | 與我們的任何候選產品或臨牀開發計劃相關的費用水平; |
| ● | 我們努力開發更多候選產品或產品的結果; |
| ● | 關於財務結果或開發時間表的估計的實際或預期變化; |
| ● | 宣佈 或預計將做出更多融資努力; |
| ● | 本公司、本公司內部人士或其他股東出售本公司普通股; |
| ● | 我們的財務業績或被認為與我們相似的公司的財務業績變化 ; |
| ● | 證券分析師的估計或建議(如果有)的變化 涉及我們的股票; |
| ● | 醫療保健支付制度結構的變化 |
| ● | 製藥和生物技術行業的市場狀況; |
| ● | 一般的經濟、工業、市場和政治條件;以及 |
| ● | 本“項目3.關鍵信息-3.風險因素”一節中描述的其他因素。 |
在過去,證券集體訴訟經常是隨着證券市場價格的下跌而對公司及其管理層提起的。這一風險與生物製藥公司尤其相關,它們在最近幾年經歷了股價的大幅波動。如果對我們提起此類訴訟,可能會導致我們或我們的管理層成員產生巨大的 成本,並將管理層的注意力和資源從我們的業務上轉移出去。
未來我們大量普通股的出售或未來出售的可能性可能會對股票價格產生不利影響 並稀釋股東權益
未來 大量出售我們的普通股,或認為此類出售將會發生,可能會導致我們普通股的市場價格下跌 。如果我們或我們的現有股東在公開市場上出售大量普通股,或市場認為可能發生此類出售,我們普通股的市場價格以及我們未來以有吸引力的條款或根本不可能通過發行股權證券籌集資金的能力可能會受到不利影響。
我們 在使用手頭現金方面擁有廣泛的自由裁量權,可能會以您不同意的方式進行投資或使用,並且 可能不會為您的投資帶來回報
截至2023年12月31日,我們擁有1280萬歐元的現金和現金等價物,以及8560萬歐元的有價證券。我們的管理層將在使用這類現金方面擁有廣泛的自由裁量權,並可能以不會改善我們的運營業績或提高我們普通股價值的方式使用這些現金。您將沒有機會影響我們關於如何使用手頭現金的決定。如果我們的管理層未能有效運用這些資金,可能會造成財務損失,損害我們的業務,導致我們的普通股價格下跌,並推遲我們的候選產品的開發。在使用之前,我們可能會將手頭的現金以不產生收入或貶值的方式進行投資。
-42-
我們 是一家外國私人發行人,因此,我們不受美國委託書規則的約束,並受《交易法》報告義務的約束 ,在某種程度上,這些義務比美國國內上市公司更寬鬆、更少發生
我們 將根據修訂後的1934年證券交易法或交易法,作為具有外國私人發行人身份的非美國公司進行報告 。由於我們符合《交易法》規定的外國私人發行人資格,因此我們不受《交易法》中適用於美國國內上市公司的某些條款的約束,包括(I)《交易法》中規範就根據《交易法》註冊的證券徵集委託、同意或授權的條款,(Ii)交易所 法案中要求內部人士就其股票所有權和交易活動以及從短時間內進行的交易中獲利的內部人士的責任提交公開報告的條款,以及(Iii)交易所法案中要求在發生指定 重大事件時向美國證券交易委員會提交包含未經審計的財務和其他指定信息的Form 10-Q季度報告 或當前的Form 8-K報告的規則。此外,外國私人發行人在每個財年結束後四個月 之前不需要提交Form 20-F年度報告,而作為加速提交者的美國國內發行人則需要在每個財年結束後75天內提交Form 10-K年報。外國私人發行人也不受《公平披露條例》的約束,《公平披露條例》旨在防止發行人選擇性披露重大信息。由於上述原因,您可能得不到為非外國私人發行人公司的股東提供的相同保護。
作為一家外國私人發行人,並在納斯達克上市要求允許的情況下,我們遵循某些母國治理實踐,而不是納斯達克的公司治理要求。
我們 是一家外國私人發行商。因此,根據納斯達克的上市要求,我們依賴母國治理要求 及其下的某些豁免,而不依賴納斯達克的公司治理要求。根據荷蘭法律 和公認的商業慣例,我們的公司章程並不規定一般適用於股東大會的法定人數要求。在這方面,我們的做法與納斯達克上市規則第5620(C)條的要求不同,後者要求發行人在其章程中規定普遍適用的法定人數,並且該法定人數不得少於已發行 有表決權股票的三分之一。雖然我們必須向股東提供股東大會的議程和其他相關文件,但 荷蘭法律沒有對徵集委託書的監管制度,而且徵集委託書在荷蘭並不是一種普遍接受的商業慣例;因此,我們的做法將與納斯達克上市規則第5620(B)條的要求有所不同。在納斯達克的上市要求允許的情況下,我們也選擇不遵守納斯達克上市規則5605(D)的要求,其中包括要求發行人有一個完全由獨立董事組成的薪酬委員會,並就薪酬顧問的獨立性做出決定;納斯達克上市規則5605(E),要求董事對董事的提名進行獨立監督 ;以及納斯達克上市規則5605(B)(2),要求發行人在董事會中擁有多數獨立董事。此外,我們已選擇不遵守納斯達克上市規則中包含的股東批准要求,在與某些事件相關的證券發行方面 ,例如收購另一家公司的股票或資產、建立或修訂員工的股權薪酬計劃、我們控制權的變更以及某些私募。在這方面,我們的做法 不同於納斯達克第5635條的要求,後者一般要求發行人在與此類活動相關的證券發行 時必須獲得股東批准。因此,您可能無法獲得受這些納斯達克要求約束的公司的股東所享有的同等保護 。
我們 預計在可預見的未來不會為我們的股本支付任何現金股息。因此,股東必須依賴資本增值(如果有的話)才能獲得投資回報
我們 從未宣佈或支付過股本現金股息。我們計劃保留我們未來的所有收益(如果有的話),為我們業務的運營、發展和增長提供資金。此外,未來任何債務或信貸協議的條款以及適用法律施加的任何限制 可能會阻止我們支付股息。因此,在可預見的未來,我們普通股的資本增值(如果有的話)將是您唯一的收益來源。尋求現金分紅的投資者不應購買我們的普通股。
見 “項目7.大股東和關聯方交易--1.大股東”。有關我們的高管、董事和主要股東及其關聯公司持有我們的已發行普通股的更多信息,請參閲本年度報告的其他部分 。
-43-
如果證券或行業分析師不發表關於我們業務的研究報告或發表不準確或不利的研究報告,我們的股價和交易量可能會下降。
我們普通股的 交易市場部分取決於證券或行業分析師發表的關於 我們或我們業務的研究和報告。我們對這些分析師沒有任何控制權。我們無法保證分析師將為我們提供服務或提供有利的 服務。證券或行業分析師可以選擇不繼續提供我們普通股的研究覆蓋, 這種缺乏研究覆蓋可能會對我們普通股的市場價格產生負面影響。如果我們確實有分析師報道, 如果一個或多個分析師下調我們的普通股評級,改變他們對我們普通股的看法,或發表不準確或不利的 關於我們業務的研究,我們的股價可能會下跌。此外,如果一名或多名分析師停止對我們公司的報道 或未能定期發佈關於我們的報告,我們可能會在金融市場上失去知名度,從而導致我們的股價或交易量下降 。
我們 使用淨營業虧損結轉和其他税務屬性的能力可能受到限制
根據《德國公司所得税法》第8c條,我們 使用NOL的能力受到限制,並且可能受到進一步限制 (Körperschaftsteuergesetz),或KStG,以及德國貿易税法第10a條(Gewerbesteuergesetz),或GewStG。 如果發生了第8c節KStG定義的合格所有權變更,且不適用豁免,則適用這些限制。一般而言, 如果在五年內將超過50%的股本或投票權直接或間接轉讓給一名股東或一組股東,則 有條件的所有權變更發生。如果交易 與股份或投票權的轉讓相當,或者如果資本增加導致各自股權發生變化,也可能發生有條件的所有權變更。 在此類符合條件的所有權變更的情況下,結轉税損將全部到期。在某種程度上,隱藏的儲備(stille 預訂)在德國應納税超過結轉的税務虧損,儘管有條件的所有權變更,它們仍可進一步使用。 如果集團內發生合格所有權變更,則在滿足某些條件的情況下,將保留結轉税款損失。 或者,在某些條件下,如果公司 自成立以來或至少自 合格所有權變更之前的第三年開始以來一直保持相同的業務運營,則可以根據申請保留税收損失結轉(堡壘).如果提出上述申請,且 所有權有條件變更後,該業務經營終止,則最新確定的税務虧損結轉(fortführungsgebundener Verlustvortrag)將會丟失。
漢堡財政法院已於2017年8月29日-2 K 245/17就第8c條第1段第2句KStG(其被取代的版本,現在:第8c段第1句KStG),即在超過50%的股份/投票權將被轉讓給新股東的情況下,沒收所有税收損失 ,提出上訴。上訴仍在審理中。目前尚不清楚聯邦憲法法院將於何時對此案做出裁決。根據德國法律文獻中的陳述,有充分的理由相信聯邦憲法法院可能得出結論,第8條第1款第2句KStG(其被取代的版本)不符合德國憲法。
截至2023年12月31日,我們有NOL結轉,用於德國公司税目的為1.96億歐元,用於貿易税目的為1.64億歐元。未來股份所有權的更改也可能觸發所有權更改,從而觸發第8c條KStG或第10a條GewStG限制。任何限制都可能導致結轉的全部税務經營虧損到期,然後才能使用 。因此,如果我們賺取淨應納税所得額,我們利用變動前淨營業虧損結轉減少德國所得税的能力可能會受到限制,這可能會導致我們未來的現金納税義務增加。
截至2023年12月31日,我們的美國子公司InflRx PharmPharmticals,Inc.有1296萬歐元或1430萬美元的NOL用於美國聯邦所得税 。轉讓或發行我們的股權可能會削弱或降低InflRx PharmPharmticals,Inc.在未來利用美國聯邦淨營業虧損結轉和某些其他税務屬性的能力。1986年修訂後的《國税法》第382節包含了一些規則,這些規則限制了一家公司利用其淨營業虧損和税收抵免結轉以及在所有權變更後數年內確認的某些固有虧損的能力。 所有權變更通常被定義為公司股票的所有權在三年滾動期間內增加50個百分點以上 的股東(直接,間接或建設性地)在相關的滾動三年期間內的任何時間,公司股票的5%或更多。如果發生所有權變更,第382條對所有權變更前NOL、抵扣和某些其他税收屬性的使用施加年度限制,以抵消所有權變更後獲得的應納税所得額。年度限額一般等於發生所有權變更的 個月有效的適用長期免税率與緊接所有權變更前的公司股票價值的乘積(受 的某些調整)。例如,這一年度限額可以進行調整,以反映前幾年任何未使用的年度限額,以及該年度某些已確認(或視為已確認)的內在損益。此外,第383節一般限制所有權變更後任何年度的税負金額,可通過所有權變更前税收抵免結轉或資本損失結轉來減少。不能保證以前的交易沒有導致該守則第382節規定的所有權變更,或者未來的交易不會導致所有權變更。即使後續交易不會導致所有權變更,也可能會大大增加我們未來所有權變更的可能性。股東出售我們的普通股 ,他們的利益可能與我們的利益不同,這可能會增加我們或我們的某個子公司 發生所有權變更的可能性。如果我們或我們的子公司已經或將要經歷所有權變更,可能會導致未來對我們的納税義務增加 。
-44-
我們 可能會在德國以外的司法管轄區徵税,這可能會增加我們的總税負
自 註冊以來,我們打算繼續在德國擁有我們有效管理的地方。因此,根據德國國家税法,我們將成為德國的税務居民。由於我們是根據荷蘭法律註冊成立的,因此根據荷蘭税法,我們也被視為荷蘭税務居民。然而,根據我們目前的管理結構和美國、德國和荷蘭的現行税法, 以及適用的所得税條約及其目前的解釋,就德意志聯邦共和國與荷蘭之間關於避免對2012年所得雙重徵税的公約或德國-荷蘭税收條約而言,我們應該只是德國的税務居民。
就德國-荷蘭税務條約而言,我們在德國的唯一税務居住權受德國-荷蘭税務條約(經不時修訂)中關於税務居住權的條款的適用。除其他國家外,《執行與税收條約有關的措施以防止税基侵蝕和利潤轉移的多邊公約》或《多邊税法》、德國和荷蘭等國也不應 影響德國-荷蘭税務條約關於税收居留的規定。
適用的税法、税務條約或其解釋可能會發生變化,包括MLI的選擇和保留。此外, 我們是否在德國擁有有效管理的地方,以及作為德國唯一的税務居民,在很大程度上是基於所有情況的事實和 程度的問題,而不是法律問題,事實和程度也可能改變。適用法律 或其解釋的變更以及適用事實和情況的變更(例如,董事會成員或 董事會會議召開地點的變更),或適用税務條約的變更,包括多邊貸款申請的變更,可能導致我們 成為德國以外司法管轄區的税務居民。因此,我們的整體有效所得税率和所得税 開支可能大幅增加,這可能對我們的業務、經營業績、財務狀況 和前景造成重大不利影響,從而可能導致我們的股價和交易量下降。
我們 認為,我們很可能在2021年、2022年和2023年為美國聯邦所得税目的的PFIC,並且我們可能在未來一個或 多個應課税年度成為PFIC。美國股東可能會在2023年以及未來 我們是PFIC的任何應納税年度受到不利的美國聯邦所得税後果的影響。
我們 認為,我們很可能在2021年、2022年和2023年成為美國聯邦所得税的PFIC,並且我們可能在一個或多個 未來應納税年度成為PFIC。此外,我們現在或將來可能直接或間接持有其他私人金融公司的股權。根據 守則,在適用有關子公司的某些審查規則後, (i)75%或以上的總收入由被動收入構成,或(ii)50%或以上的資產季度平均值 由產生或持有以產生被動收入的資產組成的任何應課税年度,我們將成為PFIC。被動收入包括股息、利息、某些非主動租金和特許權使用費以及資本收益。我們有可能在未來任何應納税年度成為PFIC ,原因之一是(i)我們擁有大量被動資產,包括可能產生被動 收入的現金和證券,(ii)為PFIC目的對我們產生非被動收入的資產進行估值,包括我們的無形資產,不確定 且可能隨時間而大幅變化;及(iii)我們的收入構成可能隨時間而大幅變化。
如果 我們在美國投資者持有普通股的任何應税年度是PFIC,則在美國投資者持有普通股的所有後續年度,我們將繼續被視為PFIC, 在美國投資者持有普通股的所有年度,即使我們不再滿足 PFIC資格的門檻要求,除非適用某些例外情況。此類美國投資者可能會受到不利的美國聯邦所得税後果的影響,包括(i)將處置所得收益的全部或部分視為普通收入,(ii)對此類收益和收取某些股息應用 遞延利息費用,以及(iii)遵守某些報告要求。
關於進一步討論,請參見“項目10。附加信息—5.税務—美國普通股持有人的重大美國聯邦所得税考慮 。
-45-
我們 可能會失去我們的外國私人發行人身份,這將要求我們遵守《交易法》的國內報告制度 ,並導致我們承擔大量的法律、會計和其他費用
We are a foreign private issuer and therefore we are not required to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act applicable to U.S. domestic issuers. If in the future we are not a foreign private issuer as of the last day of the second fiscal quarter in any fiscal year, we would be required to comply with all of the periodic disclosure, current reporting requirements and proxy solicitation rules of the Exchange Act applicable to U.S. domestic issuers. In order to maintain our current status as a foreign private issuer, either (a) a majority of our ordinary shares must be either directly or indirectly owned of record by non-residents of the United States or (b)(i) a majority of our directors and executive officers may not be United States citizens or residents, (ii) more than 50% of our assets cannot be located in the United States and (iii) our business must be administered principally outside the United States. If we were to lose this status, we would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. We may also be required to make changes in our corporate governance practices in accordance with various SEC and stock exchange rules. The regulatory and compliance costs to us if we are required to comply with the reporting requirements applicable to a U.S. domestic issuer may be significantly higher than the costs we would incur as a foreign private issuer. As a result, we expect that a loss of foreign private issuer status would increase our legal and financial compliance costs and would make some activities highly time consuming and costly. These rules and regulations could also make it more difficult for us to attract and retain qualified directors.
如果 我們支付股息,我們可能需要預扣税應支付給我們在德國和荷蘭的股份持有人的股息
We do not intend to pay any dividends to holders of our shares. However, if we do pay dividends, we may need to withhold tax on such dividends both in Germany and the Netherlands. As an entity incorporated under Dutch law any dividends distributed by us are subject to Dutch dividend withholding tax on the basis of Dutch domestic law. However, on the basis of the double tax treaty between Germany and the Netherlands, the Netherlands will be restricted from imposing dividend withholding tax if we continue to be a tax resident of Germany and our place of effective management is in Germany. However, Dutch dividend withholding tax is still required to be withheld from dividends if and when paid to Dutch resident holders of our shares (and non-Dutch resident holders of our shares that have a permanent establishment in the Netherlands to which their shareholding is attributable). As a result, upon a payment (or deemed payment) of dividends, we will be required to identify our shareholders in order to assess whether there are Dutch residents (or non-Dutch residents with a permanent establishment to which the shares are attributable) in respect of which Dutch dividend tax has to be withheld. Such identification may not always be possible in practice. If the identity of our shareholders cannot be determined, withholding of both German and Dutch dividend tax from such dividend may occur, upon a payment of dividends.
此外, 上述預扣税限制基於德國在多邊貸款倡議下的當前選擇和保留。如果德國 改變其MLI選擇和保留,我們可能無權享受德國和荷蘭之間的雙重徵税條約的任何好處, 包括預扣税限制,只要德國和荷蘭沒有就我們的税務居留權達成協議 德國和荷蘭之間的雙重徵税條約的目的,但以當局可能同意的範圍和方式 除外。因此,我們在德國 與荷蘭之間未達成此類協議的期間內分派的任何股息,都可能在德國和荷蘭繳納預扣税。
此外,荷蘭議會正在審議一項擬議的法律,即《緊急法案》有條件離職股息税(Spoedwet condition e indafrekening dividenddbelasting)如果我們不再是荷蘭税務居民,而是成為 非歐盟或歐洲經濟區成員國的司法管轄區的税務居民,當該司法管轄區不滿足某些條件時,將對某些視為分配徵收股息預扣税 (出口)税,可能具有追溯效力。在某些情況下,當股東無權獲得豁免時,我們有權 向其收回税款。
-46-
從2024年1月1日起,向低税率或非合作司法管轄區的某些相關實體分配的股息 將來可能會對股息徵收額外荷蘭預扣税
我們 沒有計劃定期支付普通股股息。然而,如果我們支付股息,根據現行荷蘭税法,我們向股份持有人支付的股息 可能會根據荷蘭股息 預扣税法(1965年後的濕潤評論), unless a domestic or treaty exemption or reduction applies; see “ITEM 10. ADDITIONAL INFORMATION - 5. Taxation - Material Dutch Tax Considerations.” As of January 1, 2024, a Dutch conditional withholding tax will be imposed on dividends paid to related entities in jurisdictions that have a corporate income tax rate below 9% (low-tax jurisdiction) or jurisdictions that are included on the EU’s blacklist of non-cooperative jurisdictions (non-cooperative jurisdictions for tax purposes). In addition, the conditional withholding tax on dividends may also apply in situations where artificial structures are put in place with the main purpose or one of the main purposes to avoid the conditional withholding tax or in the event of a hybrid mismatch. The conditional withholding tax will be imposed at the highest Dutch corporate income tax rate in effect at the time of the distribution (currently 25.8%). The conditional withholding tax on dividends will be reduced, but not below zero, by any regular Dutch dividend withholding tax withheld in respect of the same dividend payment. As such, based on the currently applicable rates, the overall effective tax rate of withholding the regular dividend withholding tax and conditional withholding tax will not exceed the highest corporate income tax rate in effect at the time of the distribution (currently 25.8%). As of January 1, 2024, the withholding tax rate on dividends paid to shareholders that are (A) entities related (格列耶德)和(B)(i)為税務目的在低税率 州或非合作司法管轄區成立,(ii)混合實體或反向混合實體,或(iii)為避免其他實體應繳税款 而介入,可從15%上調至最高公司税率(目前為25.8%)。
我們 是一家荷蘭上市有限責任公司。我們股東的權利不同於受美國司法管轄區法律管轄的公司股東的權利 ,並且可能無法以在美國司法管轄區註冊所提供的類似方式保護投資者
我們 是一家有限責任公司(Naamloze Vennootschap根據荷蘭法律組織的。我們的公司 事務受我們的公司章程和管理在荷蘭註冊成立的公司的法律管轄。然而, 無法保證荷蘭法律將來不會改變,或者它將以美國公司法原則下提供的類似方式保護投資者,這可能對投資者的權利造成不利影響。
股東的權利和董事的責任可能與受美國法律管轄的公司中股東和董事會成員的權利和義務不同。在履行職責時,荷蘭法律要求我們的執行人員和董事會 考慮我們公司、股東、員工和其他利益相關者的利益,在所有情況下都應適當 遵守合理和公平的原則。這些當事方中的某些當事方可能會有 與您作為股東的利益不同的利益,或除了您作為股東的利益之外。
我們的公司章程或荷蘭公司法的條款 可能會阻止對我們有利的收購出價,並阻止、 推遲或挫敗任何更換或罷免我們董事會成員的企圖
根據荷蘭法律,在荷蘭法律和荷蘭判例法規定的範圍內,可能並允許採取各種保護措施。我們的治理 安排包括幾項條款,這些條款可能會使收購我們公司變得更加困難或吸引力降低。 在這方面,我們的股東大會根據荷蘭法律授予一個獨立基金會或保護性基金會的權利, 根據我們與該基金會簽訂的看漲期權協議或看漲期權協議收購優先股。 認購期權協議項下的認購期權應具有連續性,可多次重複行使。
-47-
如果 保護基金會根據認購期權協議行使認購期權,則保護基金會以外的其他人持有的最多 本公司已發行股本的100%的優先股將被髮行給保護基金會。這些 優先股將被髮行給保護基金會,並有義務支付最多25%的面值。 為使保護基金會為優先股的發行價融資,保護基金會 預計將與銀行達成融資安排。作為向銀行擔保融資的替代方案,受荷蘭法律適用的限制 的限制,看漲期權協議規定,保護性基金會可要求我們提供,或促使我們的子公司 提供,為保護基金提供足夠的資金,使其能夠履行付款義務(或部分)現金 和/或從我們的利潤和/或儲備中收取相當於付款義務(或部分)的金額,以履行該 付款義務。
保護基金會的章程規定,它將不時促進和保護公司、與公司相關的業務 和公司利益相關者的利益,並抑制可能威脅 公司或與其相關的業務的戰略、連續性、獨立性和/或身份的可能影響,以至於可以認為這是對上述利益的損害。這些影響可能包括第三方收購我們相當比例的普通股,宣佈對我們普通股的主動公開收購,股東積極主義,其他 控制權集中於我們普通股或任何其他形式的不當壓力,以改變我們的戰略政策。保護性 基礎的結構應獨立於我們運行。
如果 保護性基金會行使其看漲期權,則根據此發行的優先股將根據 支付最多其面值25%的義務發行。本公司股份的投票權基於面值,由於本公司預計 本公司普通股的交易量將大大超過面值,因此,以面值25%發行的優先股可以以與本公司普通股價格相比大幅降低的價格獲得 重大投票權,因此可用作防禦 措施。這些優先股將具有比普通股更好的清算和股息優先權,並將按預先確定的比率累積現金股息 。保護性基金會預計將要求我們取消其優先股,一旦 對公司及其利益相關者的感知威脅已經消除或充分減輕或中和。然而,在遵守上述相同限制的情況下 ,保護性基金會將繼續有權在未來行使看漲期權,以應對 不時對我們、我們的業務和我們的利益相關者的利益的新威脅。
此外,我們的公司章程的某些條款可能會使第三方更難獲得我們的控制權或 改變我們的董事會。這些規定包括:一項規定,我們的董事是根據我們的董事會準備的具有約束力的 提名任命的,該提名只能由代表我們已發行股本 50%以上的三分之二多數票否決;一項規定,只有股東大會以代表50%以上的三分之二多數票才能罷免董事我們已發行股本(除非董事會提議刪除,在此情況下, 簡單多數票即可足夠);以及要求某些事項(包括對 公司章程的修訂)僅可提交股東根據董事會的建議進行表決。
我們 沒有義務也不遵守DCGC的所有最佳實踐規定。這可能會影響您作為股東的權利
我們 是一家荷蘭上市公司,有限責任(Naamloze Vennootschap),我們受DCGC約束。DCGC包含 原則和最佳實踐條款,用於規範董事會和股東(如 股東大會)之間的關係。DCGC基於"遵守或解釋"原則。因此,公司必須在荷蘭提交的年度報告中披露 是否遵守DCGC的規定。如果他們不遵守這些 條款(例如,由於納斯達克要求衝突),公司需要給出不遵守的原因。
DCGC適用於在政府認可的證券交易所上市的所有荷蘭公司,無論是在荷蘭還是其他地方,包括 納斯達克。我們不遵守DCGC的所有最佳實踐條款。這可能會影響您作為股東的權利,並且您可能 無法獲得與完全符合DCGC的荷蘭公司股東同等級別的保護。
-48-
美國民事責任索賠可能無法對我們執行
我們 是根據荷蘭法律註冊成立的,總部設在德國。我們幾乎所有的資產都位於美國境外。我們的大多數董事和執行官居住在美國境外。因此, 投資者可能無法在美國境內向此類人士送達訴訟程序,或無法在美國法院對他們 或我們執行強制執行,包括根據美國聯邦證券法民事責任條款作出的判決 。
美國和荷蘭之間沒有關於相互承認和執行民事和商事判決(仲裁裁決除外)的條約。因此,美國任何聯邦或州法院 基於民事責任作出的支付款項的最終判決(無論是否僅基於美國聯邦證券法)在荷蘭都不可執行 ,除非相關索賠在荷蘭有管轄權的法院重新提出。然而,根據現行慣例, ,荷蘭法院通常在遵守某些程序要求的前提下,在不對相關索賠的案情進行審查的情況下,作出相同判決,如果這種判決(i)是最終判決,並且是由 對相關荷蘭公司或荷蘭公司(視情況而定)確立其管轄權的法院作出的,根據國際公認的管轄權理由,(二)沒有違反適當程序原則(貝霍利克 雷赫茨普爾格), (iii)不違反荷蘭的公共政策,並且(iv)不違反(a)荷蘭法院在同一當事方之間的爭端中作出的先前判決,或(b)外國法院在同一當事方之間的爭端中作出的先前判決,涉及同一主題事項並基於同一訴因,前提是這種事先判決能夠在荷蘭得到承認。荷蘭法院可以拒絕承認和執行懲罰性賠償金或其他裁決。
此外, 荷蘭法院可以減少美國法院授予的損害賠償金額,並僅在賠償實際損失或損害所必需的情況下才承認損害賠償。美國法院在荷蘭的判決的執行和承認僅受《荷蘭民事訴訟法》的規定管轄 。基於上述情況,無法保證美國投資者 能夠執行在美國法院就民事和商業事項獲得的任何判決,包括根據美國聯邦證券作出的判決。
美國和德國沒有條約規定相互承認和執行民事和商事案件判決 。因此,美國法院作出的最終付款判決或宣告性判決,無論 是否完全基於美國證券法,都不會自動在德國得到承認或執行。如果德國法院認為美國法院不具有管轄權或判決 違反德國公共政策原則,則德國法院可以拒絕 承認和執行美國法院作出的判決。例如,判決懲罰性賠償的判決在德國通常是不可執行的 。德國法院可以減少美國法院授予的損害賠償金額,並僅在賠償實際損失或損害所必需的情況下才承認損害賠償。
此外,在德國法院針對我們、我們的董事、我們的執行官和本文中提到的專家提起的、以執行 基於美國聯邦證券法的責任的訴訟可能會受到某些限制。特別是,德國法院一般不會裁決懲罰性賠償。德國的訴訟也受與美國規則不同的程序規則的約束,包括 證據的獲取和可接受性、訴訟程序的進行和費用的分配。德國程序法沒有 規定審前文件披露,德國也不支持根據1970年《海牙證據公約》 進行審前文件披露。在德國的訴訟程序必須以德語進行,原則上,提交給法院的所有文件都必須翻譯成德語。由於這些原因,美國投資者可能難以根據美國聯邦證券法的民事責任條款,向德國法院起訴 針對我們、我們的董事、我們的執行人員 和本年度報告中提到的專家。
由於缺乏上述條約,美國投資者可能無法對我們或本文件中提到的董事、執行官或 某些專家(如果他們居住在荷蘭、德國或美國以外的其他國家或擁有資產)強制執行美國法院在民事和商事案件中獲得的任何判決,包括根據美國聯邦證券法作出的判決。
-49-
| 6. | 一般風險因素 |
一般的經濟、政治和社會狀況。
我們的業務和經營業績可能會受到金融市場中斷 、政治和監管政策的變化以及總體經濟狀況的不利影響
General economic, political and social conditions affect the United States, Europe and other global markets and our business. In particular, U.S., European and other global markets, as well as our access to financing, may be affected by factors, including economic growth or its sustainability, persistent inflation, supply chain disruptions, employment levels, work stoppages, labor shortages and labor disputes, labor costs, wage stagnation, energy prices, oil, gas and fuel prices, fluctuations or other significant changes in both debt and equity capital markets and currencies, liquidity of the global financial markets, the growth of global trade and commerce, trade policies, the availability and cost of capital and credit (including as a result of increased interest rates) and investor sentiment and confidence. Additionally, global markets may be adversely affected by the current or anticipated impact of cyber incidents or campaigns, military conflict, including the Russia-Ukraine conflict as well as the Hamas-Israel conflict and rising tensions between China and Taiwan and the relationship between China and the United States, or other geopolitical uncertainty and instability. The ongoing spread of variants of infectious diseases, such as the COVID-19 virus, may interrupt, or delay, our clinical trial activities, regulatory reviews, manufacturing activities and supply chain. The COVID-19 outbreak delayed enrollment in our clinical trials due to prioritization of hospital resources towards the outbreak or other factors, and some patients may be unwilling to enroll in our trials or be unable to comply with clinical trial protocols if quarantines impede patient movement or interrupt healthcare services, which would delay our ability to conduct clinical trials or release clinical trial results and could delay our ability to obtain regulatory approvals and commercialize our product candidates. Any sudden or prolonged market downturn in the United States or elsewhere could adversely affect our business, results of operations and financial condition, including capital and liquidity levels.
為解決環境和其他目標而採取的法律、 監管或市場措施可能會對我們的業務或運營產生負面影響
美國、歐洲和其他地方的監管 和立法機構繼續關注環境、社會和治理,或ESG, 事項,包括日益關注與氣候變化、温室氣體排放、碳税、排放交易計劃、 可持續製造、人權和公平事項有關的事項,以及與上述事項有關的披露,其中許多政策可能含糊不清, 不一致,動態或衝突。我們預計將面臨與此類新的或不斷變化的法律或監管要求相關的限制、合規成本、法律成本和費用增加。此外,遵守任何此類法律或法規要求將需要 我們投入大量時間和精力處理這些問題。此外,如果此類法律法規的解釋或應用與我們的實踐不符,我們仍可能受到處罰或潛在訴訟 。此外,我們受到媒體、股東、活動家和其他利益相關者對氣候變化、社會和可持續發展問題的關注, 這可能會對我們的聲譽或投資者信心產生負面影響。
我們 可能無法維持足夠的保險,以應對潛在訴訟或其他風險
We may not be able to maintain sufficient insurance on commercially reasonable terms or with adequate coverage levels against potential liabilities we may face in connection with potential claims, which could have a material adverse effect on our business. We may face a risk of loss from a variety of claims, including related to securities, antitrust, contracts, cybersecurity, fraud and various other potential claims, whether or not such claims are valid. Insurance and other safeguards might only partially reimburse us for our losses, if at all, and if a claim is successful and exceeds or is not covered by our insurance policies, we may be required to pay a substantial amount in respect of such successful claim. Certain losses of a catastrophic nature, such as losses arising as a result of wars, earthquakes, typhoons, terrorist attacks or other similar events, may be uninsurable or may only be insurable at rates that are so high that maintaining coverage would cause an adverse impact on our business, our investment funds and their investments. In general, losses related to terrorism are becoming harder and more expensive to insure against. Some insurers are excluding terrorism coverage from their all-risk policies. In some cases, insurers are offering significantly limited coverage against terrorist acts for additional premiums, which can greatly increase the total cost of casualty insurance for a property. As a result, we, our products and their investments may not be insured against terrorism or certain other catastrophic losses.
-50-
籌集 額外資本可能會導致股東稀釋、限制我們的運營或要求我們放棄對我們的技術 或候選產品的權利
我們 預計我們的費用可能會隨着業務的擴大而增加。如果我們通過 發行普通股、可轉換證券或其他股本證券籌集額外資本,則您的所有權權益可能會被稀釋,而這些證券的條款 可能包括清算或其他優惠以及反稀釋保護,這些條款可能會對您作為普通股股東的權利造成不利影響 。此外,債務融資(如有)可能導致固定付款義務,並可能涉及包含限制我們採取具體行動的限制性契約的協議 ,例如產生額外債務、進行資本支出 、建立留置權、贖回股份或宣佈股息,這可能對我們開展業務的能力產生不利影響。 此外,獲得融資可能需要我們的管理層花費大量時間和精力,並且可能會將不成比例的 注意力從日常活動轉移出去,這可能會對我們的管理層監督候選產品開發的能力產生不利影響 。
如果 我們通過與第三方的合作或營銷、分銷或許可安排籌集額外資金,我們可能不得不 放棄對我們的技術、未來收入來源或候選產品的寶貴權利,或以可能不利於我們 的條款授予許可。如果我們無法在需要時籌集額外資金,我們可能會被要求延遲、限制、減少或終止 產品開發或未來的商業化努力,或授予開發和銷售我們本來 更願意自己開發和銷售的候選產品的權利。
我們 面臨着巨大的競爭,這可能導致其他人比我們更成功地發現、開發或商業化產品 ,並減少或消除我們的商業機會
如果我們的競爭對手開發和商業化的產品比我們或任何未來合作者可能開發的任何產品更安全、更有效、 副作用更少或更不嚴重、更方便或更便宜, 的商業機會可能會減少或消除。我們的競爭對手也可能在我們或任何未來合作者 能夠獲得我們產品的批准之前獲得FDA或其他上市批准,這可能導致我們的競爭對手在我們或任何未來合作者能夠進入市場之前建立強大的市場地位。
我們現有的和未來潛在的競爭對手 在研發、 製造、臨牀前測試、進行臨牀試驗、獲得上市許可和上市許可產品方面的財務資源和專業知識都比我們 要多得多,並且可能能夠降低他們銷售產品的價格。製藥和生物技術行業的合併和收購 可能會導致更多的資源集中在少數競爭對手手中。規模較小或處於早期階段的公司 也可能成為重要的競爭對手,特別是被大型和成熟的 公司收購或通過與它們的合作安排。這些競爭對手還在招聘和留住合格的科學和管理人員、建立 臨牀試驗中心和臨牀試驗患者登記,以及獲取補充或必要的技術方面與我們競爭。
新產品的開發和商業化競爭激烈。我們預計,我們和任何未來合作者將面臨來自全球主要製藥公司、專業製藥公司和生物技術公司的重大 競爭, 我們或任何未來合作者將來可能尋求開發或商業化的任何候選產品。例如, 其他製藥公司可能開始開發針對與vilobelimab相同適應症的候選產品, 包括PG和嚴重COVID—19或我們可能針對的任何其他適應症。有關 我們經營的競爭環境的詳細分析,請參見“項目4。公司信息—2.業務概述—競爭"。
-51-
如果 針對我們或我們的任何合作伙伴成功提起任何產品責任訴訟,我們可能會承擔重大責任 並可能被要求限制我們候選產品的商業化
我們 面臨着與在重症患者中測試我們候選產品相關的產品責任訴訟的固有風險,如果我們的候選產品獲得監管機構批准並投入市場, 將面臨更大的風險。產品責任 索賠可能會由參加我們臨牀試驗的參與者、患者、醫療保健提供者或 使用、管理或銷售我們未來批准的任何產品的其他人向我們或我們的合作伙伴提起。如果我們不能成功地為自己辯護,以應對任何此類 索賠,我們可能會承擔重大責任。
如果 我們的任何候選產品被批准進行商業銷售,我們將高度依賴於消費者對我們的看法以及我們產品的安全性 和質量。如果我們受到與疾病相關的負面宣傳或因患者使用或誤用我們的產品或其他公司分銷的任何類似產品而導致的其他 不良反應,我們可能會受到不利影響。
雖然 我們保留產品責任保險,但該保險可能無法完全涵蓋我們可能產生的潛在責任。任何產品責任訴訟或其他程序的成本 ,即使以我們為受益方的方式解決,也可能是巨大的。如果我們將任何獲得市場批准的產品商業化,我們將需要增加 我們的保險範圍。此外,保險範圍正變得 越來越昂貴和難以獲得。如果我們無法以可接受的成本維持足夠的保險範圍,或 以其他方式防範潛在的產品責任索賠,這可能會阻止或抑制我們候選產品的開發和商業生產和 銷售,這可能會損害我們的業務、財務狀況、經營業績和前景。
我們 可能無法評估未來收購所涉及的重大風險
我們 將來可能會收購與我們的運營和客户需求互補的公司、產品和/或平臺。作為流程的一部分,我們可能會進行業務、法律和財務盡職調查,以識別和評估任何特定 交易中涉及的重大風險。儘管作出了這些努力,我們可能無法確定或評估所有此類風險。因此,可能無法實現任何給定收購的預期優勢 。如果我們未能從一項或多項收購中識別出某些重大風險,我們可能面臨 重大成本,我們的業務可能受到負面影響。
網絡 事件或IT系統中的其他故障可能導致信息被盜、數據損壞和業務運行嚴重中斷
We utilize information technology, or IT, systems and networks to process, transmit and store electronic information in connection with our business activities. As use of digital technologies has increased, cyber incidents, including deliberate cyber-attacks and attempts to gain unauthorized access to computer systems and networks, have increased in frequency and sophistication, both internal and those provided by third-party service provider. These threats pose a risk to the security of our systems and networks, the confidentiality and the availability and integrity of our data. The wars in Europe and in the Middle East, as well as the tensions between China and Taiwan may also result in heightened cybersecurity risk across our networks and platforms. We have implemented processes, procedures and internal control to mitigate cybersecurity risks and cyber intrusions and rely on industry accepted security measures and technology to securely maintain confidential and proprietary information maintained on our information systems. However, these measures, as well as our increased awareness of the nature and extent of a risk of a cyber-incident, do not guarantee that a cyber-incident will not occur or that our financial results, operations or confidential information will not be negatively impacted by such an incident, especially because cyber threats change frequently or are not recognized until launched and because cyber incidents can originate from a wide variety of sources. Similarly, there can be no assurance that our collaborators, CROs, third-party logistics providers, distributors and other contractors and consultants will be successful in protecting our clinical and other data that is stored on their systems.
-52-
如果 網絡事件發生並導致我們的運營中斷或 數據的任何破壞或丟失、損壞或不可用,則可能導致機密信息(包括商業機密、其他知識產權或 財務信息)的丟失或盜用,並嚴重擾亂我們的開發計劃和業務運營,其中任何一個都可能導致我們的研究和其他進一步開發和商業化的重大 延誤或挫折。例如, 已完成、正在進行或未來的臨牀試驗中的臨牀試驗數據丟失可能導致我們的監管審批工作延遲,並 顯著增加我們恢復或複製數據的成本。最後, 人工智能技術的使用有了重大的演變和發展,例如人工生成聊天機器人。目前,我們無法完全確定此類不斷髮展的技術對我們業務的影響或網絡安全風險。
任何 此類網絡事件或數據的破壞或丟失都可能對我們的業務和前景產生重大不利影響。此外,我們 可能會因網絡事件(包括網絡攻擊 或其他數據安全漏洞)而遭受聲譽損害或面臨訴訟或不利監管行動,並可能會產生大量額外費用以實施進一步的數據保護措施。
與數據隱私相關的 法律和監管環境正變得越來越嚴格,這可能會導致額外成本或 我們處理個人信息的方式發生變化,如果未能遵守此類法律或法規,或未能以其他方式保護我們擁有或控制的個人 數據,可能會導致罰款、訴訟或其他處罰以及聲譽損失
我們 受與隱私、數據保護和信息安全相關的法律、法規和合同義務的約束,包括(I) 2018年5月25日生效的《歐盟一般數據保護條例》,該法規對違規行為的處罰比以前的歐洲數據保護法更重,最高可處以2000萬歐元或全球年營業額4%的罰款,以及(Ii)2020年1月1日生效的《加州消費者隱私法》,其中規定了對違規行為的民事處罰,以及針對數據泄露的私人訴權,預計這將增加數據泄露訴訟。
隨着隱私、數據保護和信息安全法律的演變以及實施、解釋和應用,我們的合規成本可能會 增加,特別是在確保適當的數據保護和數據傳輸機制到位的情況下。此外,遵守此類義務和法規可能會對我們當前和計劃中的隱私和信息安全做法、我們個人數據的收集、使用、共享、保留和保護,以及我們當前和計劃中的業務活動和運營產生重大影響。不遵守此類義務或法規可能會導致罰款、訴訟或其他處罰,並對我們的聲譽造成不利影響。
如果我們對財務報告的內部控制失效,可能會導致我們財務報表中的重大錯報,導致投資者對我們報告的財務和其他公開信息失去信心,並對我們普通股的交易價格產生負面影響。
有效的財務報告內部控制對於我們提供可靠的財務報告是必要的,再加上充分的披露 控制和程序旨在防止欺詐。任何未能實施所需的新的或改進的控制措施,或在實施過程中遇到困難,都可能導致我們無法履行我們的報告義務。2002年《薩班斯-奧克斯利法案》第404條要求上市公司管理層制定和實施財務報告內部控制並評估其有效性。 如果我們未能設計和操作有效的內部控制,可能會導致我們財務報表中的重大錯報, 損害我們的收入能力,導致投資者對我們財務報表的可靠性失去信心,並使我們受到監管審查和制裁,這反過來可能損害我們普通股的市場價值。
銀行業 系統風險
我們 定期在第三方金融機構的現金餘額超過FDIC或其他類似的外國(即, 德國)存款保險限額。如果我們維持現金餘額的任何銀行或金融機構因影響銀行系統和金融市場的財務狀況而在未來進入破產程序或破產,我們獲取我們現有現金、現金等價物和投資或利用我們現有信貸額度的能力可能會受到威脅,並可能對我們的業務和財務狀況產生重大 不利影響。
-53-
項目4.關於公司的信息
| A. | 公司的歷史和發展 |
我們 是一家生物製藥公司,通過針對補體系統進行抗炎治療。我們通過應用我們的專利技術來發現、開發和商業化補體 激活因子C5a及其受體C5aR的一流、高效和特定的抑制劑。補體系統是先天免疫系統不可或缺的一部分,保護身體,例如通過識別和清除細菌、病毒和其他感染性病原體,統稱為病原體。補體末端激活,也指C5轉換酶對C5的裂解,導致C5a的釋放, 通過其受體C5aR起作用。此外,補體的最終激活,即C5a從C5的裂解,也可以通過存在於全身但不被認為是補體系統的一部分的自然產生的酶,通過外在途徑直接實現。在我們的候選治療產品中,我們針對C5a及其受體C5aR,選擇性地抑制由C5a/C5aR激活引發的各種自身免疫和其他炎症性疾病中觀察到的強大炎症反應。
我們的主要候選產品Viloblimab是一種新型靜脈注射的一流抗C5a單抗,它選擇性地與遊離C5a結合,並在多種臨牀環境中展示了修改疾病的臨牀活性和耐受性。我們正在並一直在為一系列補體介導的疾病開發Viloblimab,這些疾病具有重大的未得到滿足的醫療需求。這些包括膿皮病 壞疽,或PG,一種慢性炎症性皮膚病,我們在2023年11月開始了第三階段研究,第一名患者在2023年11月接受治療,新冠肺炎患者的危重疾病的治療,我們在2022年3月完成了第三階段研究,隨後 在2023年4月獲得食品和藥物管理局批准的EUA。
截至2023年11月,我們還在開發用於皮膚鱗狀細胞癌(CSCC)的維羅布利單抗,我們正在進行一項II期臨牀研究。我們之前也曾在其他疾病中使用viloblimab進行第二階段研究,包括化膿性汗腺炎或HS,一種慢性衰弱的全身性炎症性皮膚病和ANCA相關性血管炎,或AAV,一種罕見且危及生命的自身免疫性疾病。
我們還在開發INF904,一種口服的C5aR小分子抑制劑,我們已經完成了對健康志願者的第一階段研究,並計劃在未來啟動第二階段的臨牀研究。INF904是一種很有前途的候選產品,可用於多種炎症疾病領域,在這些領域,口服治療藥物不可用或不能滿足醫學上的需求。INF904的開發最初將針對慢性自發性蕁麻疹(CSU)和HS。
我們 還在開發IFX002,這是一種用於viloblimab的生命週期管理產品,目前處於高級臨牀前階段。
我們的法律和商業名稱是InflRx N.V,我們於2017年6月6日根據荷蘭法律註冊成立,我們的總部 已在耶拿當地法院註冊,位於德國耶拿。InflRx於2007年由Niels Riedemann教授和郭仁峯教授在德國耶拿創立,前身為InflRx GmbH。我們在美國的加工服務代理是InflRx製藥公司,位於密歇根州安娜堡南瓦格納路600號,郵編:48103。我們的主要執行辦公室和實驗室位於Winzerlaer。2,07745德國耶拿,電話:(+49)3641 508180。我們在德國的普蘭格-馬丁斯瑞德(慕尼黑)和美國密歇根州的安娜堡設有更多的辦事處,我們在那裏也有實驗室。
我們 總共僱傭了66名員工,其中22名擁有醫學博士學位。或博士度我們的管理團隊在補體研究、臨牀研究和生物製藥行業領域擁有豐富的經驗。我們的首席執行官和創始人,教授(博士)Niels Riedemann, 和我們的首席科學官兼創始人郭仁峯教授有超過20年的補體研究經驗, 廣泛發表了關於C5a及其受體的作用和功能的文章。我們的首席財務官Thomas Taapken博士在過去的19年中曾擔任過各種歐洲私營和上市生物技術公司的行政職務和董事會,在生物製藥和風險投資行業擁有超過25年的管理經驗。我們的首席醫療官Camilla Chong博士 在全球製藥行業擁有25年的臨牀開發、醫療事務、臨牀運營、 監管和藥物警戒以及新藥上市等領域的經驗。
-54-
SEC維護一個網站,其中包含有關發行人(如我們)的報告和其他信息,這些信息以電子方式與SEC存檔。該網站的 地址為www.sec.gov。我們的網站可在www.inflarx.de找到。本公司網站上的信息 未以引用方式納入本年報,閣下不應將本公司網站上的信息視為本年報的一部分。
| B. | 業務 概述 |
概述
C5a 是補體系統的中樞介質,因此是先天免疫系統的關鍵組成部分。補體系統最突出的作用 是通過幾種機制幫助身體防禦入侵的微生物,包括 快速創建炎症環境和產生直接殺死病原體和將免疫細胞募集到 感染部位的因子。補體系統的激活最終導致C5a通過從C5切割而產生。C5a通過吸引和強烈激活中性粒細胞以及通過使許多不同的細胞類型產生 促炎分子來創建 炎症環境。這類炎症通常通過幫助抵抗感染而有益於身體,但C5a的過度或不受控制的生成(因為它發生在某些疾病中)會對身體自身組織造成嚴重損害,從而導致 許多自身免疫性疾病和炎性疾病的病理生理學。
雖然 C5a在炎症中的作用模式已經得到了深入的研究和證實,開發一種 能夠完全阻斷C5a同時保留關鍵的先天防禦機制的高度特異性抗體,但膜攻擊複合物(MAC)的形成一直是一個挑戰。因此,目前還沒有批准的專門針對C5a的藥物。我們鑑定了抗體,包括 我們的先導產品候選vilobelimab,這些抗體有效且選擇性地結合C5 a在 C5 a從C5上切割後形成的構象表位,完全阻斷C5 a而不損害補體系統的重要上游功能,以及 MAC形成。
與其配體C5a不同,C5aR也可以被小分子抑制。通常認為,使用 抗體阻斷C5a提供了一種快速、完整和安全的控制C5a誘導的炎症的方法。 C5aR小分子抑制劑的優點是它可以口服給藥,從而為患者提供廣泛、長期的給藥方便,特別是用於 慢性疾病的治療。
通過 我們的內部藥物研發工作,我們確定了一種有效的C5a受體抑制劑INF904,我們認為它是一個有希望的候選藥物 。我們目前正在開發INF904,一種針對C5aR受體的口服低分子量候選藥物。我們 計劃針對補體介導的慢性自身免疫和炎症性疾病,患者需要口服小分子。
考慮到 阻斷C5a和C5aR的不同優勢,我們認為C5a和C5aR阻斷劑的開發是可能的 ,並可能有助於解決更廣泛的C5a/C5aR分子信號傳導軸相關疾病。
基於 廣泛的抗炎特性,我們目前正在開發用於多種疾病的抗C5a先導抗體和低分子量化合物 INF904。概述可在下面的管道描述中找到。
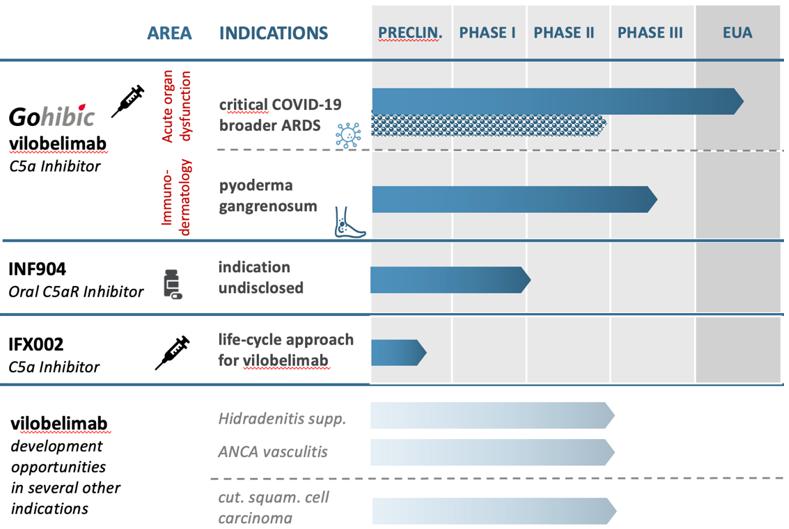
-55-
Vilobelimab 用於治療PG
我們正在開發用於治療PG的vilobelimab。 PG是一種罕見的, 慢性炎症形式的嗜酸性皮膚病,其特徵是中性粒細胞在受影響的皮膚區域的積聚。vilobelimab 被美國FDA和歐洲EMA授予用於治療PG的孤兒藥指定以及FDA的快速 指定。經過一系列與 這個FDA關於我們成功進行的II期臨牀研究的結果 以及我們的進一步開發計劃向潛在的BLA提交, 2023年, 宣佈vilobelimab治療潰瘍性前列腺癌的III期研究開始 。2023年11月,我們宣佈入組該研究的首例患者。
Vilobelimab 用於治療重度COVID—19
2023年4月,我們從FDA收到了GOHIBIC(vilobelimab)的EUA,用於治療重症、有創機械通氣 COVID—19患者。EUA得到了先前公佈的多中心III期PANAMO試驗結果的支持,該試驗證明28天全因死亡率相對降低了23.9%。隨後,2023年6月,我們根據EUA開始在美國商業化GOHIBIC(vilobelimab) 。我們的SARS—CoV—2誘導的膿毒性急性呼吸窘迫綜合徵(或ARDS)MAA申請,接受 IMV或ECMO治療維洛貝利單抗,正在接受歐洲人用藥品委員會(CHMP)的監管審查,該程序適用於歐盟所有27個成員國。為支持我們的發展,我們收到了德國聯邦政府在2021年10月至2023年6月期間授予的總計3330萬歐元 的贈款。
Vilobelimab 用於治療化膿性汗腺炎或HS
We have been developing vilobelimab for the treatment of HS. After having failed to meet our primary endpoint in an international Phase IIb study in 2019, in a post-hoc analysis of the study data we showed multiple signals of efficacy for the vilobelimab high dose group compared to the placebo group, demonstrating significant reductions in all combined inflammatory lesions, reductions of draining tunnels, or dTs, and reductions of the IHS4 score. Subsequently, we had several interactions with the FDA with the goal of agreeing on the possible design of a pivotal Phase III program for vilobelimab for the treatment of HS. Following the advice received in a Type A meeting with the FDA, in the fourth quarter of 2021, we submitted a full clinical trial protocol for the planned clinical Phase III trial of vilobelimab in HS and in January 2022 we initiated a randomized, double-blind, placebo-controlled, multi-center pivotal Phase III study to determine efficacy and safety of vilobelimab in patients with moderate to severe HS and active dTs with a modified primary clinical endpoint called m-HiSCR. Subsequently, the FDA provided conflicting advice to us, which was subsequently corrected, but in the meantime, we had halted the Phase III clinical program and are currently evaluating next steps regarding the development of vilobelimab in HS. Based on the logistical and financial effort necessary to successfully complete such a Phase III development program, we are assessing various future possible options to further this development, including collaborations with pharmaceutical partners.
Vilobelimab 用於治療ANCA相關血管炎(AAV)
我們 還開發了用於治療AV的vilobelimab。我們的vilobelimab治療AAV的臨牀開發策略首先集中於 急性疾病的AAV患者,我們認為vilobelimab有可能成功誘導緩解,並減少或消除 對高劑量皮質類固醇(HDCS)治療的需求,並提供改善的安全性特徵。我們還打算關注 誘導緩解的速度和降低腎臟替代和腎功能不全的發生率。在2021年成功完成兩項II期研究後,我們目前正在評估vilobelimab在AAV中開發的後續步驟。基於成功完成關鍵III期開發計劃所需的後勤和 財政努力,我們目前正在評估可能的 選項,以推進這一開發,包括與製藥合作伙伴的合作。
抗Ca5R 抑制劑INF904
為了擴大我們抗C5a/C5aR技術的廣度,我們還在開發INF904,這是一種靶向C5aR受體的候選產品。 在INF904中,我們發現了一種小分子C5aR抑制劑,在臨牀前研究中,該抑制劑表現出優於唯一獲批的C5aR抑制劑阿瓦可潘的潛力。與阿瓦可潘相比,INF904在動物(包括非人靈長類動物)中提供了更高的血漿暴露量,並且 在倉鼠中性粒細胞減少症模型中改善了抑制活性。此外,與阿伐可潘相反,體外實驗 顯示INF904對細胞色素P450酶3A4/5(CYP 3A4/5)的抑制顯著降低。INF904在幾種臨牀前疾病模型中顯示出抗炎治療作用的潛力。2024年1月,我們宣佈了一項在健康志願者中進行的單次和多次 遞增劑量研究的積極結果。我們計劃在未來開展II期臨牀研究。INF904是一種有前途的 候選產品,可用於炎症的幾個疾病領域,這些領域沒有口服治療藥物 ,或者儘管有其他治療方法可用,但仍存在醫療需求。
-56-
INF904治療CSU
我們還打算開發INF904用於治療CSU。CSU是一種使人衰弱且不可預測的 皮膚病,其特徵是強烈瘙癢的蕁麻疹/風團和血管性水腫。這種慢性疾病的負擔很高,並影響睡眠, 心理健康,生活質量和生產力,由於缺勤和工作。據估計,CSU影響全球約4000萬人 。據報道,C5a水平升高,C5a是肥大細胞和嗜鹼性粒細胞的主要激活物,被認為是C5a發病機制的重要貢獻者。此外,研究表明,CSU中補體激活(包括C5a)可 導致組胺釋放。目前的治療是有限的,並且在相當大比例的患者中存在顯著的未滿足需求。
INF904治療HS
我們還打算開發INF904治療HS。HS是一種慢性、複發性、使人衰弱的 嗜中性粒細胞驅動的炎症性疾病,可持續多年,並極大地影響生活質量;其特徵是膿腫、 結節和引流隧道,可爆發並引起疤痕。INF904抑制已知的C5a誘導的中性粒細胞活化 和免疫細胞組織蓄積效應,包括組織損傷機制的產生(酶釋放和氧化自由基形成) 以及誘導NETosis——被認為參與HS進展和引流通道形成的機制。現有C5a/C5aR產品的臨牀證據 也支持阻斷該通路可減少病變計數。已知在相當多的病例中,患者對已批准的抗TNF—α或抗IL 17藥物治療的反應會隨着時間的推移而減弱;這些患者需要使用新機制進行治療 。
抗C5a 抗體IFX002
我們 還在開發用於治療慢性炎症性疾病的IFX 002。IFX002是一種高效抗C5a抗體,與vilobelimab結合 相同的C5a蛋白結構域,但與vilobelimab相比,其人源化等級更高,藥代動力學特性也改變 。IFX002目前處於臨牀前開發階段。鑑於IFX002的剩餘專利壽命較長,我們認為IFX002是 vilobelimab的生命週期管理產品。
Vilobelimab 用於治療皮膚鱗狀細胞癌(cSCC)
We have been developing vilobelimab for the treatment of PD-1 / PD-L1 inhibitor resistant / refractory locally advanced or metastatic cSCC. We have been recruiting patients in two independent arms, vilobelimab as monotherapy and in combination with pembrolizumab. The main objectives of the trial were to assess the safety and antitumor activity of vilobelimab monotherapy and to determine the maximum tolerated or recommended dose, safety and antitumor activity in the combination arm in order to evaluate and establish the safety of vilobelimab in cSCC patients. The initial treatment responses of the study were encouraging. However, in view of the recent emergence of new alternative treatments for cSCC and the recommendation by our U.S. and international clinical experts to change course and to study additional patients with a higher dose of vilobelimab as monotherapy in a larger cohort, we have decided to cease the development in cSCC for the time being to prioritize our efforts and to reallocate our resources towards the development of our orally available C5aR inhibitor INF904. Nonetheless, patients who are currently in treatment will be treated for up to 24 months according to the protocol. However, we will not enroll any new patients in the study and we will close clinical sites in which no patients are being treated. Our decision to wind down this clinical study does not preclude us from considering the development of vilobelimab or INF904 in this or similar oncological indications in the future.
有關我們的技術或開發計劃的更多信息,請參閲下面的詳細信息。
我們的技術
C5a 是補體系統的中樞介質,因此是先天免疫系統的關鍵組成部分。補體系統最突出的作用 是通過幾種機制幫助身體防禦入侵的微生物,包括 快速創建炎症環境和產生直接殺死病原體和將免疫細胞募集到 感染部位的因子。補體系統的激活最終導致C5a通過從C5切割而產生。C5a通過吸引和強烈激活中性粒細胞以及通過使許多不同的細胞類型產生 促炎分子來創建 炎症環境。
補體系統和C5a/C5aR軸作為免疫系統中關鍵成分的作用以及控制的需要
補體級聯由大約30種相互作用的蛋白質組成,形成先天免疫系統的關鍵組成部分。這個 系統保護身體,例如通過識別和清除細菌、病毒和其他傳染性病原體(統稱為 )。補體系統的激活會導致一系列類似酶的反應,這些反應產生直接 殺死病原體並將免疫細胞募集到感染部位的因子。這種激活可以通過三種主要途徑觸發:經典 途徑、甘露糖結合凝集素或MBL途徑和替代途徑。任何途徑的激活都將導致C3的裂解 和C5轉化酶的形成。末端補體激活,也稱為C5的切割,可通過 這些C5轉化酶實現。此外,終末補體激活也可以通過外源途徑通過存在於全身但不被認為是補體系統的一部分的天然存在的酶直接實現。
-57-
C5的裂解 導致C5a和C5b的生成,這兩種具有不同生物活性的分子。C5a是一種強炎症放大器 ,通過結合兩種不同的受體C5aR和C5L2發揮其生物學功能。另一方面,C5 b與C6、C7、C8和許多C9分子組裝形成MAC,這是一種重要的內在防禦機制,使微生物的膜變得可滲透,導致其解體或溶解。
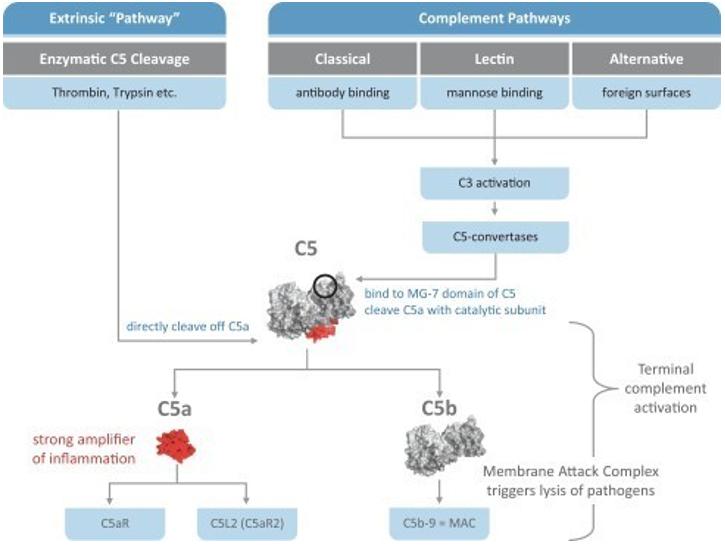
補體系統關鍵功能概述
補體系統在先天免疫應答中發揮許多關鍵功能,例如:
| ● | 快速 創造一個煽動性的環境。促炎分子的產生,例如 作為C5a,優化了酶促和其他過程可以對 微生物的這些炎症性疾病包括髮熱或釋放 嗜中性粒細胞的攻擊性酶和氧自由基。 |
| ● | 溶解 微生物通過MAC的形成。快速的第一線防禦機制 導致入侵微生物的細胞膜形成孔,導致 他們的解體。 |
| ● | 橋 適應性免疫系統。此功能由 的激活產品提升 C3,稱為C3b,可標記顆粒並使其可見,並更容易被 處理 免疫刺激細胞。然後,這些細胞將這些顆粒呈遞給B細胞,B細胞反過來 產生針對顆粒的抗體,導致靶向清除。此機制 需要幾周時間才能完全生效。 |
| ● | 間隙 死亡細胞顆粒的數量。補體系統還用於各種其他目的,包括 從體內清除死細胞顆粒。此功能尤為重要 因為未清除的細胞顆粒被認為可能誘導抗體的產生 抗正常細胞和組織,導致自身免疫性炎症反應和疾病。 |
需要 進行控制
補體 激活是一把雙刃劍:由C5a 驅動的促炎反應的快速作用和相對非特異性功能以及通過MAC形成裂解微生物通常受到非常嚴格的控制。然而,系統的不適當激活 會迅速將其從有益的防禦系統轉變為不受控制的炎症反應。C5a在 某些疾病狀態下的不受控制的活動可在體內產生炎症環境,導致組織損傷並促進促炎性T細胞自身免疫反應。據信,由此產生的組織損傷對許多急性 以及慢性炎性和自身免疫性疾病的疾病進展至關重要,特別是在發作期。這方面的例子包括狼瘡病, 炎症性腸病和嗜中性粒細胞驅動的疾病。
儘管 MAC作為一種快速的第一線防禦機制發揮作用,但MAC的形成也會導致某些 疾病中我們身體細胞的損傷。正常情況下,人體細胞和組織通過防止 MAC形成的表面抑制劑而免受MAC介導的裂解。然而,在陣發性夜間血紅蛋白尿素(PNH)中,患者的細胞缺乏在細胞表面保持MAC抑制劑的能力 ,導致對MAC相關細胞裂解的極端敏感性。此外,患有涉及 腎內皮細胞的疾病的患者,如非典型溶血性尿毒症綜合徵和某些形式的腎小球腎炎,也常常出現 與MAC相關的損害。在這些非常罕見的疾病中阻斷MAC形成可以挽救生命。
-58-
雖然 阻斷MAC形成在某些情況下可能是有益的,但實質上阻斷MAC形成也可能導致 對危及生命的感染易感性。例如,服用阻斷MAC形成的藥物(如市售抗體依庫珠單抗)的患者必須接種腦膜炎球菌病免疫,後者也具有副作用的風險。因此,當阻斷補體介導的損傷時,希望保持MAC 形成完整,其中不受控制的炎症反應 ,尤其是C5a,已被描述為損傷的關鍵驅動因素。
我們 認為,C5a是導致許多炎性疾病中組織損傷的關鍵炎性介質,因此代表了具有巨大治療潛力的非常有意義的 藥物靶點。因此,我們自成立以來已經進行了大量研究,以生成僅靶向C5 a的高度 特異性抗體,同時保留MAC形成完整,從而為該有吸引力的 靶點提供理想的治療方法。
C5激活機制
C5可以由許多細胞產生,包括各種器官的上皮細胞、T細胞和其他免疫活性細胞。終末C5激活 不需要激活三種補體途徑和相關C5轉化酶的形成。其他酶也可以直接切割和激活C5,使得功能活性C5 a可以在完全不存在其他補體組分的情況下產生。例如,在細胞培養物中不存在其他補體因子的情況下,肺上皮細胞可在刺激後生成C5,而肺巨噬細胞可切割並激活C5,導致C5 a的生成。本實施例説明C5可以被激活並且C5a 可以獨立於補體途徑產生。
我們 進一步證明,在存在依庫珠單抗的情況下,C5的直接酶切不受抑制,依庫珠單抗是一種已知的C5抑制劑, 可結合C5的MG—7結構域,並阻止C5轉化酶接合和結合C5。這項研究表明,C5a從C5上直接酶切的機制不會被C5抑制劑(如依庫珠單抗)阻斷。我們的研究進一步證明 ,接受足夠劑量的依庫珠單抗的患者仍可能顯示血漿C5a水平升高,這意味着像依庫珠單抗這樣的C5抑制劑 無法完全阻斷和控制C5a信號通路。因此,在其發揮關鍵促進作用的疾病中,我們認為直接靶向C5a可能會產生有意義的治療益處。
C5a及其在疾病和炎症中的作用
C5a 是一種74個氨基酸的小蛋白,其生化和免疫學特性已在科學文獻中得到充分的記載。C5a通過吸引和強烈激活中性粒細胞以及通過使許多不同的 細胞類型產生促炎和炎症相關分子來創造炎症環境。雖然這可以通過優化防禦環境來幫助身體對感染作出強烈而迅速的反應,但在各種疾病中,不受控制的C5a生成會導致身體組織受損。因此,我們認為控制和限制體內C5a的生成可以防止過度激活的C5a免疫反應的負面影響 。
C5a 與至少兩個獨立的受體—C5aR和C5L2(有時稱為C5aR2)快速相互作用。C5aR和C5L2作為C5a引發效應的大信號庫。C5aR已被充分表徵為信號傳導受體,可在各種疾病環境中幾乎任何細胞中強烈上調 。儘管不太瞭解,但C5L2也已被證明促進炎症,並通過促進由未受控制的C5a引起的不良反應或AE, 對各種實驗性疾病的結局產生負面影響。重要的是,各種其他補體激活產物(例如,C3a、C3a—desArg和C4a)與C5L2結合並引起與C5a引起的效應不同的 效應。因此,通過使用vilobelimab實現的特異性阻斷C5a將僅消除C5a 介導的效應。
C5a在嗜中性粒細胞驅動的炎症性疾病中的作用
在炎症反應中,C5a是炎症的加速劑或“助推劑”。C5a的這一作用延伸到廣泛的 響應,包括以下機制:
| ● | C5a 促進許多不同細胞因子的生成,如IL—8、IL—6、IL 17、TNF—α和 其他在各種細胞類型以及血液中; |
| ● | C5a 誘導免疫活性細胞的細胞信號級聯發生複雜變化,導致 其他已知信號刺激的信號轉導發生了改變,並且經常增強, 如Toll樣受體信號傳導; |
| ● | C5a 影響T細胞反應並引起促炎反應,導致生成 其他促炎細胞因子;和 |
| ● | C5a 能夠誘導血管表面的粘附分子表達,導致 中性粒細胞粘附到血管內壁並通過血管遷移到 感染部位。 |
-59-
當 C5a與中性粒細胞上的受體結合時,它們被強烈激活,並通過被稱為趨化的過程移動到損傷或感染的來源,產生氧自由基和活化酶,這兩者被認為是人體細胞和組織損傷的主要原因 。此外,C5a被認為可誘導中性粒細胞細胞外陷阱(NET)的形成和 中性粒細胞經歷某種形式的細胞死亡,同時形成稱為Netosis的NET,據信這會引起組織中的額外炎症 和損傷。鑑於這一中心功能,C5a是一種強大的工具,當不適當地激活時,它能夠促進對身體的損害,最終導致器官功能障礙和衰竭。
通過觀察中性粒細胞表面標誌物CD 11b的上調(證明中性粒細胞激活的既定方法)來評估中性粒細胞 激活。在2013年和2014年與 雅典大學的一名研究者合作進行的一項研究中,我們發現當添加重組人C5a或添加化膿性汗腺炎患者血漿時,作為中性粒細胞活化標誌物的CD 11b在來自 健康志願者的新鮮人全血中顯著增強。Vilobelimab, 我們的高特異性抗C5a抗體,完全抑制了添加HS血漿引起的中性粒細胞活化,表明 C5a可能是受該疾病影響的患者血漿中的關鍵介質,導致中性粒細胞活化。

新鮮人全血中的流式細胞術檢測顯示血液中性粒細胞上的CD11b增加作為中性粒細胞活化的標誌物:重組 人C5a強烈激活全血中的人中性粒細胞(huPP—ctr +20 nM rhC5a),其可通過添加vilobelimab完全阻斷 (上圖中的"IFX—1")(huPP—ctr +20 nM rhC5a +20 nM vilobelimab)(空心白色柱)。來自兩個不同HS 患者的血漿(pat088和pat092)也激活全血中的人中性粒細胞,該效應可通過添加 vilobelimab(中間和深灰色條)而完全阻斷,因此表明HS患者血漿中的C5a是關鍵的中性粒細胞激活因子。
人類的各種 慢性炎症和自身免疫性疾病的特徵是發作期,在此期間會發生大量的組織損傷。 考慮到C5a的多種促炎功能,在慢性炎性疾病中阻斷C5a可能對T細胞功能產生積極影響 ,對疾病的炎症狀態進行全面控制,並對中性粒細胞產生強大的抗炎作用,這 可能會減少發作期的組織損傷。多個國際研究小組已經在各種炎症動物模型中證明,阻斷C5a/C5aR信號傳導軸導致炎症減少、器官功能改善和臨牀終點的良好結局 ,包括改善死亡率、疾病嚴重程度或損傷評分。
C5a 還被描述為通過下調調節性T細胞和促進 促炎性T細胞應答而平衡T細胞應答的潛在幹擾因子。2013年發表在Nature Immunology和Journal of Experimental Medicine上的研究表明,阻斷小鼠中的C5a/C5aR信號傳導軸可恢復調節性T細胞功能,抑制誘導的自身免疫性疾病的進展。因此,C5a是治療與T細胞失衡相關的自身免疫性疾病和慢性炎性疾病的潛在藥物靶點。
C5a在癌症生長和轉移性疾病中的作用
Different cancer cells have been found to generate their own C5a when cultured in vitro in the absence of any other complement factors or intact complement pathways. This result is possible because cancer cells produce C5, together with enzymes to directly cleave C5, thereby generating functionally active C5a. Recent research suggests that C5a contributes to cancer growth and metastatic disease, with multiple mechanisms proposed in the literature to explain this phenomenon. C5a appears to be associated with the recruitment and activation of myeloid-derived suppressor cells, or MDSCs, in tumors. Activating MDSCs suppresses the important T-cell-mediated mechanisms that usually inhibit tumor growth. Recently published findings in Cancer Cell in 2018 confirmed this mode of action that has been suggested in earlier published work. In addition, C5a generates a microenvironment favorable for tumor growth by increasing angiogenesis and enhancing the expression of the checkpoint molecule PDL1, as well as other mediators that enable tumor growth. These and other existing data may explain why combined therapy of anti-PD-1/PD-L1 and C5a blockade has been shown to effectively reduce tumor growth and metastasis in a pre-clinical mouse model.
-60-
C5aR作為治療幹預的潛在靶點的作用
兩種 C5aR(也稱為C5aR1或CD 88)和C5aR2(也稱為C5L2或GPR77)介導C5a的生物學活性。 C5aR的激活已得到廣泛認可,而C5aR 2的激活作用仍不太清楚 ,最近的科學工作表明對C5aR 1激活具有潛在的調節作用。在膿毒症的動物模型中,抗C5a治療 改善了炎症反應的發展並改善了存活率。此外,實驗證據表明,C5aR信號傳導的阻斷 類似地改善了膿毒症動物的存活率。最後,C5aR拮抗劑在涉及補體激活的許多炎性疾病模型中顯示出優異的治療效果 。
與其配體C5a不同,C5aR可以被小分子抑制。2021年10月,口服C5aR拮抗劑avacopan在美國獲得 上市批准,作為成人重度活動性ANCA相關血管炎(特別是 MPA和GPA)的預防性治療,與包括糖皮質激素在內的標準療法聯合使用。
一般認為,使用抗體阻斷C5a提供了一種快速、完整和安全的控制C5a誘導的炎症的方法。 C5aR低分子量化合物抑制劑的優點是可以口服給藥,從而為患者提供廣泛、長期的 給藥方便。
我們的 專有抗C5a/C5aR技術和候選產品
我們的 抗C5a技術
儘管 C5a在促進不同疾病中的炎症和相關組織和器官損傷方面具有很好的特徵,但目前還沒有上市的 靶向C5a的藥物。基於在該領域超過17年的研究,我們認為靶向C5a的挑戰是完全 在其自然環境中的生物學功能,並保持MAC形成完整。我們相信,我們專有的抗C5a 技術使我們能夠通過發現C5a上的新表位或結合位點來克服這一挑戰。我們認為,這種構象 表位僅在C5a從C5分子上切割後形成,這表明C5a的三維結構在C5釋放後發生變化 ,產生了僅存在於遊離C5a分子上的新表位。這允許僅在 遊離C5a從C5上裂解後與遊離C5a結合,從而允許阻斷C5a同時保持MAC形成完整。我們認為,這代表了終末補體C5a抑制領域的突破 ,並且在治療C5a驅動的疾病(如PG和嚴重COVID—19、cSCC、HS、AAV等)時,這可能特別有價值。
我們鑑定了抗體,包括我們的主要候選產品viloblimab,它能有效和選擇性地與C5a從C5裂解後形成的構象表位 結合,完全阻斷C5a,而不會影響補體系統的重要上游功能以及MAC的形成。我們打算利用我們專有的抗C5a技術 來發現和開發治療方法,以解決一系列具有重大未滿足需求的補體介導的疾病。
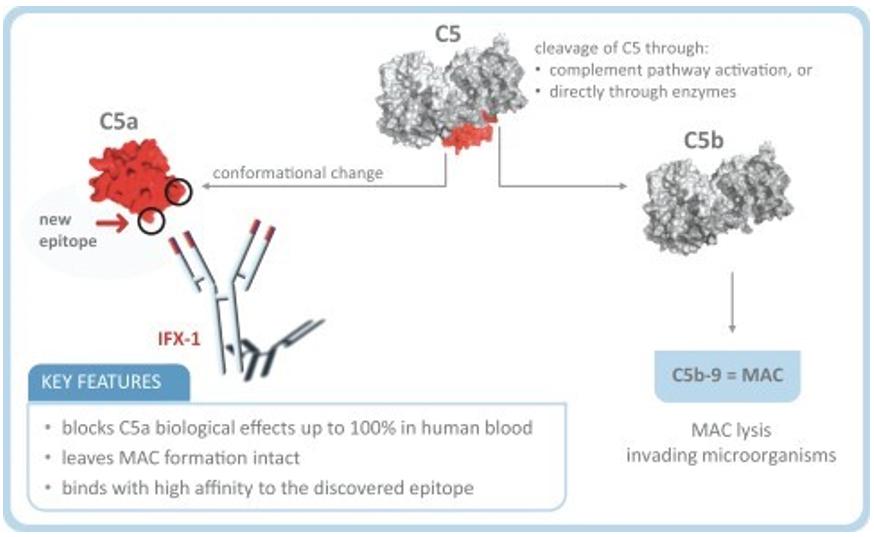
C5a分子表面的構象表位允許產生針對 C5a的高度特異性封閉抗體。
-61-
我們的抗C5a單抗具有以下特性:
| ● | 完全免疫阻斷和抑制C5a誘導的效應:人體具有豐富的產生C5a的能力,並通過其兩個受體C5aR和C5L2誘導炎症效應。因此,我們的抗C5a抗體旨在: |
| o | 產生對C5a分子的完全免疫阻斷,以實現有效的治療。 缺乏這一特性的抗體或抑制劑可能會給C5a留下“信號缺口”,在疾病環境中,可能足以產生強烈的促炎作用。這種信號間隙將限制沉默C5a/C5aR和C5a/C5L2信號軸以達到預期的治療效果的能力; |
| o | 結合與C5a具有高親和力的 ,以抵消分子與其兩個受體C5aR和C5L2的快速相互作用,C5aR和C5L2廣泛存在於人體絕大多數細胞類型 上,並可在各種疾病情況下上調。 |
| ● | 對MAC形成的影響有限:抑制血液中MAC形成的C5阻斷分子 增加了由被包裹的細菌(如腦膜炎球菌)引起的危及生命的感染的風險。因此,保持MAC結構完整可能在C5a驅動的疾病中提供顯著的優勢。 |
我們 認為,所有這些特徵對於任何靶向C5a的藥物都是必要的,以獲得臨牀上有意義的藥理性能 ,用於治療由C5a引起的疾病,如PG、嚴重新冠肺炎、口腔鱗癌或其他疾病。此外,我們認為C5a引起的疾病可能不能有效地被補體抑制方法有效地靶向,因為補體抑制方法不能特異性和完全阻斷C5a。這些方法,如阻斷補體途徑驅動的C5裂解或抑制C5上游的補體途徑,具有以下兩個基本缺點。
| ● | 無法在不直接靶向的情況下完全阻斷C5a:C5a可以在完全缺乏補體途徑的情況下由各種酶通過C5激活而生成。例如,在實驗環境中,用C5抑制劑eculizumab阻斷補體C5轉換酶驅動的切割並不能阻止酶促C5的直接激活和C5a的生成。這可能解釋了為什麼在有效服用eculizumab的患者中,升高的C5a水平仍然可以測量到。因此,不直接結合和抑制C5a的非特異性方法可能無法 完全阻斷其影響。 |
| ● | 缺少 控制C5a信號傳導能力:C5a受體大量存在於 在某些疾病中可以強烈而迅速地上調 states.因此,即使C5a水平較低,受體也會產生大量的"信號 “sink”為甚至少量的C5a提供了豐富的能力來傳輸信號。 因此,為了實現完全控制,需要採用完全阻斷靶向C5a方法 過度C5a誘導的信號傳導事件可能在高度急性炎症中特別重要 設置. |
Vilobelimab 作為同類抗C5a單克隆抗體
我們的 主要候選產品vilobelimab是一種靜脈給藥的單克隆抗C5a抗體。它基於我們專有的抗C5a 技術,是第一個進入臨牀開發的C5a單克隆抗體。Vilobelimab的區別在於其能夠:
| ● | 完全 抑制C5a誘導的信號傳導和衍生的生物學功能,其能力證明 完全防止人全血中C5a誘導的中性粒細胞活化;和 |
| ● | 離開 MAC形成完整,通過檢測完整補體途徑驅動的MAC證明 在紅細胞上形成,導致這些細胞的溶解。 |
-62-
我們 在健康志願者中完成了一項安慰劑對照、單中心I期研究,並完成了兩項雙盲、安慰劑對照、 在兩種急性護理適應症、早期膿毒性器官功能障礙和複雜心臟手術中進行的多中心IIa期研究。我們還完成了 HS中的IIa期和IIb期臨牀研究、兩項AAV II期研究、PG中的IIa期研究和一項II/III期臨牀研究 重症COVID—19機械通氣患者。
在 所有已完成的研究中,觀察到vilobelimab的耐受性良好。在兩種急性護理適應症中進行的安慰劑對照、多中心IIa期研究表明,vilobelimab可阻斷C5a,具有較高的統計學顯著性(p值
為了 確定數據是否具有統計學意義,我們使用"p值",它表示隨機機會 可以解釋結果的概率。FDA在評價臨牀試驗結果時,使用報告的統計學指標(包括 通過p值作為有效性證據標準測量的統計學顯著性)來評價候選產品安全性和有效性的報告證據。如果沒有特別説明,我們使用常規的5%或更低的p值(p
基於我們迄今為止完成的臨牀試驗以及EpiScreen的結果 離體免疫原性T細胞應答試驗, 我們認為vilobelimab在給藥後引發免疫應答的風險較低。免疫原性試驗使用了來自21名供體的 外周血單核細胞,並測試了在引入vilobelimab後,有多少供體細胞顯示出CD 4 + T細胞應答 離體.應答率超過10%(或超過21/21)意味着適用蛋白質被視為 免疫原性高風險,而應答率低於10%意味着蛋白質被視為低風險。vilobelimab的試驗結果 顯示,與對照組(使用 A33抗體)相比,21名供體中的0名供體具有T細胞應答率,對照組的應答率為30%。此外,根據與我們在HS中進行的IIb期臨牀試驗有關的ADA檢測試驗,10%的患者在研究期間的任何時間都有ADA。只有一名受試者存在ADA與 任何特定AE模式相關,表明症狀可能與ADA的存在或出現導致免疫反應的任何ADA相關。
我們 目前正在評估vilobelimab在各種疾病適應症中的應用。迄今為止,在所有正在進行和已完成的臨牀研究中,我們從未 觀察到會對vilobelimab作為治療藥物候選者的既定安全性產生懷疑的效應。
我們 還將繼續評估vilobelimab在其他疾病環境中的開發潛力,我們認為抗C5a抗體 可以成功開發為上市治療。
C5aR小分子抑制劑的開發
兩種 C5aR(也稱為C5aR1或CD 88)和C5aR2(也稱為C5L2或GPR77)介導C5a的生物學活性。 C5aR的激活已被廣泛認可的促炎作用,而C5aR 2的激活仍不太清楚, 最近的科學工作表明C5aR激活具有潛在的調節作用。C5aR是一種G蛋白偶聯受體,主要 由粒細胞表達,特別是在疾病條件下,廣泛存在於各種組織和其他免疫細胞類型中,介導 C5a的病理生理效應。
在膿毒症動物模型中,抗C5a治療改善了炎症反應的發展並改善了生存率。此外, 實驗證據表明,C5aR信號傳導的阻斷類似地提高了膿毒症動物的存活率。與其配體 C5a不同,C5aR也可以被低分子量化合物抑制。
-63-
低 分子量C5aR拮抗劑已在涉及補體激活的炎性 疾病的許多體外和體內動物模型中顯示出優異的治療效果。C5aR的低分子量抑制劑的優點是它可以口服給藥, 從而為患者提供廣泛、長期的給藥方便,特別是用於治療慢性疾病。通過對這些小分子C5aR拮抗劑在C5a/C5aR軸激活誘導的疾病中的適當 臨牀研究,可以確定這些藥物的安全性和 有效性。
Avacopan 是第一種口服C5aR拮抗劑,在美國已獲得上市批准,作為成人重度活動性 ANCA相關血管炎(特別是MPA和GPA)的輔助治療,與標準治療(包括糖皮質激素)聯合使用。
通過 我們的內部藥物研發工作,我們確定了一種有效的C5a受體抑制劑INF904,我們認為它是一個有希望的候選藥物 。我們目前正在開發INF904,一種針對C5aR受體的口服低分子量候選藥物。我們 計劃針對補體介導的慢性自身免疫和炎症性疾病,患者需要口服小分子。
鑑於阻斷C5a和C5aR的不同優勢,我們認為C5a和C5aR阻斷劑的開發是可能的,並且 可能有助於解決更廣泛的C5a/C5aR分子信號軸相關疾病。
我們的臨牀前和臨牀開發計劃
Vilobelimab 用於治療PG
我們正在開發用於治療PG的vilobelimab。 確切的病理生理學機制尚不完全清楚,但據推測,炎性細胞因子的產生以及中性粒細胞的激活和功能障礙導致皮膚無菌炎症。PG通常表現為疼痛的膿皰疹或丘疹,主要發生在下肢,可迅速進展為極其疼痛的擴大潰瘍。相關症狀包括髮燒、身體不適、體重減輕和肌肉疼痛。PG通常會對患者的生活造成毀滅性的影響,因為劇烈的疼痛和明顯的運動障礙 取決於病變的位置。PG的確切流行情況尚不清楚,但據估計,美國和歐洲有多達51,000名患者 受到這種疾病的影響。
已知的疾病類型有4種:潰瘍性(經典變種,這是我們的研究重點)、大皰性(非典型)、膿皰性、 和植物性(淺表、肉芽腫)。潰爛變異型是PG最常見和典型的形式,病變主要發生在下肢。
目前在美國或歐洲還沒有批准用於治療PG的藥物。當地批准的唯一治療方法是阿達利單抗,它已在日本獲得批准,但沒有在其他國家獲得批准。目前還沒有建立在PG對照研究基礎上的護理標準。但是,由於與疾病相關的高度醫療需求,某些藥物在醫療實踐中被用作受影響患者的治療嘗試 。這些藥物包括某些口服藥物,如免疫抑制劑,包括有時也同時使用的環孢素或皮質類固醇,以及外用藥物,如他克莫司和其他藥物。最後,靜脈注射腫瘤壞死因子-α抑制劑,如英夫利昔單抗或阿達利單抗或其他生物藥物也被用作治療嘗試,儘管沒有正式的監管批准。
2019年2月,我們啟動了一項開放標籤的多中心IIa期探索性研究,在加拿大、美國和加拿大招募了19名中度到重度PG患者。居住在美國的國家和 波蘭。本研究的目的是評估三種不同劑量的維羅布利單抗在該患者羣體中的安全性和有效性,並在PG治療的註冊第三階段研究中確定未來開發維羅布利單抗的合適劑量。
-64-
2021年4月,該研究達到了19名患者的招募目標。2021年10月,我們公佈了研究的初步結果。在第三個劑量隊列中,每週2400 mg,7名患者中有6名臨牀緩解,PGA評分 為≤1,這反映了目標潰瘍的關閉。在第三次給藥隊列中,所有患者的C5a水平在基線 時升高,在開始使用Viloblimab治療後持續抑制。
未發現與劑量相關的不良反應。總體而言,觀察到的聲發射譜與潛在疾病是一致的。
所有患者的最終結果於2022年3月在美國皮膚病學會AAD年會上由梅奧診所皮膚科副教授Afsaneh Alavi醫學博士在一次口頭晚間會議上公佈。報告的最終結果顯示,在2400毫克的最高劑量隊列中, 存在劑量依賴效應,證實了初步結果,7名患者中有6名患者臨牀緩解(醫生全球評估,或PGA,得分≤1),並在該劑量隊列中目標潰瘍關閉。第七位患者(PGA評分4分)略有改善,目標潰瘍面積縮小50%以上。在隨訪期內,除一名患者外,所有患者的潰瘍在治療完成兩個月後仍保持關閉狀態,並在最後一次服藥後觀察到持續抑制C5a長達20天。
根據這些結果,Viloblimab被美國FDA授予治療PG的孤兒藥物稱號Nited 個州而歐洲的EMA以及快車道的指定是FDA。此外,我們還宣佈,我們在2022年6月與FDA就PG的第三階段開發計劃進行了卓有成效的第二階段結束會議。2023年1月,我們宣佈了計劃中的第三階段研究的設計細節,該研究使用Viloblimab治療潰瘍性PG。
The Phase III study is designed to enroll patients in the United States, Europe and selected countries in other regions. The study design is based on detailed feedback and recommendations from the FDA Division of Dermatology and Dentistry and was developed in close collaboration with the Company´s advisors from the United States, Europe and other regions. The multi-national, randomized, double-blind, placebo-controlled Phase III trial has two arms: vilobelimab (2,400mg every other week) plus a low dose of corticosteroids and placebo plus the same low dose of corticosteroids. In both arms, corticosteroid treatment will be initiated on day one and will be tapered off within the first eight weeks of the treatment period. The primary endpoint of the study will be complete closure of the target ulcer at any time up to 26 weeks after initiation of treatment. Treatment will be discontinued for patients whose disease progresses or fails to improve at defined time points during the study. The study has an adaptive trial design with an interim analysis blinded for the sponsor and investigators (but unblinded for the independent data safety monitoring committee), which is planned upon enrollment of approximately 30 patients, divided equally between the two arms of the study. The interim analysis with a set of predefined rules will take into account the then-observed difference in complete target ulcer closure between the two arms and will then determine whether the trial sample size will be adapted or whether the trial should be stopped due to futility. The enrollment period is projected to last at least two years, and its overall period will depend on the total trial size after sample size adaptation.
2023年11月,我們宣佈註冊 第一個病人在 的線索。
Vilobelimab 用於治療重症、有創機械通氣COVID—19患者
嚴重 COVID—19的特徵是嚴重的肺部炎症和凝血激活,在 患者在重症監護室時,經常需要機械通氣。儘管已確定廣泛使用皮質類固醇和其他抗炎藥,但重症、有創機械通氣 COVID—19患者的死亡率和發病率較高。疾病結局不佳 與補體系統激活有關,特別是C5a/C5aR分子信號軸。其他病毒性肺病的實驗研究 表明C5 a是一種強效過敏毒素,可將中性粒細胞和單核細胞吸引至感染部位 ,從而導致組織損傷、內皮細胞分泌和微血栓形成。小鼠研究還表明,阻斷C5a/C5aR1分子信號軸可限制受損器官中骨髓細胞的浸潤,並防止過度的肺部炎症和內皮素。
-65-
儘管 在最近的COVID—19大流行中廣泛使用了抗SARS—CoV—2疫苗,疾病管理也有所改善,包括使用了抗IL 6抗體或JAK抑制劑等免疫調節劑、糖皮質激素和抗凝治療,但 重症患者、插管患者和機械通氣患者的死亡率一直保持在50%以上的水平。根據美國疾病控制和預防中心(CDC)的數據,2023年全年美國每週有450至3800起與COVID—19相關的死亡病例,https://covid.cdc.gov/covid-data-tracker/#trends_weeklydeaths_select_00 )這顯示醫療方面仍有需要有效治療這些病人。
2020—2023年COVID—19大流行期間,很明顯,無法獲得治療重症或重症COVID—19患者的有效療法,或尚未對該適應症進行測試。為了努力為這一醫療緊急情況做出貢獻, 基於我們現有的關於C5a在病毒誘導性肺炎中的作用的臨牀前研究,我們決定在患有嚴重進展性肺炎的重症COVID—19患者中啟動vilobelimab的臨牀開發 項目。
臨牀 開發
2020年3月,我們啟動了一項隨機、開放標籤多中心試驗II/III期臨牀開發項目,在患有嚴重進展性肺炎的重症COVID—19患者中使用vilobelimab。在研究的II期部分,我們評估了vilobelimab治療加最佳 支持治療與單獨使用最佳支持治療相比長達28天。氧合指數 (定義為PaO2/FiO2比值)自基線至第5天的相對變化(%)與其他臨牀參數一起評估,作為主要終點,直至第28天。在本研究中, 患者被隨機分配至兩個治療組,A組(最佳支持治療)和vilobelimab組(單獨使用最佳支持治療)或B組(單獨使用最佳支持治療)。 主要終點是氧合指數(PaO2/FiO2)自基線至第5天的相對百分比變化。
On June 17, 2020, we announced results from the Phase II part of the study. A total of 30 patients were randomized in the trial, and 15 patients were treated in each arm: vilobelimab plus best supportive care or best supportive care alone. Over a treatment period of 28 days, patients in the vilobelimab arm received a maximum of seven doses of 800 mg vilobelimab intravenously on separate days. At randomization, 18 patients were intubated (60%), and 12 patients (40%) had other oxygen supply. A higher number of patients with two or more comorbidities associated with increased COVID-19 mortality were reported in the vilobelimab treatment group compared to best supportive care group. Relative change in the oxygenation index at day 5 showed no differences between treatment groups. However, vilobelimab treatment was associated with a lower 28-day all-cause mortality when compared to the best supportive care group, along with trends in disease improvement, as evidenced by fewer patients experiencing renal impairment assessed by estimated glomerular filtration rates, more patients showing reversal of blood lymphocytopenia and a greater lowering of lactate dehydrogenase concentrations. In vilobelimab-treated patients, pulmonary embolisms reported as SAEs occurred less compared to the best supportive care arm. Also, a temporary increase of D-dimer levels, as potential expression of induction of blood clot lysis, was detected in the first days after initiation of vilobelimab treatment. Twenty-eight-day all-cause mortality in the vilobelimab treatment group was 13% (2 out of 15) versus 27% (4 out of 15) in the control group. In the best supportive care group, four patients died of COVID-19-induced multi-organ failure, and three of them had pulmonary embolisms reported as a SAE. In the vilobelimab arm, one patient died after an acute ventilator tube complication (leakage) and one patient with a history of severe chronic obstructive pulmonary disease died of pulmonary failure.
兩組間SAE 發生率相當,但vilobelimab 治療組報告為SAE的肺栓塞發生率顯著較低。在審查安全性數據後,獨立數據安全性監測委員會建議將試驗繼續進入 III期部分。
在 研究的第三階段部分, 從2020年9月至2021年10月,我們在歐洲各研究中心入組了369名機械通氣COVID—19患者,泌尿外科聯合會南美洲和 其他地區。患者以1:1的比例隨機接受vilobelimab或安慰劑;大多數患者接受標準治療(97%使用糖皮質激素, 98%使用抗血栓藥物)。主要終點是28天全因死亡率;關鍵次要終點包括器官支持評估和疾病改善。
-66-
2022年3月,我們宣佈成功完成了對COVID—19患者進行機械通氣的II/III期PANAMO研究的III期部分,並顯示28天全因死亡率相對降低了23. 9%(p = 0. 094)。在試驗過程中,根據監管機構 的建議,我們變更了主要終點的統計分析方法。原始方案 指定了非分層Cox迴歸分析,最終統計分析計劃指定了研究中心分層分析,旨在 説明隨機化時患者的研究中心分層。原始方案規定的分析將導致 p值為0.027(統計學顯著性),而研究中心分層Cox迴歸導致p值為0.094(無統計學顯著性 )。此外,28天死亡率的預先指定邏輯迴歸分析得出的p值為:
監管 活動
2022年9月,在2022年夏季舉行的B類會議上,我們與FDA進行了鼓勵性互動之後,宣佈了EUA的申報。此外,我們還獲得了FDA的快速通道指定,用於治療重症、插管、機械通氣的COVID—19患者。
2023年4月,FDA發佈了GOHIBIC(vilobelimab)的EUA,用於在接受IMV或ECMO後 48小時內啟動治療住院成人COVID—19。
2023年7月,我們向EMA提交了接受IMV或ECMO的SARS—CoV—2誘導膿毒性ARDS的上市許可申請(MAA)。 2023年8月,EMA驗證了MAA。這意味着,CHMP目前正根據適用於歐盟所有27個成員國的集中 程序對申請進行監管審查。
為了 實現全面商業規模併成功實現產品未來的全部市場潛力,我們還希望獲得 Gohibic(vilobelimab)的全面市場批准。因此,我們計劃提交BLA,以獲得Gohibic(vilobelimab) 在我們的COVID—19適應症中的完全批准,將來可能用於其他病毒引起的急性呼吸窘迫情況的類似適應症。2023年10月,為了進一步努力獲得BLA,我們與 FDA舉行了令人鼓舞的C類會議。在那次會議上,FDA表示願意與我們合作,確定BLA 的開發途徑,用於更廣泛的ARDS標籤。為了實現這一目標,我們需要在更廣泛的 ARDS環境中進行額外的控制良好且充分把握度的研究,以證明vilobelimab的安全性和有效性。會議期間,我們討論了此類 試驗的不同選擇,包括潛在試驗設計、患者人羣和試驗把握度方面。
我們 正在積極評估並朝着下一步努力,以便在更廣泛的ARDS環境中實現此類試驗,目前正在探索不同的 資金選擇,包括政府撥款以及與第三方的合作。
商業化
我們 打算在包括美國和歐洲在內的主要市場尋求全面的營銷授權。為此,我們可能會聘請銷售和營銷方面的專家 ,並在內部和/或在外部服務提供商的潛在協助下構建必要的商業和物流基礎設施。同時,我們還打算尋找合作伙伴來支持我們的商業化,例如在選定地區建立合作伙伴關係,如果獲得EUA,還可能在其他地區建設商業基礎設施。
2023年6月,我們開始在美國將GOHIBIC(Viloblimab)商業化,用於治療住院的成人新冠肺炎,在接受IMV或ECMO後48小時內啟動。我們與Cencora Inc.或Cencora(前身為amerisourceBergen Corp.)的某些子公司簽訂了協議。作為我們的美國分銷商,並根據EUA向美國醫院客户提供GOHIBIC(Viloblimab)供 訂購。Cencora提供冷藏、冷鏈配送服務、庫存管理和二次貼標/包裝等服務。為了支持我們的商業努力,我們已經並將繼續聘請在醫院市場醫療產品商業化方面擁有相關經驗的美國 專家,包括在銷售、銷售運營、營銷、市場準入、分銷、醫療事務等領域。此外,我們還在內部和外部服務提供商的幫助下擴展必要的基礎設施,包括IT系統、供應鏈、財務報告系統和庫存管理系統。
-67-
隨着我們擴大我們的商業努力,我們繼續加強我們的商業戰略計劃,進一步擴大我們的銷售隊伍和醫療事務團隊,準備針對醫療保健提供者和其他利益相關者的相關宣傳和醫學教育材料, 完善我們的醫療事務戰略,以提高醫學界對EUA的認識,並啟動我們的銷售活動。
在2023年7月和隨後的2023年10月,美國國立衞生研究院公佈了新冠肺炎患者的治療指南,並隨後更新了指南。美國國立衞生研究院的指南規定,沒有足夠的證據建議或反對使用維洛布利單抗治療危重新冠肺炎患者。這一中立的NIH指南建議對GOHIBIC(Viloblimab)的商業採用產生了負面影響,因為許多醫療保健提供者,尤其是醫院,在起草處方和下新訂單時依賴NIH治療指南。我們認為,美國國立衞生研究院的分析沒有考慮到食品和藥物管理局在批准EUA時考慮的積極因素,我們正在努力向國家衞生研究院和醫學界其他人提供儘可能多的科學證據,以期達成國家衞生研究院指南委員會的立場與FDA發表的詳細審查的一致,從而重新考慮 他們對患有Gohibic(Viloblimab)的危重新冠肺炎患者的治療建議。
政府資助
2021年10月,我們宣佈,我們從德國教育和研究部和德國衞生部獲得了高達4,370萬歐元的贈款,以支持我們開發用於治療重症新冠肺炎患者的Viloblimab。由於我們的研發計劃隨後發生了變化,而且在贈款的時間範圍內預計成本較低,因此我們接到通知,可供我們使用的金額為4,140萬歐元。這筆贈款的結構是報銷某些預先指定的費用的80%,這些費用與維羅貝利單抗的臨牀開發和製造有關。資助期於2023年6月30日結束。在截至2023年12月31日的資助期內,我們總共收到3330萬歐元,用於支持我們的活動,支持我們將維洛貝卡 開發為治療重症新冠肺炎患者的新治療劑,並建立商業規模的生產 工藝,以確保能夠向更廣泛的人羣提供此類治療。
維洛布單抗治療口腔鱗狀細胞癌
皮膚鱗狀細胞癌,或CSCC,是第二常見的皮膚癌。CSCC的發病率隨着太陽曝曬和年齡的增加而增加,皮膚和頭髮白皙的人更容易受到影響。美國每年報告的CSCC病例約為200,000至400,000例,估計高達每年100萬例。 歐洲的估計數字因地理位置而異,從北歐每年約30例,10萬人中約10例,到南歐約10例,10萬人中約10例。CSCC的發病率在全球範圍內呈上升趨勢。然而,進展型和轉移型的CSCC很少見。而晚期和轉移型CSCC的治療緩解率程序性死亡蛋白-1,或PD-1,/程序性死亡配體-1,或PD-L1,抑制劑被認為在50%的範圍內,患者 經常復發,耐藥/難治患者通常預後非常差。
CSCC局部復發或轉移的可能性因原發灶的病理變化和部位而異,CSCC轉移的風險約為2-5%。有區域淋巴結轉移的晚期CSCC患者的10年生存率不到20%,有遠處轉移的患者不到10%。患有疾病的患者遠處轉移的中位生存期不到2年。
我們還在開發Viloblimab,用於治療PD-1/PD-L1抑制劑耐藥/難治、局部晚期或轉移性CSCC。CSCC是第二常見的皮膚癌。CSCC的發病率隨着累積陽光暴露量和年齡的增加而增加,皮膚和頭髮白皙的人更容易受到影響。CSCC的局部復發或轉移的可能性因病理變化和原發灶的位置而異,CSCC的轉移風險約為2%-5%。晚期CSCC的10年生存率在區域淋巴結受累時不到20%,在遠處轉移時不到10%。
-68-
在這項於2021年4月啟動的研究中,我們招募了兩個獨立的組的患者,單獨使用維洛布利單抗(組A)和維羅布利單抗聯合培布羅利單抗(組B)。該試驗的主要目標是評估維洛布利單抗的安全性和抗腫瘤活性,確定與培布羅利單抗聯合治療的最大耐受量或推薦劑量,以及評估聯合治療組在CSCC患者中的安全性和抗腫瘤活性。
截至本研究日期,有10名患者進入A組,他們在第1、4、8和15天接受800毫克的維洛利單抗磨合劑量,隨後從第22天開始每兩週注射1,600毫克維洛利單抗。2023年7月對10名患者進行了A組的中期分析,評估了A組的治療反應。中期療效分析顯示,1例患者完全緩解(CR),1例患者根據方案和《實體瘤療效評價標準》(RECIST)繼續病情穩定。這位CR患者仍在接受治療。
在對研究A組的前三名患者進行了五週的治療後,成功地完成了安全性評估,也開始了B組的登記。
在B組中,截至本研究的日期,已有3名患者在研究的第一劑量隊列中接受治療(從第22天起,在第1、4、8、15天和之後每兩週靜脈滴注400毫克維羅布利單抗,從第22天起每兩週靜脈滴注800毫克,此外,在第8天和此後每6周靜脈滴注400毫克培布羅珠單抗)。6名患者接受下一個高劑量(第二)劑量的治療(在第1、4、8和15天靜脈滴注維羅貝利單抗600 mg,從第22天開始每兩週靜脈滴注1,200 mg,第8天和此後每6周靜脈滴注400 mg培溴利珠單抗 )。在第三個劑量隊列中,6名患者按計劃的最高劑量進行治療(在第1、4、8和15天靜脈滴注維羅貝利單抗800毫克,從第22天起每兩週靜脈滴注1600毫克,第8天和此後每6周再靜脈滴注400毫克培布羅珠單抗)。每一次劑量遞增都是根據建議進行的,並由外部臨牀顧問組成的獨立指導委員會在審查安全數據後進行。到目前為止,總共有15名患者參加了B組試驗(三個劑量隊列中的3+6+6)。在進行第二階段的ARM B研究之前,在最近與我們的專家組討論中評估的中期療效數據顯示,第二個隊列的一名患者部分緩解,第三個隊列的一名患者部分緩解,他們仍在接受治療。
單次給藥組A的 治療應答和聯合給藥組B的初步觀察結果令人鼓舞。 然而,鑑於最近出現了新的cSCC替代治療方法,以及我們的美國和國際專家建議改變治療方案,並在更大的隊列中研究更多患者使用更高劑量的vilobelimab作為單藥治療,我們 決定暫時停止cSCC的發展。雖然我們仍有興趣進一步瞭解vilobelimab在該腫瘤適應症中的潛在單抗作用,但進一步的研究將需要大量資源,並顯著延長正在進行的臨牀項目的 時間軸。因此,在2023年11月,我們決定優先考慮我們的工作,並重新分配我們的資源 用於開發我們的口服C5aR抑制劑INF904。
根據方案,目前正在接受治療的患者 將接受長達24個月的治療。但是,我們不會招募任何新患者 參與本研究,我們將關閉沒有患者接受治療的臨牀研究中心。我們決定終止本臨牀研究, 並不妨礙我們考慮將來在這種或類似腫瘤適應症中開發vilobelimab或INF904。
Vilobelimab 用於治療HS
HS 是一種慢性衰弱的全身性皮膚病,會導致毛囊疼痛性炎症,最明顯的是腋窩、腹股溝和生殖器區域。這種疾病的臨牀特徵包括非常痛苦的炎性結節、膿腫或膿腫, 通常打開並釋放有氣味的炎性液體。在更慢性的疾病中,患者會出現dt(以前 稱為引流瘻管或竇),最終導致某些區域的瘢痕形成和相關功能殘疾。 HS患者主要遭受疼痛和嚴重不適,這些疼痛和嚴重不適是由不斷形成的膿液引起的,通常需要使用 繃帶和尿布,從而導致社交孤立。HS嚴重影響患者的生活質量。HS通常出現在患者生命的第二和第三個十年,並經常發展為終身衰弱的慢性疾病。
vilobelimab的目標患者人羣是顯示中度至重度疾病的HS患者。在美國,我們 估計中度至重度HS的患病率高達200,000例,儘管最近的出版物表明患病率更高。 在歐洲,據信受影響的患者數量也更多,在氣候較暖的國家,HS的患病率和發病率較高。
-69-
HS患者接受的(但未經批准的)治療標準包括局部、口服或靜脈內抗生素治療,以及手術, 這些治療通常只能暫時緩解症狀。HS被認為是一種全身性自身免疫性疾病,有許多 的潛在病因,包括遺傳學。中性粒細胞被認為具有潛在的疾病促進作用,以及在自身免疫性疾病中常見的某些 細胞因子和介質,如TNF—α、IL—17、IL—1等。C5a促進炎症介質 ,並且是中性粒細胞的強激活劑,這是我們在 HS患者中研究C5a阻斷藥物候選物vilobelimab的基礎。我們確定,患有HS的患者表現出顯著的全身補體激活, 血漿C5a和其他標誌物濃度升高。
在美國和歐洲,唯一批准的治療HS的藥物是阿達木單抗,一種腫瘤壞死因子—α或TNF—α抑制劑。 雖然它為部分中重度HS患者提供了臨牀獲益,但大約50%或更多的患者 對阿達木單抗治療無效。因此,HS患者的醫療需求仍然很高。
化膿性汗腺炎臨牀反應或HiSCR評分已開發用於評估臨牀試驗中HS治療的有效性 。當觀察到炎性病變計數(包括膿腫和炎性 結節或AN)減少≥ 50%時,患者定義為HiSCR應答者。同時,與基線相比,不應觀察到平均值或dt的增加。HiSCR 是主要終點,用於支持FDA和EMA批准阿達木單抗治療HS患者。
與HiSCR的二分法性質相反,IHS 4評分旨在以連續 的方式對嚴重程度進行評分並跟蹤治療反應,作為HiSCR的替代方案。然而,IHS 4評分尚未用作HS後期臨牀研究的主要終點 。該複合評分以一分加權最波動的炎性結節,以兩分加權,以 四分加權。
我們 一直在開發用於治療HS的vilobelimab。最初,我們在一項IIa期、單中心開放標籤 研究中對12例重度HS患者進行了評價,這些患者對既往治療嘗試部分無效。試驗結果表明,在8周治療結束時, 75%的患者和12周試驗觀察期結束時,有83%的患者出現了HiSCR反應 ,證明瞭候選產品具有改善疾病效果的初步臨牀證據。結果還表明 vilobelimab給藥耐受性良好,未檢測到藥物相關不良事件,未觀察到輸注相關過敏或 速發過敏反應。
隨後, 我們完成了一項更大規模的多中心、國際IIb期研究(SHINE),以確定維洛貝利單抗在中度至重度HS患者中的療效和安全性。該試驗是一項隨機、雙盲和安慰劑對照、多中心研究,包括5個劑量組,包括 一個安慰劑組。在16周的安慰劑對照雙盲期後,每例患者再接受維洛貝利單抗開放標籤治療 28周,以評估長期療效和安全性。本研究的主要目的是評價通過 第16周的HiSCR評分作為主要終點評估的劑量反應信號。
2019年6月,我們宣佈了國際SHINE IIb期研究的最高結果,其中我們在第16周使用HiSCR未能達到我們的主要終點 。這項隨機化、雙盲、安慰劑對照、多中心研究在北美和歐洲9個國家的40多個研究中心共招募了179名患者,分別來自 4個活性劑量組和1個安慰劑組。雖然最高劑量(每 兩週1200 mg)導致HiSCR降低45.5%,但安慰劑對HiSCR終點的應答達到47.1%。安慰劑組和維洛貝利單抗治療組之間治療後出現的AE未檢測到差異 。
在 2019年7月發表的後續事後分析中,我們顯示了維洛貝利單抗高劑量組與安慰劑組相比的多個療效信號,證明所有合併炎性病變顯著減少,dt計數減少,IHS 4評分減少 。例如,在第16周時,與安慰劑相比,高劑量維洛貝利單抗 組中可以觀察到dt計數相對於基線的統計學顯著降低(平均值:—63.3% vs—18.0%;p = 0.0359;所有患者基線時至少有一個dt)。
-70-
In 2021 we submitted a Special Protocol Assessment, or SPA, to the FDA for the Phase III HS program for vilobelimab HS, suggesting IHS4 as the primary efficacy endpoint, which was subsequently declined by the FDA. The FDA agreed to the dosing regimen in the protocol but did not agree with the assessment of the primary endpoint using IHS4. We later held a Type A meeting with the FDA to align on the Phase III study design and a proposed new primary endpoint instead of IHS4. The discussion focused on reaching consensus on the overall study population and the primary endpoint measure. In September 2021, we announced the outcome of this meeting in which the FDA was supportive of the proposed pivotal study program focusing on patients with active dTs. The FDA also supported a new primary efficacy endpoint that would include measuring the reduction of all three inflammatory lesions associated with HS - inflammatory nodules, abscesses and dTs, called m-HiSCR (modified HiSCR). A m-HISCR responder is defined as, relative to baseline, at least a 50% reduction of ANdT count and at least a 50% reduction of dT count. The FDA provided advice on how to implement, name and validate the meaningfulness of the m-HiSCR for the intended patient population, especially since a reduction in dT count is not adequately captured by the HiSCR. Following the advice received in the Type A meeting, we submitted a full clinical trial protocol for the planned clinical Phase III trial of vilobelimab in HS patients with actively draining disease to the FDA. Upon submission of study protocol for review, we received no comments from FDA within the 30-day and 60-day review periods.
2022年1月,我們啟動了一項隨機、雙盲、安慰劑對照、多中心關鍵性III期研究,以確定vilobelimab在中重度HS和活動性dt患者中的療效 和安全性,其中m—HiSCR為主要終點。2022年2月, 在收到FDA的建議函後,我們暫停了研究,該函指出FDA建議將HiSCR作為III期試驗的主要 終點。FDA建議是在我們提交方案近三個月後提供的,與 之前舉行的A類會議上向我們提供的FDA建議形成對比。然而,FDA沒有發佈臨牀暫停。2022年3月,FDA更正了對我們的建議。在更正的建議函中,FDA表示,與其2022年2月的建議函相反,FDA 不再建議使用HiSCR作為所選患者人羣的主要終點,而是給出了與 實施m—HiSCR終點相關的建議。隨後,我們停止了III期臨牀項目,目前正在評估關於在HS中開發vilobelumab的後續步驟 。
2022年2月,我們還舉辦了一次虛擬研發活動,其中我們披露了對 IIb期SHINE數據的m—HiSCR的事後分析(如下所示)。
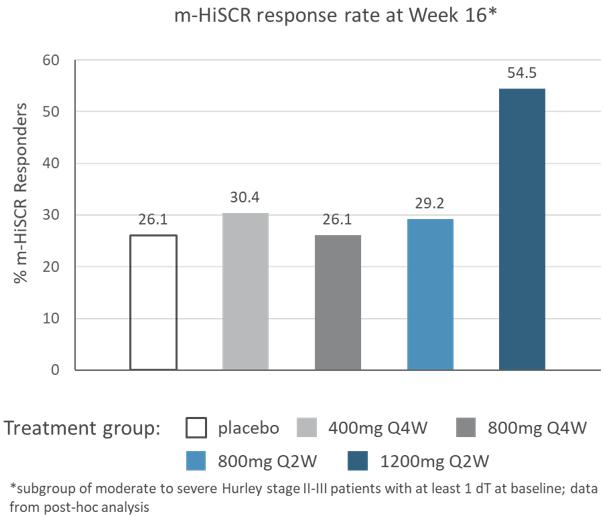
數據與IIb期SHINE研究中,僅在高 劑量組中實現了dt計數顯著降低的事實一致。
基於 成功繼續並完成HS中vilobelimab的III期開發項目所需的後勤和財政努力,我們目前正在評估未來進一步開發的方案,包括與潛在的製藥合作伙伴。
-71-
Vilobelimab 用於治療抗中性粒細胞胞質抗體(ANCA)相關血管炎
ANCA 相關性血管炎,或AAV是一種罕見的,危及生命的自身免疫性疾病,具有複發性,特徵為壞死性血管炎, 血管炎症。該疾病的特徵是危及生命的發作期,影響腎功能和其他 器官,導致器官功能障礙和衰竭,除非適當治療,否則這是一種潛在的致命結局。AAV主要影響 與抗中性粒細胞胞質抗體(ANCA)相關的小血管。它包括三種疾病實體:GPA或肉芽腫病 伴多血管炎(稱為韋格納肉芽腫病);MPA或顯微鏡下多血管炎;EGPA或嗜酸性肉芽腫病 伴多血管炎(稱為Churg—Strauss綜合徵)。
AAV 被指定為孤兒疾病,在美國和歐洲分別影響約40,000名和75,000名患者。 此外,據報道,AAV在美國和歐洲每年分別有4,000名和7,500名新患者。
由於這種疾病具有危及生命的特點,當出現耀斑時,迅速誘導緩解至關重要。誘導緩解的治療方法不同於維持治療。目前誘導緩解的治療方案使用大劑量皮質類固醇或HDCS與利妥昔單抗或環磷酰胺的組合。此外,文冠果最近已獲得批准,可能會被添加到這些療法中。長期的HDCS治療對患者來説伴隨着顯著的副作用和額外的危及生命的風險。
C5a對AAV的促病作用已經確立。C5a對中性粒細胞的啟動作用可能是導致血管內皮細胞中性粒細胞相關損傷的重要因素。此外,與AAV緩解期患者相比,急性AAV疾病患者血漿中補體激活參數顯著升高。在實驗性AAV病小鼠模型中,研究表明,雖然C5aR缺乏會導致疾病活動性降低,但C6缺乏並不會導致這種改善, 這表明MAC的形成可能在這種疾病中不起主要作用。然而,需要更多的研究來證實這一結論。
我們 也一直在開發用於治療AAV的Viloblimab。我們針對AAV的Viloblimab的臨牀開發策略首先將重點放在急性AAV患者身上,我們相信Viloblimab有可能成功誘導緩解,減少或消除對大劑量皮質類固醇(HDCS)治療的需求,並提供更好的安全性。我們還打算將重點放在誘導緩解的速度和降低腎臟替換率和腎功能障礙上。
2021年5月,我們宣佈了一項隨機、三盲、安慰劑對照的II期臨牀研究的TOPLINE結果,該研究使用Viloblimab來評估對中到重度AAV患者的療效和安全性,其中19名患者在美國的中心登記。所有三組患者都接受了由利妥昔單抗或環磷酰胺組成的標準護理劑量治療,並被隨機分為三組,分別接受小劑量維洛昔單抗聯合標準劑量糖皮質激素、大劑量維羅布單抗聯合標準劑量糖皮質激素或安慰劑與標準劑量糖皮質激素聯合治療。這項研究的主要終點是在24周時每個治療組至少經歷一次治療急性腦梗塞的受試者的數量和百分比 。這項研究的關鍵次要終點是伯明翰脈管炎活動評分(BVAS)在第16周下降50% 這一公認的終點已在以前的AAV研究中使用,並伴隨着臨牀緩解。總體而言,維洛利單抗是安全的,耐受性良好,因為觀察到的治療-緊急不良反應反映了疾病和SOC治療。與基線相比,16周達到臨牀反應的患者比例定義為BVAS減少50%(且任何身體系統都沒有惡化),臨牀緩解定義為BVAS=0。儘管這項試驗的樣本量很小,而且很難解釋沒有統計意義的結果,但所有三個治療組的患者在第16周都表現出了強烈的反應,在整個研究過程中,與SOC加安慰劑相比,接受SOC加維羅布利單抗治療的患者在不同的時間點有更多的臨牀緩解。
In November 2021, we announced topline results from a randomized, double-blind, placebo-controlled Phase II clinical study with vilobelimab to evaluate efficacy and safety in patients with moderate to severe AAV, in which 54 patients were enrolled at centers in Europe. The primary endpoint of the study was a 50% reduction in BVAS at week 16. Secondary efficacy endpoints being analyzed include clinical remission, evaluation of the Vasculitis Damage Index, or VDI, reduction of glucocorticoid toxicity index, or GTI, several relevant biomarkers like glomerular filtration rate, and patient reported outcomes. The study was conducted in two parts. In part 1, patients were randomized to receive either vilobelimab plus a reduced dose of glucocorticoids, or placebo plus a standard dose of glucocorticoids. In part 2 of the study, patients were randomized to receive either vilobelimab plus placebo, glucocorticoids or placebo plus a standard dose of glucocorticoids. Patients in both arms received standard of care immunosuppressive therapy, consisting of rituximab or cyclophosphamide. The study achieved its principal objective, demonstrating comparable clinical response of vilobelimab to standard of care, while significantly reducing the need for glucocorticoid (GC) treatment in this life-threatening indication. Clinical response as well as remission were achieved in comparably high rates in all three arms: clinical response at week 16 was observed in 16 out of 18 (88.9%) evaluable patients in the group receiving vilobelimab alone; in 22 out of 23 (95.7%) patients receiving SDGC; and in 10 out of 13 (76.9%) patients in the vilobelimab + RDGC group. The GTI composite score at week 16 was substantially lowered in the vilobelimab only group (mean value of 0.8) when compared to the SDGC group (mean value of 44.9) and the vilobelimab + RDGC group (mean value of 26.1). Assessment of the VDI at week 16 suggested comparable values between groups with the vilobelimab only group showing the lowest value: vilobelimab only group (1.0), SDGC group (1.5) and vilobelimab + RDGC group (1.9). eGFR, a secondary endpoint of the study, demonstrated no medically meaningful changes in all three arms. The vilobelimab only group had the lowest number of reported treatment-emergent AEs, as well as related treatment-emergent AEs.
-72-
我們 目前正在評估在AAV中開發vilobelimab的後續步驟,並考慮在確定後續步驟之前與監管機構討論 美國和歐盟研究的數據。基於成功完成關鍵III期開發計劃所需的後勤和財政努力 ,我們目前正在評估進一步開發的不同方案, 包括與潛在製藥合作伙伴合作。
vilobelimab的其他 臨牀和臨牀前開發
除了 PG、重度COVID—19、cSCC、HS和AAV,我們在上述章節中描述的適應症,我們一直在探索推進 vilobelimab在其他炎症和慢性補體介導的自身免疫性疾病適應症中的臨牀開發的可能性, 存在良好的臨牀前或臨牀概念證明,並且C5a已被證明是關鍵的疾病促進因子 或者在鑑定出類似的機制,如嗜中性粒細胞驅動的影響皮膚和/或其他器官的系統性疾病的情況下。
INF904作為口服給藥抑制C5aR的低分子量分子
Inhibition of the C5a/C5aR axis provides strong anti-inflammatory effects in a variety of diseases. Blockade of C5a using highly specific antibodies, such as vilobelimab, may offer a fast, effective, and safe way to control C5a-induced inflammation. In addition to this approach, inhibition of C5aR by oral small molecules may provide the ease of administration required for effective long-term treatment for more chronic inflammatory diseases. To expand the breadth of our anti-C5a/C5aR technologies, we are also developing INF904, an oral, small molecule drug candidate that targets the C5aR receptor. C5aR, a G-protein-coupled-receptor expressed primarily by granulocytes, mediates the pathophysiological effects of C5a. In INF904, we discovered a small molecule C5aR inhibitor that in pre-clinical studies has shown potential for superior characteristics to the only approved C5aR inhibitor, avacopan. INF904 has provided higher plasma exposure in animals, including non-human primates, and improved inhibitory activity in a hamster neutropenia model compared to avacopan. Furthermore, in contrast to avacopan, in vitro experiments showed INF904 has substantially less inhibition of the cytochrome P450 3A4/5 (CYP3A4/5) enzymes, which play an important role in the metabolism of a variety of drugs, including glucocorticoids. No obvious toxicological findings, even in the highest dose groups tested in required GLP toxicity analyses, were identified. INF904 demonstrated potential for anti-inflammatory therapeutic effects in several preclinical disease models.
所有 IND啟用研究(包括某些GLP毒理學研究)均已完成,我們於2022年11月至2024年1月進行了一項I期單次和多次遞增劑量臨牀研究。
2023年9月,我們宣佈了一項隨機、雙盲、安慰劑對照 INF904 I期試驗的單次遞增劑量或SAD部分的頂線結果。
I期首次人體試驗的 SAD部分入組了6個不同劑量組的62名健康志願者(從3 mg到240 mg),他們 被隨機分配接受INF904或安慰劑。對60 mg給藥組檢測了不同的藥物濃度。主要 目的是評估空腹條件下單次遞增劑量的安全性和耐受性。次要終點包括幾個 藥代動力學或PK參數,還探索了INF 904對來自治療志願者的血樣中C5a誘導的中性粒細胞活化的影響 。
結果表明,INF904在接受治療的患者中耐受性良好, 3 mg—240 mg單次給藥時沒有引起關注的安全性信號。與安慰劑組相比,INF904治療患者中不良事件(AE)的總體百分比更低 ,並且在任何劑量水平下均未觀察到嚴重或重度AE。未報告與INF904給藥相關的AE。
-73-
受試者血漿樣本中INF904 PK的 分析顯示,持續暴露於INF904,達到最大濃度6小時,或t最大值. INF904血漿水平與全身暴露量(AUC)成劑量比例最後的)且與最大濃度幾乎成劑量比例 (C最大值)在研究中使用的劑量範圍內。在30 mg劑量下,INF904達到C最大值289 ng/ml,AUC最後 為5197 h.ng/ml,分別比 上市對照藥物avacopan發表的I期數據高約3倍和10倍。
30 mg或更高劑量的INF904單次給藥實現了≥ 90%的C5a誘導受試者血漿樣本中中性粒細胞活化標誌物CD11b上調 離體給藥後24小時。當加入12.6nM重組C5a作為刺激物時,在該測定中實現了這種抑制,C5a濃度可在患有嚴重炎症性病症的患者中觀察到,例如免疫皮膚病、化膿性汗腺炎,或在危及生命的炎症期間(例如,重症COVID—19患者 或膿毒症患者)。因此,INF904抑制人血漿中C5a誘導的中性粒細胞活化達到了在疾病相關C5a水平下有效控制C5aR的設定目標。
2024年1月,我們宣佈了多劑量遞增(MAD)的頂線結果,這是INF904隨機、雙盲、安慰劑對照 I期試驗的一部分。PK和藥效學或PD參數證實了我們在 研究SAD部分觀察到的有利數據,這為INF904的同類最佳潛力提供了支持。INF904耐受性良好,在整個試驗劑量範圍內,受試者重複給藥後未發生安全性不良事件 。
在隨機化、雙盲、安慰劑對照I期試驗的MAD部分,24名受試者接受了14天的INF904多次給藥,劑量為30 mg/天一次,或QD、30 mg/天兩次,或BID或90 mg BID。該研究的主要目的是評估 重複給藥的安全性和耐受性。將幾個PK參數作為次要終點進行了分析,並在離體試驗中探索了給藥方案 對參與者血樣中C5 a誘導中性粒細胞活化的影響。
I期研究MAD部分的 安全性分析表明,在整個 劑量範圍內,INF904在受試者中耐受良好,沒有引起關注的安全信號。INF904治療受試者中AE的總體百分比為77.8%, 低於安慰劑組中觀察到的83.3%。在任何劑量水平下均未觀察到嚴重或重度AE。
PK特徵分析 顯示潛在目標AUC0-12h, C最大值在30 mg BID給藥14天內迅速達到谷值。INF904暴露量隨着劑量高達90 mg BID而進一步成比例增加。即使參與者在空腹狀態下攝入藥物,也證明瞭這些結果 ,這表明不需要食物來達到潛在的治療藥物水平 。
PD特徵分析 顯示,在體外激發試驗中,在14天給藥期內,對於所有試驗劑量,INF904對C5a誘導的中性粒細胞活化的阻斷活性達到等於或高於90%,其中將生理和疾病相關水平的C5a添加到試驗參與者提供的血樣中 。
與此同時,我們在商業上可行的INF904配方的開發方面取得了進展,我們計劃在2024年底將其引入第二階段開發。
我們 目前正在進行額外的必要臨牀前研究,包括長期慢性毒理學研究,以便 長期給予INF904治療慢性炎性疾病。我們目前計劃在2024年底啟動一項短期給藥II期研究 ,隨後在2025年啟動一項長期給藥II期研究。我們最初計劃開發INF904用於治療兩種初始免疫皮膚病學適應症:HS和CSU。
-74-
IFX002作為後續抗C5a單克隆抗體候選產品
為了 擴大我們抗C5a技術的廣度,我們還開發了用於治療慢性炎症適應症的IFX 002。 IFX002是抗C5a技術的一個進步。它是一種高效抗C5a抗體,具有更高的人源化等級和改變的 藥代動力學特性,目前正在臨牀前開發。
IFX002是一種可注射的候選產品,其血漿半衰期比VILOBLIMAB更長,這使得它可能更適合治療病情不那麼嚴重或更接近疾病發病的慢性炎症適應症。IFX002與Viloblimab具有相同的作用機制,因為它具有高度特異性地阻斷C5a的潛力,但設計的劑量方案可能更適合於慢性治療。此外,IFX002結合了與Viloblimab相同的遊離C5a表位,具有類似的選擇性。 IFX002的臨牀前開發部分得到了德國政府的撥款支持。IFX002將保持性能相關的 特性,以完全阻止C5a誘導的生物效應,同時保持MAC形成的完整性。我們認為IFX002具有治療各種慢性炎症性疾病的潛力,並可能受益於更適合慢性治療的給藥方案。考慮到IFX002的剩餘專利壽命很長,我們還認為 IFX002是viloblimab的生命週期管理產品。
管道
我們 打算利用我們在補體領域的專業知識以及我們的專有技術,通過開發專注於補體介導的自身免疫和炎症性疾病的多樣化渠道,保持我們在抗C5a/抗C5aR領域的領先地位,這些疾病具有高度未得到滿足的需求。根據我們授予的最新專利,我們專有的抗C5a/抗C5aR技術的權利 目前預計將至少延長到2041年。
下圖 彙總了有關我們當前候選產品渠道的關鍵信息和開發狀況:
我們的戰略
我們的 目標是保持並進一步提升我們在抗C5a/抗C5aR補體領域的領先地位,為市場提供一流的自身免疫和抗炎治療 。為了實現這一目標,我們希望執行下述戰略。
| ● | 在PG中提升 viloblimab。基於肯定結束的開放標籤IIa期研究,在收到FDA提供的與臨牀試驗設計相關的建議後,我們正在進行第三期關鍵臨牀計劃。 |
-75-
| ● | 繼續進行INF904的臨牀開發。我們已經對我們的C5aR拮抗劑INF904進行了單劑量和多劑量遞增 劑量研究,目前正在進行其他所需的非臨牀研究,以進入患者的II期試驗。 |
繼續 以優化viloblimab的製造流程。我們建立了經過充分驗證的Viloblimab生產流程,並建立了信譽良好的CDMO,目標是滿足質量標準,以獲得監管部門對此類流程的批准。我們正在 在德國建立藥品成品的最終制造(即“填充和完成”),並正在 考慮將藥物物質的製造工藝從中國轉移到德國或其他國家。
評估用於HS、CSCC和AAV的viloblimab的 開發選項。由於自行開展這些開發計劃所需的資源,我們決定停止這些開發計劃後,我們目前正在評估有關在HS、CSCC和AAV中開發Viloblimab的選項。 根據成功完成這些 適應症中的每個關鍵階段開發計劃所需的後勤和財務努力,此類選項包括與製藥合作伙伴的潛在合作。
| ● | 尋求IFX002的進一步開發,為潛在的臨牀開發做準備。 我們正在開發IFX002作為一種半衰期比VILOBLIMAB更長的注射劑,使其適用於病情不太嚴重或更接近發病時間的慢性炎症指徵。基於專利壽命可能超過2040年,我們將該項目 視為viloblimab的生命週期管理,並正在進行臨牀前開發工作 ,以更接近臨牀開發的可能開始。 |
| ● | 通過充分利用我們在補體和炎症研究方面的專有技術和專業知識,鞏固並繼續擴大我們在抗C5a/抗C5aR領域的領導地位的廣度 。我們打算在內部或與合作伙伴合作,繼續發現和開發有潛力的治療方法,以解決補體介導或免疫應答 介導的具有重大未滿足需求的廣泛適應症。為了實現這一目標,我們繼續通過位於密歇根州安娜堡的發現部門來補充我們的研發活動 ,我們正在進一步擴大我們的知識產權組合和業務開發能力。 |
根據授予的EUA單獨或與製藥合作伙伴合作將 GOHIBIC(Viloblimab)商業化。我們正在將用於嚴重新冠肺炎的Gohibic(Viloblimab)在美國獨立商業化,並評估在歐洲獨立或與潛在合作伙伴合作將該藥物商業化的選項。我們採用了有針對性的商業基礎設施,通過在美國這些核心市場治療患有這種疾病的患者的卓越中心,促進獲得維羅貝利單抗。在美國和歐洲以外,我們可以單獨或與其他公司合作,尋求Viloblimab for Severe新冠肺炎的批准和商業化。對於其他適應症,我們打算獨立或通過與其他各方合作開發和商業化viloblimab。
將Viloblimab推向市場批准用於嚴重新冠肺炎:繼續歐洲藥品管理局對MAA的批准流程,並向FDA提交完整的BLA 。
| ● | 探索將viloblimab的應用擴展到相關疾病的可能性。如果我們 在美國或歐洲獲得監管部門批准在重症新冠肺炎中使用viloblimab,我們打算探索將該標籤擴展到我們過去為其生成臨牀前數據的其他關鍵 護理適應症的可能性。最值得注意的是,我們可能會考慮進行更多的研究,可能會與政府機構或商業合作伙伴合作,將該標籤擴展到病毒誘導的ARDS產品。 |
我們的知識產權
我們 旨在通過尋求和維護 旨在涵蓋我們的產品候選和組合物及其使用方法、製造它們的方法、相關治療靶點和相關治療方法以及對我們的業務具有商業重要性的任何其他發明的美國和外國專利,來保護我們的候選產品和其他具有商業重要性的抗C5a和C5aR專利技術。我們還依靠商業祕密、技術訣竅和其他知識產權來保護我們業務中不受專利保護或我們認為不適合專利保護的方面。此外,我們的目標是通過尋求和維護美國和外國商標註冊來保護我們的商標、服務標記和商號。我們的成功將在很大程度上取決於我們是否有能力獲得和維護此類專利和其他專有保護,保護和執行我們的專利,保護我們的商業祕密,並在不侵犯、挪用或以其他方式侵犯任何專利或其他知識產權的情況下運營我們的業務 ,包括任何第三方的專有權利。有關更多信息,請參閲標題為“項目3.關鍵信息 -3.風險因素-與我們的知識產權有關的風險”一節。
-76-
截至2023年12月31日,我們擁有10項已批准的美國專利和5項未決的美國非臨時專利申請,34項外國專利,包括在88個國家和地區驗證的4項歐洲專利和2項在16個國家驗證的歐亞專利,以及84項外國專利申請,包括8項歐洲專利申請和3項歐亞專利申請,涵蓋C5a和C5aR抑制劑及其相關使用方法。
截至2023年12月31日,我們與Viloblimab、IFX002和INF904相關的專利組合摘要如下。
截至2023年12月31日,我們擁有四項已頒發的美國專利,涵蓋阻斷C5a的抗體成分及其在涉及急性或慢性炎症的疾病患者中使用 阻斷C5a誘導的生物效應,這將包括 在其範圍內HS和AAV。此外,我們擁有20項已頒發的外國專利,包括兩項已在74個國家和9個國家驗證的已授權歐洲專利 和一項在9個國家驗證的歐亞專利;兩項待決的外國專利申請,包括一項正在申請的歐洲專利; 涵蓋阻斷C5a的抗體的成分及其在治療各種涉及急性或慢性炎症的疾病中的用途,這將包括HS和AAV,並且根據適用專利的管轄權, 具體涵蓋此類抗體在治療缺血和再灌注相關損傷、急性肺損傷和肺炎等疾病中的使用。
已頒發的美國和外國專利預計將於2030年到期,不包括任何額外的專利期調整或專利期延長 。如果獲得批准,未決的美國和外國專利申請預計將於2030年到期,不包括任何額外的專利期限調整或專利期限延長。
截至2023年12月31日,我們擁有兩項授權的美國專利和四項授權的外國專利,包括一項在三個國家和地區驗證的歐洲專利和一項外國專利申請,其中包括使用某些結合部分(如抗體)來抑制C5a治療病毒性肺炎。
如果頒發 ,美國和外國的專利預計將於2035年到期,不包括任何額外的專利期限調整或專利 期限延長。
截至2023年12月31日,我們擁有三項已授權的美國專利,一項未決的美國非臨時專利申請,八項已授權的外國專利,包括一項在12個國家驗證的歐洲專利和一項在八個國家驗證的歐亞專利,25項未決的外國專利申請,包括三項未決的歐洲專利申請,涉及使用C5a活性抑制劑,例如維洛布利單抗,用於治療HS和其他皮膚、中性粒細胞炎症性疾病。
已頒發的美國和外國專利預計將於2038年到期,不包括任何額外的專利期調整或專利期延長 。
截至2023年12月31日,我們擁有一項美國專利申請和16項外國專利申請,其中包括一項歐洲專利申請 ,涉及一種改進的C5a特異性抗體。
如果頒發 ,美國和外國專利預計將於2041年到期,不包括任何額外的專利期限調整或專利 期限延長。
截至2023年12月31日,我們擁有一項未決的美國非臨時專利申請和16項外國專利申請,包括 一項歐洲專利申請,涉及使用C5a活性抑制劑,例如Viloblimab,用於治療冠狀病毒 疾病。
如果 頒發,基於PCT申請的美國和外國專利預計將於2040年到期,不包括任何額外的專利期限調整或專利期限延長。
截至2023年12月31日,我們擁有一項已授權的美國專利,一項未決的美國非臨時專利申請,以及18項外國專利申請,包括一項涉及C5aR抑制劑的歐洲專利申請。
-77-
已頒發的美國和外國專利預計將於2040年到期,不包括任何額外的專利期調整或專利期延長 。
截至2023年12月31日,我們擁有一項美國專利申請和一項國際專利申請,涉及C5a活性抑制劑和護理標準抑制劑在治療感染性肺炎和傳染性ARDS中的使用。
如果頒發 ,美國和外國的專利預計將於2043年到期,不包括任何額外的專利期限調整或專利 期限延長。
截至2023年12月31日,我們擁有兩項歐洲專利申請,涵蓋使用C5aR抑制劑治療各種疾病。
如果頒發 ,美國和外國的專利預計將於2043年到期,不包括任何額外的專利期限調整或專利 期限延長。
截至2023年12月31日,我們在美國和29個國家和地區擁有兩個商標“GOHIBIC”和“Vilway si”的商標註冊 ,其中包括23個歐洲國家和地區,以及7個藥品和服務領域的外國商標申請。
截至2023年12月31日,我們在美國擁有兩個用於治療炎症性、炎症相關、腫瘤和神經疾病的藥物製劑領域的商品和服務的“InflRx”商標註冊。 在美國以外,截至2023年12月31日,我們在30個國家/地區擁有“InflRx”的商標註冊。
UPC
除波蘭、西班牙和克羅地亞外,歐盟27個成員國中已有24個成員國加入了《統一專利法院協定》,該協定規定了單一專利或以上專利,並導致了統一專利法院的成立。UPCA已於2023年6月1日生效。24個成員國中有17個在《聯合國兒童權利公約》生效時完全批准了該公約,並從一開始就參加了該公約。其餘七個歐盟成員國預計將在未來幾年內加入。自UPCA開始以來,傳統的歐洲專利可以在UPC集中執行,但也可能在UPC面臨中央撤銷程序,除非它被選擇退出新法院系統的管轄權。
UPC在與歐洲專利、單一專利、為此類專利所涵蓋的產品頒發的補充保護證書和歐洲專利申請相關的民事訴訟方面擁有專屬管轄權(詳情請參閲《美國專利保護法》第32條)。因此,除非選擇退出,否則UPC不僅有權審理與UPS有關的案件,還有權審理與歐洲傳統專利申請 和專利(下稱“歐洲專利(S)”)有關的案件。UPC在一個國家的決定對參與UPCA的所有其他州 具有約束力。然而,該決定對沒有參加UPCA的歐盟成員國以及不是歐盟成員國的EPC締約國(例如英國和瑞士)沒有任何法律約束力,因此 沒有資格參與新系統。
一般認為,在評估已授予專利的有效性和侵權方面,UPC對專利持有人都是友好的,專利持有人在UPC而不是在歐洲專利局和/或國家無效法院或民事法院強制執行和捍衞其專利可能是有益的。然而,目前還沒有UPC的判例法允許預測UPC 在決定中心撤銷訴訟或撤銷反訴時可能如何適用可專利性標準,或者它將如何解釋中心侵權訴訟中的索賠。鑑於這些不確定性,一方面是任何新制度所固有的,另一方面是歐洲專利局成熟的反對做法,以及國家無效和侵權訴訟,我們認為 從UPC選擇退出所有歐洲專利申請和授予的歐洲專利,並 遵循UPC的判例法來確定UPC制度的利弊是一種謹慎和值得推薦的方法。如果UPC似乎對有效性和/或侵權做出了比歐洲專利局和國家法院更有利的裁決,則應重新考慮是否加入之前已選擇退出的一項或多項歐洲專利或專利申請。
-78-
我們的協作 協議
共同開發 協議與史泰森(北京)生物製藥有限公司(北京德豐瑞生物科技有限公司(BDB)的前身)
2015年12月,我們與北京德豐瑞生物科技有限公司 簽訂了一項共同開發協議,即共同開發協議,用於北京德豐瑞生物技術有限公司在中國銷售的候選藥物的開發。 根據協議,我們授予北京德豐瑞生物科技有限公司獨家、不可轉讓的許可,允許其僅在中國生物科技有限公司的候選藥物BDB-001和BDB-002以及與特定靶點結合或相互作用的分子中開發和商業化使用Viloblimab細胞系和相關知識產權。或靶標結合分子。
根據協議,我們有權從包含BDB-001或BDB-002的BDB產品的淨銷售額中獲得中位數百分比的版税。我們保留在中國研發和製造Viloblimab和IFX002的權利,僅用於在中國以外的地方將 產品商業化,並將在中國的Viloblimab和IFX002細胞系用於非商業目的。在我們獲得中國以外的監管機構批准使用Viloblimab或IFX002進行產品商業化的範圍內,如果我們沒有尋求監管部門批准我們使用Viloblimab或IFX002作為指示 ,並且我們沒有尋求監管部門批准中國提供相同或基本上類似的指示的BDB-001或BDB-002, 通過向BDB提供書面通知,我們可以選擇尋求監管部門批准將中國相關指示 中的該等產品商業化。如果我們行使這項權利,我們將被要求按此類產品的淨銷售額支付BDB中位數個位數百分比的版税。
根據共同開發協議,BDB有權使用viloblimab細胞系生產抗C5a抗體,即BDB-001。BDB-001只能由BDB在中國的中國進行商業化,InflRx不直接參與BDB-001的開發,這仍然是BDB的唯一責任。根據共同開發協議,InflRx擁有中國以外的所有全球商業權利,擁有開發BDB-001所產生的任何和所有發現。為了支持BDB 開發BDB-001,在2020年,InflRx允許BDB在西班牙、印度、印度尼西亞和孟加拉國進行BDB-001的臨牀研究。然而,InflRx仍然是中國之外的BDB-001的所有商業權的唯一所有者,包括在BDB正在進行臨牀試驗的國家。北控無權尋求營銷授權,也無權在中國之外對北控-001進行商業開發。維洛布單抗不是北京開發銀行在中國進行臨牀試驗的產品。相反,它是BDB自己的抗體,稱為BDB-001。
此外,我們保留在中國之外將含有BDB-001和BDB-002的產品商業化的權利,對於這些適應症,我們 選擇不商業化Viloblimab或IFX002。在我們行使這項權利的範圍內,我們將被要求按我們這類產品的淨銷售額支付BDB較低的個位數 百分比的版税。
Bdb 必須毫不拖延地通知我們它對靶標結合分子進行的測試。如果任何此類測試的結果與目標的結合或相互作用都令北京開發銀行和我們滿意,北京開發銀行必須將該結果通知我們,並可在通知後 後六個月內行使選擇權,開始將中國成功測試的目標結合分子商業化。在BDB行使這種選擇權的範圍內,BDB將被要求為含有 此類靶標結合分子的產品的淨銷售額支付較低的個位數百分比版税。BDB還允許我們利用中國以外的任何靶向結合分子,或者,如果BDB沒有在相同或基本上類似的適應症中尋求監管批准,則在中國。在我們行使此類權利的範圍內,我們將被要求按此類產品的淨銷售額支付BDB低至中個位數百分比的版税。
2021年11月,我們與BDB和Staidson(北京)生物製藥有限公司(簡稱Staidson)簽訂了共同開發協議的第二份附錄。根據第二份增編,BDB作為Staidson的全資附屬公司,將共同開發協議及其下的所有權利和義務轉讓給Staidson。
-79-
2022年12月,我們修改了與Staidson的現有聯合開發協議,以支持Staidson監管部門努力 批准其候選專利藥物BDB-001用於治療中國的新冠肺炎。根據修正案,我們將在中國治療新冠肺炎的BDB-001淨銷售額的基礎上獲得10%的特許權使用費。我們向Staidson授予了在 中國中使用的獨家許可,以獲得我們有關Viloblimab的某些臨牀、製造和監管數據,以支持和促進監管 向中國國家醫療產品管理局(簡稱NMPA)提交BDB-001用於治療重症新冠肺炎患者的申請。根據現有的共同開發協議,斯泰德森正在開發BDB-001,用於治療中國的嚴重新冠肺炎和其他炎症性疾病。該協議繼續有效,除非提前終止。經雙方同意,本協議可終止,或在收到違約通知後30天內未得到糾正的另一方違約時,由另一方終止。此外,如果任何一方對終止方對根據協議許可給非終止方的任何知識產權的所有權提出質疑,或發生某些破產或資不抵債事件,任何一方均可終止協議。
在修訂共同發展協議的同時,吾等亦與Staidson的聯屬公司及根據香港法律成立的有限責任公司Staidson Hong Kong Investment Company Limited訂立購股協議,根據該協議,Staidson Hong Kong Investment Company Limited以每股5.00美元的價格向吾等購入總值250萬美元(230萬歐元)的普通股,從而出售500,000股股份。購股協議還包括一項選擇權,根據該選擇權,施泰森香港投資有限公司可酌情購買額外普通股,總金額為額外750萬美元。後續購買的期權將在施泰德森獲得中國對BDB-001的監管批准的12個月週年紀念日到期。該等後續投資將以每股5.00美元或較該等後續投資截止日期前15個交易日的加權平均股價溢價20%的價格進行。
臨牀 與默克公司的試驗協作和供應協議。
在2020年3月20日,我們與Merck&Co.,Inc.(在美國和加拿大以外稱為MSD)簽訂了一項臨牀試驗協作和供應協議,以評估Viloblimab和默克的抗PD-1療法KEYTRUDA的組合®1 (Pembrolizumab)用於CSCC患者。根據協議條款,我們使用兩個Viloblimab 手臂進行第二階段臨牀研究,其中一個使用Pembrolizumab。2023年11月,我們宣佈在CSCC中停止開發viloblimab,以優先考慮其他 計劃。根據協議,目前仍在接受治療的患者將接受長達24個月的治療;但不會有新的患者加入研究,目前沒有患者正在接受治療的臨牀站點將被關閉。
銷售 和市場營銷
2023年6月,我們開始在美國將GOHIBIC(Viloblimab)商業化,用於治療住院的成人新冠肺炎,在接受IMV或ECMO後48小時內啟動。關於商業化的開始,我們與Cencora的某些子公司簽訂了 協議,作為集團的美國分銷商,並根據EUA向美國醫院客户提供GOHIBIC(Viloblimab) 供訂購。Cencora提供冷藏、冷鏈配送服務、庫存管理和二次貼標/包裝等服務。
為了支持我們的商業努力,我們已經並將繼續聘請在醫院市場醫療產品商業化方面具有相關經驗的美國專家,包括在銷售、銷售運營、市場營銷、市場準入、分銷、醫療事務和其他領域。此外,在內部和外部服務提供商的協助下,我們正在擴展必要的基礎設施,包括IT系統、供應鏈、財務報告系統和庫存管理系統。
隨着我們擴大我們的商業努力,我們繼續加強我們的商業戰略計劃,進一步擴大我們的銷售隊伍和醫療事務團隊,準備針對醫療保健提供者和其他利益相關者的相關宣傳和醫學教育材料, 完善我們的醫療事務戰略,以提高醫學界對EUA的認識,並進一步推動我們的銷售努力。
| 1 | Keytruda®是默克·夏普·多姆公司的註冊商標,默克公司是默克公司的子公司,位於美國新澤西州凱尼爾沃斯。 |
-80-
2023年,我們自成立以來首次確認產品銷售收入為63,089歐元。報告的收入 是對美國最終客户(醫院)的銷售。
在截至2023年12月31日的12個月內,集團產生了400萬歐元的銷售和營銷費用。這些費用主要由100萬歐元的人事費用和190萬歐元的外部服務組成,用於分銷Gohibic。集團於2023年4月獲得歐盟協議後,開始了商業化活動。在此之前,未發生任何銷售和營銷費用 。
如果獲得適用監管機構的批准,我們 還打算通過採用有針對性的商業基礎設施,通過在這些核心市場治療PG的卓越中心來促進對viloblimab的使用,從而獨立地在美國和歐洲推動用於PG的viloblimab的商業化。我們相信,這樣的組織將能夠解決醫生社區的問題,他們是治療正在為其開發Viloblimab和任何其他候選產品的患者羣體的關鍵專家。該組織的職責 將包括制定與經批准的產品有關的教育計劃,並與PG和任何其他相關醫學領域的關鍵專家建立關係。還將評估與擁有成熟商業基礎設施的較大組織協作的選項。
我們 也可以考慮在其他適應症中實現viloblimab的商業化,或者獨立地將我們其他開發的產品商業化 。不過,我們也在考慮與較大的公司建立潛在的合作伙伴關係,這些公司在不同領域擁有更成熟的基礎設施和更多的財務資源,包括在銷售和營銷方面。
製造業
我們 目前不擁有或運營用於生產臨牀或商業批量候選產品的製造設施。 我們打算依靠現有的第三方合同製造商來生產我們的產品,並打算招聘更多具有 經驗的人員來管理生產我們的候選產品和其他候選產品或我們未來可能開發的產品的第三方合同製造商。此外,我們還聘請了德國、美國和其他 國家的其他第三方製造商進行與潛在銷售相關的活動,例如,在美國和其他地方對我們批准的任何產品進行包裝和標籤。我們持有製造和進口許可證,並通過內部運行關鍵的免疫釋放檢測來參與viloblimab的藥物產品釋放程序,使我們只能釋放證明必要的、 預先指定的高生物阻斷活性的藥物產品批次。因此,我們負責監督整個製造過程,並與我們合格的人員一起發佈 最終的灌裝成品。
競爭
生物製藥行業的特點是技術快速進步,競爭激烈,並高度重視專利產品 。雖然我們相信我們的技術、知識、經驗和科學資源為我們提供了競爭優勢,但我們面臨着來自許多不同來源的潛在競爭,包括主要的製藥、專業製藥和生物技術公司、學術機構和政府機構以及公共和私人研究機構。我們成功開發和商業化的任何候選產品都將與現有療法和未來可能推出的新療法競爭。
壞疽膿皮病的競爭
目前主要市場上還沒有批准用於治療PG的藥物。當地批准的唯一治療方法是阿達利單抗,它已在日本獲得批准,但沒有在其他國家根據當地進行的小型臨牀試驗獲得批准。然而,由於與疾病相關的醫療需求很高,某些藥物在常規醫療實踐中被用作受影響患者的治療嘗試。這些藥物包括某些口服藥物,如免疫抑制劑,包括環孢素或皮質類固醇,或抗生素,如氨苯碸。此外,局部應用他克莫司在某些情況下也會使用。最後,還使用靜脈注射的腫瘤壞死因子-α抑制劑,如英夫利昔單抗或阿達利單抗或其他生物藥物,儘管沒有正式的監管批准。
-81-
截至本文發佈之日,據我們所知,正在進行的臨牀試驗中的其他治療方法包括:
| ● | 口服巴利替尼,一種Janus激酶-1和Janus激酶-2,或JAK1/JAK2抑制劑,目前正在進行一項開放標籤的第二階段概念驗證研究 |
| ● | 靜脈注射白介素36受體或白介素36R的單抗目前正在進行二期開放標記單臂研究 |
| ● | 口服去克拉維替尼,一種Janus激酶-2或JAK2抑制劑,目前正在對10名患者的I期單臂進行研究 |
| ● | 皮下注射Canakinumab,一種抗IL-1β或IL-1β的單抗,用於24名PG亞型PG患者的II期臨牀試驗,這是一種化膿性不育性關節炎壞疽膿皮病和痤瘡綜合徵(PAPA)綜合徵 |
此外, 以下開發已終止、完成或放棄,並且在最近 年中未進入註冊第三階段試驗或在以前的臨牀試驗中失敗:
| ● | 在三項臨牀III期試驗(共招募16、15和9名患者)和一項II期研究(共招募8名患者)中, 給予吉伐珠單抗(一種抗白細胞介素—1 β或IL—1 β)單克隆抗體 |
| ● | 在一項入組10名患者的臨牀II期研究中, 注射bermekimab,一種抗白細胞介素—1 α或IL—1 α單克隆抗體 |
| ● | 在一項臨牀II期試驗中,入組 4名患者後,Subject 給予ixek珠單抗(一種抗白細胞介素—17 α或IL—17 α)單克隆抗體 |
| ● | 在一項臨牀II期試驗中, 口服給予etramod,(APD 334),一種選擇性鞘氨醇—1—磷酸,或S1P—1受體調節劑, 入組兩名患者 |
| ● | 皮下注射 烏司奴單抗(一種靶向白細胞介素—12或IL—12和白細胞介素—23或IL—23的共享p40亞基的單克隆抗體), 在一項單例患者病例報告研究中描述了在接受慢性免疫抑制治療的患者中成功治療 |
| ● | 在一項單患者病例報告研究中,在其他全身治療失敗後,Subject給予了阿基諾尤單抗(一種抗白細胞介素—17 α或IL—17 α單克隆抗體) |
如果 獲批用於治療PG,vilobelimab將可能面臨來自目前使用的治療方法的競爭,如糖皮質激素、 環孢菌素或其他免疫抑制療法,如阿達木單抗、英夫利西單抗或其他療法。
治療重症、有創機械通氣COVID—19患者的競爭
如果 獲批用於治療重症、有創機械通氣COVID—19患者,維洛貝利單抗將面臨 目前使用或批准的治療藥物的競爭,如皮質類固醇、白細胞介素—1或IL—1、抑制劑阿那白素、IL—6抑制劑(如託珠單抗)、JAK抑制劑(如巴里西替尼)和抗血栓治療。鑑於COVID—19大流行導致了對有效治療的高醫療需求 ,許多不同的治療實體和靶點已經或仍在評估,以治療該患者人羣 。雖然在特定患者人羣中進行臨牀試驗是能夠 獲得該特定患者人羣治療的監管批准的先決條件,但已在其他 患者人羣中進行了多項臨牀試驗(例如,住院患者,而不是我們的目標亞組重症患者,有創機械通氣患者 ),並且這些試驗的結果已經部分地外推到我們的目標人羣中。這些治療均未接受 EUA,我們認為,為了獲得BLA的全面批准,這些治療需要在隨機 對照臨牀試驗中顯示臨牀療效。因此,我們認為競爭將主要由為預期用途人羣開發的產品所面臨。
-82-
目前或之前正在研究或完成的重度COVID—19治療,包括重症機械通氣患者的治療,包括:
| ● | 口頭 在204名住院患者中,服用了一種微管破壞劑沙比扎布林,完成了一項III期研究 2019冠狀病毒病患者發生急性呼吸窘迫綜合徵或死亡的高風險,減少了55% 在第60天的死亡率。2022年11月9日FDA肺過敏藥物諮詢委員會 以8票對5票的投票結果表明,沙比扎布林的已知和潛在益處並不超過已知的。 和沙比扎布林的潛在風險。2023年3月2日,試驗申辦方Veru Pharmaceuticals Inc. FDA宣佈拒絕為sabizabulin授予EUA。 |
| ● | 內部操作 給予nangibotide,一種合成肽和骨髓中的第一類觸發受體 細胞—1或TREM—1抑制劑在COVID—19 ICU患者中的隨機對照II期試驗中。 本研究的結果顯示,28天全因死亡率相對降低了43% 分析的患者羣體。 |
| ● | 內部操作 給予依庫珠單抗(一種C5的單克隆抗體抑制劑)已處於開放標籤狀態 在接受持續氣道正壓通氣的COVID—19感染患者中進行的II期試驗, 或者CPAP呼吸機支持在本次非隨機化研究中,只有10名患者接受了依庫珠單抗治療 與52名患者進行比較。依庫珠單抗安全且耐受性良好,但未顯示 這對降低這一相對較小的患者死亡率產生了重大影響。無額外 已經報道了在該患者羣體中使用依庫珠單抗的研究。 |
| ● | 內部操作 在患者II期研究中給予ravulizumab(一種C5單克隆抗體抑制劑) 患有新型冠狀病毒嚴重肺炎、急性肺損傷或急性呼吸窘迫綜合徵 後,本研究已停止 中期分析,沒有結果。 |
| ● | 內部操作 在研究者發起的一種抗C5aR抗體Avdoralumab(IPH5401), 一項在COVID—19重度肺炎患者中與安慰劑進行的雙盲、隨機化II期研究。 入組了208名患者,該項目在試驗結束後於2021年7月停止 沒有達到主要終點。 |
| ● | 內部操作 給予asunercept,CD95—Fc融合蛋白,特異性結合並有效 在438名患有 的患者中進行的雙盲、隨機II期研究中, 2021年10月完成的重度COVID—19,顯示出對某些結局的療效 措施雖然尚未發佈完整數據集,但asunercept嘗試在 中進一步開發 第三階段研究,636名患者。2023年8月18日,研究終止,到期 只有34名患者被招募。 |
| ● | 內部操作 AMY—101,一種靶向補體因子C3的環肽,在II期臨牀中 評估因COVID—19感染導致的ARDS患者安全性和有效性的試驗 沒有達到其主要療效終點。 |
| ● | 內部操作 給予APL—9,一種特異性靶向C3的蛋白質,補體中的一種中心蛋白質 在一項治療嚴重COVID—19患者的I/II期研究中, 在獨立數據監測委員會進行中期分析後,未發現有意義的減少 可以觀察到APL—9治療患者的總死亡率,因此 研究提前結束。 |
cSCC中的競爭
如果 批准用於程序性死亡—1、或PD—1和程序性死亡配體—1、或PD—L1抑制劑、耐藥/難治、局部晚期或 轉移性cSCC,維洛貝利單抗將面臨目前使用的治療藥物如表皮生長因子受體或EGFR、 抑制劑如西妥昔單抗、化療藥物如順鉑、多柔比星、紫杉烷、吉西他濱、甲氨蝶呤和5—氟尿嘧啶的競爭, 或5—FU,以及局部應用的產品,如咪喹莫特或替巴尼布林,即使其中一些治療並不全部批准 用於該適應症。
-83-
此外,FDA批准了兩種PD—1抑制劑用於治療局部晚期或轉移性cSCC。Pembrolizumab(一種靶向 PD—1的單克隆抗體)適用於無法通過手術或放射治療治癒的複發性或轉移性cSCC,以及cemiplumab(一種靶向PD—1的單克隆抗體 )適用於不適合治療性 手術或放射治療的患者的轉移性cSCC或局部晚期cSCC。
目前正在調查的其他 治療方法包括:
| ● | 在一項已完成的註冊使能試驗中,內注射cocibelimab (一種靶向PD—L1的單克隆抗體),顯示總緩解率 或ORR為54. 8%,於2023年1月提交給FDA,FDA於2023年3月接受。2023年12月,FDA發佈了一份完整 回覆函,原因是第三方CMO發現。儘管如此,該藥物預計將在2024年獲得批准 |
| ● | 在一項治療不可切除cSCC的II期研究中, 內注射avelumab(一種靶向PD—L1的單克隆抗體)聯合根治性放療 |
| ● | 在 晚期cSCC的II期隨機試驗中, 內注射西妥昔單抗(一種靶向EGFR的單克隆抗體)與avelumab(一種靶向PD—L1的單克隆抗體) |
| ● | 在兩項II期試驗中,單藥治療nivolumab(一種靶向PD—1的單克隆抗體),並在II期研究中與ipilimumab(一種靶向細胞毒性T淋巴細胞相關蛋白4或CTLA—4的單克隆抗體)聯合給藥 |
| ● | 在晚期罕見腫瘤(包括轉移性cSCC)的II期研究中,口服 cobimetinib(一種絲裂原活化蛋白激酶或MEK的小分子抑制劑)與atezulizumab(一種靶向PD—L1的單克隆抗體)聯合使用 |
| ● | 在患有無法治癒的晚期/轉移性表達MAGE—A3的實體瘤(包括cSCC)患者中進行的I/II期試驗中, 使用表達黑色素瘤抗原A3或MAGE—A3或MG1MA3的Maraba病毒載體,作為單藥治療和 在肌內注射抗腫瘤疫苗(含轉基因MAGE—A3)或AdMA3後作為加強治療 |
| ● | 在多項I期和II期研究中, 瘤內注射溶瘤病毒載體talimogene laherparepvec與帕尼單抗(一種靶向EGFR的單克隆抗體)(以及 與靶向PD—1或PD—L1的其他抗體聯合)治療難治性 和/或晚期cSCC |
| ● | 在一項I期多中心開放標籤 I/Ib期研究中,靜脈內給予nanrilkefusp alfa(一種IL—15超激動劑)或與pembrolizumab聯合給藥,以評價SO—C101在複發性/難治性、晚期/轉移性cSCC中的安全性和初步療效 |
| ● | 在兩項II期研究中,靜脈內 輸注ASP—1929光免疫療法(單藥治療或與派姆單抗聯合治療) 原發性或複發性局部cSCC。2023年10月,由於缺乏療效,該試驗已停止 |
| ● | 在一項探索性I/II期研究中, 瘤內注射外泌體(CDK—002或exSTING),包括幾種實體瘤(如cSCC) |
競爭 HS
直到 2023年,在美國和 歐洲,唯一獲批和上市的用於治療中度至重度HS患者的全身給藥產品是阿達木單抗,一種腫瘤壞死因子—α或TNF—α抑制劑。2023年,發佈了另外兩種 藥物的III期結果,結果如下:
| ● | 皮下注射 的阿基諾尤單抗,一種IL—17 α單克隆抗體已提交併獲得批准 |
| ● | 貝美珠單抗(一種阻斷IL—17 A/F的單克隆抗體)給藥 已完成III期研究,目前正在 註冊過程中 |
-84-
如果 我們在HS中開發並獲得vilobelimab的批准,我們將面臨來自目前批准的治療藥物(如阿達木單抗、 瑞克諾單抗和bimek珠單抗(如獲得批准))、局部治療(包括克林黴素、間苯二酚等)、病灶內應用的皮質類固醇、口服抗生素(如四環素、克林黴素、利福平、甲硝唑、頭孢菌素、 氨苯碸等)的競爭。此外,正在研究一系列外科手術、激光和放療手術,並用於治療HS。最後,我們可能面臨來自目前正在開發的其他候選產品的競爭,這些候選產品可能在我們之前獲得 HS批准。
之前已經或目前正在研究和開發其他的系統性給藥候選產品,以治療HS 的作用機制不同:
| ● | 申請人 正在II期臨牀研究中研究SAR442970(一種抗TNF—OX40L納米抗體)。 84例中重度HS患者的試驗 |
| ● | 口頭 在 中,正在研究給予SAR 444656(一種低分子量IRAK4降解劑) 99例中重度HS患者的II期臨牀試驗 |
| ● | 申請人 正在研究amlitelimab(SAR445229)(一種完全人源OX40L結合抗體)給藥 在一項針對84名中重度HS患者的II期臨牀試驗中 |
| ● | 口頭 在II期研究中正在研究給予託法替尼(一種IFN信號傳導阻滯劑) 在47例唐氏綜合徵患者中,評價該藥物治療各種疾病的療效 皮膚病,包括HS |
| ● | 口頭 給予低分子量JAK—1抑制劑povorcitinib(INCB 54707), 在1560例中重度HS患者中進行了兩項III期臨牀研究 |
| ● | 口頭 給予Avacopan(一種低分子量C5aR抑制劑)完成了II期研究 2021年,435名中重度HS患者 |
| ● | 申請人 給予bermekimab(一種靶向IL 1—α的單克隆抗體),完成了三個階段 II臨牀研究共337例中重度HS患者 |
| ● | 申請人 目前正在研究給予izokibep,一種IL—17A的選擇性抑制劑, IIb期研究(180例患者)和III期研究(250例患者) |
| ● | 申請人 施用sonelokimab(ALX 0761,M1095),由單價 組成的三價納米抗體 特異於人白細胞介素IL—17A、IL—17F和人血清白蛋白的駱駝衍生納米抗體 VHH,完成了一項234例患者II期臨牀試驗,其中包括活動性、中度至 重度HS |
| ● | 申請人 目前正在使用lutik珠單抗,一種靶向IL—1 α/β的單克隆抗體 在200例抗TNF失敗的中度至重度HS患者中進行的II期研究中進行了調查 治療以及對生物療法初治的患者 |
| ● | 申請人 施用iscalumab(CFZ—533),一種非消耗性抗CD40抗體,正在測試 在中度至重度HS患者中開展的200例患者II期探索性研究平行 與其他實驗性治療,包括LYS006、MAS825、雷米替尼(LOU064)和ianalumab一起使用 (VAY736) |
| ● | 口頭 給予LYS006,白三烯A4水解酶或LTA4H的選擇性抑制劑, 在一項針對中度至重度HS患者的200例患者的II期探索性研究中進行了測試 與其他實驗性治療並行,包括iscalumab(CFZ—533)、MAS825、remigurinib (LOU064)和ianalumab(VAY736) |
| ● | 申請人 施用MAS825、T細胞免疫球蛋白和粘蛋白結構域3或TIM—3抑制劑正在 在一項針對中度至重度HS患者的200例患者的II期探索性研究中進行了測試 與其他實驗性治療並行,包括iscalumab(CFZ—533)、LYS006、remigratiinib (LOU064)和ianalumab(VAY736) |
-85-
| ● | 口頭 給予雷米替尼(LOU 064)、低分子量Burton酪氨酸激酶或BTK, 正在一項針對中度腫瘤患者的200例患者的II期探索性研究中進行測試 與其他實驗性治療(包括iscalumab(CFZ—533))並行治療嚴重HS, LYS066、MAS825和ianalumab(VAY736) |
| ● | ****下 給予ianalumab(VAY736),一種全人抗BAFF—R單克隆抗體, 在一項針對中度至重度HS患者的200例患者的II期探索性研究中進行了測試 與其他實驗性治療並行,包括iscalumab(CFZ—533),LYS 066,MAS825 和雷米替尼(LOU064) |
| ● | 申請人 給予eltrekibart(LY3041658),一種靶向和中和幾個 含有ELR肽基序的CXC家族人趨化因子目前正在 在一項II期臨牀試驗中評估了350例中度至重度患者 HS |
| ● | 口頭 給予upadacitinib(Janus激酶抑制劑)完成了II期臨牀研究 對68例中重度HS患者進行了研究, |
| ● | 內部操作 施用spesolimab、白細胞介素—36或IL—36受體(IL 1RL2/IL 1RAP)靶向抗體, 目前正在為HS患者開發,並在 52例 |
| ● | 申請人 並靜脈注射imsidolimumab(ANB 019),一種抑制功能的抗體 白細胞介素36受體(IL—36R)在149名患者中完成了II期臨牀試驗 為了探討HS受試者對imsidolimumab的免疫應答, |
| ● | 專題 ruxolitinib,低分子量JAK 1和JAK 2抑制劑,配製為 目前正在II期研究1.5%乳膏,93名HS患者 |
| ● | 口服PTM-001,一種未披露作用模式的實驗性候選藥物,目前正在對50名中到重度HS患者進行II期臨牀研究 |
| ● | 三種不同的口服新型激酶抑制劑在194名患者的II期探索性研究中進行了平行測試,這些患者患有中度到重度HS。2022年報道的結果表明,安慰劑和試驗性劑量方案PF 06826647和PF 06650833之間的差異沒有統計學意義,而PF 06700841顯示出優於安慰劑 |
| ● | 口服奧司司特,一種磷酸二酯酶-IV或PDE-IV抑制劑,目前正在對24名患者進行臨牀試驗,以評估口服奧司司特治療成人輕、中、重度HS的有效性和安全性。 |
| ● | 靜脈注射針對IL-17受體或IL17R的單抗溴鋁單抗,完成了治療中度HS的II期臨牀研究 |
我們 認為以下正在接受臨牀調查的候選產品暫時不會構成競爭威脅,或者 根本不會:
| ● | 口服IL-8B受體拮抗劑RIST4721已經在33名HS患者的II期臨牀研究中進行了測試,由於安全發現,該研究已終止 |
| ● | 申請人 給予利桑珠單抗,靶向白細胞介素—23 A或IL—23 A的單克隆抗體, 在243名患者的臨牀II期試驗中進行了研究(於2021年完成)—差異 安慰劑和利桑單抗試驗劑量方案之間無統計學意義 |
| ● | 內部操作 並皮下注射guselkumab,一種靶向IL—23的單克隆抗體,完成 在184例中重度HS患者中開展的II期臨牀試驗(已於2020年完成) —guselkumab安慰劑和試驗劑量方案之間的差異不具有統計學意義 顯著 |
-86-
| ● | 內部操作 已經在三個II期研究了阿那白素(一種IL—1受體拮抗劑)的治療 TNF—α治療難治性患者的試驗(已於2017年完成) |
| ● | 口頭 已在II期研究了阿普斯特、磷酸二酯酶—IV或PDE—IV給藥 在20名中度至重度HS患者中進行的試驗(於2017年完成) |
| ● | 申請人 給予CJM 112後,靶向IL—17A和IL—17A/F的單克隆抗體完成了 在66名中度至重度慢性HS患者中開展的II期研究(已完成 2016年,沒有達到主要結果) |
| ● | 申請人 給予MEDI8968,一種選擇性抗 的研究性單克隆抗體候選藥物 已在221例中重度HS患者的II期臨牀試驗中研究了IL—1R (2014年參加了比賽,沒有達到其主要結果) |
| ● | 口頭 給予zunsemetinib(ATI—450),絲裂原激活蛋白或MAP,激酶激活 蛋白激酶2或MAPKAPK2或MK 2抑制劑目前正在IIa期進行研究 對95例患者進行臨牀研究,以探討治療效果, 嚴重HS。該研究沒有達到其主要終點 |
| ● | 在20名中重度HS患者中,皮下注射 烏司奴單抗(一種靶向IL—12和IL—23共享p40亞基的單克隆抗體)完成了II期臨牀試驗 (該試驗於2014年完成) |
ANCA相關血管炎的競爭
如果 被批准用於治療AAV,vilobelimab將可能面臨來自目前使用的治療方法的競爭,包括低分子 C5aR—抑制劑avacopan(FDA於2021年10月批准用於該適應症)、皮質類固醇、硫唑嘌呤、甲氨蝶呤、環孢菌素、 黴酚酸酯和利妥昔單抗。當前誘導急性疾病的AAV患者緩解的標準治療是通過利妥昔單抗或硫唑嘌呤與HDCS聯合使用 來完成的。利妥昔單抗已獲批並上市用於該適應症,標籤擴展研究 正在進行中。維持緩解的治療包括小劑量皮質類固醇、甲氨蝶呤、黴酚酸酯和美羅華。美泊珠單抗是一種靶向白介素—5或IL—5的單克隆抗體,也被FDA批准用於治療成人中的一種稱為EGPA的AAV。
| ● | 在一項I期研究中,在24名患有多種自身免疫性 疾病(包括AAV)的患者中,對KYV—01(一種全人抗CD19 CAR T細胞療法)進行了研究 |
| ● | 在一項30例PR 3患者II期研究中,研究了腔內給予Obinutumab(一種抗CD 20單克隆抗體)——AAV患者 |
| ● | 在一項對10名患者進行的AAV患者進行I期研究中, 注射MT 2990(一種靶向白細胞介素—33的單克隆抗體) |
| ● | 在一項140名患者的III期臨牀研究中,注射 一種靶向白細胞介素—5的單克隆抗體或IL—5受體或美泊珠單抗(靶向 IL—5的單克隆抗體),在一項針對一種類型的AAV,EGPA的臨牀研究中,注射 |
| ● | 在66名複發性、非重度EGPA患者中進行的III期臨牀研究中, 注射阿巴西普(一種靶向CTLA—4的單克隆抗體) |
| ● | 在160例複發性或難治性EGPA患者的III期臨牀試驗中, 內給藥depemokimab |
| ● | 口服NS—229(一種JAK—1抑制劑)正在進行一項II期臨牀試驗,其中有45名EGPA患者 |
-87-
口服C5aR抑制劑對INF904的競爭
我們 於2024年1月下半年完成了單次和多次遞增劑量I期項目,其中口服C5aR抑制劑INF904。 本計劃可能證明瞭長期有效治療慢性炎性疾病所需的假定良好PK和安全性特徵以及給藥的易用性 。通過臨牀和 非臨牀開發的不同階段開發低分子量候選藥物是一個耗時且成本密集的過程。截至本報告日期,據我們所知,Avacopan是 目前唯一獲批用於治療AAV的口服C5aR抑制劑。如果我們達到市場批准階段,我們可能 已經遇到了各種競爭產品,可能不會在市場上僅次於Avacopan。即使上市,INF904未來可能面臨來自其他口服小分子的競爭。
據我們所知,正在積極開發的最先進的口服C5aR抑制劑是ACT—1014—6470。在一項已完成的I期臨牀 試驗中,開發該產品的公司報告了健康受試者和腎損害患者的安全性數據。 然而,ACT—1014—6470 II期開發的目標適應症和開發狀態尚未公開披露 。
此外, 目前並一直有幾個候選產品處於臨牀前和早期臨牀開發階段。這些包括低分子量 化合物、環肽和其他類別的候選藥物。據我們所知,除Avacopan外,這些候選藥物均未在III期註冊試驗中成功測試,也未在任何監管機構接受審查。至少有10—15種不同的 C5aR抑制劑被提及處於臨牀前和早期臨牀開發的不同階段,但近年來沒有 關於其各自的開發進展的更新,因此我們假設大多數開發項目同時 暫停或終止。
在終末補體抑制領域與治療藥物的競爭
有幾家臨牀或商業階段公司專注於用生物分子(包括單克隆 抗體)抑制C5aR。
C5a/C5aR1信號通路在各種炎症性疾病中起着重要作用。已經發現,C5a不僅可以通過C5a轉化酶的常規補體激活途徑(經典的、凝集素的、替代的)產生,而且還可以通過各種酶(例如,凝血酶和纖溶酶)。據報道,通過 酶促途徑產生的C5a不受上游補體阻滯劑(包括C5阻滯劑如依庫珠單抗)的影響。因此,控制 並完全阻斷人體中C5a誘導的信號傳導,因此保證了通過直接阻斷C5a或C5aR1的靶向方法。
目前有幾種C5a和C5aR抑制劑處於活躍臨牀開發的不同階段:
| ● | Avdoralumab (IPH5401)是一種抗C5aR1抗體,目前正在研究不同炎症性疾病。一項評價Avdoralimab在COVID—19重度肺炎患者中 安全性和療效的II期臨牀試驗在試驗的所有三個隊列中均未達到其主要終點 。該化合物目前尚未積極開發。大皰性類天皰瘡患者的II期研究正在進行中, |
| ● | STSA—1002是一種抗C5a人源化抗體,目前正處於ARDS患者的Ib/II期臨牀試驗中。 | |
| ● | AON—D21是一種抗C5a L—適體,目前正處於IIa期臨牀開發,用於重症肺炎插管患者。在健康志願者中進行的單次遞增劑量研究中,AON—D21被證明是安全的且耐受性良好。2022年年中完成了多次劑量遞增研究。 |
| ● | MOR210是一種抗C5aR抗體,是一種針對C5aR 1的新型人抗體。在I期試驗中研究了MOR210作為複發性或難治性晚期實體瘤的治療方法。 |
| ● | VIS954, 一種抗C5aR抗體在I期SAD研究中在健康成人蔘與者中進行了研究。 |
-88-
更廣泛地 ,在終末補體空間,目前有幾種獲批藥物,包括用於治療陣發性夜間血紅蛋白尿症(PNH)和非典型溶血性尿毒症綜合徵(aHUS)的依庫珠單抗和ravulizumab、用於治療全身性重症肌無力(gMG)的zilicoplan、用於治療地理性關節炎(GA)的Avaccapad pego。此外,還有幾個開發 項目可開發用於其他適應症的C5抑制劑,包括Hoffmanm—La Roche AG、Akari Therapeutics Plc.,Alnylam Pharmaceuticals, Inc.,Regeneron Pharmaceuticals,Inc.和諾華公司。
除了 C5和C5a,還有針對C5上游補體抑制的臨牀階段公司,如C3、因子D和凝集素途徑的組分 。這些方法也可能導致血液中C5a生成的降低。該領域的公司包括 Apellis Pharmaceuticals,Inc.,UCB S.A.,阿斯利康公司和Omeros公司等。此外,還有許多其他 公司開發靶向終末補體因子及其受體的臨牀前候選藥物。
摘要
影響我們候選產品(如果獲得批准或授權)成功的關鍵競爭因素可能是其功效、 安全性、給藥便利性、價格和市場接受程度,以及我們或我們的合作伙伴的營銷能力、 競爭水平以及政府和其他第三方支付方的補償。
如果我們的競爭對手開發和商業化的產品比我們可能開發的任何產品更安全、更有效、 副作用更少或更不嚴重、更方便或更便宜,我們 的商業機會可能會減少或消除。我們的競爭對手 的產品獲得FDA或其他監管機構批准的速度可能比我們獲得批准的速度更快,這可能導致 我們的競爭對手在我們能夠進入市場之前就建立了強大的市場地位。此外,即使我們的候選產品 獲得上市和銷售批准,它們也可能無法獲得醫生、患者、第三方支付方和醫療界其他人的充分市場認可,包括如果醫生不願意將患者從現有療法中轉換(例如阿達木單抗 治療HS)。見"項目3。關鍵信息—3.風險因素—與我們候選產品的發現、開發 和商業化相關的風險—即使我們的候選產品獲得了上市批准, 它可能無法達到醫生、患者、第三方支付方和醫療界其他人的市場接受程度,而這是商業成功所必需的 ,在這種情況下,我們可能無法產生顯著收入或盈利。"
政府 法規和產品批准
所有主要藥品市場的政府 當局廣泛監管(除其他事項外)醫藥產品的研發、測試、製造、包裝、儲存、記錄保存、標籤、廣告、促銷、分銷、營銷和進出口(如我們正在開發的產品)。雖然我們最初的重點將在美國和歐洲,但我們打算在其他國家和地區(如加拿大或日本)開發和 為我們的產品尋求上市許可,以及 遵循領先權威的市場(如巴西或韓國)。在美國、歐洲 和其他國家獲得監管批准的過程以及隨後遵守適用的法規和法規,需要花費大量 時間和財政資源。
FDA 批准流程
我們當前的所有候選產品 均受FDA在美國的監管,無論是生物製品或生物製品, ,還是新化學實體或NCE。FDA對生物製品和NCE進行了廣泛的上市前和上市後監管。《公共衞生服務法》(簡稱PHSA)、《聯邦食品、藥品和化粧品法》(Federal Food,Drug and Cosmetic Act)以及其他聯邦和州的法規和法規,除其他事項外,管轄生物製品和NCE的研究、開發、測試、製造、儲存、記錄保存、批准、標籤、推廣和營銷、分銷、 批准後監測和報告、取樣以及進出口。未遵守適用的美國 要求可能使公司受到各種行政或司法制裁,例如FDA拒絕批准待決BLA 或NDA、撤回批准、臨牀擱置、警告信、產品召回、產品扣押、全部或部分暫停 生產或分銷、禁令、罰款或民事或刑事處罰。
PHSA強調了對無法精確定義屬性的產品進行生產控制的重要性。PHSA還授權FDA 在存在對公眾健康危險的情況下立即暫停許可證,在產品短缺和關鍵的公共衞生需要時準備或採購 產品,並授權創建和執行法規,以防止傳染病在美國和州際的傳入或傳播。
-89-
BLA 和保密協議申請和批准
FDA在新的生物製品或NCE在美國上市前所需的 過程漫長、昂貴,而且本身就不確定。在美國,生物製品和NCE的開發通常涉及臨牀前實驗室和動物試驗、向FDA提交IND(必須在臨牀試驗開始之前生效)以及充分和受控的臨牀試驗,以 為尋求FDA批准的每個適應症確定生物製品或NCE的安全性、純度和效力(安全性和有效性)。開發數據以滿足FDA的上市前審批要求通常需要多年時間,實際所需時間可能會因產品或疾病的類型、複雜性和新穎性而有很大不同。
臨牀前研究包括對製成的藥物物質或活性藥物成分以及配方藥物或藥物產品的純度和穩定性進行實驗室評估,以及體外和動物研究,以評估候選藥物在人體內的安全性和活性,並建立治療使用的理論基礎。臨牀前研究的實施受聯邦法規和要求的約束,包括GLP法規。臨牀前試驗的結果與製造信息、分析數據、任何可用的臨牀數據或文獻以及臨牀試驗計劃等一起,作為IND的一部分提交給FDA。在IND提交後,一些長期的臨牀前測試,如生殖不良事件和致癌性的動物測試,可能會繼續 。
IND必須在美國臨牀試驗開始前生效。在開始人體臨牀試驗之前,要求在提交每個IND之後有30天的等待期。如果FDA在這30天內既沒有對IND發表評論,也沒有對IND提出質疑,IND中提議的臨牀試驗可能會開始。
臨牀試驗涉及在合格的研究人員的監督下,將正在研究的新藥或生物製劑應用於健康志願者或接受調查的患者。臨牀試驗必須(I)符合聯邦法規,(Ii)符合良好臨牀實踐或GCP,這是一項旨在保護患者權利和健康並定義臨牀試驗發起人、管理者和監督者角色的國際標準,以及(Iii)根據詳細説明試驗目標、用於監測安全性的參數和要評估的有效性標準的方案進行。涉及對美國患者進行測試和後續方案修訂的每個方案都必須作為IND的一部分提交給FDA。
如果FDA認為臨牀試驗未按要求進行或對臨牀試驗對象構成不可接受的風險,則FDA可隨時下令暫時或永久中止臨牀試驗,或施加其他制裁。臨牀試驗受試者的研究方案和知情同意信息也必須提交給機構審查委員會(IRB)批准。IRB還可以因未能遵守IRB的要求而要求暫時或永久停止現場的臨牀試驗,或者可以施加其他條件。研究贊助商還可以隨時以各種理由暫停臨牀試驗,包括確定受試者或患者面臨不可接受的健康風險 。
用於支持BLA或NDA以獲得上市批准的臨牀試驗通常分三個連續階段進行,但這三個階段可能會重疊或合併。在第一階段,候選藥物首先被引入到健康的人體受試者或患者中,並進行測試以評估其藥代動力學或PK、特性、藥理作用、與增加劑量相關的副作用,以及如果可能的話,評估有效性的早期證據。對於一些用於嚴重或危及生命的疾病的產品,如癌症治療,必須在目標患者羣體中進行初步的人體測試。第二階段通常涉及在有限和特定的患者人羣中進行試驗,以確定生物製劑對特定適應症、劑量耐受性和最佳劑量的有效性,並確定常見的不良反應和潛在的安全風險。如果候選藥物在第二階段評估中證明瞭有效性和可接受的安全性,則進行第三階段試驗,以獲得更多 患者的臨牀有效性和安全性的額外信息,這些患者代表未來的預期使用人羣,通常是在地理上分散的臨牀試驗地點。這些 第三階段臨牀試驗旨在建立足夠的數據,以證明該產品的有效性和安全性 ,以使FDA能夠評估生物或NCE的總體益處-風險關係,併為藥物的標籤提供足夠的信息。在美國境外根據當地適用法律在類似的符合GCP的條件下進行的試驗也可能被FDA接受,以支持產品許可。
-90-
研究藥物臨牀試驗的贊助商 必須公開披露某些臨牀試驗信息,包括詳細的試驗設計 和政府公共數據庫中的試驗結果。這些要求受特定時間表的限制,適用於FDA監管產品的大多數受控臨牀試驗。
在完成所需的臨牀測試後,將準備BLA(用於生物)或NDA(用於NCE)並提交給FDA。在美國開始銷售該產品之前,需要FDA審查 並獲得BLA或NDA的批准。BLA或NDA必須包括 所有臨牀前、臨牀和其他測試的結果,以及與產品的藥理、化學、製造和控制相關的數據彙編,並必須根據這些結果證明產品的安全性和有效性。BLA或NDA還必須 包含大量製造信息。準備和提交BLA或NDA的成本是相當高的。根據聯邦法律,提交大多數BLA或NDA還需繳納高額的申請使用費和年度計劃使用費,這筆費用總計可能達數百萬美元,而且通常每年都會增加。
FDA自收到BLA或NDA之日起有60天的時間,根據該機構關於申請是否足夠完整、允許進行實質性審查的門檻確定,決定是否接受申請備案。一旦提交的申請被接受,FDA就開始進行深入的審查。FDA已同意在審查BLAS和NDA時的某些績效目標。大多數標準審查候選藥物的此類申請都會在申請被接受提交之日起10個月內進行審查。儘管FDA經常達到其用户付費績效目標,但如有必要,它可以延長這些時間期限,並且其審查可能不會及時進行 。FDA通常將新藥或出現安全性或有效性難題的藥物的申請提交給諮詢委員會(通常是包括臨牀醫生和其他專家的小組)進行審查、評估,並就是否應批准申請提出建議。FDA不受諮詢委員會建議的約束,但它經常遵循這樣的建議。在批准BLA或NDA之前,FDA通常會檢查一個或多個臨牀地點,以確保符合GCP。此外,FDA將檢查製造藥物的一個或多個設施。FDA不會批准該產品,除非它證實符合cGMP標準令人滿意,並且BLA或NDA包含的數據提供了確鑿的證據,證明該藥物在所研究的適應症中是安全有效的。
在FDA評估了BLA或NDA和製造設施之後,它會發出一封批准信或一封完整的回覆信。 一封完整的回覆信通常會列出提交中的不足之處,並可能需要大量的額外測試或信息 以便FDA重新考慮申請。如果或當這些缺陷已在重新提交BLA或NDA時得到FDA滿意的解決 ,FDA將簽發批准信。FDA承諾將根據所包含的信息類型,在兩到六個月內審查此類重新提交的申請。FDA的批准永遠不能得到保證,如果不符合適用的監管標準,FDA可能會拒絕批准BLA或NDA。
根據PHSA,FDA可以批准BLA或NDA,如果它確定產品是安全、純淨和有效的,並且將生產該產品的設施符合旨在確保其繼續安全、純淨和有效的標準。批准函授權該藥物的商業銷售,並提供特定適應症的具體處方信息。對藥物的批准可能比申請中要求的限制多得多,包括對特定疾病和劑量或使用適應症的限制, 這可能會限制產品的商業價值。FDA還可能要求在產品標籤中包含某些禁忌症、警告或預防措施 。此外,作為BLA或NDA批准的條件之一,FDA可能要求風險評估和緩解戰略,或REMS,以幫助確保藥物的好處大於潛在風險。REMS可以包括藥物指南、醫療保健專業人員的溝通計劃和確保安全使用的要素,或ETASU。ETASU可包括但不限於處方或配藥的特殊培訓或認證、僅在特定情況下的配藥、特殊監控和患者登記簿的使用。對REMS的要求或與藥物一起使用的配套診斷可能會對該藥物的潛在市場和盈利能力產生重大影響。此外,作為批准的條件之一,產品批准可能需要大量的批准後測試和監督,以監測藥物的安全性或有效性。一旦獲得批准,如果沒有遵守監管標準或在初始營銷後發現問題,產品審批可能會被撤回。
-91-
BLA或NDA獲得批准後,產品也可能需要經過正式批放行。作為製造過程的一部分,製造商 在放行分銷前,必須對每批產品進行某些測試。如果產品需要 FDA的正式批放行,則製造商向FDA提交每批產品的樣品以及放行方案 ,其中應顯示該批產品的生產歷史摘要以及製造商對該批產品進行的所有檢測結果。FDA還可能在放行批次供製造商分銷之前對某些產品(如病毒疫苗)進行某些確證性測試。此外,FDA還開展與產品安全性、純度、效價和有效性相關的監管標準相關的實驗室研究。藥品批准後,製造商必須解決出現的任何安全問題, 可能會被召回或停止生產,並接受定期檢查。
緊急 使用授權
The FDA can facilitate the availability and use of medical countermeasures needed during public health emergencies via EUA. When The United States Secretary of Health and Human Services, or HHS Secretary, declares that an EUA is appropriate, FDA may authorize unapproved medical products or unapproved uses of approved medical products to be used in an emergency to diagnose, treat or prevent serious or life-threatening diseases or conditions caused by chemical, biological, radiological or nuclear threat agents. For example, in January 2020, the HHS Secretary determined that a public health emergency, or PHE, existed (and has subsequently extended the declaration on numerous occasions,) that had a significant potential to affect national security or the health and security of U.S. citizens due to the emergence and spread of COVID-19. Based on this determination, the HHS Secretary also declared that circumstances existed justifying EUA of certain medical products. The EUA declaration is distinct from the PHE declaration. As long as the applicable EUA declaration and determination remains in effect, an EUA may remain in effect beyond the duration of the PHE declaration if all other statutory conditions are met. Even if the EUA declaration remains in effect, the FDA may revoke the EUA at any time if certain circumstances or criteria are met. We have been granted the EUA for GOHIBIC (vilobelimab), for the treatment of COVID-19 in hospitalized adults when initiated within 48hours of receiving IMV or ECMO.
報告cGMP合規性的不良 事件
FDA批准BLA或NDA後,需要提交不良事件 並提交定期報告。FDA還可能要求進行上市後 檢測,稱為IV期檢測、REMS和監督,以監測已批准產品的效果,或者可能對 批准設置可能限制產品分銷或使用的條件。此外,生產、包裝、貼標、儲存和分銷 程序在批准後必須繼續符合現行cGMP。藥品製造商及其某些分包商必須 向FDA和某些州機構註冊其機構。向FDA註冊的實體需要接受FDA的定期 突擊檢查,在此期間,FDA檢查生產設施以評估其是否符合cGMP。因此,製造商 必須繼續在生產和質量控制領域花費時間、金錢和精力,以保持符合cGMP。如果公司未能遵守監管標準,如果在首次上市後遇到問題,或者如果後來發現之前 未被識別的問題,監管機構可以撤銷產品批准、要求產品召回或通過標籤變更或產品移除實施銷售限制。
孤兒 藥品名稱
Under the Orphan Drug Act, the FDA may grant orphan drug designation to biologics or NCEs intended to treat a rare disease or condition-generally a disease or condition that affects fewer than 200,000 individuals annually in the United States. Orphan drug designation must be requested before submitting a BLA or NDA. After the FDA grants orphan drug designation, the generic identity of the drug candidate and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not necessarily convey any advantage in, or shorten the duration of, the regulatory review and approval process. The first BLA or NDA applicant to receive FDA approval for a particular product to treat a particular disease with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the United States for that product, for that indication. During the seven-year exclusivity period, the FDA may not approve any other applications to market the same drug for the same disease, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition or, a drug that is otherwise the same drug which may be clinically superior. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the BLA or NDA application user fee.
-92-
快速 磁道指定
快速跟蹤是一個旨在促進藥物開發和加快審查的流程,以治療嚴重疾病並滿足未滿足的 醫療需求。這樣做的目的是為了讓患者更早地獲得重要的新藥。Fast Track針對的病情範圍很廣。 判斷病情是否嚴重是一個判斷問題,但通常是基於藥物是否會對生存、日常功能等因素產生影響,或者如果不進行治療,病情會從較輕的病情發展為更嚴重的情況的可能性。滿足未滿足的醫療需求被定義為在不存在的情況下提供一種療法,或者提供一種可能比現有療法更好的療法。任何正在開發的治療或預防目前沒有治療方法的疾病的藥物都是針對未得到滿足的需求。如果有可用的治療方法,快速通道藥物必須顯示出某些優於現有治療方法的優勢, 例如:顯示出卓越的療效、對嚴重結果的效果或改善對嚴重結果的效果;避免可用治療的嚴重副作用 ;在早期診斷可改善結果的情況下改進對嚴重情況的診斷;降低可用治療的臨牀顯著毒性(常見並導致中斷治療或解決新出現或預期的公共衞生需求的能力)。獲得快速通道指定的藥物符合以下部分或全部條件: 更頻繁地與FDA開會討論藥物的開發計劃,並確保收集支持藥物批准所需的適當數據;FDA更頻繁地就擬議臨牀試驗的設計和生物標記物的使用等事項進行書面溝通;如果滿足相關標準,則有資格獲得加速批准和優先審查;滾動審查,這意味着製藥公司可以提交其BLA或NDA的完整部分供FDA審查,而不是等到 NDA的每個部分完成後才可以審查整個申請。BLA或NDA審查通常在製藥公司向FDA提交整個申請之前不會開始。快速通道指定必須由製藥公司申請。該請求可以在藥物開發過程中的任何 時間發起。FDA將審查申請,並在60天內根據藥物是否滿足嚴重情況下未滿足的醫療需求做出決定。一旦一種藥物獲得快速通道指定,在整個藥物開發和審查過程中,鼓勵FDA和製藥公司之間及早和頻繁的溝通。溝通的頻率確保了問題和問題得到快速解決,通常會導致患者更早地批准和訪問藥物。
我們 已獲得美國維羅布利單抗PG適應症的孤兒藥物地位和快速通道稱號。我們還在美國獲得了新冠肺炎的快速通道稱號。根據其他適應症中的維羅貝利單抗研究結果和可用數據,我們可能會申請美國的孤兒藥物地位。
加速了 審批
FDA制定了加速審批計劃,以允許更早地批准治療嚴重疾病的藥物,並基於替代終點滿足未滿足的 醫療需求。替代終點是一種標記物,如實驗室測量、放射圖像、體徵或其他被認為可以預測臨牀益處但本身並不是臨牀益處的衡量標準。使用替代終點可以大大縮短獲得FDA批准之前所需的時間。製藥公司仍需進行研究以確認預期的臨牀益處。這些研究被稱為第四階段驗證性試驗。如果驗證性試驗顯示該藥物確實提供了臨牀益處,則FDA將按傳統方式批准該藥物。如果驗證性試驗 沒有顯示該藥物提供臨牀益處,FDA已制定監管程序,可能導致該藥物從市場上下架 。
優先級 審核
1992年,根據《處方藥使用者法》(PDUFA),FDA同意了改善藥品審查時間的具體目標,並建立了兩級審查時間制度--標準審查和優先審查。優先審查指定意味着FDA的目標是在6個月內對申請採取行動(與標準審查的10個月相比)。優先審查指定將把全部 注意力和資源集中到對藥物申請的評估上,這些藥物申請如果獲得批准,與標準申請相比,將顯著改善治療、診斷或預防嚴重疾病的安全性或有效性。顯著的改善可通過以下例子來證明:在治療、預防或診斷疾病方面的有效性提高的證據;消除或大幅減少治療限制藥物反應;記錄的患者依從性的提高,預計將導致嚴重結果的改善;或在新的亞羣中的安全性和有效性的證據。FDA對每項申請的審查指定做出決定。但是,申請人可以明確要求優先複審。它不影響臨牀試驗期的 長度。FDA在收到BLA或NDA後60天內通知申請人優先審查指定。將一種藥物指定為“優先”並不改變批准的科學/醫學標準或所需證據的質量。
-93-
SPA 流程
SPA 是一個過程,公司可以要求與FDA會面,就某些臨牀試驗、臨牀 研究或動物研究的設計和規模達成協議,以確定它們是否充分滿足了支持 上市批准的研究的科學和監管要求。SPA協議表明FDA同意 總體方案設計的特定關鍵要素的充分性和可接受性(例如,入選標準、劑量選擇、終點和計劃分析),用於支持未來上市申請的研究。這些要素對於確保根據方案進行的試驗可被視為充分 且控制良好的研究,可支持上市批准至關重要。對這些問題的反饋可以確保 規劃後期發展戰略時的充分性。但是,SPA協議並不表明FDA同意每個方案細節 。SPA協議的存在並不保證FDA將提交(接受)BLA或NDA,或者結果足以支持批准。
其他 醫療保健法律和合規要求
在 美國,我們的活動可能受到聯邦、州和地方當局以及FDA的監管, 包括醫療保險和醫療補助服務中心、美國衞生與公眾服務部的其他部門(例如,監察長辦公室),美國司法部和司法部內的個別美國檢察官辦公室, 和州和地方政府。
歐盟 批准流程
EMA是歐盟的一個分散的科學機構。它協調中央授權的 藥品的評價和監測。它負責對歐盟上市許可申請進行科學評估,以及制定 技術指南並向申辦者提供科學建議。EMA通過由成員國提名的由歐盟各地約4500名專家組成的網絡來分散其藥物的科學評估 。EMA利用了來自歐盟成員國40多個國家主管部門或NCA的資源。Paul Ehrlich Institute(PEI)是德國的NCA之一,負責監管抗體產品等。
在歐盟, 關於藥品批准的流程大致遵循與美國相同的流程,通常 涉及圓滿完成以下各項:
| ● | 臨牀前 實驗室試驗、動物研究和製劑研究均按照 適用的歐盟藥物非臨牀研究質量管理規範法規; |
| ● | 提交 提交給臨牀試驗申請或每份試驗CTA的相關國家監管機構 在人類中,在每個國家,患者 計劃入學; |
| ● | 性能 充分且控制良好的臨牀試驗,以確定該藥物的安全性和有效性 每個建議適應症的產品; |
| ● | 提交 向上市許可申請或MAA的相關主管部門提交, 包括支持安全性和有效性的數據以及生產的詳細信息 臨牀開發中產品的成分和建議的標籤; |
| ● | 滿意 相關國家主管部門完成對生產設施的檢查 或設施,包括第三方的設施,在那裏生產產品,以評估 遵守嚴格執行的cGMP; |
| ● | 潛力 對生成數據的非臨牀和臨牀試驗中心進行稽查,以支持 MAA;及 |
| ● | 審核 並在進行任何商業營銷之前獲得MAA相關主管部門的批准, 產品的銷售或發貨。 |
-94-
臨牀前研究
臨牀前測試包括對產品的化學成分、配方和穩定性進行實驗室評估,以及在動物實驗中評估毒性的研究,以評估產品的質量和潛在的安全性和有效性。進行臨牀前試驗和用於試驗的化合物配方必須符合相關的國際、歐盟和國家立法、法規和指南。臨牀前試驗結果與相關生產信息和分析數據一起作為CTA的一部分提交。
臨牀試驗批准
《臨牀試驗法規(歐盟)第536/2014號》於2022年1月31日廢除了《臨牀試驗指令2001/20/EC》,並從2023年1月31日起適用於所有新的臨牀試驗。所有正在進行的試驗必須在2025年1月31日之前過渡到新的進程。《臨牀試驗條例》協調了整個歐盟的臨牀試驗流程和監督。贊助商通過臨牀試驗信息系統(CTI)提交一份申請,要求批准在幾個歐洲國家進行臨牀試驗。臨牀試驗的評估、授權和監督是歐盟成員國和歐洲經濟區(EEA)國家的責任。CTIS申請必須有研究藥品檔案(IMPD)、研究方案以及《臨牀試驗條例》和其他適用指南文件規定的進一步支持信息。CTIS程序有預先確定的時間表,根據研究階段的不同,時間表可能會有所不同。該流程包括監管機構要回答的一輪或多輪問題或要滿足的請求的時間表。
第 (EU)第536/2014號法規包括透明度要求(在歐盟數據庫中主動發佈臨牀試驗數據)以及關於在臨牀試驗完成時、期間和之後發佈提交文件的時間的相關規則。透明度要求適用於臨牀試驗申請或CTA中包含的文件、CTI中提供的檔案,以及在試驗生命週期中提交的所有臨牀試驗信息,但與質量相關的 文檔、財務安排和一些與監督相關的信息除外。
研究用藥品的製造和進口到歐盟必須持有適當的授權,並且必須根據cGMP進行。
營銷 授權應用程序
在歐盟成員國銷售產品的授權是通過以下四種程序之一進行的:集中授權程序、相互承認程序、分散程序或國家程序。由於我們的產品是基於抗體的生物製品或NCEs,屬於集中式程序,因此這裏僅介紹此程序。
集中 授權流程
某些 藥品,包括通過生物技術開發的藥品,必須通過集中授權 上市授權程序審批。集中授權程序下的成功申請將獲得歐盟委員會的營銷授權,該授權在所有歐盟成員國以及其他歐洲經濟區成員國(即挪威、冰島和列支敦士登)自動有效。EMA和歐盟委員會負責管理集中授權程序。
在集中授權程序下,CHMP是代表EMA就人類產品的安全性、有效性和質量發表意見的科學委員會。CHMP由每個成員國的國家禁毒當局提名的專家組成,其中一人被任命為評估協調報告員,並可能得到委員會另一名成員擔任聯合報告員的協助。批准後,報告員(S)將在產品的整個生命週期內繼續對其進行監測。CHMP被要求在收到有效申請後210天內發表意見,但當需要要求申請人澄清或提供進一步的佐證數據時,時鐘會停止 。這一過程很複雜,需要與成員國監管當局和一些專家進行廣泛的 磋商。程序完成後,將生成一份歐洲公共評估報告,簡稱EPAR。如果CHMP得出結論認為藥品的質量、安全性和有效性得到了充分的證明,它將採取積極的意見。CHMP的意見將發送給歐盟委員會,歐盟委員會將該意見作為其決定是否授予營銷授權的基礎。如果意見是否定的,則提供關於得出這一結論的理由的信息。
-95-
政策 0070旨在促進EMA決策和臨牀數據的透明度,管理EMA如何通過集中程序收集、審查和發佈由申請者和營銷授權持有人(或MAHS)提交的臨牀數據。
在藥品獲得授權並投放市場後,必須對與其質量、安全性和有效性有關的所有方面進行審查,這是維持上市授權的條件。如果未能遵守營銷授權的條件,可能會受到制裁。 在極端情況下,授權可能被撤銷,導致產品退出銷售。
加速了 評估流程
當從公共衞生的角度,特別是從治療創新的角度,提交人用藥物的上市授權申請時,申請人可根據(EC)726/2004號條例第14條第(9)款要求加速評估程序。根據加速評估程序,CHMP必須在收到有效申請後150天內 發佈意見,但必須停止計時。我們認為,我們的候選產品目前正在或可能在未來開發的一些疾病適應症 符合這一規定,我們將適當利用這一規定 。
有條件的 審批
根據法規(EC)726/2004第14條第(7)款,如果一種藥物能夠滿足未滿足的醫療需求,如果其即時供應符合公眾健康利益,則可根據通常要求的不完整的臨牀數據獲得有條件的上市授權,但須遵守對授權持有人施加的具體義務。EMA將每年對這些具體義務進行審查。應向公眾公佈這些義務的清單。此類授權的有效期為 一年,可續展。
授權和續訂期限
營銷授權最初有效期為五年,然後可根據EMA或授權成員國的主管當局對風險-收益餘額進行重新評估而續簽。為此,營銷授權持有人應 向EMA或主管當局提供關於質量、安全和有效性的文件的合併版本,包括自授予營銷授權以來引入的所有變種,至少在營銷授權失效前 六個月。一旦續簽,上市授權將無限期有效,除非歐盟委員會或主管當局基於與藥物警戒有關的正當理由決定繼續進行一次為期五年的續簽。任何授權 如果沒有在授權後三年內將藥品實際投放到歐盟市場(如果是集中式程序)或授權成員國的市場上,則將失效(所謂的日落條款)。
孤兒 藥品名稱
法規 (EC)141/2000規定,如果申辦者能夠確定,藥物應被指定為孤兒藥:
| ● | 這 用於診斷、預防或治療危及生命或慢性疾病 在歐洲聯盟,每10,000人中影響不超過5人的衰弱性疾病 當提出申請時,或預期用於診斷、預防或治療歐盟內危及生命、嚴重衰弱或嚴重慢性疾病 ,且如果沒有獎勵,該藥物在歐盟的銷售不太可能產生足夠的 回報以證明必要的投資是合理的;以及 |
| ● | 這 目前尚無令人滿意的診斷、預防或治療方法 已在歐盟獲得授權,或如果存在此類方法, 藥物對那些受這種疾病影響的人有很大的好處。 |
-96-
法規 (EC)847/2000規定了指定孤兒藥的標準。在提交產品上市批准申請之前, 可隨時申請指定為孤兒產品。孤兒藥的上市許可導致 10年的市場獨佔期,這意味着在相同適應症中,任何類似藥品都不能獲得批准。但是,如果在第五年結束時,確定產品不再符合 孤兒藥指定的標準,例如,由於產品利潤充足,不足以證明繼續具有市場獨佔性,則此 期限可縮短至六年。 此外,在某些特定情況下,可以根據個別情況授予市場獨佔權的減損,例如 上市許可持有人的同意、無法供應足夠數量的產品或類似藥品證明"臨牀相關 優效性"。根據法規(EC)141/2000指定為孤兒藥的藥品有資格享受歐盟和成員國提供的獎勵,以支持孤兒藥的研究、開發和供應。
如果 根據法規(EC)141/2000指定為孤兒藥的藥品的MAA包含了按照商定的PIP進行的所有研究的結果 ,並且隨後在授予的上市許可中包含了相應的聲明,則 10年的市場獨佔期將延長至12年。
我們 在歐盟已獲得vilobelimab PG適應症的孤兒藥地位。根據vilobelimab在其他適應症中的研究結果和可用 數據,我們也可能申請歐洲的孤兒藥狀態用於這些適應症。
監管 數據保護
在不影響 工商產權保護法的情況下,新藥品的上市許可將享受 8 + 2 + 1年的監管保護期。
該 制度包括8年的監管數據保護期加上10年的同時市場獨佔權,如果在這10年的前8年內,上市許可持有人獲得了一種或多種新治療適應症的批准 ,在批准前的科學評估期間,與現有療法相比,確定會帶來 顯著的臨牀益處。根據現行規則,第三方可在首次批准後8年開始引用參考產品的臨牀前 和臨牀數據,但第三方可在僅10年(或11年)後上市參考產品的仿製藥 。
其他 國際法規
除了美國和歐洲的法規外,還有各種外國法規管理藥品的臨牀試驗、商業銷售和 分銷。批准過程因國家而異,批准時間可能比FDA或歐盟委員會批准所需的時間更長或 。
醫藥 保險範圍、定價和報銷
FDA和其他政府機構批准的產品的覆蓋範圍和報銷狀態存在很大 不確定性。我們產品的銷售 部分取決於第三方支付方(包括美國政府醫療計劃(如Medicare和Medicaid)、商業私人和公共醫療保險公司以及管理式醫療機構)為此類產品提供覆蓋範圍和建立 充分報銷水平的程度。確定付款人是否將為產品提供承保的過程可以 與設置付款人將在承保 批准後為藥品產品支付的價格或報銷率的過程分開。第三方支付方越來越多地質疑收費價格,審查醫療必要性,審查醫療產品和服務的成本效益,並實施控制以管理成本。第三方付款人可能會將承保範圍限制在獲批清單或處方集上的特定藥品,其中可能不包括特定適應症的所有獲批產品。
-97-
為確保 批准銷售的任何藥品的承保範圍和報銷,除 獲得FDA或其他類似監管批准所需的費用外,公司可能需要進行昂貴的 藥物經濟學研究,以證明產品的醫療必要性和成本效益。儘管如此,候選產品可能不被視為 醫療必要或成本效益。此外,付款人決定為藥品提供保險並不意味着 將批准適當的報銷率。此外,一個付款人決定為藥品產品提供保險 並不保證其他付款人也將為藥品產品提供保險。第三方報銷可能不足以 維持足夠高的價格水平,以實現產品開發投資的適當回報。
控制醫療成本也已成為聯邦、州和外國政府的優先事項,藥品價格也一直是這一努力的重點。各國政府對實施成本控制計劃表現出了極大的興趣,包括價格控制、對報銷的限制和非專利產品的替代要求。採取價格控制和成本控制措施,以及在擁有現有控制和措施的司法管轄區採取更嚴格的政策,可能會進一步限制我們的淨收入和業績。承保政策和第三方報銷費率可能隨時發生變化。即使一家公司或其協作者獲得監管批准的一個或多個產品獲得了有利的承保和報銷 狀態,未來也可能實施不太有利的承保政策和報銷費率。
在 美國境外,確保我們產品的充分覆蓋和支付將面臨挑戰。處方藥的定價 在許多國家受到政府控制。與政府機構的定價談判可能遠遠超出產品獲得 監管上市批准的範圍,可能要求我們進行臨牀試驗,比較 我們的產品或產品與其他可用療法的成本效益。進行此類臨牀試驗可能會很昂貴,並導致 我們的商業化努力的延遲。
In the European Union, pricing and reimbursement schemes to restrict the range of drug products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use vary widely from country to country. Some countries provide that drug products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of a particular drug product to currently available therapies. EU member states may also require approval of a specific price for a drug product or, instead, may adopt a system of direct or indirect controls on the profitability of the company placing the drug product on the market. Other EU member states allow companies to fix their own prices for drug products but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription drugs, has become intense. As a result, increasingly high barriers are being erected to the market entry of new pharmaceutical products. In addition, in some countries, cross-border imports from low-priced markets exert competitive pressure that may reduce pricing within a country. Any country that has price controls or reimbursement limitations for drug products may not allow favorable reimbursement and pricing arrangements.
C.組織結構
InflaRx N.V.有兩家直接全資子公司,InflaRx GmbH和InflaRx Pharmaceuticals,Inc.,分別列於附件8.1 中。
D.財產、廠房和設備
我們的總部位於德國耶拿,根據一份將於2025年12月到期的租約,我們在那裏佔用了約8,000平方英尺的辦公和實驗室空間。此外,根據一份將於2027年5月到期的租約,我們在德國普蘭格-馬丁斯里德(慕尼黑附近)租用了約13,700平方英尺的辦公空間。此外,根據2026年4月到期的租約,我們還在美國密歇根州安娜堡租用了辦公和實驗室空間。
-98-
項目 4A。未解決的員工意見
不適用 。
項目 5.經營和財務回顧及展望
| A. | 經營業績 |
您 應該閲讀以下對我們的財務狀況和經營結果的討論和分析,以及我們合併財務報表中的信息 及其附註。
以下討論基於我們根據國際會計準則理事會發布的《國際財務報告準則》編制的財務信息,這些財務信息在 重大方面可能與美國和其他司法管轄區公認的會計原則不同。以下討論包括涉及風險、不確定性和假設的前瞻性陳述。由於許多因素的影響,我們的實際結果可能與這些前瞻性陳述中預期的結果大不相同,其中包括“第3項.關鍵信息-3.‘風險因素’和前瞻性陳述”中描述的那些因素。
有關截至2022年12月31日的年度與截至2021年12月31日的年度的綜合業績、分部業績以及流動資金和資本資源的詳細信息,請參閲“項目5.經營和財務回顧與展望“在公司2022年年度報告Form 20-F中,該信息以引用方式併入本文。
概述
我們 是一家生物技術公司,是抗炎療法的先驅,專注於應用我們專有的抗C5a和C5aR技術 來發現、開發和商業化補體激活因子C5a 及其受體C5aR的一流、有效和特定的抑制劑。C5a是一種強大的炎症介質,參與了多種自身免疫性和其他炎症性疾病的進展。我們的主要候選產品Viloblimab是一種新型靜脈注射的一流抗C5a單抗,它選擇性地與遊離C5a結合,並在多種臨牀環境中展示了修改疾病的臨牀活性和耐受性 。2023年4月,我們從FDA獲得了用於GOHIBIC(維洛利單抗)的EUA,用於治療危重、侵入性機械通氣的新冠肺炎患者。隨後,在2023年6月,我們開始在美國將GOHIBIC(Viloblimab)商業化。 我們還在開發用於治療壞疽膿皮病(PG)的viloblimab,這是一種慢性炎症性皮膚病,我們 目前正在進行第三階段研究。除了PG之外,我們還一直在開發Viloblimab,以生成大量補體介導的疾病的概念驗證數據,這些疾病具有重大的未滿足的醫療需求。為此,我們之前已經在HS(一種慢性衰弱的全身性炎症性皮膚病)、ANCA相關性血管炎(或AAV)和CSCC(一種罕見的危及生命的自身免疫性疾病)中進行了 第二階段研究,因為進一步的臨牀開發將需要大量資源,並顯著延長正在進行的臨牀計劃的時間表 。我們決定優先考慮我們的努力,並重新分配我們的資源,用於開發我們的口服C5aR抑制劑 INF904,這是一種針對C5aR受體的口服小分子候選藥物。我們計劃針對補體介導的慢性自身免疫和炎症條件,這些情況下口服小分子是患者的首選給藥途徑。我們最近在健康志願者中完成了I期單劑量上升劑量(SAD)和多劑量上升劑量(MAD)研究,在該研究中,我們能夠確認INF904的良好安全性以及卓越的藥代動力學和藥效學特徵。我們目前正在進行其他所需的臨牀前研究,包括長期的慢性毒理學研究,以實現長期劑量的INF904治療慢性炎症性疾病。我們還在開發IFX002,這是一種用於viloblimab的生命週期管理產品。
自2007年12月我們成立以來,我們將幾乎所有的資源都投入到了建立公司、籌集資金、開發我們專有的抗C5a/C5aR技術、識別和測試潛在的候選產品以及對我們的主要候選產品viloblimab和其他候選產品IFX002和INF904進行臨牀試驗上。到目前為止,我們只產生了最低限度的產品收入,到目前為止,我們主要通過在公開發行和私募中發行證券以及各種贈款的其他收入來為我們的運營提供資金,包括德國聯邦政府在2021年10月至2023年6月期間為某些研究和開發活動授予的贈款。截至2023年12月31日,我們擁有1280萬歐元的現金和現金等價物,以及8560萬歐元的有價證券。此外,截至2023年12月31日,作為2021年10月至2023年6月期間授予我們的贈款的一部分,我們總共收到了3330萬歐元 ,用於支持我們新冠肺炎臨牀開發的發展。
-99-
2020年7月8日,我們向美國證券交易委員會提交了一份關於 公司證券要約和出售的表格F—3登記聲明,或2020年—貨架登記聲明。我們還向美國證券交易委員會提交了一份招股説明書補充文件,內容涉及一項上市計劃 ,該計劃規定根據與SVB Leerink LLC簽訂的銷售協議 ,隨時間推移,我們的股票銷售額最高為5000萬美元。在2023財年,我們根據其上市計劃發行了3,235,723股普通股,產生了 1440萬歐元或1570萬美元的淨收益。截至2023年12月31日,以及在整個上市計劃期間,我們共發行了5,803,931股普通股,導致我們的淨所得款項總額為2620萬歐元。 上市計劃的期限於2023年7月8日到期。
通過 2023年4月的承銷公開發行,我們以每股4. 25美元的價格出售和發行了總計10,823,529股普通股,每股面值為0. 12歐元,其中1,411,764股是根據承銷商行使超額配售權而出售的。扣除承銷折扣和其他 發行費用後,本次發行的淨收益為3,870萬歐元。
2023年6月30日,我們提交了2023—登記聲明,根據該聲明,公司在2023財年未發行普通股或主動發售計劃 。該等證券的首次發售價總額將不超過2.5億元。
在 2023財年,我們共發行並註冊了120,257股普通股,原因是前僱員行使股票期權權 。所有購股權已根據二零一七年長期激勵計劃授出。其中14,930份購股權已於二零二二年十二月行使,105,327份購股權已於二零二三財政年度行使。普通股的面值為每股0. 12歐元。98,754股普通股以每股1.85美元的價格出售,21,503股普通股以每股3.35美元的價格出售。
截至2023年12月31日,我們的累計赤字為2.861億歐元。自成立以來,我們每 年都產生重大淨經營虧損,預計在可預見的將來,我們將繼續產生可比或不斷增加的淨經營虧損。我們的 淨虧損可能會因季度和年而大幅波動。我們預計,如果我們:
| ● | 展開 我們在美國為GOHIBIC(vilobelimab)所做的商業努力,通過增強我們的商業能力 戰略計劃,進一步擴大我們的銷售隊伍和醫療事務團隊,準備相關 針對醫療保健提供者和其他利益相關者的宣傳和醫學教育材料, 完善我們的醫療事務戰略,以提高醫療人員對EUA的認識 社區,並擴大我們的銷售和營銷努力; |
| ● | 繼續 在美國和歐洲開展vilobelimab的監管活動,包括 嚴重的COVID—19、PG和潛在的各種其他適應症; |
| ● | 繼續 生產vilobelimab,同時遵守適用的監管標準,用於臨牀 商業需求; |
| ● | 繼續 通過臨牀開發推進vilobelimab,包括PG和其他潛在的 適應症; |
| ● | 評估 並繼續進行INF904的II期臨牀開發; |
| ● | 啟動 繼續開展研究項目和開發活動,包括IFX002的開發; |
| ● | 積極 尋求確定更多研究項目和更多候選產品; |
| ● | 維護、擴大和保護我們的知識產權組合; |
| ● | 僱用 並保留人員,例如研發、法規事務、業務 開發和商業運營、製造和供應鏈管理等; 和 |
| ● | 招致 作為上市公司運營的額外成本,包括擴大我們的運營, 行政、財務和管理團隊。 |
-100-
| B. | 財務 業務概覽 |
| 1. | 收入 |
2023年6月,我們開始在美國商業化GOHIBIC(vilobelimab)。關於商業化的開始, 本集團與Cencora Inc.的某些子公司簽訂了協議。(原名:AmerisourceBergen Corp.)作為 集團的美國分銷商,並向美國醫院客户提供GOHIBIC(vilobelimab)。Cencora提供冷藏、 冷鏈配送服務、庫存管理和二次標籤/包裝等服務。
2023年,我們自成立以來首次從產品銷售中實現收入,共計6.3萬歐元。報告的收入 為最終客户(醫院)的銷售額。向分銷商的銷售並不構成盈利過程的完成,因此不會 導致根據國際財務報告準則第15號確認本公司的收入。
| 2. | 成本 銷售 |
截至2023年12月31日止的12個月內確認的銷售成本 為50萬歐元,與GOHIBIC(vilobelimab)在美國的收入 以及庫存減記有關。
在這些期間銷售的產品的銷售成本 不包括材料成本,因為這些材料的相關成本 是在FDA於2023年4月批准GOHIBIC(vilobelimab)EUA之前在之前期間發生的。這些材料在產生期間被記錄為"研究 和開發費用"。
截至2023年12月31日止十二個月的 銷售成本主要包括將在 預期銷售前到期的存貨減記。
| 3. | 其他 收入 |
2021年10月,我們宣佈獲得德國教育和研究部 和德國衞生部最多4370萬歐元的贈款,以支持維洛貝利單抗的某些研發活動,用於治療重症COVID—19患者。由於我們的研發計劃隨後發生變化,以及在資助期限內預計的成本減少, 我們接到通知,我們可獲得的最高總金額已降至4140萬歐元。該補助金的結構是償還 與vilobelimab臨牀開發和生產相關的某些預先指定費用的80%。在2021年10月1日至2023年6月30日期間, 為符合條件的活動頒發了該補助金。在補助金期間,截至2023年12月31日,我們總共收到了3330萬歐元,用於支持我們開發vilobelimab作為一種新型治療藥物治療重症新型冠狀病毒的活動—19名患者,並建立商業規模的生產工藝, 確保能夠為更廣泛的人羣提供此類治療。
| 4. | 銷售 及市場推廣開支 |
銷售 和營銷費用主要包括:
| ● | 用於分發GOHIBIC以構建的外部 服務必要的商業和物流基礎設施,包括外部銷售專業人員; |
| ● | 營銷活動 ; |
| ● | 與員工相關的費用,包括工資、福利和基於員工在組織中的角色的股票薪酬費用;以及 |
| ● | 專業的 服務費,使GOHIBIC能夠在美國使用。 |
銷售和市場營銷費用總額為400萬歐元。這些費用主要包括100萬歐元的人事費用,100萬歐元的法律和諮詢費用,以及190萬歐元的GOHIBIC分銷外部服務費用。集團從商業化開始 a在2023年4月歐盟協議獲得批准時開展的活動。在此之前,未發生任何銷售和營銷費用 。
-101-
| 5. | 研發費用 |
研究和開發費用主要包括:
| ● | 根據與CRO、合同製造組織或CDMO、顧問和代表我們進行研發、臨牀前和臨牀活動的獨立承包商的協議而產生的費用 ; |
| ● | 與員工相關的費用,包括工資、福利和基於員工在組織中的角色的股票薪酬費用;以及 |
| ● | 與保護和維護我們的知識產權有關的律師的專業費用。 |
我們在2023年的總研發費用高於我們在2022年的支出,也高於我們在2021年的支出。隨着我們推進PG中的viloblimab的第三階段開發,並啟動INF904的第二階段開發,預計2024年的成本將增加(與2023年相比)。預計2024年及未來期間研發費用的增加將主要與以下關鍵計劃和活動有關:
維洛布利單抗.
我們預計,隨着我們在PPG中進行第三階段臨牀藥物研究的進展,與2023年上半年相比,我們與維羅貝利單抗相關的費用將在2024年左右增加。此外,由於在美國、歐洲和其他地方準備和提交viloblimab的完整市場授權,我們正在並預計還將產生 費用。我們還可能考慮在更多的適應症中開發Viloblimab。此外,我們還產生了與臨牀試驗材料的製造和完成最終建立商業規模生產的活動相關的費用 。
| ● | INF904. 我們還在開發INF904,這是一種針對C5aR受體的候選產品。 我們預計推進INF904的臨牀和非臨牀開發將產生額外的成本。具體地説,我們預計將通過開發新配方、對幾種動物進行長期毒理學研究以及啟動第二階段臨牀試驗來產生費用。我們計劃在補體介導、慢性自身免疫和炎症情況下對INF904進行研究 在口服低分子化合物可能對患者有優勢或患者需要的情況下,並且口服給藥是醫學上的首選給藥方式。 |
| ● | IFX002。我們 還在開發用於治療慢性炎症適應症的IFX002。IFX002是一種高效的抗補體C5a抗體,人源化程度更高,與維洛布單抗相比, 藥代動力學特性發生了改變,目前正處於臨牀前開發階段。 該計劃的費用主要包括工資,CRO進行臨牀前測試的成本 以及生產臨牀前材料的成本。 |
| ● | 其他 開發計劃。我們的其他研發費用與我們對其他候選產品的臨牀前研究和發現活動有關,這些費用主要包括工資、臨牀前化合物的生產成本和支付給CRO的成本。 |
在2023年和2022年,我們分別產生了4100萬歐元和3750萬歐元的研發費用。我們的研究和開發費用可能因研究和開發活動的時間不同而有很大差異,包括臨牀試驗啟動和潛在登記的時間。
我們 按所發生的費用來支付研發費用。我們根據對完成特定任務的進度的評估,確認某些開發活動的成本,如臨牀前研究和臨牀試驗。我們使用我們的 供應商提供給我們的信息,例如患者登記或臨牀站點激活,以瞭解所接受的服務和花費的努力。研發活動是我們業務模式的核心。
-102-
我們的候選產品能否成功開發具有很大的不確定性。目前,我們無法合理估計完成任何候選產品開發所需的工作的性質、時間 和估計成本,或可能開始現金淨流入的期間(如果有的話)。這是由於與開發藥物相關的許多風險和不確定性,包括:
| ● | 臨牀試驗或我們的候選產品產生否定或不確定的結果,包括未能證明統計意義。 |
| ● | 我們的臨牀試驗、非臨牀試驗和其他相關活動的範圍、進度、結果和成本; |
| ● | 延遲 與預期試驗地點或預期CRO就可接受的臨牀試驗合同或臨牀試驗方案達成或未能達成協議,其中的條款可以 進行廣泛的協商,並可能在不同的CRO和審判 地點之間存在較大差異; |
| ● | 為我們的候選產品和我們可能開發的任何產品製造臨牀用品和建立商業用品的成本 ; |
| ● | 第三方承包商未能及時遵守法規要求或履行其對我們的合同義務 ,或根本不遵守; |
| ● | 我們所追求的候選產品的數量和特點; |
| ● | 不良副作用或其他意想不到的特徵,導致我們或我們的調查人員、監管機構或機構審查委員會暫停或終止試驗; |
| ● | 潛在的 監管機構要求的額外安全監測或其他研究; |
| ● | 監管審批的成本、時間和結果; |
| ● | 審批所需的試驗次數; |
| ● | 患者隨訪時間; |
| ● | 建立銷售、營銷和分銷能力的成本和時間;以及 |
| ● | 我們可能建立的任何協作、許可和其他安排的條款和時間,包括據此支付的任何里程碑和版税。 |
與我們可能開發的Viloblimab、IFX002或任何其他候選產品相關的這些變量中的任何一個的結果發生變化 可能意味着與開發該候選產品相關的成本和時間的重大變化。
| 6. | 一般費用和管理費用 |
我們的一般和行政費用主要包括:
| ● | 與員工相關的費用,包括工資、福利和基於員工在組織中的角色的股票薪酬費用; |
| ● | 保險 包括董事和高級職員責任保險費在內的費用; |
| ● | 與研發活動無關的審計師專業費用和諮詢費; |
| ● |
| ● | 設施費用、差旅費用、通信費用和辦公費用。 |
我們 預計未來我們的一般和管理費用將隨着業務的擴展而增加,並且我們會產生與上市公司運營相關的額外成本 。這些與上市公司相關的成本主要涉及額外的人員、額外的法律費用、審計費、董事和高級管理人員的責任保險費以及與投資者關係相關的成本。
-103-
| 7. | 運營結果 |
集團面臨歐元與美元之間的匯率風險。由於本公司以美元登記發行各種普通股,本集團持有大量現金、現金等價物及美元有價證券。這可能會對我們的運營業績產生實質性影響。
下面的數字來自我們的合併財務報表,包括在本文其他地方。下面的討論應與這些合併財務報表一起閲讀,並通過參考它們來對其整體進行限定。
2023年和2022年12月31日終了年度比較
| 2023 | 2022 | 變化 | ||||||||||
| (歐元) | ||||||||||||
| 收入 | 63,089 | — | 63,089 | |||||||||
| 銷售成本 | (532,262 | ) | — | (532,262 | ) | |||||||
| 毛利 | (469,173 | ) | — | (469,173 | ) | |||||||
| 銷售和市場營銷費用 | (4,001,299 | ) | — | (4,001,299 | ) | |||||||
| 研發費用 | (41,024,131 | ) | (37,526,090 | ) | (3,498,041 | ) | ||||||
| 一般和行政費用 | (12,628,756 | ) | (14,869,564 | ) | 2,240,808 | |||||||
| 其他收入和支出(淨額) | 13,215,264 | 20,157,788 | (6,942,524 | ) | ||||||||
| 息税前虧損 | (44,908,096 | ) | (32,237,866 | ) | (12,670,230 | ) | ||||||
| 淨財務業績 | 2,240,566 | 2,753,255 | (512,689 | ) | ||||||||
| 税前虧損 | (42,667,529 | ) | (29,484,611 | ) | (13,182,918 | ) | ||||||
| 所得税費用 | — | — | — | |||||||||
| 當期虧損 | (42,667,529 | ) | (29,484,611 | ) | (13,182,918 | ) | ||||||
| 外幣折算操作的匯兑差異 | 125,085 | 4,206,810 | (4,081,725 | ) | ||||||||
| 全面損失總額 | (42,542,444 | ) | (25,277,801 | ) | (17,264,643 | ) | ||||||
收入
| 2023 | 2022 | 變化 | ||||||||||
| (歐元) | ||||||||||||
| 收入 | 63,089 | — | 63,089 | |||||||||
| 總計 | 63,089 | — | 63,089 | |||||||||
2023年6月,我們開始在美國將GOHOBIC(Viloblimab)商業化。關於商業化的開始, 我們與某些子公司簽訂了協議Cencora Inc.(前身為amerisourceBergen Corp.)作為集團在美國的分銷商,並讓GOHIBIC(Viloblimab)可供美國醫院客户訂購。 Cencora提供冷藏、冷鏈配送服務、庫存管理和二次標籤/包裝等服務。
2023年,我們自成立以來首次確認了產品銷售收入。報告的收入是對最終客户的銷售 (醫院)。向分銷商銷售並不構成完成對客户收益過程的履約義務,因此,不會導致根據IFRS 15確認公司收入。
-104-
銷售成本
| 2023 | 2022 | 變化 | ||||||||||
| (歐元) | ||||||||||||
| 銷售成本 | 532,262 | — | 532,262 | |||||||||
| 總計 | 532,262 | — | 532,262 | |||||||||
在截至2023年12月31日的12個月內確認的銷售成本與GOHBIC(Viloblimab)在美國的收入有關。 在此期間銷售的產品的銷售成本不包括材料成本,因為這些材料的相關成本是在授予GOHIBIC(Viloblimab)EUA之前的 期間發生的。這些材料的製造成本在發生期間記為研究和開發費用。
最初的產品批次後來在庫存中資本化,是用前幾年製造的材料生產的。
銷售 和營銷費用
| 2023 | 2022 | 變化 | ||||||||||
| (歐元) | ||||||||||||
| 第三方費用 | 1,851,158 | — | 1,851,158 | |||||||||
| 人員費用 | 1,040,587 | — | 1,040,587 | |||||||||
| 律師費和諮詢費 | 1,054,971 | — | 1,054,971 | |||||||||
| 其他費用 | 54,581 | — | 54,581 | |||||||||
| 銷售和營銷費用總額 | 4,001,299 | — | 4,001,299 | |||||||||
在截至2023年12月31日的12個月中,我們產生了400萬歐元的銷售和營銷費用。這些費用主要由100萬歐元的人事費用、100萬歐元的法律和諮詢費用以及1.9歐元的外部服務組成,用於GOHIBIC的分銷 。
研發費用
| 2023 | 2022 | 變化 | ||||||||||
| (歐元) | ||||||||||||
| 第三方費用 | 31,802,983 | 28,543,503 | 3,259,480 | |||||||||
| 人員費用 | 6,776,853 | 6,957,866 | (181,013 | ) | ||||||||
| 其他費用 | 2,444,295 | 2,024,721 | 419,574 | |||||||||
| 總計 | 41,024,131 | 37,526,090 | 3,498,042 | |||||||||
與截至2022年12月31日的年度的相應成本相比,截至2023年12月31日的年度的研究和開發費用增加了350萬歐元,這主要是由於第三方材料和製造(CDMO)成本增加以及臨牀試驗增加了190萬歐元。
-105-
一般費用和管理費用
| 2023 | 2022 | 變化 | ||||||||||
| (歐元) | ||||||||||||
| 人員費用 | 5,392,905 | 7,125,798 | (1,732,893 | ) | ||||||||
| 法律、諮詢和審計費用 | 3,239,809 | 3,104,624 | 135,185 | |||||||||
| 其他費用 | 3,996,042 | 4,639,142 | (643,100 | ) | ||||||||
| 總計 | 12,628,756 | 14,869,564 | (2,240,809 | ) | ||||||||
一般 及行政開支由截至二零二二年十二月三十一日止年度的1490萬歐元減少220萬歐元至截至二零二三年十二月三十一日止年度的1260萬歐元。這一減少的部分原因是人員費用減少170萬歐元, 受基於股份的薪酬支出減少的推動。其他費用減少60萬歐元,主要原因是 D & O保險費用降低。
淨 財務結果
| 2023 | 2022 | 變化 | ||||||||||
| (歐元) | ||||||||||||
| 利息收入 | 3,804,827 | 608,679 | 3,196,148 | |||||||||
| 利息支出 | (16,538 | ) | (23,303 | ) | 6,765 | |||||||
| 租賃負債利息 | (19,090 | ) | (21,947 | ) | 2,857 | |||||||
| 淨利息結果 | 3,769,199 | 563,429 | 3,205,770 | |||||||||
| 外匯收入 | 5,529,389 | 6,924,697 | (1,395,308 | ) | ||||||||
| 外匯費用 | (7,371,261 | ) | (4,482,399 | ) | (2,888,862 | ) | ||||||
| 淨匯兑結果 | (1,841,872 | ) | 2,442,298 | (4,284,170 | ) | |||||||
| 其他財務結果 | 313,240 | (252,471 | ) | 565,711 | ||||||||
| 淨財務業績 | 2,240,566 | 2,753,256 | 512,690 | |||||||||
截至2023年12月31日的年度淨財務業績減少50萬歐元至220萬歐元,而截至2022年12月31日的年度財務業績為270萬歐元 。這一總體淨減少主要是由於與截至2022年12月31日的年度相比,外匯兑換結果減少了430萬歐元,來自有價證券的利息收入增加了320萬歐元。
-106-
2022年和2021年12月31日終了年度比較
| 2022 | 2021 | 變化 | ||||||||||
| (歐元) | ||||||||||||
| 研發費用 | (37,526,090 | ) | (35,697,935 | ) | (1,828,155 | ) | ||||||
| 一般和行政費用 | (14,869,564 | ) | (11,984,722 | ) | (2,884,842 | ) | ||||||
| 其他收入和支出(淨額) | 20,157,788 | 47,840 | 20,109,948 | |||||||||
| 息税前虧損 | (32,237,866 | ) | (47,634,816 | ) | 15,396,950 | |||||||
| 淨財務業績 | 2,753,255 | 2,004,757 | 748,498 | |||||||||
| 税前虧損 | (29,484,611 | ) | (45,630,059 | ) | 16,145,448 | |||||||
| 所得税費用 | — | — | — | |||||||||
| 當期虧損 | (29,484,611 | ) | (45,630,059 | ) | 16,145,448 | |||||||
| 外幣折算操作的匯兑差異 | 4,206,810 | 6,777,061 | (2,570,251 | ) | ||||||||
| 全面損失總額 | (25,277,801 | ) | (38,852,998 | ) | 13,575,197 | |||||||
研發費用
| 2022 | 2021 | 變化 | ||||||||||
| (歐元) | ||||||||||||
| 第三方費用 | 28,543,503 | 28,247,081 | 296,422 | |||||||||
| 人員費用 | 6,957,866 | 5,941,813 | 1,016,053 | |||||||||
| 其他費用 | 2,024,721 | 1,509,041 | 515,680 | |||||||||
| 總計 | 37,526,090 | 35,697,935 | 1,828,155 | |||||||||
截至2022年12月31日止年度,研究及開發費用較截至2021年12月31日止年度增加1. 8百萬歐元。 此增加主要是由於薪酬相關成本增加了100萬歐元,主要是由於基於股份的薪酬支出增加了90萬歐元 。
一般費用和管理費用
| 2022 | 2021 | 變化 | ||||||||||
| (歐元) | ||||||||||||
| 人員費用 | 7,125,798 | 6,500,680 | 625,118 | |||||||||
| 法律、諮詢和審計費用 | 3,104,624 | 2,065,423 | 1,039,201 | |||||||||
| 其他費用 | 4,639,142 | 3,418,619 | 1,220,523 | |||||||||
| 總計 | 14,869,564 | 11,984,722 | 2,884,842 | |||||||||
一般 及行政開支由截至二零二一年十二月三十一日止年度的12. 0百萬歐元增加2. 9百萬歐元至截至二零二二年十二月三十一日止年度的14. 9百萬歐元。這一增長部分歸因於 基於股份的薪酬支出增加了90萬歐元。截至2022年12月31日止年度,法律、諮詢和審計費用及其他費用增加了100萬歐元至 ,主要是由於我們首次遵守2002年《薩班斯—奧克斯利法案》第404(b)條的審計師認證要求,因此在加強內部控制環境方面產生的諮詢和法律費用增加 。其他費用增加120萬歐元主要是由於D & O保險費用增加。
-107-
淨 財務結果
| 2022 | 2021 | 變化 | ||||||||||
| (歐元) | ||||||||||||
| 利息收入 | 608,679 | 109,391 | 499,288 | |||||||||
| 利息支出 | (23,303 | ) | (10,714 | ) | (12,589 | ) | ||||||
| 租賃負債利息 | (21,947 | ) | (14,055 | ) | (7,892 | ) | ||||||
| 淨利息結果 | 563,429 | 84,622 | 478,807 | |||||||||
| 外匯收入 | 6,924,697 | 5,569,836 | 1,354,862 | |||||||||
| 外匯費用 | (4,482,399 | ) | (3,605,701 | ) | (876,699 | ) | ||||||
| 淨匯兑結果 | 2,442,298 | 1,964,135 | 478,163 | ) | ||||||||
| 其他財務結果 | (252,471 | ) | (44,000 | ) | (208,471 | ) | ||||||
| 淨財務業績 | 2,753,256 | 2,004,757 | 748,498 | |||||||||
與截至2021年12月31日的財年相比,截至2022年12月31日的財年淨財務業績增加了70萬歐元。這一總體淨增長主要是由於與截至2021年12月31日的年度相比,外匯收入和支出淨增加50萬歐元,有價證券利息收入增加50萬歐元。
| C. | 流動性 和資本資源 |
| 1. | 現金需求和流動資金來源概覽 |
自 成立以來,由於研發活動和G&A成本,我們發生了嚴重的運營虧損。截至2023年12月31日和2022年12月31日的年度,我們的淨虧損分別為4,270萬歐元和2,950萬歐元。我們現金的主要用途 用於營運資金、運營租賃和一般企業用途。
我們的主要資金來源是出售我們股票的收益,包括我們的首次公開募股和後續發行。此外,在2021年,我們從德國聯邦政府獲得了一筆撥款,在2021年10月至2023年6月期間,我們獲得了3330萬歐元。授權期於2023年6月30日結束。從歷史上看,我們一直能夠通過配售股票來通過股權融資獲得現金來滿足我們的資本需求。
於2023年4月透過包銷公開發售,本公司共發售及發行普通股10,823,529股,其中1,411,764股是根據承銷商行使超額配股權而售出。普通股以每股4.25美元的價格出售 ,面值為每股0.12歐元。在扣除250萬歐元(280萬美元)的承銷折扣後,此次發行的收益為3910萬歐元(4320萬美元)。其他發行費用為40萬歐元,導致此次發行的淨收益總額為3870萬歐元。
2023年6月30日,我們向美國證券交易委員會提交了關於要約和出售公司證券的F-3表格(2023年-註冊聲明),該表格於2023年7月11日生效。根據本招股説明書,該公司可能提供和出售的證券的初始發行價合計不超過2.5億美元。本公司於2023財政年度內並無根據《2023-註冊聲明》 發行普通股,亦未啟動任何主動發售計劃。
我們的營運資金不包括2023年或2022年的任何債務。
-108-
截至2023年12月31日,我們的現金和現金等價物達到1280萬歐元(2022年:1630萬歐元)。截至2022年12月31日,我們還持有價值8560萬歐元(2022年:6720萬歐元)的有價證券。我們的現金和現金等價物主要包括銀行存款賬户中以美元和歐元計價的現金。我們的有價證券包括由具有投資級信用評級(BBB+至AAA)的金融機構發行的報價債務證券。我們的現金存放在國際評級機構評估的信用評級同樣高的銀行。
我們 預計在不久的將來,我們將主要通過現金和現金等價物以及出售有價證券的收益來為我們未來的運營和營運資金需求提供資金。
| 2. | 現金流動--2023年和2022年12月31日終了年度比較 |
下表彙總了我們截至2023年12月31日和2022年12月31日的綜合現金流量表:
| 2023 | 2022 | |||||||
| (歐元) | ||||||||
| 用於經營活動的現金淨額 | (37,812,966 | ) | (33,742,817 | ) | ||||
| 淨現金(用於投資活動)/來自投資活動 | (17,696,616 | ) | 19,358,095 | |||||
| 融資活動的現金淨額 | 52,986,269 | 1,937,459 | ||||||
| 期初的現金和現金等價物 | 16,265,355 | 26,249,995 | ||||||
| 現金和現金等價物的匯兑(損失)/收益 | (974,099 | ) | 2,462,622 | |||||
| 期末現金和現金等價物 | 12,767,943 | 16,265,355 | ||||||
淨額 經營活動中使用的現金
在所有期間使用現金的主要原因是我們的淨虧損(根據非現金費用和營運資本組成部分的變化進行調整)。
經營活動所用現金淨額 從截至2022年12月31日的 年度的3380萬歐元增加至截至2023年12月31日止年度的3780萬歐元,主要是由於德國聯邦政府補助金確認的收入減少、存貨大量生產 以及GOHIBIC的營銷和銷售活動支出增加(vilobelimab),由於2023年該產品開始商業化,這是第一次記錄。
用於投資活動的現金淨額
截至2023年12月31日止年度,投資活動使用的現金淨額為1770萬歐元,原因是購買淨額高於銷售有價證券所得款項。在截至2022年12月31日的上一年度,出售有價證券產生了1940萬歐元現金。
融資活動現金淨額
融資活動產生的現金淨額 由截至2022年12月31日止年度的190萬歐元 增加至截至2023年12月31日止年度的5300萬歐元,主要由於通過2023年4月承銷公開發售及 在2023年7月8日到期前利用市場融資進行股份發行。
-109-
| 3. | 合同 義務和承諾 |
下表載列了截至2023年12月31日的運營費用和來自合同義務的資本支出。
| 按期間到期的付款 | ||||||||||||||||||||
| 總計 | 不足 1年 | 在1之間 和3年 | 在3之間 和5年 | 多過 5年 | ||||||||||||||||
| (歐元) | ||||||||||||||||||||
| 經營合同或服務項下的非合同(CRO、CDMO)承諾和其他合同義務: | 18,233,216 | 13,926,289 | 4,265,136 | 41,791 | — | |||||||||||||||
| 合同租賃債務 (包括資本化租賃) | 1,158,363 | 397,942 | 760,421 | — | — | |||||||||||||||
| 總計 | 19,391,579 | 14,324,231 | 5,025,557 | 41,791 | — | |||||||||||||||
我們 與CRO和臨牀研究中心簽訂合同,以進行臨牀試驗,專業顧問提供專家建議, 與其他供應商(如CDMO)簽訂合同,用於臨牀供應品生產或正常業務過程中的其他服務。這些合同通常 可在30至180天內通知終止。除此最短期限外,這些合同還要求為已經開始的服務支付全額 。在上表中,不可避免的合同義務的金額假設合同於2023年12月31日終止,然後將持續約30至180天。
合同 租賃債務
合同 租賃責任主要包括根據與我們的辦公室租賃有關的不可撤銷租賃協議支付的款項。 我們位於德國耶拿的場地的租賃期將於2025年12月到期。我們位於德國Planegg—Martinsried的場地的租賃期將於2027年5月到期。本公司位於美國密歇根州安娜堡之物業之租期將於二零二六年四月屆滿。
未來資本支出的資金需求
我們 相信,我們現有的現金和現金等價物以及金融資產將使我們能夠至少在未來24個月內滿足當前業務計劃下的運營費用和資本支出 需求。
We anticipate that our expenses will increase in the next years in connection with our ongoing activities. In particular, we anticipate that we might expand our sales and marketing efforts for GOHIBIC(vilobelimab) in the United States, we might advance our Phase III clinical development program with vilobelimab in PG, pursue the further clinical development for INF904 and will advance preparation of necessary submission documents for additional regulatory submissions to the EMA and for a full BLA submission to the FDA beyond the received the EUA as granted by the FDA in April 2023 for GOHIBIC (vilobelimab). We will also explore clinical development of vilobelimab in several other indications. We also plan to continue clinical development of INF904 and to initiate Phase II clinical trials once we selected the appropriate indications. We also plan to continue preclinical development of IFX002. We plan to initiate new research and preclinical development efforts. If clinical data is supportive, we may seek marketing approval for any product candidates that we successfully develop. Additionally, we will validate our manufacturing process for vilobelimab to be able to apply for marketing authorization and to be able to provide commercial grade product. In addition, if we obtain marketing approval for any of our product candidates, we expect to incur significant commercialization expenses related to establishing sales, marketing, distribution and other commercial infrastructure to commercialize such products. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce, or eliminate our research and development programs or future commercialization efforts.
-110-
在 我們能夠產生大量有意義的產品收入之前,我們希望通過 股權發行、債務融資、基於版税的融資、未來合作、戰略聯盟、許可協議和 政府補助等多種方式來滿足我們的現金需求。在我們通過出售股權或可轉換債務證券籌集額外資本的情況下,我們現有股東的利益 將被稀釋,這些證券的條款可能包括對 您作為普通股股東的權利產生不利影響的投票權或其他權利。債務融資(如果可用)可能涉及協議,其中包括限制或限制 我們採取具體行動的能力,例如產生額外債務、進行資本支出或宣佈股息。如果我們通過與第三方的額外合作、戰略聯盟或許可協議籌集 資金,我們可能不得不放棄 對我們的技術、未來收入來源、研究項目或候選產品的權利,或者以可能不利於我們 的條款授予許可。通過政府補助金獲得的資金可能要求我們在這些司法管轄區的不利條件下提供產品,如果得到監管機構的批准。
| D. | 研發、專利和許可證等。 |
參見 "項目4。公司信息—2.業務概述—我們的知識產權。"
| E. | 趨勢 信息 |
除本年報表格20—F其他地方披露的情況外, 我們不知道截至2023年12月31日止年度的任何趨勢、不確定性、需求、承諾、 或事件合理可能對我們的淨收入、收入、 盈利能力、流動性或資本資源產生重大不利影響,或導致所披露的財務信息不一定指示未來的經營成果或財務狀況(見"項目5,業務及財務回顧及展望”)。
| F. | 關鍵 會計估計 |
我們的合併財務報表是按照國際會計準則委員會發布的國際財務報告準則編制的。在編制我們的合併財務報表時,我們對我們的會計政策的應用做出判斷、估計和假設,這些政策會影響報告的資產、負債、收入和費用的金額。我們的關鍵會計判斷和估計不確定性的來源載於附註B.2。我們的綜合財務報表,包括在本年度報告的其他部分。
-111-
第 項6.董事、高級管理人員和員工
| A. | 董事 及高級管理層 |
董事會和高級管理團隊
下表列出了截至本年報日期有關我們董事會和高級管理層的信息。
| 名字 | 職位 | 年齡 | 第一年 InflaRx GmbH, InflaRx N.V. 或InflaRx pharmaceuticals Inc. (視乎情況而定) | ||||||
| 尼爾斯·裏德曼 | 董事執行總裁兼首席執行官 | 52 | 2007 | ||||||
| 郭仁峯 | 執行董事兼首席科學官 | 53 | 2007 | ||||||
| 託馬斯·塔普肯 | 首席財務官 | 58 | 2020 | ||||||
| 張嘉美 | 首席醫療官 | 58 | 2023年(7月起) | ||||||
| 德瓦爾·歐卡羅爾 | 高級副總裁,全球監管事務與合規負責人 | 58 | 2023年(4月起) | ||||||
| 尼古拉斯·富爾皮烏斯 | 董事非執行董事兼董事會主席 | 50 | 2007 | ||||||
| 理查德·布魯德尼克 | 非執行董事董事 | 67 | 2019 | ||||||
| 馬克·庫布勒 | 非執行董事董事 | 49 | 2015 | ||||||
| 安東尼·吉布尼 | 非執行董事董事 | 53 | 2021 | ||||||
| 黑格·赫爾斯特羅姆 | 非執行董事董事 | 58 | 2023年(4月起) | ||||||
Mark Kubler和Anthony Gibney被任命為董事會成員的任期將於2024年到期, Richard Brudnick、Nicolas Fulpius、Renfeng Guo、Niels Riedemann和Hege Hellstrom被任命為董事會成員的任期將於2026年到期。
女士 Derval O'Carroll於2023年4月晉升為高級副總裁,並通過晉升有效地成為我們高級管理團隊的一員。 Camilla Chong女士於2023年7月加入公司擔任首席醫療官。
除非 另有説明,否則我們董事、高級管理層和主要員工目前的辦公地址為InflaRx N.V.,Winzerlaer Strasse 2,07745耶拿,德國。
以下是我們董事、高級管理層和主要員工的業務經驗的簡要總結。每位董事的任期反映了這些董事在InflaRx GmbH董事會的任期。
非執行 董事
Nicolas Fulpius,主席. Nicolas Fulpius是InflaRx的聯合創始人之一,自2007年成立以來一直擔任董事會主席。Nicolas長期活躍於美國和歐洲的風險投資領域,曾為Lombard Odier Immunology基金、Ultreia Capital以及Affentranger Associates的合夥人工作,從本質上講,Nicolas已經成為一名企業家:他創建、開發了生物技術、清潔技術和ICT領域的幾家公司,並幫助投資 。最近,Fulpius先生還擔任過Veltigroup的首席執行官、Swisscom的首席執行官 和Swisscom Ventures投資委員會成員。2020年,Nicolas Fulpius共同創立了瑞士領先的ICT 服務公司之一Ansam Group,他擔任首席執行官兼董事長。Nicolas Fulpius擁有瑞士聖加侖大學的MBA學位,以及美國斯坦福大學的工程學碩士學位。
-112-
Richard Brudnick. Richard Brudnick目前擔任Prime Medicine,Inc.的首席商務官,基因編輯領域的領導者 加入Prime Medicine之前,Brudnick先生曾擔任Codiak Biosciences的首席商務官兼戰略主管,Codiak Biosciences是 外泌體治療領域的領導者。在Codiak之前,Brudnick先生是Bioverativ,Inc.業務開發和聯盟管理執行副總裁 ,他在2016年幫助創立的公司在Bioverativ於2018年3月被賽諾菲收購之前,Brudnick先生領導了 業務開發工作,在罕見血液疾病領域建立了重要的管道,包括收購、多產品合作 以及其他科學合作和許可。Brudnick先生在Bioverativ從Biogen分拆出來時加入了Bioverativ,在近15年的時間裏,他發起、領導並完成了一些交易,這些交易導致了該公司的幾個上市產品和後期 生產線,包括Tecfidera、Spinraza、Leqembi及其與三星的生物仿製藥合資企業。Brudnick先生還擔任一家區域性 藥品分銷業務的首席執行官,他將該業務出售給了一位戰略買家;與人共同創辦了兩家公司;並擔任 貝恩公司的戰略顧問。
Mark Kubler. Kubler先生自2015年以來一直擔任董事會的董事。Kubler先生一直是GIG Ltd.的合夥人,自2012年以來,該公司在瑞士和馬耳他設有辦事處。他曾在WWM AG和Jobydu AG的董事會任職,這兩個公司的總部都位於瑞士。Kubler先生曾於2003年至2010年擔任一傢俬募股權控股公司的董事總經理兼公司祕書。 2003年之前,他在國際投資銀行和精品店擔任過各種職務。Kubler先生擁有商業和經濟學碩士學位,以及瑞士聖加侖大學的法律碩士學位。
安東尼·吉普尼 was formerly the Chief Business and Strategy Officer at Iveric Bio, overseeing the business development and corporate strategy for the retina-focused, biotechnology company, through the closing of the sale of Iveric to Astellas for $5.9 billion in 2023. Prior to Iveric, Mr. Gibney was the CFO and CBO at FogPharma, driving the business development and finance functions of the company. Mr. Gibney served as the Chief Business Officer of Achillion Pharmaceuticals, Inc., where he was responsible for corporate and portfolio strategy, business development and corporate communications and led the successful sale of Achillion to Alexion in 2020. Before Achillion, Tony Gibney was a life sciences-focused investment banker for 24 years. From 2009 through 2017, he served as a managing director and co-head of the biotechnology investment team for Leerink Partners LLC, where he was a senior leader of Leerink’s biopharmaceutical investment banking franchise. From 1999 to 2009, he worked as a managing director at Merrill Lynch Inc. and executed a variety of significant financing and M&A transactions for various biotechnology companies. From 1993 to 1999, Mr. Gibney was an investment banker at Lehman Brothers in the firm’s Healthcare Investment Banking Group. He graduated with distinction from Yale University in 1993 with a B.A. in History and Economics.
赫克 赫爾斯特倫目前是Advicenne的首席商務官,Advicenne是一家法國製藥公司,專門從事腎臟病的 創新治療的開發。她自2019年起擔任Vivesto AB和Camurus AB的非執行董事會成員,這兩家公司都是瑞典上市公司 ,她也是兩家公司的審計委員會成員。她是投資和諮詢公司Belnor BV的創始人兼董事總經理。Hellstrom女士在銷售、市場營銷、戰略開發、 商業化、合作伙伴聯盟和執行管理方面擁有超過30年的經驗。從2013年到2018年,她在瑞典生物製藥公司Sobi擔任歐洲、中東、北非和俄羅斯總裁,領導了血友病和代謝疾病等罕見疾病的幾次上市。在Sobi之前,她在Genzyme工作了11年,擔任過從比荷盧總經理到歐洲、拉丁美洲和日本亞太地區腎臟和內分泌業務負責人的職務。Genzyme被賽諾菲收購後,她繼續擔任賽諾菲心血管 產品全球副總裁。在Genzyme之前,她在Baxter Healthcare工作了13年。赫爾斯特羅姆夫人擁有理學學士學位,生物醫學實驗室 挪威奧斯陸城市大學的科學家。
執行 董事
Niels Riedemann,首席執行官兼創始人。 Riedemann教授是我們的聯合創始人之一,自2007年成立以來一直擔任我們的首席執行官 。Riedemann教授在生物技術行業和藥物開發方面擁有超過15年的經驗,在補體免疫學研究方面擁有超過20年的經驗。他於2007年創立了InflaRx,自公司成立以來一直擔任首席執行官 。他在公司的多輪私人和公共融資中發揮了重要作用,並領導了2017年納斯達克IPO的負責人。他是InflaRx多項國際授權核心專利的發明人。 作為醫生,他被任命為副主任(萊登·奧貝拉爾茨特),他在德國耶拿市弗里德里希·席勒大學(Friedrich Schiller University,Jena,2015年)領導了一個有50張牀位的大學重症監護病房超過6年。在此之前,他於2007年在MHH(德國漢諾威醫學院)完成外科獎學金後獲得了董事會證書 作為普通外科醫生,他還獲得了 他的博士學位(相當於博士學位)。在那裏,他仍然持有兼職教授(APL教授)。他在美國密歇根大學做了三年博士後 研究員,直到2003年。他在德國弗萊堡阿爾伯特路德維希大學(ALU)和美國斯坦福大學接受醫學訓練,畢業後獲得醫學博士學位(相當於醫學博士學位)。1998年,ALU。他的研究獲得了 多個國家和國際獎項。他獲得了廣泛的校外資助,並在高排名期刊上發表了60多篇同行評審的科學論文。他曾擔任兩個大型 科學政府資助項目的董事會和科學顧問委員會成員。他目前擔任Bio—Deutschland健康政治工作組的聯合主席, 他擔任德國敗血癥基金會董事會成員。
-113-
郭仁峯,首席科學官兼創始人。 郭仁峯教授於2007年共同創立了InflaRx。自成立以來,他一直負責InflaRx的科學開發,擔任全職CSO。郭教授利用其在抗體研究和炎症方面的專業知識,將 高效的藥物開發研究團隊集合在一起,以尖端技術為基礎建立了一個重點突出的管道。他的早期研究導致了InflaRx的主要藥物vilobelimab的發現。他繼續是其他 管道藥物開發的推動力,也是InflaRx知識產權組合的關鍵發明者。郭教授獲得醫學博士學位。畢業於中國白求恩醫學院,並在密歇根大學安娜堡分校Peter Ward教授的實驗室進行博士後研究。在2001年開始在密歇根大學擔任初級和高級教員後,他目前是兼職研究副教授。郭教授在癌症、傳染病和炎症研究領域發表了80多篇高影響力的同行評審論文。
高級 管理層
Thomas Taapken,首席財務官。 Taapken博士於2020年加入InflaRx擔任首席財務官。他在生命科學領域的高級 管理職位和風險投資者方面擁有超過25年的經驗。他曾擔任Medigene AG的首席財務官(在德國上市),任Epigenomics AG首席執行官兼首席財務官(在德國上市),他領導公司 努力獲得FDA對公司主導產品的監管批准,並監督其隨後進入美國市場的 ,並擔任Biotie Therapies的首席財務官(芬蘭上市,現為Acorda Therapeutics)及其前身 公司。在此之前,他曾在德國風險投資公司(Deutsche Venture Capital,DVC)和Burrill & Co.擔任過七年的風險投資者。Taapken博士的職業生涯始於Hoechst AG(現為賽諾菲)。他擁有博士學位。在柏林工業大學學習有機化學專業,還在哥廷根大學學習經濟學、化學和物理學。Taapken博士自2017年起擔任 Scibase AB的董事會成員,自2019年起擔任lmcyse SA的董事會主席,自2021年起擔任memo therapeutics AG的董事會成員,直至 2023年底。
Camilla Chong,首席醫療官。 Camilla Chong博士於2023年7月加入擔任首席醫療官或CMO。她是一名在全球製藥行業擁有25年經驗的醫生。她成功領導了臨牀開發、醫療事務、 臨牀操作、監管和藥物警戒等方面的團隊。她的豐富經驗還包括在協和麒麟、葛蘭素史克、輝瑞和Teva擔任高級領導職務,在不同地區開發和推出新藥 。Chong博士在英國倫敦大學學院皇家自由醫院醫學院獲得醫學學位。她持有藥學文憑 ,是藥學院(MFPM)的成員。
Derval O'Carroll,法規事務和質量保證部高級副總裁. O'Carroll女士於2022年加入InflaRx擔任副總裁兼法規事務主管。她在法規事務方面擁有超過30年的經驗,過去19年擔任高級管理職位 ,為全球產品開發和商業化活動做出了戰略貢獻。她之前曾擔任過 罕見病公司Amryt Pharmaceuticals全球監管和質量副總裁五年,在罕見病公司Travere擔任 法規事務高級總監兩年,在Real Regulatory Regulatory Regulatory Regulatory Consultancy執行顧問11年,她曾為國際客户參與多項藥物開發項目。她在多個 崗位(包括藥物、器械和體外診斷產品)方面積累了豐富的經驗。O'Carroll女士在全球領先的創新新產品註冊活動方面有着良好的記錄,並在指導團隊通過FDA和EMA監管機構 進行授權前、授權和複雜的上市後承諾方面經驗豐富。她擁有生物化學碩士學位和都柏林大學學院的工商管理碩士學位。
-114-
| B. | 補償 |
董事和高級管理人員的薪酬
截至2023年12月31日止年度, 所有身份服務的 總薪酬(包括應計或支付給我們的高級管理層或高級管理層的實物利益)共計5,291,128歐元。於二零二三年,我們向董事會或董事會及高級管理層授出購股權以購買1,263,500股普通股。
截至2023年12月31日止年度,我們非執行董事就各種身份提供的服務而累計或支付的報酬總額(包括實物利益)為606,677歐元。於2023年,我們向 非執行董事授出購股權以購買157,500股普通股。
我們 參見附註C.10。董事、高級管理層及非執行董事以股份為基礎之付款。
| 尚未行使的股票期權 截至 1月1日, 2023 | 股票期權 授予 2023 | 庫存 選項 行使 2023 | 股票期權 傑出的 截至 十二月三十一日, 2023 | 加權 平均值 鍛鍊 價格 歐元 | 加權 平均值 剩餘 合同 生活 | |||||||||||||||||||
| 2023年未行使的股票期權 | ||||||||||||||||||||||||
| 高級管理層,包括執行董事 | ||||||||||||||||||||||||
| 首席執行官尼爾斯·C·裏德曼教授 | 2,289,714 | 548,000 | 2,837,714 | 1.75 | 6.24 | |||||||||||||||||||
| 郭仁峯教授,社會服務總監 | 1,814,197 | 430,500 | 2,244,697 | 1.82 | 6.39 | |||||||||||||||||||
| 首席財務官託馬斯·塔普肯 | 397,002 | 110,000 | 507,002 | 1.88 | 7,24 | |||||||||||||||||||
| Camilla Chong,首席營銷官 | 0 | 150,000 | 150,000 | 3.60 | 9.52 | |||||||||||||||||||
| 德瓦爾·奧卡羅爾,監管事務和合規部全球負責人高級副總裁 | 20,000 | 25,000 | 45,000 | 2.22 | 9.00 | |||||||||||||||||||
| 4,520,913 | 1,263,500 | 5,784,413 | ||||||||||||||||||||||
| 非執行董事 | ||||||||||||||||||||||||
| 尼古拉斯·富爾皮烏斯, 審計委員會主席和成員 | 119,065 | 45,000 | 164,065 | 1.85 | 6.82 | |||||||||||||||||||
| 安東尼·吉布尼 審計委員會主席 | 58,085 | 30,000 | 88,085 | 1.88 | 7.50 | |||||||||||||||||||
| 理查德·布魯德尼克 審計委員會委員 | 74,850 | 30,000 | 104,850 | 1.85 | 7.24 | |||||||||||||||||||
| 馬克·庫布勒 審計委員會委員 | 98,172 | 30,000 | 128,172 | 1.73 | 6.0 | |||||||||||||||||||
| Hege Hellstrom(從2023年4月開始) | - | 22,500 | 22,500 | 3.87 | 9.4 | |||||||||||||||||||
| 4,871,085 | 1.398,500 | 6,292,085 | ||||||||||||||||||||||
我們 根據荷蘭法律制定了有關董事薪酬的政策。董事會根據薪酬政策確定董事的薪酬,但有一項諒解,即執行董事不參與有關確定執行董事薪酬的決策過程 。
截至2023年12月31日,我們沒有為我們的高級經理或董事提供養老金、退休或類似福利的準備金或應計金額 。
-115-
退還政策
根據這項政策,如果我們的財務報表因重大不遵守財務報告標準或由於欺詐活動而被重述,董事會有權追回 授予高管的任何獎勵薪酬,該薪酬超出了根據重述財務業績應獲得的薪酬。追回條款 適用於基於績效的獎金、基於股權的薪酬以及與實現特定財務目標或非財務目標相關的任何其他薪酬。退款政策規定了確定何時可以啟動退款的程序、時間表和標準,以及計算和收回金額的流程。
追回政策在我們的公司治理原則中披露,並受薪酬委員會和董事會的監督。我們相信,這一政策的實施反映了我們致力於保持強有力的公司治理做法,並提升股東價值。
管理 和董事服務協議
我們 與我們的每一名執行管理團隊成員,包括我們的兩名執行董事 簽訂了管理服務協議,該協議在我們完成首次公開募股或這些經理加入本公司時生效。管理服務協議包含針對我們和由 股東大會任命的執行董事的終止通知期。所有管理服務協議都規定,經理或執行董事(視情況而定)可在發生緊急原因(德里根德·裏登),恕不另行通知。如果董事高管 不再擔任董事高管,但仍受僱擔任本公司高管僱員,則董事高管將無權獲得任何合同遣散費或解僱費。相反,我們將與董事高管簽訂僱傭協議,其中可能包括與管理服務協議中提供的薪酬條款基本相似的條款。 管理服務協議包含終止後的限制性約定,包括永久保密以及終止後的競業禁止和競業禁止約定。
此外,吾等與各非執行董事訂立函件協議,協議於完成首次公開招股或該等董事獲股東大會委任為本公司董事會成員時生效。如果非執行董事董事被免職、辭去董事會職務或該董事在董事會的任期屆滿而不再擔任非執行董事,則董事協議可以終止,恕不另行通知。 此外,每份函件協議都規定了補償,包括年度現金費用、年度股權授予、董事會委員會成員年費和董事會委員會主席年費 。此外,信件協議還包含一項永久的保密公約。
基於共享 的薪酬計劃
2016年計劃
根據2016年股票期權計劃的條款和條件,或2016年計劃,我們向董事、高級管理人員和關鍵員工授予了認購普通股的權利。
2016年計劃下的所有未償還期權獎勵在首次公開募股結束時自動歸屬。
與 在我們首次公開發行之前進行的企業重組相結合,根據 2016年計劃授予的所有未償還獎勵或以其他方式轉換為InflaRx N.V.普通股可行使的獎勵將受 2016年計劃條款的約束。
-116-
2017年計劃
在完成首次公開募股的同時,我們制定了一項新的綜合計劃,即2017年長期激勵計劃,或 2017年計劃,旨在通過增強我們吸引、留住和激勵預期對我們作出重要貢獻的 個人的能力,促進我們股東的利益。2017年計劃管理自我們首次公開發行結束之日起及 之後的股權激勵獎勵的發行。根據2017年計劃授出的股權激勵 獎勵可供發行的普通股初步最高數目初步等於2,341,097股普通股。於2021年1月1日及其後每個歷年的1月1日,根據2017年計劃授出的股權激勵獎勵,將有相當於上一年度12月31日已發行普通股總數4%的額外股份數量(或董事會釐定的任何較低股份數量)可供發行。
2020年7月16日舉行的股東周年大會批准了對2017年計劃的修訂,自2021年1月1日起生效:
| ● | 增加 公司資本中可供 根據2017年計劃發行,自2021年1月1日起,將其發行額增至公司發行額的4%(從3%開始) 發行在外的普通股(截至上一年的12月31日確定); 和 |
| ● | 刪除 2017年計劃中的某些限制,這將允許委員會管理 2017年計劃及董事會(i)降低任何購股權及/或股份的每股行使價 根據2017年計劃發行的增值權,或採取任何其他視為"重新定價"的行動 以及(ii)取消任何購股權和/或股份增值權以換取現金 或根據2017年計劃授予的其他獎勵,在任何情況下,未經 公司股東。 |
計劃 管理. 2017年計劃由董事會任命的長期激勵委員會(LTI)管理,該委員會由不少於三名董事組成。
資格. 股權激勵獎勵可授予我們的員工、非員工董事、顧問或其他顧問,以及由我們將來可能收購的公司授予的 股權補償獎勵的持有人。
獎項. 2017年計劃項下的股權激勵獎勵可以以股票期權、股票增值權、限制性股票、 限制性股票單位、業績獎勵或其他股份獎勵的形式授予。購股權和股票增值權的行使價格 由計劃委員會決定,但不低於相關普通股於授出日期的公平市值。
歸屬. 2017年計劃下股權激勵獎勵項下的授予條件將載於適用的獎勵文件。 然而,在下文所述若干情況下的加速規定的限制下,獎勵(替代獎勵除外)可能 不會在授出日期一週年前全部歸屬,但2017年計劃項下可供發行的最多5%的股份可能提供替代歸屬條件除外。
服務終止 和控制權變更.如果參與者終止僱用,計劃委員會可酌情決定股權激勵獎勵的行使、結算、歸屬、支付或沒收程度。如果 公司控制權發生變化(定義見2017年計劃),任何當時的繼承人或存續公司可以繼續未償還的 獎勵,或轉換或取代該等獎勵,以換取與繼承人或存續公司股票有關的獎勵或權利, 在這種情況下,如果參與者被繼承人或尚存法團無"原因"或"良好 理由"終止(在每種情況下,定義見2017年計劃)在控制權變更後24個月內,參與者持有的所有股權激勵獎勵 將立即歸屬。如果在公司控制權發生變化後,未繼續發放或轉換任何未償還的獎勵,則該等獎勵將立即歸屬,期權和股票增值權將完全可行使。在 控制權變更時,計劃委員會可酌情采取若干其他行動,包括加速 任何股權激勵獎勵的歸屬,或終止或取消任何股權激勵獎勵以支付現金。
-117-
保險 和賠償
我們的現任和未來董事(以及董事會指定的其他高級管理人員或員工)享有本公司章程中的賠償條款。這些條款賦予受補償人就履行職責中的行為或不作為向我們追回金額的權利,包括訴訟費用和他們被勒令支付的任何損害賠償。但是,對於被認為構成惡意、重大疏忽、故意魯莽和/或可歸因於該受補償人的嚴重過失的行為或不作為, 我們無權獲得賠償。此外,在首次公開招股完成後,我們與我們的董事和高管簽訂了協議,以在法律允許的範圍內最大限度地保障他們免受費用和責任的影響。除某些例外情況外,這些協議還規定對相關費用進行賠償,包括律師費、判決書、罰金、罰款和這些個人在任何訴訟或訴訟中產生的和解金額。除了這樣的賠償外,我們還為我們的董事提供董事和高級管理人員責任保險。
鑑於根據證券法規定,董事或控制吾等的人士可根據上述條文對根據證券法產生的責任作出彌償,美國證券交易委員會已獲告知,該等彌償違反證券法所述的公共政策,因此不可強制執行。
符合納斯達克上市要求
We are a foreign private issuer. As a result, in accordance with Nasdaq listing requirements, we comply with certain home country governance requirements rather than complying with certain Nasdaq corporate governance requirements. In accordance with Dutch law and generally accepted business practices, our Articles of Association do not provide quorum requirements generally applicable to general meetings of shareholders in the United States. To this extent, our practice varies from the requirement of Nasdaq Listing Rule 5620(c), which requires an issuer to provide in its bylaws for a generally applicable quorum, and that such quorum may not be less than one-third of the outstanding voting stock. Although we must provide shareholders with an agenda and other relevant documents for the general meeting of shareholders, Dutch law does not have a regulatory regime for the solicitation of proxies and the solicitation of proxies is not a generally accepted business practice in the Netherlands, and thus our practice will vary from the requirement of Nasdaq Listing Rule 5620(b). As permitted by the listing requirements of Nasdaq, we also opted out of the requirements of Nasdaq Listing Rule 5605(d), which requires an issuer to have a compensation committee that, among other things, consists entirely of independent directors and makes determinations regarding the independence of any compensation consultants, Nasdaq Listing Rule 5605(e), which requires an issuer to have independent director oversight of director nominations, and Nasdaq Listing Rule 5605(b)(2), which requires an issuer to have a majority of independent directors on its board. In addition, we opted out of shareholder approval requirements for the issuance of securities in connection with certain events such as the acquisition of stock or assets of another company, the establishment of or amendments to equity-based compensation plans for employees and certain private placements. To this extent, our practice varies from the requirements of Nasdaq Listing Rule 5635, which generally requires an issuer to obtain shareholder approval for the issuance of securities in connection with such events. For an overview of our corporate governance principles, see “ITEM 10. ADDITIONAL INFORMATION - 2. Memorandum and articles of association.”
| C. | 董事會 做法 |
董事會
董事會由六名成員組成,直至2023年4月26日舉行的年度股東大會,以及七名成員組成,自該日期起,在本報告的剩餘期間 ,其中兩名成員為執行董事。我們的執行董事和董事會主席的任期最初為四年,我們的其他非執行董事的任期最初為三年,在每種情況下, 直至其繼任者獲得正式任命、辭職或免職為止。在這些任期屆滿後,我們的董事可以被提名連任,任期視董事會認為適當。有關董事首次任命的年份和任期屆滿日期,請參見'A.董事和高級管理人員”。
-118-
納斯達克的 董事會多元化規則
納斯達克的 董事會多元化規則於2021年8月6日獲得SEC批准,是一項披露標準,旨在鼓勵公司實現最低限度的董事會多元化 ,併為利益相關者提供關於公司當前董事會組成的一致、可比的披露。 納斯達克的董事會多元化規則要求在納斯達克上市的公司使用 標準化模板公開披露董事會層面的多元化統計數據。
董事會 多樣性矩陣(截至2024年3月20日) 至 由外國發行人填寫(其主要執行辦事處位於美國境外)外國私人發行人 | ||||
| 主要執行辦公室的國家/地區 | 德國 | |||
| 外國 私人發行商 | 是 | |||
| 根據母國法律,披露信息是被禁止的 | 不是 | |||
| 導向器總數 | 7 | |||
| 女性 | 男性 | 非二進制 | 沒有透露性別嗎 | |
| 第一部分:性別認同 | ||||
| 董事 | 1 | 6 | 0 | 0 |
| 第二部分:人口統計背景 | ||||
| 在本國司法管轄區任職人數不足的個人 | 1 | |||
| LGBTQ+ | 0 | |||
| 沒有透露人口統計背景嗎 | 6 | |||
董事會於2021年12月採納了多元化政策,並於2023年10月修訂為多元化與包容政策, 該政策已在公司網站上公佈。本政策規定了多元化和包容性方面,包括我們與董事會組成多元化有關的目標。我們相信多樣性包含接受和尊重,認識到每個人都是獨一無二的。我們致力於支持、重視和利用整個公司和董事會的組成 的多樣性。
董事會 委員會
審計委員會
審計委員會目前由Richard Brudnick先生、Nicolas Fulpius先生、Anthony Gibney先生和Mark Kuebler先生組成。安東尼·吉普尼先生是審計委員會的主席。審計委員會協助董事會監督我們的會計和財務 報告過程以及對我們的財務報表的審計。此外,審計委員會還直接負責 對我們獨立註冊會計師事務所的任命、薪酬、保留和監督工作提出建議。 董事會已確定,審計委員會的每一名成員均滿足《交易法》第10A—3條規定的"獨立性"要求,且每一名成員均具備"審計委員會財務專家"的資格,該術語在 證券交易委員會規則中定義。審計委員會受符合適用納斯達克規則的章程管理,該章程已在我們的網站上公佈 。
審計委員會的職責包括:
| ● | 推薦 (四)向股東大會委任獨立核數師; |
| ● | 任命、補償、保留和監督任何會計師事務所, 編制或出具審計報告或執行其他審計服務的目的; |
| ● | 預審批 我們的獨立審計師在 之前提供的審計服務和非審計服務 核數師受聘提供該等服務; |
| ● | 評估 獨立審計師的資格、業績和獨立性,以及陳述 至少每年向全體監事會報告其結論; |
| ● | 查看 並與董事會和獨立審計師討論審計計劃, 以及我們的年度經審計財務報表和季度財務報表, 提交各自的年度和季度報告; |
-119-
| ● | 監督 內部審計職能的有效性和完整性,確保其獨立性, 客觀性,遵守既定政策和程序; |
| ● | 查看 我們遵守法律法規,包括主要法律法規措施 並審查針對我們的任何重大訴訟或調查,這些訴訟或調查可能涉及材料 對我們的財務報表的影響; |
| ● | 查看 內部審計結果,包括內部審計的設計和運作的有效性 控制; |
| ● | 查看 我們的道德準則的運作和遵守情況; |
| ● | 查看 我們的運作是否符合我們的投資政策,規範所有現金投資 關於可用現金金額的投資和現金維護的決策 公司所有實體的投資組合,包括所有貨幣兑換交易; |
| ● | 查看 風險管理系統的運作;及 |
| ● | 審批 或批准任何關聯人交易(定義見我們的關聯人交易 政策)根據我們的相關人員交易政策並審查潛在的 涉及董事的利益衝突 |
審計委員會應在審計委員會的一名或多名成員認為必要時舉行會議,但無論如何至少每季度舉行一次會議。 審計委員會每年至少與我們的獨立會計師舉行一次會議,而我們的執行董事不在場。
薪酬委員會
薪酬委員會由Richard Brudnick先生、Nicolas Fulpius先生和Mark Kubler先生組成。Nicolas Fulpius先生是薪酬委員會主席。薪酬委員會協助董事會確定董事的薪酬。 委員會向董事會建議,以確定每位董事的薪酬。根據SEC和納斯達克的規則, 薪酬委員會成員的獨立性標準得到了更高的標準,包括禁止從我們那裏獲得 標準董事費以外的任何薪酬。根據納斯達克上市要求,我們選擇退出納斯達克上市規則5605(d),該規則要求薪酬委員會完全由獨立董事組成。薪酬委員會 受我們網站上發佈的章程管轄。
薪酬委員會的職責包括:
| ● | 識別, 審查和批准公司目標和與高管薪酬相關的目標 高級職員及董事; |
| ● | 分析 可變薪酬部分的可能結果以及它們可能如何影響 我們的行政人員的薪酬; |
| ● | 決定 第 行中每個執行官薪酬的任何長期激勵部分 並審查我們的執行官薪酬和福利 一般政策; |
| ● | 準備 定期向董事會提交薪酬報告; |
| ● | 查看 並評估我們員工薪酬政策和做法產生的風險,以及 任何此類風險合理可能對我們造成重大不利影響;及 |
| ● | 保留 或從薪酬顧問、法律顧問或其他顧問處獲得建議,作為 薪酬委員會認為有必要或適當履行其職責。 |
-120-
提名 和公司治理委員會
提名和公司治理委員會由Nicolas Fulpius先生、Hege Hellstrom女士和Mark Kubler先生組成。Nicolas Fulpius 先生是提名和公司治理委員會主席。提名和公司治理委員會協助董事會 根據董事會制定的標準確定有資格成為董事會成員的個人,並制定我們的公司治理原則。根據納斯達克上市要求的允許,我們 選擇退出納斯達克上市規則5605(e),該規則要求獨立董事監督董事提名。提名和公司 治理委員會受我們網站上發佈的章程管理。
提名和公司治理委員會的職責包括:
| ● | 準備 並審查董事會的甄選標準和任命程序; |
| ● | 通過考慮任何多樣性和包容性標準,審查 董事會的規模和組成,並提交關於董事會組成概況的建議 ; |
| ● | 領導 董事會進行自我評估,以確定董事會及其委員會是否有效運作; |
| ● | 考慮到任何多樣性和包容性標準,準備 並審查董事繼任計劃;以及 |
| ● | 提交 任命或重新任命董事的建議。 |
| D. | 員工 |
截至2023年12月31日,我們有66名員工,其中包括22名醫學博士。或博士度 截至2022年12月31日,我們有48名員工,其中包括18名醫學博士。或博士度截至2021年12月31日,我們共有59名員工,其中18名醫學博士。 或博士度
| E. | 共享 所有權 |
參見 "項目7。主要股東及關聯方交易—1.大股東”。
| F. | 披露 登記人追討錯誤判給的補償的訴訟 |
截至2023年12月31日 ,我們沒有根據我們的回扣政策進行需要收回錯誤授予的基於激勵的補償的會計重述。截至2023年12月31日,沒有因將保單應用於先前的重報而收回的錯誤裁定的基於激勵的 補償的未償餘額。我們的退款政策載於本年報附件97—1。
-121-
項目 7.大股東和關聯方交易
| A. | 少校 股東 |
下表呈列有關截至2023年12月31日我們普通股實益擁有權的信息:
| ● | 我們所知的每一個 個人或一組關聯人,實益擁有我們發行在外普通股的5%或5%以上(截至 該股東向美國證券交易委員會提交的附表13G文件之日); |
| ● | 我們的每一位 董事和高級管理人員;以及 |
| ● | 所有 董事和高級管理人員作為一個整體。 |
The number of ordinary shares beneficially owned by each entity, person or director is determined in accordance with the rules of the SEC, and the information is not necessarily indicative of beneficial ownership for any other purpose. Under such rules, beneficial ownership includes any ordinary shares over which the individual has sole or shared voting power or investment power or to receive the economic benefit of ownership of the shares, as well as any ordinary shares that the individual has the right to acquire within 60 days of December 31, 2023 through the exercise of any option, warrant or other right. The percentage of shares beneficially owned is computed on the basis of 58,883,272 ordinary shares outstanding as of December 31, 2023. Ordinary shares that a person has the right to acquire within 60 days of December 31, 2023 are deemed outstanding for purposes of computing the percentage ownership of the person holding such rights, but are not deemed outstanding for purposes of computing the percentage ownership of any other person, except with respect to the percentage ownership of all directors and senior management as a group. Except as otherwise indicated, and subject to applicable community property laws, the persons named in the table have sole voting and investment power with respect to all ordinary shares held by that person. All shareholders have similar voting rights. As of December 31, 2023, 16,818,972 ordinary shares, representing approximately 28.6% of our issued and outstanding ordinary shares, were held by 20 U.S. record holders.
此 表基於我們指定的高級管理人員、董事和主要股東提供的信息,以及提交給SEC的附表13D和 13G。已發行普通股百分比按截至2023年12月31日的58,883,272股已發行普通股計算 。個人有權在2023年12月31日起60天內收購的普通股,為計算持有該等權利的人的所有權百分比,被視為已發行,但為計算其他人的所有權百分比,不被視為已發行。
除非 另有説明,下表所列各實益擁有人的地址由InflaRx N.V.轉交,Winzerlaer Str. 2,07745耶拿,德國。
| 普通 共享 實益擁有 | ||||||||
| 數 | 百分比 | |||||||
| 5%的股東 | ||||||||
| 實體 Suvretta Capital Management LLC(1) | 5,733,910 | 9.7 | % | |||||
| 董事 及高級管理層 | ||||||||
| 尼爾斯·裏德曼(2) | 3,851,622 | 6.1 | % | |||||
| 郭仁峯(3) | 3,951,841 | 6.3 | % | |||||
| 託馬斯·塔普肯(4) | 455,502 | * | ||||||
| 卡米拉 Chong,2023年7月的官員 | 0 | 0 | % | |||||
| 德瓦爾·歐卡羅爾(5) | 25,000 | * | ||||||
| 尼古拉斯·富爾皮烏斯(6) | 631,986 | 1.1 | % | |||||
| 理查德·布魯德尼克(7) | 154,850 | * | ||||||
| 馬克·庫布勒(8) | 1,088,187 | 1.8 | % | |||||
| 安東尼·吉布尼(9) | 98,085 | * | ||||||
| Hege Hellstrom,導演,2023年4月 | 0 | 0 | % | |||||
| 全部 董事和高級管理人員作為一個團體(10人) | 10,257,073 | 14.8 | % | |||||
| * | 表示 實際擁有少於總髮行在外普通股1%。 |
| (1) | 作為 根據截至2023年4月12日的附表13G備案,Aaron Cowen擁有 的實益所有權 憑藉其作為Suvretta Capital Management LLC的控制人的角色。 的地址 Suvretta Capital Management LLC地址:540 Madison Avenue,7這是樓層,紐約州,紐約州 約克10022 |
-122-
| (2) | Consists of (a) 1,068,908 ordinary shares, (b) 404,040 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2016 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $3.35 per share), which shall expire on November 18, 2031, (c) 126,005 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the Series B financing at an exercise price of €0.0012 per share, (d) 689,253 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $3.35 per share), which shall expire on December 13, 2025, (e) 5,409 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $3.35 per share), which shall expire on November 20, 2026, (f) 350,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $5.14 per share), which shall expire on January 4, 2031, (g) 112,007 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $2.99 per share), which shall expire on July 1, 2031, (h) 548,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $4.13 per share), which shall expire on January 11, 2032, and (i) 548,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $2.37 per share, which shall expire on January 24, 2033. |
| (3) | Consists of (a) 1,762,144 ordinary shares, (b) 336,672 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2016 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $3.35 per share), which shall expire on November 18, 2031, (c) 623,610 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $3.35 per share), which shall expire on December 13, 2025, (d) 5,409 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $3.35 per share), which shall expire on November 20, 2026, (e) 275,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $5.14 per share), which shall expire on January 4, 2031, (f) 88,006 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $2.99 per share), which shall expire on July 1, 2031, (g) 430,500 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $4.13 per share), which shall expire on January 11, 2032, and (h) 430,500 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $2.37 per share, which shall expire on January 24, 2033. |
| (4) | Consists of (a) 3,500 ordinary shares, (b) 150,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $4.83 per share), which shall expire on September 17, 2028, (c) 50,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $5.14 per share), which shall expire on January 4, 2031, (d) 32,002 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $2.99 per share), which shall expire on July 1, 2031, (e) 110,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $4.13 per share), which shall expire on January 11, 2032, and (f) 110,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $2.37 per share, which shall expire on January 24, 2033. |
| (5) | 包含 25,000股普通股,根據行使期權, 根據2017年計劃以每股2.37美元的行使價發行,這將 2033年1月24日到期。 |
| (6) | Consists of (a) 467,921 ordinary shares, (b) 34,464 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $3.35 per share), which shall expire on December 13, 2025, (c) 30,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $5.14 per share), which shall expire on January 4, 2031, (d) 9,601 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $2.99 per share), which shall expire on July 1, 2031, (e) 45,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $4.13 per share), which shall expire on January 11, 2032, and (f) 45,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $2.37 per share, which shall expire on January 24, 2033. |
-123-
| (7) | Consists of (a) 50,000 ordinary shares, (b) 18,450 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $3.35 per share), which shall expire on February 4, 2027, (c) 20,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $5.14 per share), which shall expire on January 4, 2031, (d) 6,400 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $2.99 per share), which shall expire on July 1, 2031, (e) 30,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $4.13 per share), which shall expire on January 11, 2032, and (f) 30,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $2.37 per share, which shall expire on January 24, 2033. |
| (8) | Consists of (a) 960,015 ordinary shares, (b) 7,308 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the Series B financing at an exercise price of €0.0012 per share, (c) 34,464 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $3.35 per share), which shall expire on December 13, 2025, (d) 20,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $5.14 per share), which shall expire on January 4, 2031, (e) 6,400 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $2.99 per share), which shall expire on July 1, 2031, (f) 30,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $4.13 per share), which shall expire on January 11, 2032, and (g) 30,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $2.37 per share, which shall expire on January 24, 2033. |
| (9) | Consists of (a) 10,000 ordinary shares, (b) 11,667 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $22.75 per share), which shall expire on February 7, 2026, (c) 16,418 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $2.99) per share, which shall expire on July 1, 2031, (d) 30,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $1.86 per share (after re-pricing on April 13, 2022 from $4.13 per share), which shall expire on January 11, 2032, and (e) 30,000 ordinary shares that may be acquired pursuant to the exercise of options which were issued pursuant to the 2017 Plan at an exercise price of $2.37 per share, which shall expire on January 24, 2033. |
-124-
| 1. | 重要 大股東所有權變動 |
2020年7月8日,我們提交了《2020—貨架註冊聲明》。我們還向美國證券交易委員會提交了一份招股説明書補充文件,內容涉及上市計劃 ,該計劃規定根據與 SVB Leerink LLC簽訂的銷售協議,隨時間推移,我們的股票銷售額最高為5000萬美元。
截至 2023年12月31日,我們在上市計劃的整個期限內共發行了5,803,931股普通股,導致 我們獲得了總計2620萬歐元的所得款項淨額,其中3,235,723股普通股,導致我們獲得了1440萬歐元的所得款項淨額 是在2023財年發行的。市場計劃的期限已於二零二三年七月八日屆滿。
2023年4月,我們在承銷的公開發行中共出售併發行了10,823,529股普通股,其中1,411,764股是根據承銷商行使超額配售權而出售的。普通股以每股4.25美元的價格出售 ,面值為每股0.12歐元。扣除承銷折扣後,本次發行的收益為3910萬歐元。
2023年6月30日,我們向美國證券交易委員會提交了一份F—3表格,即2023年登記聲明,有關 公司證券的要約和出售,該文件於2023年7月11日生效。本公司根據本招股説明書可能發售和出售的 證券的總首次發行價將不超過2.5億美元。本公司於2023財政年度內未根據2023年登記聲明發行 普通股,亦未啟動任何主動發售計劃。
在2023財年,我們共發行和登記了120,257股因前員工行使股票期權而產生的普通股 。所有股票期權都是根據2017年計劃授予的。其股票期權已於2022年12月行使了14,930份 ,2023財年行使了105,327份。普通股的面值為每股0.12歐元。 98,754股普通股以每股1.85美元的價格出售,21,503股普通股以每股3.35美元的價格出售。
Suvretta Capital Management LLC及其附屬實體截至2023年12月31日提交了附表13G,聲明持有5,733,910股 ,從而持有公司9.7%的股份。
| B. | 相關的 方交易 |
以下是我們自2023年1月1日以來與我們的任何高級管理人員、董事和持有我們超過5%普通股的 持有者進行的關聯方交易的描述:
| 1. | 賠償協議 |
我們 與我們的董事和高級管理層簽訂了賠償協議。賠償協議和我們的 協會章程要求我們在法律允許的最大程度上賠償我們的董事。關於這些賠償協議的説明,見“第6項.董事、高級管理人員和僱員--2.賠償--保險和賠償”。
| 2. | 與Staidson及其附屬公司的協議 |
2022年12月21日,InflRx GmbH與Staidson(Beijing)BiopPharmticals Co.,或Staidson(簡稱Staidson)簽訂了共同開發協議的第三份附錄,日期為2015年12月28日(共同開發附錄)。根據共同開發附錄的條款,InflRx GmbH將獲得新冠肺炎在中國的BDB-001淨銷售額(定義見共同開發附錄)的10%的特許權使用費。 InflRx GmbH已授予Staidson用於中國的獨家許可,允許InflRx GmbH的某些臨牀、製造和 監管文件有關維羅布單,以支持和便利向中國國家醫療產品管理局提交BDB-001用於治療重症新冠肺炎患者的監管備案。
關於共同開發附錄,本公司於2022年12月21日與Staidson Hong Kong Investment Company Limited簽訂了購買協議。根據收購協議,本公司向Staidson Hong Kong Investment Company Ltd.出售500,000股普通股,每股面值0.12歐元,每股價格5.00美元,總購買價為2,500,000美元。 根據購買協議的條款,Staidson Hong Kong Investment Company Ltd.可根據本公司的選擇權,以總購買價7,500,000美元額外購買 股份,但須受若干條件規限。普通股。
| 3. | 專家和法律顧問的興趣 |
不適用 。
-125-
第 項8.財務信息
| A. | 合併 報告和其它財務信息 |
| 1. | 財務 報表 |
參見 "項目18。財務報表”,其中包含我們根據國際財務報告準則—國際會計準則理事會編制的經審計財務報表。
| 2. | 法律程序 |
我們不時會捲入日常業務過程中出現的法律訴訟。我們相信,如果這些 程序的結果被裁定為不利的,將不會對我們的財務狀況造成重大不利影響。在 此處包含的經審計和批准的財務報表所涵蓋的期間內,我們沒有成為對我們的財務狀況造成重大不利影響的訴訟的當事方,也沒有支付任何損害賠償。未來的任何訴訟都可能導致鉅額成本,並分散 管理層和員工的注意力。我們無法保證未來訴訟不會對我們的財務狀況產生重大不利影響 。關於與法律訴訟有關的某些風險的其他討論,見"項目3。關鍵信息— 3.風險因素”。
| 3. | 除法 和股利政策 |
我們 從未就普通股支付或宣佈任何現金股息,我們預計在可預見的將來不會就普通股支付任何現金股息。我們打算保留所有可用資金和任何未來收益,以資助我們業務的發展和擴展 。根據荷蘭法律,我們只能在股東權益(本徵性變應原)超過 實繳股本和已繳股本加上荷蘭法律或本公司公司章程規定的儲備金之和。 在遵守這些限制的情況下,未來任何派付股息的決定將由董事會酌情決定,並將 取決於多個因素,包括我們的經營業績、財務狀況、未來前景、合同限制、 適用法律施加的限制以及董事會認為相關的其他因素。
| B. | 重要 變化 |
關於我們業務中重大變化的討論,請參見“項目4。公司信息—1.公司的歷史 和發展。"
-126-
第 項9.報價和列表
| A. | 產品 以及清單細節 |
不適用 。
| B. | 分銷計劃 |
不適用 。
| C. | 市場 |
我們的 普通股於2017年11月8日開始在納斯達克全球精選市場交易,代碼為"IFRX"。
| D. | 出售 股東 |
不適用 。
| E. | 稀釋 |
不適用 。
| F. | 發行費用 |
不適用 。
-127-
第 項10.其他信息
| A. | 股份 資本 |
不適用 。
| B. | 備忘錄和公司章程 |
我們的 股東採納了附件1.1所載的公司章程。2023年3月22日向SEC提交的截至2022年12月31日的年度表格20—F年度報告。
我們 在本年度報告中引用了我們F—3註冊聲明 (文件編號333—273058)中包含的公司章程的描述,該聲明最初於2023年6月30日向SEC提交,並於2023年7月11日生效,經修訂。該等描述 概述了目前有效的公司章程細則的某些條款。
於回顧期內有效的 公司組織章程已於2021年8月25日在股東周年大會上獲得通過,並可在我們的網站上查閲。
| C. | 材料 合同 |
除 本年度報告(包括附件)中另行披露的情況外,我們目前並沒有,而且在過去兩年中也沒有 任何重大合同(在正常業務過程中籤訂的合同除外)的當事方。
| D. | Exchange 控制 |
不適用 。
| E. | 税收 |
以下摘要描述了某些美國聯邦收入、荷蘭和德國對我們普通股所有權和處置的税務後果 。本摘要基於美國、荷蘭和德國税法及其相關法規 ,截至本協議之日,這些法規可能會發生變化。
-128-
重要 普通股美國持有人的美國聯邦所得税考慮因素
以下描述了擁有和 處置普通股對美國持有人(定義如下)的重大美國聯邦所得税後果。它沒有列出可能與特定人士決定持有普通股有關的所有税務考慮因素。
本 部分僅適用於持有普通股作為美國聯邦所得税資本資產的美國持有人。此外, 它沒有列出所有可能與美國持有人的特定情況相關的美國聯邦所得税後果,包括替代最低税收後果、被稱為醫療保險 貢獻税的《法典》條款的潛在應用以及適用於美國持有人受特殊規則約束的税務後果,例如:
| ● | 某些 金融機構; |
| ● | 經銷商 或使用按市值計價的税務會計方法的證券交易者; |
| ● | 人員 持有普通股作為套期保值交易的一部分、跨接、清洗銷售、轉換 交易或其他綜合交易或進行推定銷售的人員 就普通股而言; |
| ● | 美國聯邦所得税的本位幣不是美元的人員。 |
| ● | 實體 為美國聯邦所得税目的分類為合夥企業或其他相關實體; |
| ● | 免税 實體,包括“個人退休賬户”或“羅斯IRA”; |
| ● | 人員 擁有或被視為擁有我們10%或以上股份(通過投票或價值); |
| ● | 人員 直接或間接獲得我們的股份與服務的履行有關; |
| ● | 人員 受《守則》第451(b)條約束的人;或 |
| ● | 人員 持有與在美國境外進行的貿易或業務有關的普通股 states. |
如果 為美國聯邦所得税目的被分類為合夥企業的實體持有普通股,則美國聯邦所得税 對合夥企業的待遇將取決於合夥企業的地位和合夥企業的活動。持有普通股 的合夥企業和此類合夥企業的合夥人應就擁有和處置普通股的特定美國聯邦所得税後果諮詢其税務顧問。
本 部分基於《法典》、行政公告、司法裁決、最終、臨時和擬議財政部法規, 以及德國和美國之間的所得税條約和荷蘭和美國之間的所得税條約 (如適用,並根據上下文要求"條約"),所有截至本協議之日,其中任何一項可能會發生變化或 不同的解釋,可能具有追溯效力。無法保證IRS將同意 在此討論中表達的觀點,或者法院不會支持IRS在訴訟中提出的任何質疑。我們尚未獲得, 我們也不打算獲得美國國税局關於以下摘要中所作陳述和得出的結論的裁決。
"美國持有人"是指就美國聯邦所得税而言,為普通股實益擁有人,有資格 享受本條約利益的持有人,並且:
| ● | 美國公民或個人居民; |
| ● | 公司,或其他應作為公司徵税的實體,根據或根據美國、美國任何州 或哥倫比亞特區的法律創建或組織; |
-129-
| ● | 遺產或信託,其收入無論其來源如何,均須繳納美國聯邦所得税:或 |
| ● | 信託,如果美國法院可以對信託的管理行使主要監督,並且一個或多個美國人被授權 控制信託的所有實質性決定。 |
美國 持有人應諮詢其税務顧問,瞭解在其特定情況下擁有和處置普通股的美國聯邦、州、地方和非美國税務後果。特別是,由於我們的集團包括一家美國子公司InflaRx Pharmaceuticals, Inc.,因此,根據現行法律,我們的子公司InflaRx GmbH被視為受控制的外國公司(無論 我們是否被視為受控制的外國公司),擁有或被視為擁有10%或以上股份的任何美國持有人 建議(通過投票或價值)諮詢其税務顧問,瞭解“子部分F收入”和 “全球無形低税收入”的潛在應用我們普通股的投資規則
| 1. | 税務 的分佈來 |
As discussed above under “ITEM 8. FINANCIAL INFORMATION - 1. Consolidated statements and other financial information - 1.3 Dividends and dividend policy,” we do not currently expect to make distributions on our ordinary shares. In the event that we do make distributions of cash or other property, subject to the PFIC rules described below, distributions paid on ordinary shares, other than certain pro rata distributions of ordinary shares, will be treated as dividends to the extent paid out of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles). For so long as we are treated as a PFIC with respect to a U.S. Holder (or were treated as a PFIC with the respect to the U.S. Holder in the preceding taxable year), dividends paid to certain non-corporate U.S. Holders will not be eligible for taxation as “qualified dividend income.” To the extent we are not treated as a PFIC with respect to a U.S. Holder and were not treated as a PFIC with the respect to the U.S. Holder in the preceding taxable year (if for example in future years we cease to meet the threshold requirements for PFIC status and the U.S. Holder initially acquires our ordinary shares in a year in which we are not treated as a PFIC and we are not so treated thereafter or we were a PFIC with respect to a U.S. Holder for a year during which a U.S. Holder holds ordinary shares but the U.S. Holder makes a valid deemed sale or deemed dividend election under the applicable Treasury regulations with respect to its ordinary shares), for so long as our ordinary shares are listed on Nasdaq or another established securities market in the United States or we are eligible for benefits under the Treaty, dividends paid to such a U.S. Holder that is not a corporation would generally be eligible for taxation as “qualified dividend income” if certain other requirements are met, which is generally taxable at rates not in excess of the long-term capital gain rate applicable to such U.S. Holders. The amount of a dividend will include any amounts withheld by us in respect of German or Dutch income taxes. Subject to the PFIC rules described below, the amount of the dividend will be treated as foreign-source dividend income to U.S. Holders and will not be eligible for the dividends-received deduction available to U.S. corporations under the Code and dividends will be included in a U.S. Holder’s income on the date of the U.S. Holder’s receipt of the dividend. The amount of any dividend income paid in euros will be the U.S. dollar amount calculated by reference to the exchange rate in effect on the date of actual or constructive receipt, regardless of whether the payment is in fact converted into U.S. dollars at that time. A U.S. Holder may have foreign currency gain or loss if the dividend is converted into U.S. dollars after the date of receipt. Generally, gain or loss, if any, resulting from currency exchange fluctuations during the period from the date the dividend is paid to the date such payment is converted into U.S. dollars will be treated as U.S. source ordinary income or loss.
受適用限制的約束,從普通股股息中扣除的德國或荷蘭所得税税率不超過本條約規定的税率 ,將有資格從美國持有人的美國聯邦所得税負債中扣除。超過本條約適用税率的德國或荷蘭預扣税款 將不符合美國持有人的聯邦所得税負債的抵免條件。 管理外國税收抵免的規則很複雜,美國持有人應諮詢其税務顧問,瞭解 在其特定情況下外國税收的可信賴性。美國持有人可以在計算其應納税收入時扣除外國税款,包括 任何德國或荷蘭所得税,以代替申請外國税收抵免,但須遵守美國法律的一般適用限制。 扣除外國税款而不是申請外國税款抵免的選擇適用於應課税年度內支付或應計的所有外國税款。參見 "項目3。關鍵信息—3.風險因素—與我們的普通股和我們作為上市公司的地位有關的風險 如果我們支付股息,我們可能需要預扣税應支付給我們在德國和荷蘭的股份持有人的股息。"
| 2. | 銷售 或其他處置普通股 |
根據 下文所述的PFIC規則,出售或以其他方式處置普通股所實現的收益或損失將為資本收益或 損失,如果美國持有人持有普通股超過一年,則將為長期資本收益或損失。 收益或損失的金額將等於美國持有人在出售普通股中的税基與出售時實現的金額之間的差額,在每種情況下均以美元確定。
-130-
| 3. | PFIC 規則 |
We believe it is likely that we were a PFIC for U.S. federal income tax purposes in 2021, 2022 and 2023, and we may be a PFIC in one or more future taxable years. In addition, we may, now or in the future directly or indirectly, hold equity interests in other PFICs (any such PFIC, a “Lower-tier PFIC”). Under the Code, generally a non-U.S. corporation will be a PFIC for any taxable year in which, after the application of certain look-through rules with respect to subsidiaries, either (i) 75% or more of our gross income consists of passive income or (ii) 50% or more of the average quarterly value of our assets consists of assets that produce, or are held for the production of, “passive income.” For purposes of the above calculations, we will be treated as if we hold our proportionate share of the assets of, and receive directly our proportionate share of the income of, any other corporation in which we directly or indirectly own at least 25%, by value, of the shares of such corporation. Passive income includes, among other things, dividends, interest, certain non-active rents and royalties, and capital gains. It is also possible that we will be a PFIC in any future taxable year because, among other things, (i) we currently own a substantial amount of passive assets, including cash and securities that may give rise to passive income, (ii) the valuation of our assets that generate non-passive income for PFIC purposes, including our intangible assets, is uncertain and may vary substantially over time, and (iii) the composition of our income may vary substantially over time. Accordingly, there can be no assurance that we will not be a PFIC for any taxable year. If we are a PFIC for any year during which a U.S. Holder holds ordinary shares, we would continue to be treated as a PFIC with respect to that U.S. Holder for all succeeding years during which the U.S. Holder holds ordinary shares, even if we ceased to meet the threshold requirements for PFIC status, unless under certain circumstances the U.S. Holder makes a valid deemed sale or deemed dividend election under the applicable Treasury regulations with respect to its ordinary shares.
根據 歸屬規則,假設我們是一家PFIC,美國持有人將被視為擁有其在任何較低層PFIC中的按比例股份,並且 將根據以下段落中所述的規則繳納美國聯邦所得税:(i)較低層PFIC的某些分配 ;(ii)較低層PFIC的股份處置,在每種情況下,猶如美國持有人直接持有該等股份, 即使美國持有人尚未收到該等分派或處置所得款項。
If we were a PFIC for any taxable year during which a U.S. Holder held ordinary shares (assuming such U.S. Holder has not made a timely mark-to-market election, as described below), gain recognized by a U.S. Holder on a sale or other disposition (including certain pledges) of the ordinary shares, or an indirect disposition of shares of a Lower-tier PFIC, would be allocated ratably over the U.S. Holder’s holding period for the ordinary shares. The amounts allocated to the taxable year of the sale or other disposition and to any year before we became a PFIC would be taxed as ordinary income. The amount allocated to each other taxable year would be subject to tax at the highest rate in effect for individuals or corporations, as appropriate, for that taxable year, and an interest charge would be imposed on the amount allocated to that taxable year. Further, to the extent that any distribution received by a U.S. Holder on its ordinary shares (or a distribution by a Lower-tier PFIC to its shareholder that is deemed to be received by a U.S. Holder) exceeds 125% of the average of the annual distributions on the ordinary shares received during the preceding three years or the U.S. Holder’s holding period, whichever is shorter, that distribution would be subject to taxation in the same manner as gain, described immediately above.
A U.S. Holder can avoid certain of the adverse rules described above by making a mark-to-market election with respect to its ordinary shares, provided that the ordinary shares are “marketable.” Ordinary shares will be marketable if they are “regularly traded” on a “qualified exchange” or other market within the meaning of applicable Treasury regulations. Our ordinary shares will be treated as “regularly traded” in any calendar year in which more than a de minimis quantity of the ordinary shares is traded on a qualified exchange on at least 15 days during each calendar quarter. Nasdaq, on which the ordinary shares are currently listed, is a qualified exchange for this purpose. If a U.S. Holder makes the mark-to-market election, it will recognize as ordinary income any excess of the fair market value of the ordinary shares at the end of each taxable year over their adjusted tax basis, and will recognize an ordinary loss in respect of any excess of the adjusted tax basis of the ordinary shares over their fair market value at the end of the taxable year (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). If a U.S. Holder makes the election, the U.S. Holder’s tax basis in the ordinary shares will be adjusted to reflect the income or loss amounts recognized. Any gain recognized on the sale or other disposition of ordinary shares in a year when we are a PFIC will be treated as ordinary income and any loss will be treated as an ordinary loss (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). U.S. Holders should consult their tax advisers regarding the availability and advisability of making a mark-to-market election in their particular circumstances.
此外,為了避免適用上述規則,出於美國聯邦所得税目的而持有PFIC股票的美國個人 可以選擇將PFIC和PFIC持有股權的每個PFIC視為合格的選擇 基金(任何該等選舉、優質教育基金選舉),而該等私人金融機構提供進行該等選舉所需的資料 。為了進行這種選擇,美國人必須為每個PFIC進行QEF選舉,方法是在美國人及時提交的美國聯邦所得税申報表 中附上 每個PFIC單獨正確填寫的IRS表格8621, 一般是該實體被視為PFIC的第一個納税年度。美國持有人通常 可以單獨選擇延遲支付QEF規則下未分配收入的税款,但如果延遲支付,任何 此類税款都需要支付利息。
-131-
If a United States person makes a QEF Election with respect to a PFIC, the United States person will be currently taxable on its pro rata share of the PFIC’s ordinary earnings and net capital gain (at ordinary income and capital gain rates, respectively) for each taxable year that the entity is classified as a PFIC and will not be required to include such amounts in income when actually distributed by the PFIC. If a U.S. Holder makes a QEF Election with respect to us, any distributions paid by us out of our earnings and profits that were previously included in the U.S. Holder’s income under the QEF Election will not be taxable to the U.S. Holder. A U.S. Holder will increase its tax basis in its ordinary shares by an amount equal to any income included under the QEF Election and will decrease its tax basis by any amount distributed, if any, on the ordinary shares that is not included in its income. In addition, a U.S. Holder will recognize capital gain or loss on the disposition of ordinary shares in an amount equal to the difference between the amount realized and its adjusted tax basis in the ordinary shares. U.S. Holders should note that if they make QEF Elections with respect to us and Lower-tier PFICs, if any, they may be required to pay U.S. federal income tax with respect to their ordinary shares for any taxable year significantly in excess of any cash distributions, if any, received on the shares for such taxable year. U.S. Holders should consult their tax advisers regarding making QEF Elections in their particular circumstances.
此外,如果我們是PFIC,或就特定美國持有人而言,我們在 支付股息的納税年度或上一個納税年度被視為PFIC,則向某些非公司美國公司支付股息的優惠股息率。 持有人不適用。
如果 美國持有人在我們作為PFIC的任何一年內擁有普通股,則該美國持有人必須提交年度報告,其中包含美國財政部在IRS表格8621(或任何後續表格)中可能要求的與我們有關的 信息,與該年度美國持有人的 聯邦所得税申報表一起提交,除非有關該表格的説明中另有規定。
美國聯邦所得税規則與私人金融公司有關非常複雜。強烈建議美國持有人諮詢其税務顧問,瞭解 PFIC地位對購買、所有權和處置我們普通股的影響, 投資於PFIC對他們的影響(以及任何較低級別的PFIC)、關於我們普通股的任何可用選擇以及IRS信息 有關購買的報告義務,擁有和處置PFIC普通股。
國税局已完成《財政部條例》的定稿,解決了與確定外國公司是否為PFIC以及 美國股東是否持有PFIC股票有關的各種問題,併發布了擬議的《財政部條例》,解決了與確定外國公司是否為PFIC有關的各種問題。這些財務條例和擬議的財務條例(如最終確定)可能會影響 我們在未來任何一年是否為PFIC。您應諮詢您的税務顧問,瞭解本《財政部條例》或擬議的《財政部條例》對確定我們的PFIC地位可能產生的影響(如有)。
| 4. | 信息 報告和備份預扣 |
在美國境內或通過某些美國—相關金融中介機構須 進行信息報告,並可能須接受後備預扣税,除非(i)美國持有人是一家公司或其他豁免收件人 或(ii)在後備預扣税的情況下,美國持有人提供正確的納税人識別號並證明其 不受後備預扣税的約束。
向美國持有人支付的任何後備預扣金額將被允許作為持有人的美國聯邦所得税債務的抵免 ,並有權獲得退款,前提是所需信息及時提供給美國國税局。
| 5. | 信息 關於外國金融資產的報告 |
某些 作為個人和某些實體的美國持有人可能被要求報告與我們普通股權益有關的信息, 但有某些例外情況(包括某些美國金融機構賬户中持有的普通股除外)。 美國持有人應諮詢其税務顧問,瞭解其是否有義務報告與其所有權和普通股處置有關的信息 。
-132-
| 6. | 材料 荷蘭税務考慮 |
一般信息
以下是收購、持有和出售我們普通股的某些重大荷蘭税務後果的一般摘要。 本摘要無意描述可能與普通股持有人或潛在 持有人相關的所有可能的税務考慮因素或後果,也無意處理適用於所有類別投資者的税務後果,其中某些 (如信託或類似安排)可能會受到特殊規則的約束。鑑於其一般性質, 應相應謹慎對待本一般摘要。在本摘要涉及現行荷蘭税法下的法律結論的範圍內,並 根據其包含的限制條件,它代表了NautaDutilh N.V.的意見,我們的荷蘭特別檢察官股份持有人或潛在的 持有人應就其 特定情況下投資股份的税務後果諮詢自己的税務顧問。以下討論僅供一般參考。
為了 本討論的目的,假設我們是德國國家税法規定的德國税務居民,因為我們打算 從我們的註冊開始,並在持續的基礎上,在德國擁有我們的有效管理場所。參見風險因素"我們可能 在德國以外的司法管轄區徵税,這可能會增加我們的總税收負擔。"
請 注意,本摘要不描述以下方面的荷蘭税務注意事項:
| ● | 持有人 如果持有人為個人,則其合夥人 或其直系親屬(包括寄養子女), 有重大利益(Aanmerkelijk Belang)或被視為重大權益(虛構 貝朗省)根據2001年荷蘭所得税法(濕墨噴 2001).一般而言,公司證券持有人被視為持有 在該公司的重大權益,如果該持有人是單獨的,或者如果是個人, 與他或她的伴侶(如2001年荷蘭所得税法案中的定義)一起,直接 或間接持有(i)已發行及未發行資本總額的5%或以上的權益 該公司或某一類別已發行及未發行股本的5%或以上 (ii)直接或間接取得該等權益的權利; 或(iii)該公司的某些利潤分享權,涉及該公司的5%或以上 年利潤及/或公司清盤所得的5%或以上。一個被視為 如果在公司中的重大權益(或部分權益)已經 已被處置或被視為已被處置,在不承認的基礎上; |
| ● | 持有人 如果該等持有人持有的股份有資格或有資格作為參與,則該等持有人持有的股份 (正在開發)1969年《荷蘭企業所得税法》(Wet op de vennootschapsbelasting 1969).一般而言,納税人持股5%或以上 公司名義繳足股本(或在某些情況下,投票權) 有資格參與。持有人也可以參與,如果該持有人沒有 持有5%或以上的股權,但相關實體(法定定義的術語)參與其中 或持有股份的公司為關聯實體(法定定義的術語); |
| ● | 持有人 股份或從股份中獲得的任何利益的個人 是或被視為(就業)活動或服務的報酬 由此類持有人或與此類持有人有關的某些個人執行,無論是在 或者在僱傭關係之外,從經濟上講,為持有人提供 與相關工作活動或服務有關的某些福利(定義見 2001年《荷蘭所得税法》;以及 |
| ● | 養老金 基金、投資機構(財政支持正在安裝),豁免投資 機構(Vrijsterelde BeleggingsInstellingen)(定義見荷蘭公司 1969年所得税法)和其他實體,全部或部分不受或 在荷蘭免徵企業所得税,以及免税的實體 從其居住國的企業所得税中扣除,該居住國為 歐盟的另一個國家、挪威、列支敦士登、冰島或任何其他國家, 荷蘭同意按照國際標準交換信息。 |
-133-
除 另有説明外,本摘要僅涉及荷蘭國家税務立法和已公佈的法規,其中荷蘭 和荷蘭法律分別指荷蘭王國位於歐洲的部分及其法律,在本報告之日生效 和已公佈的判例法(荷蘭最高法院,Hoge Raad der Nederlanden))直至此日期,不影響 在稍後日期提出(或生效)和/或實施的任何修訂,無論是否具有追溯力。適用的 税法或其解釋可能會發生變化,或者相關事實和情況可能會發生變化,這些變化可能會影響本節的內容 ,本節的內容將不會更新以反映任何此類變化。
本 討論僅供一般參考之用,並非税務建議或與收購、持有和出售我方股份有關的所有荷蘭税務後果的完整描述。本公司股份的持有人或潛在持有人應根據其特定情況諮詢其税務顧問 ,瞭解與收購、持有和出售本公司股份有關的税務後果。
股息 預提税金
We are incorporated under the laws of the Netherlands, and therefore a Dutch tax resident for Dutch domestic tax law purposes, including the Dutch Dividend Withholding Tax Act 1969. As such, we are required to withhold Dutch dividend withholding tax at a rate of 15% from dividends distributed by us (which withholding tax will not be borne by us but will be withheld by us from the gross dividends paid on the shares). We are however also treated as a German tax resident for German domestic tax law purposes, since our place of effective management is located in Germany. As long as we continue to have our place of effective management in Germany, and not in the Netherlands, under the convention between the Federal Republic of Germany and the Netherlands for the avoidance of double taxation with respect to taxes on income of 2012, we will be considered to be exclusively tax resident in Germany. Consequently, the Netherlands will be restricted to impose Dutch dividend withholding tax on dividends distributed by us (we will not be required to withhold Dutch dividend withholding tax). This exemption from withholding does not apply to dividends distributed by us to a holder of our ordinary shares who is resident or deemed to be resident in the Netherlands for Dutch income tax purposes or Dutch corporation tax purposes or to a holder of our ordinary shares that is neither resident nor deemed to be resident of the Netherlands if the ordinary shares are attributable to a Dutch permanent establishment of such non-resident holder, in which events the following applies. See Risk Factor “If we ever pay dividends, we may need to withhold tax on such dividends payable to holders of our shares in both Germany and the Netherlands.”
-134-
股息 由我們分配給居住在荷蘭或被視為居住在荷蘭的個人和法人實體,用於荷蘭的 税務目的("荷蘭居民個人"和"荷蘭居民實體"視情況而定)或 我們的普通股持有人,這些普通股既不是荷蘭居民也被視為荷蘭居民,如果普通股歸屬於 該非居民持有人的荷蘭永久機構須按15%的税率繳納荷蘭股息預扣税。
"分派股息"一詞包括,除其他外:
| ● | 分佈 現金或實物、推定分配和實繳資本的償還 未確認為荷蘭股息預扣税目的; |
| ● | 清算 收益、贖回股份的收益或我們回購股份的收益 或我們的子公司或其他附屬實體,但此類收益超過 就荷蘭股息而言確認的這些股份的平均實繳資本 預扣税,除非在回購的情況下,適用特定的法定豁免; |
| ● | 一個 等於已發行股份面值或增加股份面值的金額, 在以下情況下,為荷蘭語目的確認的貢獻似乎不存在 股息預扣税,已徵收或將徵收;及 |
| ● | 部分 償付實繳資本,為荷蘭股息預扣税目的確認, 如果我們有淨利潤(祖韋爾風),除非持有人 在股東大會上,已事先決議償還該等款項,且面值 相關股份的價值已通過修正案減少相等金額 我們的公司章程。 |
荷蘭 居民個人和荷蘭居民實體通常可以從其所得税或 公司所得税負債中扣除荷蘭股息預扣税。如果股份歸屬於該非居民持有人的荷蘭永久機構,則上述規定同樣適用於非居民持有人的普通股持有人。
-135-
根據 反制"股息剝離"的立法,如果股息的接收者不是受益所有人,則拒絕減免、減免、抵免或退還荷蘭股息預扣税 被拒絕(這是一件非常重要的事情1965年荷蘭股息預扣税法(1965年後的濕潤評論).該立法通常針對以下情況: 股東保留其在股份中的經濟利益,但通過與另一方的交易減少股息的預扣税成本。這些規則不要求股息接受者知道發生了股息剝離交易 。荷蘭財政國務祕書的立場是,本立法引入的受益所有權定義 也將適用於雙重徵税公約。自2024年1月1日起,荷蘭股息預扣税的抵銷、豁免 和減少或退還適用更嚴格的規則,以解決抵銷、豁免、減少 或退還索賠可能符合荷蘭税法或雙重徵税公約的文字,但違背股息剝離規則的基本意圖或精神 的情況,正如立法者所認為的。此外,在某些情況下,與股息剝離 和受益所有人身份相關的案件中,舉證責任已從税務檢查員轉移到提出抵銷、 減少或退還或免除荷蘭股息預扣税的索賠人。此外,對於在受監管市場上交易的股票,包括 普通股,已編纂為在確定有權獲得股息的人時使用記錄日期。
有條件 股息預扣税(截至2024年1月1日)
此外, 不能排除,我們分配給某些非荷蘭居民的相關實體的股息在某些特定情況下(見下文)將須繳納荷蘭有條件預扣税,無論 我們在德國有有效管理地,因此根據德國國家 税法,我們是德國的税務居民。自2024年1月1日起,我們向相關實體分派的股息將徵收荷蘭有條件預扣税 (格列耶德)居住在某些列出的司法管轄區或在濫用安排的情況下(所有這些都在2021年荷蘭預扣税法的含義內; 濕支氣管鏡2021).股息的荷蘭有條件預扣税將按分派時有效的荷蘭 最高企業所得税税率(2024年:25. 8%)徵收。股息的荷蘭有條件預扣税 將通過就同一股息分配預扣税而減少,但不低於零。 因此,根據現行適用税率,預扣定期荷蘭股息預扣税 (如上所述)和股息的荷蘭有條件預扣税的整體實際税率將不會超過分派時有效的最高企業所得税税率 (2024年:25. 8%)。
收入和資本利得税
荷蘭 居民實體
從荷蘭居民實體持有的股份中獲得或被視為獲得的任何利益,包括 出售股份時實現的任何資本收益,一般將按19%的税率繳納荷蘭企業所得税,税率為200,000歐元以下的應納税利潤,税率為25.8%(2024年的税率和等級)。
荷蘭 居民個人
如果 股份持有人是荷蘭居民個人,則從普通股中獲得或被視為獲得的任何利益均應按 累進所得税税率(最高税率為49.5%, 2024),如果:
| i. | 普通股歸屬於企業,該企業的持有人從該企業獲得。 一部分利潤,無論是作為一個企業家(代名詞)或作為一個 有一個共同權利的淨值(一種新的治療方法--驅蟲藥), 不作為股東,如荷蘭所得税法案2001所定義);或 |
| 二、 | 普通股持有人被視為就該等股份進行活動 這超越了普通資產管理(Normaal,Actief Vermogensbeheer)或派生 應作為其他活動的利益徵税的股份利益(結果 烏特高等工廠). |
-136-
儲蓄和投資的税收
如果 上述條件(i)和(ii)不適用於荷蘭居民個人,則普通股將根據儲蓄和投資制度繳納 年度荷蘭所得税(墨水瓶裏的墨水).只有 荷蘭居民個人當年的淨投資資產超過法定門檻值時才徵税(Heffingvrij Vermogen). 本年度的淨投資資產是投資資產的公允市值減去負債的公允市值 於相關歷年1月1日(參考日期; 基準面). 普通股實現的實際收入或資本收益本身無需繳納荷蘭所得税。
荷蘭居民個人在此制度下徵税的資產和負債,包括普通股,分配到 以下三類:(a)銀行儲蓄(班克特戈登)、(B)其他投資(比齊廷根王朝),包括 普通股,及(c)負債(舒爾登)。本年度的應課税利益(這是一件非常重要的事情。) 等於(x)視為總回報除以銀行儲蓄、其他投資及負債的總和,以及(y) 銀行儲蓄、其他投資及負債的總和減去法定門檻的乘積,並按36%的統一税率徵税(2024年税率)。
適用於其他投資(包括普通股)的 視為回報率設定為2024年日曆年度的6.04%。如果普通股 持有人不能充分證明此類交易是出於税務以外的原因,則在相關日曆年1月1日前後三個月期間執行的交易 在適用於銀行儲蓄、其他投資和負債的推定回報 百分比之間進行仲裁,則將忽略該交易。
現行荷蘭儲蓄和投資所得税制度在荷蘭最高法院 的裁決後在荷蘭税法中實施(霍格·拉德)2021年12月24日(ECLI:NL:2021:1963)(“決定”)。在該判決中,荷蘭最高法院 裁定,根據推定回報對儲蓄和投資徵税的(舊)制度在特定情況下可能違反 《歐洲人權公約第一議定書》第1條以及《歐洲人權公約》("歐洲人權公約")第14條。荷蘭最高法院正在審理一項新的法庭程序,質疑 當前儲蓄和投資的税收制度是否符合該決定。2023年9月18日(ECLI:NL:PHR:2023:655),總檢察長瓦特爾得出結論,新的税收制度不符合該決定,除了對銀行儲蓄徵税外,因為該制度 ,簡而言之,仍然基於推定的回報而不是實際回報,因此,該制度違反了歐洲委員會人權法案。 荷蘭最高法院預計將於2024年年中做出裁決。此外,2023年9月8日,前內閣公佈了一項法律提案 ,根據資產積累制度的實際回報為基礎,為儲蓄和投資建立新的税收制度,即"實際 回報箱3法案"(濕潤的WEKELIJK Rendbox 3)。擬議的制度預計最早將於2027年1月1日生效。然而,這取決於新內閣向荷蘭議會提交最終的法律提案。
建議普通股持有人 諮詢他們自己的税務顧問,以確保根據相關時間適用的荷蘭税務規則 對普通股徵税。
荷蘭非居民
我們普通股的 持有人既不是荷蘭居民實體也不是荷蘭居民個人,將不會就普通股項下的任何付款或出售普通股時實現的任何收益或損失繳納 荷蘭税,條件是:
| i. | 這樣 持有人在企業或視為企業(定義見 荷蘭所得税法和1969年荷蘭企業所得税法),其中全部或部分, 在荷蘭有效管理或通過常設機構進行, 在荷蘭被視為永久性機構或常駐代表,以及 普通股歸屬於哪個企業或企業的一部分;以及 |
| 二、 | 在 如果該持有人是個人,則該持有人不會在 關於超出普通資產管理範圍的普通股,荷蘭 (Normaal,Actief Vermogensbeheer)且不從普通股中獲益 在荷蘭的其他活動的收益應納税(結果 韋爾克扎姆赫登). |
-137-
禮品 和遺產税
荷蘭居民
贈與或遺產税將在荷蘭就贈與方式轉讓普通股或在贈與時或該持有者去世時在荷蘭居住或被視為居住在荷蘭的普通股持有人 產生。
荷蘭非居民
對於非荷蘭居民或被視為荷蘭居民的普通股持有人以贈與方式轉讓我們的普通股或其去世,將不會產生荷蘭贈與税或遺產税,除非 個人在贈與之日既不是荷蘭居民也不被視為荷蘭居民的個人贈送股票,該個人在贈與之日起180天內去世,同時在荷蘭居住或被視為居民。
就荷蘭贈與税和遺產税等而言,如果持有荷蘭國籍的人在贈與之日或其去世前10年內的任何時間一直居住在荷蘭,則該人將被視為荷蘭居民 。此外,就荷蘭贈與税等而言,如果非荷蘭國籍的人在贈與之日前12個月內的任何時間居住在荷蘭,則該人將被視為荷蘭居民。適用的税收條約可以凌駕於被視為居留的地位。
此外,對於荷蘭贈與和遺產税而言,根據條件先例作出的贈與被視為在滿足該條件先例的瞬間 作出。如果在捐贈人死亡後符合先例,贈與被視為在捐贈人死亡時作出。
其他 税和關税
持有本公司普通股的持有人將不會就持有或出售普通股的任何代價支付任何荷蘭增值税及荷蘭登記税、印花税或任何其他類似文件税或税項。
以下是收購、持有和出售我們普通股的某些重大荷蘭税務後果的一般摘要。 本摘要無意描述可能與普通股持有人或潛在 持有人相關的所有可能的税務考慮因素或後果,也無意處理適用於所有類別投資者的税務後果,其中某些 (如信託或類似安排)可能會受到特殊規則的約束。鑑於其一般性質, 應相應謹慎對待本一般摘要。在本摘要涉及現行荷蘭税法下的法律結論的範圍內,並 根據其包含的限制條件,它代表了NautaDutilh N.V.的意見,我們的荷蘭特別檢察官股份持有人或潛在的 持有人應就其 特定情況下投資股份的税務後果諮詢自己的税務顧問。以下討論僅供一般參考。
為了 本討論的目的,假設我們是德國國家税法規定的德國税務居民,因為我們打算 從我們的註冊開始,並在持續的基礎上,在德國擁有我們的有效管理場所。參見風險因素"我們可能 在德國以外的司法管轄區徵税,這可能會增加我們的總税收負擔。"
請 注意,本摘要不描述以下方面的荷蘭税務注意事項:
| ● | 持有人 如果持有人為個人,則其合夥人 或其直系親屬(包括寄養子女), 有重大利益(Aanmerkelijk Belang)或被視為重大權益(虛構 貝朗省)根據2001年荷蘭所得税法(濕墨噴 2001)。一般而言,一家公司的證券持有人被認為持有該公司的大量權益,如果該持有人單獨持有該公司的重大權益,或者就個人而言,則被視為與其合夥人(如2001年荷蘭所得税法所定義)一起、直接或間接地持有該公司的大量權益。持有(I)該公司已發行及已發行股本總額的5%或以上的權益,或該公司某類別股份已發行及已發行股本的5%或以上的權益。(Ii)直接或間接獲得該等權益的權利;或(Iii)與公司年度利潤的5%或以上及/或公司清算收益的5%或以上有關的該公司的某些利潤分享權。如果一家公司的重大權益(或其部分)已在非確認基礎上被處置或被視為已被處置,則可能產生被視為 重大權益; |
-138-
| ● | 持有人 如果該等持有人持有的股份有資格或有資格作為參與,則該等持有人持有的股份 (正在開發)1969年《荷蘭企業所得税法》(Wet op de vennootschapsbelasting 1969).一般而言,納税人持股5%或以上 公司名義繳足股本(或在某些情況下,投票權) 有資格參與。持有人也可以參與,如果該持有人沒有 持有5%或以上的股權,但相關實體(法定定義的術語)參與其中 或持有股份的公司為關聯實體(法定定義的術語); |
| ● | 持有人 股份或從股份中獲得的任何利益的個人 是或被視為(就業)活動或服務的報酬 由此類持有人或與此類持有人有關的某些個人執行,無論是在 或者在僱傭關係之外,從經濟上講,為持有人提供 與相關工作活動或服務有關的某些福利(定義見 2001年《荷蘭所得税法》;以及 |
| ● | 養老金 基金、投資機構(財政支持正在安裝),豁免投資 機構(Vrijsterelde BeleggingsInstellingen)(定義見荷蘭公司 1969年所得税法)和其他實體,全部或部分不受或 在荷蘭免徵企業所得税,以及免税的實體 從其居住國的企業所得税中扣除,該居住國為 歐盟的另一個國家、挪威、列支敦士登、冰島或任何其他國家, 荷蘭同意按照國際標準交換信息。 |
除 另有説明外,本摘要僅涉及荷蘭國家税務立法和已公佈的法規,其中荷蘭 和荷蘭法律分別指荷蘭王國位於歐洲的部分及其法律,在本報告之日生效 和已公佈的判例法(荷蘭最高法院,Hoge Raad der Nederlanden))直至此日期,不影響 在稍後日期提出(或生效)和/或實施的任何修訂,無論是否具有追溯力。適用的 税法或其解釋可能會發生變化,或者相關事實和情況可能會發生變化,這些變化可能會影響本節的內容 ,本節的內容將不會更新以反映任何此類變化。
本 討論僅供一般參考之用,並非税務建議或與收購、持有和出售我方股份有關的所有荷蘭税務後果的完整描述。本公司股份的持有人或潛在持有人應根據其特定情況諮詢其税務顧問 ,瞭解與收購、持有和出售本公司股份有關的税務後果。
股息 預提税金
We are incorporated under the laws of the Netherlands, and therefore a Dutch tax resident for Dutch domestic tax law purposes, including the Dutch Dividend Withholding Tax Act 1969. As such, we are required to withhold Dutch dividend withholding tax at a rate of 15% from dividends distributed by us (which withholding tax will not be borne by us but will be withheld by us from the gross dividends paid on the shares). We are however also treated as a German tax resident for German domestic tax law purposes, since our place of effective management is located in Germany. As long as we continue to have our place of effective management in Germany, and not in the Netherlands, under the convention between the Federal Republic of Germany and the Netherlands for the avoidance of double taxation with respect to taxes on income of 2012, we will be considered to be exclusively tax resident in Germany. Consequently, the Netherlands will be restricted to impose Dutch dividend withholding tax on dividends distributed by us (we will not be required to withhold Dutch dividend withholding tax). This exemption from withholding does not apply to dividends distributed by us to a holder of our ordinary shares who is resident or deemed to be resident in the Netherlands for Dutch income tax purposes or Dutch corporation tax purposes or to a holder of our ordinary shares that is neither resident nor deemed to be resident of the Netherlands if the ordinary shares are attributable to a Dutch permanent establishment of such non-resident holder, in which events the following applies. See Risk Factor “If we ever pay dividends, we may need to withhold tax on such dividends payable to holders of our shares in both Germany and the Netherlands.”
-139-
股息 由我們分配給居住在荷蘭或被視為居住在荷蘭的個人和法人實體,用於荷蘭的 税務目的("荷蘭居民個人"和"荷蘭居民實體"視情況而定)或 我們的普通股持有人,這些普通股既不是荷蘭居民也被視為荷蘭居民,如果普通股歸屬於 該非居民持有人的荷蘭永久機構須按15%的税率繳納荷蘭股息預扣税。
"分派股息"一詞包括,除其他外:
| ● | 分佈 現金或實物、推定分配和實繳資本的償還 未確認為荷蘭股息預扣税目的; |
| ● | 清算 收益、贖回股份的收益或我們回購股份的收益 或我們的子公司或其他附屬實體,但此類收益超過 就荷蘭股息而言確認的這些股份的平均實繳資本 預扣税,除非在回購的情況下,適用特定的法定豁免; |
| ● | 一個 等於已發行股份面值或增加股份面值的金額, 在以下情況下,為荷蘭語目的確認的貢獻似乎不存在 股息預扣税,已徵收或將徵收;及 |
| ● | 部分 償付實繳資本,為荷蘭股息預扣税目的確認, 如果我們有淨利潤(祖韋爾風),除非持有人 在股東大會上,已事先決議償還該等款項,且面值 相關股份的價值已通過修正案減少相等金額 我們的公司章程。 |
荷蘭 居民個人和荷蘭居民實體通常可以從其所得税或 公司所得税負債中扣除荷蘭股息預扣税。如果股份歸屬於該非居民持有人的荷蘭永久機構,則上述規定同樣適用於非居民持有人的普通股持有人。
根據 反制"股息剝離"的立法,如果股息的接收者不是受益所有人,則拒絕減免、減免、抵免或退還荷蘭股息預扣税 被拒絕(這是一件非常重要的事情1965年荷蘭股息預扣税法(1965年後的濕潤評論).該立法通常針對以下情況: 股東保留其在股份中的經濟利益,但通過與另一方的交易減少股息的預扣税成本。這些規則不要求股息接受者知道發生了股息剝離交易 。荷蘭財政國務祕書的立場是,本立法引入的受益所有權定義 也將適用於雙重徵税公約。
有條件 股息預扣税(截至2024年1月1日)
此外, 不能排除,我們分配給某些非荷蘭居民的相關實體的股息在某些特定情況下(見下文)將須繳納荷蘭有條件預扣税,無論 我們在德國有有效管理地,因此根據德國國家 税法,我們是德國的税務居民。自2024年1月1日起,我們向相關實體分派的股息將徵收荷蘭有條件預扣税 (格列耶德)居住在某些列出的司法管轄區或在濫用安排的情況下(所有這些都在2021年荷蘭預扣税法的含義內; 濕支氣管鏡2021).股息的荷蘭有條件預扣税將按分派時有效的荷蘭 最高企業所得税税率(二零二二年:25. 8%)徵收。股息的荷蘭有條件預扣税 將通過就同一股息分配預扣税而減少,但不低於零。 因此,根據目前適用的税率,預扣定期荷蘭股息預扣税 (如上所述)和股息的荷蘭有條件預扣税的整體實際税率將不會超過分派時生效的最高企業所得税税率 (2022年:25. 8%)。
-140-
收入和資本利得税
荷蘭 居民實體
從荷蘭居民實體持有的股份中獲得或被視為獲得的任何利益,包括 出售股份時實現的任何資本收益,一般將按19%的税率繳納荷蘭企業所得税,税率為200,000歐元以下的應納税利潤,税率為25.8%(2023年的税率和等級)。
荷蘭 居民個人
如果 股份持有人是荷蘭居民個人,則從普通股中獲得或被視為獲得的任何利益均應按 累進所得税税率(2023年最高税率為49.5%)納税,如果:
| i. | 普通股歸屬於企業,該企業的持有人從該企業獲得。 一部分利潤,無論是作為一個企業家(代名詞)或作為一個 有一個共同權利的淨值(一種新的治療方法--驅蟲藥), 不作為股東,如荷蘭所得税法案2001所定義);或 |
| 二、 | 普通股持有人被視為就該等股份進行活動 這超越了普通資產管理(Normaal,Actief Vermogensbeheer)或派生 應作為其他活動的利益徵税的股份利益(結果 烏特高等工廠). |
對儲蓄和投資徵税
如果 上述條件(i)和(ii)不適用於荷蘭居民個人,則普通股將根據儲蓄和投資制度繳納 年度荷蘭所得税(墨水瓶裏的墨水).只有 荷蘭居民個人當年的淨投資資產超過法定門檻值時才徵税(Heffingvrij Vermogen). 本年度的淨投資資產是投資資產的公允市值減去負債的公允市值 於相關歷年1月1日(參考日期; 基準面)。普通股計入投資資產。 本年度的應納税利益(這是一件非常重要的事情。)按32%的統一税率徵税(2023年税率)。普通股的實際收入 或已變現的資本利得本身無需繳納荷蘭所得税。
本年度的應税福利計算如下:
| i. | 荷蘭居民個人在此制度下徵税的資產和負債,包括 普通股分配到以下三個類別:(a)銀行儲蓄, (b)其他投資,包括普通股,及(c)負債。 |
| 二、 | 返回(租借)計算如下 (the返回是在一個最小值nihil): |
| ● | a 1月銀行儲蓄和現金實際金額的公平市場價值的視為回報率 相關歷年的1個;加上 |
| ● | a 其他投資實際金額的公平市價的推定回報,包括 有關歷年1月1日的普通股;減去 |
| ● | a 有關歷年的一月一日減去法定負債限額(夢境). |
| 三、 | 回報率(%)(回報率)計算如下: |
| ● | 通過 將上述(ii)項下計算的回報除以當年的淨投資資產 荷蘭居民個人;乘以100。 |
| 四、 | 應課税基數(貝爾根地區格隆斯拉格斯帕倫)計算如下: |
| ● | 荷蘭居民個人當年的淨投資資產;減去 |
| ● | 適用法定門檻。 |
| v. | 年內應納税利益等於根據上文第(iv)項計算的應納税基數乘以 按上文第(iii)項計算的回報率百分比計算。 |
-141-
於 本報告日期,上述(ii)項下所述不同投資類別的推定回報率暫定為: (a)0.01%、(b)5.69%及(c)2.46%。2023年的最終百分比將在2024年的頭幾個月公佈,並將 追溯至2023年1月1日。如果普通股持有人不能充分證明此類交易是出於 税務原因以外的原因,則相關歷年 1月1日前後三個月期間內實施的交易 將忽略不計,以在適用於銀行儲蓄、其他投資和負債的推定回報率百分比之間進行仲裁。
非荷蘭居民
我們普通股的 持有人既不是荷蘭居民實體也不是荷蘭居民個人,將不會就普通股項下的任何付款或出售普通股時實現的任何收益或損失繳納 荷蘭税,條件是:
| i. | 這樣 持有人在企業或視為企業(定義見 荷蘭所得税法和1969年荷蘭企業所得税法),其中全部或部分, 在荷蘭有效管理或通過常設機構進行, 在荷蘭被視為永久性機構或常駐代表,以及 普通股歸屬於哪個企業或企業的一部分;以及 |
| 二、 | 在 如果該持有人是個人,則該持有人不會在 關於超出普通資產管理範圍的普通股,荷蘭 (Normaal,Actief Vermogensbeheer)且不從普通股中獲益 在荷蘭的其他活動的收益應納税(結果 韋爾克扎姆赫登). |
禮品 和遺產税
荷蘭居民
贈與或遺產税將在荷蘭就贈與方式轉讓普通股或在贈與時或該持有者去世時在荷蘭居住或被視為居住在荷蘭的普通股持有人 產生。
非荷蘭居民
對於非荷蘭居民或被視為荷蘭居民的普通股持有人以贈與方式轉讓我們的普通股或其去世,將不會產生荷蘭贈與税或遺產税,除非 個人在贈與之日既不是荷蘭居民也不被視為荷蘭居民的個人贈送股票,該個人在贈與之日起180天內去世,同時在荷蘭居住或被視為居民。
就荷蘭贈與税和遺產税等而言,如果持有荷蘭國籍的人在贈與之日或其去世前10年內的任何時間一直居住在荷蘭,則該人將被視為荷蘭居民 。此外,就荷蘭贈與税等而言,如果非荷蘭國籍的人在贈與之日前12個月內的任何時間居住在荷蘭,則該人將被視為荷蘭居民。適用的税收條約可以凌駕於被視為居留的地位。
此外,對於荷蘭贈與和遺產税而言,根據條件先例作出的贈與被視為在滿足該條件先例的瞬間 作出。如果在捐贈人死亡後符合先例,贈與被視為在捐贈人死亡時作出。
其他 税費
持有本公司普通股的持有人將不會就持有或出售普通股的任何代價支付任何荷蘭增值税及荷蘭登記税、印花税或任何其他類似文件税或税項。
-142-
7. 材料德國税務注意事項
以下部分描述了在購買、持有或轉讓公司股份時相關的重大德國税務考慮因素。公司在德國擁有其唯一的管理地點,因此,有資格作為受 德國無限企業所得税約束的公司;但是,由於公司的税務居留權取決於 公司管理和控制地點的未來事實,因此德國無限企業所得税責任可能會在未來發生變化。 本節未列出可能與股東相關的所有德國税務方面。該部分基於自本文件之日起適用的德國税法。應注意的是,法律可能會在本年度報告之日後發生變更,且 此類變更可能具有追溯效力。
德國購買、擁有和轉讓股份的實質税務原則如下所述。本節並非 旨在全面或完整地分析或列出購買、擁有或處置股份的所有潛在税務影響 ,也未列出可能與特定人士決定收購普通股 相關的所有税務考慮因素。以下所有內容可能會有所更改。此類更改可追溯適用,並可能影響下文 所述後果。本節不涉及任何外國賬户税收合規法(或FATCA)方面。
建議股東 就德國税法在其特定情況下的適用問題,特別是 就獲得股息和資本收益預扣税減免所需遵守的程序,諮詢自己的税務顧問(卡皮特拉格斯圖爾) 以及關於雙重徵税條約條款的影響,以及根據任何州、 當地或其他外國司法管轄區的法律產生的任何税務後果。出於德國税務目的,股東可能包括個人或實體,其 對股份不具有法定所有權,但股份歸屬於其,其依據是該個人或實體擁有 股份的實益權益,或依據特定的法定條款。
此 部分不構成特定税務建議。本公司股份的潛在購買者應根據其特定情況諮詢其 税務顧問,瞭解購買、擁有和處置股份的税務後果。
股息税
股息預扣税
從公司分配給股東的股息 須繳納預扣税,但須遵守某些豁免(例如,從税收權益賬户償還資本 (Einlagekonto鐵板),如進一步描述的。預扣税税率為25%外加 5.5%的團結附加費(團結一致)按普通股股東大會批准的股息總額的26.375%。預扣税由國內或國外信貸或金融服務機構的國內分支機構(Kredit-and FinanzdienstleistungsInstitute),由國內 證券交易公司(《華爾街日報》發表聲明稱,)或國內證券交易銀行(inländische Wertpapierhandelsbank)保管和管理股份,並將股息支付或貸記,或將股息支付給 外國代理人,或由證券託管銀行(WertPapiersamMelbank),如果股息由該證券託管銀行(稱為股息支付代理)分配給外國代理,則股票將被委託進行集體託管。如果股票不是以集體存款的形式存放在股息支付機構,公司有責任 扣繳税款並將其匯至主管税務機關。
無論股息分配是否以及在多大程度上應按股東的級別徵税,也無論股東是居住在德國或外國的人,都將徵收和預扣此類預扣税。
-143-
在 向2011年11月30日修訂的歐盟指令2011/96/EU第2條或以另一個歐盟成員國為註冊地的歐盟母子公司指令第2條所指的公司分配股息的情況下,如果滿足更多先決條件,將根據請求免除預扣税(Freistellung im Steuerabzugsverfahren)。 這也適用於分配給位於歐盟另一個成員國的常設機構的股息, 母公司或德國居民的母公司,如果公司的參與與這個 常設機構有效相關。適用歐盟母子公司指令的關鍵前提是股東直接持有公司股本至少10%至少一年。
如果德國 已與股東居住國簽訂雙重徵税條約,且股東並未將其股票作為德國常設機構或固定營業地資產的一部分,或作為已在德國指定常駐代表的企業資產持有,則根據雙重徵税條約,對分配給其他外國居民股東的 預扣税予以減免。預扣税的減免是按程序批准的,包括團結附加費在內的預扣總額與根據適用的雙重徵税條約中規定的税率確定的納税義務之間的差額(除非符合其他條件,否則為15%)由德國税務行政當局應請求退還(聯邦中央税務局(德國聯邦儲備銀行),位於波恩-布埃爾的主要辦公室,以及D-53225波恩)。
在 法定所在地和有效管理地點不在德國的公司收到股息的情況下,如果公司因此不是德國的納税居民,則可退還已扣除和匯出的預扣税的五分之二,而不需要 滿足根據歐盟母子公司指令或雙重徵税條約所要求的所有退税先決條件,或者如果德國和股東居住國之間沒有締結雙重徵税條約。
為了根據雙重徵税條約或前述外國公司選項獲得退税,股東 必須提交一份完整的退税表格(可在聯邦中央税務局(www.bzst.de)以及德國大使館和領事館獲得)以及扣繳税款證明(卡皮特拉悲劇性飲食)由代扣代繳税款的機構出具。
根據歐盟母子公司指令或雙重徵税條約以及上述退還預扣税的選項(受雙重徵税條約保護或不受雙重徵税條約保護),是否可以獲得預扣税豁免取決於是否滿足了 某些額外的先決條件。適用的預扣税減免只有在德國反避税規則(或“指令優先”或“條約優先”)的前提條件下,特別是德國所得税法 50D節, 3段(Einkommensteuergesetz),都得到了滿足。
如果(I)適用的雙重徵税條約規定減税導致適用税率低於15%,以及(Ii)股東不是直接持有公司股本至少10%的公司,並且在其居住國的收入和利潤須繳納税款而不獲豁免,則上述預扣税減免(或豁免)將受到進一步限制。在這種情況下,減少(或免徵)預扣税必須滿足以下三個累積的 先決條件:(I)股東必須有資格成為公司股票的實益所有人,最短持有期為在股息到期日前45天和之後45天內連續45天;(Ii)股東必須承擔最低持股期內與公司股票相關的至少70%的價值變化風險,不得直接或間接對衝,以及(Iii)不得要求股東直接或間接將股息全部或大部分補償給第三方。然而,如果股東在收到股息後至少一年內一直是本公司股份的實益擁有人,則這些進一步的先決條件不適用。此外,限制預提税額的特別規則不適用於在評估期內總股息收入不超過20,000歐元或在收到股息後 已連續至少一年成為公司股票實益所有者的股東。
對於並非通過常設機構持有股份的居住在德國境外的個人或公司股東徵税(Betriebsstätte) 在德國或作為業務資產(Betriebsvermögen),其常駐代表(斯坦迪格·維特雷特) 已在德國被任命,剩餘的和已支付的預扣税(如有)是最終的(即,不退還),並解決股東在德國的有限税務責任。對於個人或企業股東,在德國納税居民(例如, 居住地、住所、註冊辦事處或管理地點位於德國的股東)持有其股份作為商業資產,以及 對於通過德國常設機構持有其股份或作為商業資產 已在德國指定常駐代表的股東,預扣税(包括團結附加費) 可從股東在德國的個人所得税或公司所得税負債中扣除。超過此類納税義務的任何預扣税 (包括團結附加費)將予以退還。對於在德國擁有 公司股份作為私人資產的納税居民個人股東,預扣税是最終税(阿比格爾通斯圖爾),但須遵守以下部分所述的例外情況 。
-144-
根據 關於預扣税抵免限制的特殊規則,預扣税抵免必須符合以下三個累積 先決條件:(i)股東必須有資格作為本公司股份的實益擁有人,在股息到期日之前45天和之後45天的期間內,(ii)股東必須在最短持有期內承擔至少70%的與本公司股份有關的價值變動風險,而不直接或間接對衝,及(iii)股東不得被要求全部或大部分直接或間接補償向第三方支付的股息 。如果不滿足所有三個先決條件,則股息徵收的預扣税的五分之三不得計入股東(公司)所得税負債,但可根據申請從相關評估期間的 股東税基中扣除。由於免税而未扣除任何預扣税 的股東收到股息總額,但沒有資格享受全額税收抵免,因此 必須相應通知主管當地税務局,並必須支付扣除的預扣税金額。
税務 德國納税居民持有公司股份作為私人資產的股東的股息收入
對於 居住在德國並持有公司股份作為私人資產的個人股東,股息 須繳納統一税率税,該税率由實際預扣的預扣税(阿比格爾通斯圖爾).因此,股息收入 將按25%的統一税率徵税,另加5.5%的團結附加費(總計26.375%)和教堂税(基爾琴斯圖爾) 如果股東因個人情況而須繳納教會税。通過預扣税的方式扣除教會税的自動程序將適用於須繳納教會税的股東,除非股東已提交阻止通知(Sperrvermerk) 與德國聯邦税務局(有關具體税率(包括教會税)的計算細節將與相關股東的個人税務顧問討論 )。除每年一筆過儲蓄津貼外(斯佩爾--鮑什背叛) 最高1,000歐元(個人申請人)或最高2,000歐元(已婚夫婦和根據註冊 合夥關係法的合夥人(Gesetz über die Eingetragene Lebenspartnerschaft)共同備案),私人個人股東將無權從其股息收入中扣除與資本投資有關的費用。
股息收入所欠的所得税由股息支付代理人預扣的預扣税支付。但是,如果統一 税導致比私人股東的個人税率更高的税收負擔,私人股東可以選擇 按其個人所得税率徵税。在這種情況下,最終預扣税將從所得税中扣除 。然而,根據德國税務當局和法院裁決,私人股東仍然無權從其收入中扣除與資本投資有關的費用。該選擇權只能適用於在相關評估期內統一收到的所有資本投資的資本收入 ,已婚夫婦以及根據 註冊合夥企業法共同備案的合夥人只能共同行使該選擇權。
從統一税率税中扣除 (通過在來源處預扣税來滿足)(阿比格爾通斯圖爾)可適用於 持有一家公司至少25%股權的股東和持有該公司至少1%股權、以專業身份為該公司工作並在上述公司的經濟活動中具有重大影響力的股東。 在這種情況下,將股份作為業務資產持有的獨資企業同樣適用於此規則(見下文“--居住在德國的股東將持有本公司股票作為業務資產徵税--(Ii)獨資企業”)。
-145-
股東股利收入的徵税 居住在德國的居民將公司股票作為企業資產徵税
如果股東將公司股票作為業務資產持有,股息收入的徵税取決於股東是公司、獨資企業還是合夥企業。
(A) 個公司
股息 公司股東的收入免徵公司所得税,前提是公司實體在支付股息的日曆年初直接持有公司股本至少10%的股份。就本條而言,在一個日曆年的過程中獲得至少10%的參與被視為發生在該日曆年開始時。參與公司股東通過合夥企業持有的公司股本,包括共同創業(Mitunternehmerschaften),只能按公司股東在相關合夥企業的資產中的權益份額的比例按比例分配給該公司股東。然而,5%的免税股息 在税務方面被視為不可扣除的業務費用,因此需要繳納企業所得税(外加團結附加費)和貿易税;即 免税95%。與收到的股息相關的業務支出可完全抵扣税款。
出於貿易税收的目的,整個股息收入都要繳納貿易税(即 在確定貿易應税收入時必須加上免税股息),除非公司股東在相關納税申報期開始時至少持有公司註冊股本的15%(刺五加草)。在間接參與的情況下通過合作伙伴關係 請參閲下面的“合作伙伴關係”部分。
如果股利在股本中的持股比例低於10%,則股息應按15%的適用企業所得税税率外加5.5%的税率徵税。 股息的團結性附加費和貿易税(税率取決於公司股東所在的城市)。
如果公司的股票按照德國商法典第340E條的 含義作為交易組合資產持有,則適用特殊的 法規,取消95%的免税。德國商報)由(I)信貸機構(KreditInstitute)、(Ii) 保安機構(WertPapierInstitute),(iii)金融服務機構(金融機構),或 (iv)德國銀行法含義內的金融企業(Kreditwesengesetz),如果信貸機構、擔保機構或金融服務機構直接或間接持有該金融企業50%以上的股份,以及如果股份歸屬於資本投資,則由人壽保險公司、健康保險公司或養老基金持有,產生全部應納税所得。
(b) 獨資業主
對於 居住在德國的個人所有者(個人),持有股份作為企業資產,股息須遵守部分收入規則 (泰林昆夫特費爾法倫).因此,只有(i)60%的股息收入將按其個人所得税率加上5.5%的團結附加費和教會税(如適用)徵税,以及(ii)60%的與股息收入有關的業務開支可用於税務目的扣除。此外,如果股份作為GewStG定義的德國永久機構的業務資產持有,則股息收入將完全繳納貿易税,除非股東在相關評估期開始時持有公司註冊股本的至少15%。根據適用的市政貿易税率和股東的個人税務情況,徵收的貿易税將有資格從 股東的個人所得税債務中扣除 。
(c) 夥伴關係
如果 股份由合夥企業持有,則合夥企業本身無需繳納公司所得税或個人所得税(除非根據《德國公司所得税法》第1a節的 選項適用於合夥企業作為公司納税)。 在這方面,企業所得税或個人所得税(以及教會税,如果適用)以及團結附加費 僅在合夥人的層面上就其相關部分利潤徵收,並取決於其個人情況。
如果 合夥人是公司,股息收入將繳納公司所得税加團結附加費(見"—(i)公司")。
如果 合夥人是獨資經營者(個人),股息收入將受部分收入規則的約束(見“—(ii)獨資經營者”)。
-146-
股息收入須繳納合夥企業層面的交易税(前提是合夥企業有責任繳納交易税),除非 合夥企業在相關評估期開始時持有公司註冊股本的至少15%, 在這種情況下,股息收入可免徵交易税。對於合夥企業的法人股東,沒有關於股息徵税的明確法律規定 。然而,如果股息 屬於該等公司合夥人的股份,則將對股息的5%徵收貿易税,而該等公司合夥人的股份應佔該等股息(按查看基準 ),因為該部分股息將被視為不可扣除的業務開支。
如果 合夥人是個人,則根據適用的市政貿易税率和個人税收情況,在合夥企業層面支付的貿易税 部分或全部從合夥人的個人所得税債務中扣除。
如果公司是合夥人,則將適用於《德國銀行法》所指信貸機構、 證券機構、金融服務機構或金融企業的交易組合資產的特殊規定(Kreditwesengesetz) 或人壽保險公司、健康保險公司或養老基金(見"—(i)公司")。
因此, 合夥企業層面的實際貿易税(如果有的話)取決於合夥企業的持股配額和合夥人的性質 (例如個人或公司)。
德國境外納税居民股東股息收入的税收
對於 德國境外的外國個人或公司股東,其納税居民不通過德國的常設機構持有股份, 或作為已在德國指定常駐代表的商業資產,扣除的預扣税(可能 通過雙重徵税條約或國內税法下的税收減免而減少,例如與歐盟母公司子公司指令有關) 是最終的(即,不可退還),並解決股東在德國的有限税務責任,除非股東有權 申請預扣税退税或免税。
相比之下,德國境外的個人或企業股東通過在德國的永久 機構持有公司股份或作為商業資產(已在德國指定了永久代表)持有公司股份的個人或企業股東應遵守 適用於(如上所述)持有股份作為商業資產的德國居民股東的相同規則。預****r}預扣税款(包括團結附加費)從股東在德國的個人所得税或公司所得税 負債中扣除。
8. 資本利得徵税
預扣税 資本利得
對於 居住在德國的個人股東(個人),持有股份作為私人資產,出售股份時實現的資本收益 需繳納最終預扣税。因此,如果股東因其個人情況需要繳納教會税,則資本利得將按25%的統一税率徵税,外加5.5%的團結附加費(總計26.375%)和教會税。通過預扣税的方式扣除教會税的自動程序將適用於 須繳納教會税的股東,除非股東已提交阻止通知(Sperrvermerk)與德國聯邦税務局 協商(具體税率(包括教會税)的計算細節將與相關股東的個人税務顧問 討論)。應課税資本收益乃通過從出售所得款項中扣除股份收購成本及 與出售直接相關的開支計算。除此之外,除了每年一次過 儲蓄津貼(斯佩爾--鮑什背叛)最多€1,000 (for個人申報者)或最高為€2,000 ( 已婚夫婦和根據登記合夥法的合夥人,Gesetz über die Eingetracene 生活夥伴)共同備案),私人個人股東將無權從其資本收益中扣除與資本投資有關的費用。
-147-
税收 德國納税居民持有股份作為私人資產實現的資本利得
對於居住在德國的 個人股東(個人),持有股份作為私人資產,出售 股份實現的資本收益需要繳納最終預扣税。因此,如果股東因個人情況而須繳納教會税,資本利得將按25%的統一税率徵税,另加5.5%的團結附加費(總計26.375%)和教會税。 除非 股東已提交阻止通知(Sperrvermerk)與德國聯邦税務局(有關具體税率(包括教會税)的計算 的細節將與相關股東的個人税務顧問討論)。應納税 資本收益是通過從 出售所得款項中扣除股份收購成本和與出售直接相關的費用計算的。除此之外,除每年一次過儲蓄津貼外(備件—Pauschbetrag)最高801歐元(個人申請人)或最高1,602歐元(已婚夫婦和根據登記合夥法的合夥人 (Gesetz über die Eingetragene Lebenspartnerschaft)共同備案),私人個人股東將無權 從其資本收益中扣除與資本投資有關的費用。
如果 與私人股東的個人所得税率相比,單一税導致更高的税收負擔,私人股東 可以選擇按其個人所得税率徵税。在這種情況下,預扣的預扣税(包括團結附加費) 將從所得税中扣除。然而,根據德國税務當局的規定,私人股東仍然無權從其收入中扣除與資本投資有關的費用。該選擇權僅可適用於 在相關評估期間內統一收到的來自資本投資的所有資本收入,已婚夫婦以及根據註冊合夥企業法共同備案的合夥人 只能共同行使該選擇權。
出售股份所產生的資本損失只能與同一日曆年內出售股份 或其他股份公司股份所產生的其他資本收益相抵銷。不可能用其他收入(如業務 或租金收入)和其他資本收入抵銷整體虧損。這些損失將結轉,並將在未來年份與出售股份公司股份所得的正資本收益相抵消。
如果股份的賣方或在無償轉讓的情況下,其法定前身在出售前五年內的任何時間直接或間接持有至少1%的公司註冊股本, 最終預扣税將不適用。在這種情況下,資本利得受部分收益規則的約束。因此,只有(i)60%的資本收益將按其個人 個人所得税率加上5.5%的團結附加費和教會税(如適用)徵税,(ii)60%的與資本收益相關的業務開支 可用於税務目的扣除。預扣税(包括團結附加費)將計入 股東在德國的個人所得税負債。
税收 德國納税居民持有公司股份作為業務資產實現的資本收益
如果 股東持有股份作為企業資產,則處置該股份所實現的資本收益的徵税取決於 各股東是公司、獨資經營者還是合夥企業:
(A) 個公司
公司股東出售股份時實現的資本收益 一般免除公司所得税和貿易税。但是, 免税資本利得的5%被視為税務目的不可扣除的業務費用,因此需繳納 企業所得税(加上團結附加費)和貿易税;即,免税95%。與 資本收益有關的業務費用完全可以免税。
因出售股份或其他股份價值減值而產生的資本損失不可扣税。利潤減少 還定義為在貸款或證券由股東 或其關聯方或對上述人員有追索權的第三方授予且股東 直接或間接持有公司25%以上註冊股本的情況下,與貸款或證券有關的任何損失。
如果股份由信貸機構、證券機構、金融服務 機構或《德國銀行法》定義的金融企業作為交易組合資產持有,則適用特殊 法規(Kreditwesengesetz)以及人壽保險公司、健康保險公司或養老基金(見"—(i)公司")。
-148-
(b) 獨資業主
如果 股份由獨資所有者持有,則在出售股份時實現的資本收益受部分收入規則的約束。 因此,只有(i)60%的資本收益將按個人所得税率加上5.5%的團結附加費 和教堂税(如適用)徵税,以及(ii)60%的與股息收入相關的業務開支可用於税務目的扣除。 此外,如果股份作為GewStG定義的德國常設機構的商業資產持有,則60%的資本收益需要繳納貿易税。根據適用的市政貿易税率和個人 納税情況,徵收的貿易税部分或全部從股東的個人所得税債務中扣除。
(c) 夥伴關係
在 股份由合夥企業持有的情況下,合夥企業本身不需要繳納公司所得税或個人所得税, 也不需要繳納團結附加費(和教堂税),因為合夥企業符合德國税務目的的透明度(除非根據 《德國企業所得税法》第1a節的選擇,合夥企業作為公司納税)。在這方面, 公司所得税或個人所得税以及團結附加費(以及教會税,如果適用)僅在合夥人的層面 就其相關利潤部分徵收,並視其個人情況而定。
如果 合夥人是一家公司,則資本利得將繳納公司所得税加團結附加費(參見"—(i) 公司")。如果合夥企業的相關利潤 在合夥企業層面不繳納貿易税,則將在合夥企業層面額外徵收貿易税。但是,對於企業所得税和貿易税,上述95%免税 規則適用。
如果合夥人是獨資經營者(個人),則資本收益受部分收入規則的約束(見"—(ii)獨資經營者")。
此外,如果合夥企業有責任繳納貿易税,則60%的資本收益在合夥企業層面繳納貿易税, 合夥企業為個人,5%的資本收益繳納貿易税,合夥企業為公司。 但是,如果合夥人是個人,則根據適用的市政貿易税率和個人税收情況,在合夥企業層面支付的貿易 税將從合夥人的個人所得税負債中扣除。
關於公司合夥人,如果它們被信貸機構、證券 機構、金融服務機構或《德國銀行法》定義的金融企業或人壽保險公司、 健康保險公司或養老基金作為交易組合資產持有,則適用特殊法規。
(D) 對居住在德國境外的股東實現的資本利得税徵税
資本 德國境外的股東税務居民出售股份時實現的收益應繳納德國税,條件是:(I)公司股票作為常設機構的業務資產持有,或作為已在德國指定的常駐代表的業務資產持有,或(Ii)股東或在無償轉讓的情況下,其法律前身在出售前五年內的任何時間直接或間接持有公司至少1%的股本。 在這些情況下,對於居住在德國的股東,資本利得通常遵守與上述相同的規則。然而, 如果公司是公司的股東,則不清楚是否適用5%的税(請參閲“-公司- 對持有公司股票的德國居民所實現的資本利得税作為業務資產徵税”) 或者資本利得税是否完全免除德國税。
然而, 除了上文(I)所述的情況外,與德國締結的一些雙重徵税條約規定完全免徵德國的税收。
-149-
遺產税和贈與税
以繼承或捐贈方式將公司股份轉讓給他人的 需繳納德國繼承税和贈與税(埃爾布沙夫特和申孔斯特)如果:
| i. | 繼承人、捐贈人、繼承人、受贈人或者其他受益人在轉移時在德國有住所、住所、註冊辦事處或者管理地, 或連續五年未在國外居留且未在德國居留的德國公民; |
| 二、 | (不論個人情況如何)股份由死者或捐贈人作為企業資產持有,在德國設有常設機構或指定常駐代表;或 |
| 三、 | (無論 個人情況如何)至少10%的股份直接或間接 由贈與人或贈與人本人或與關聯方共同持有( 1第2款《外國税法》)。 |
特殊的 法規適用於符合條件的德國公民,他們既沒有在德國居住,也沒有在德國有住所,但在低税收司法管轄區 ,以及前德國公民,也會導致遺產税和贈與税。德國加入的少數關於遺產税和贈與税的雙重徵税條約規定,只有在(I) 的情況下才徵收德國遺產税和贈與税,在有某些限制的情況下, 在(Ii)的情況下徵收德國遺產税和贈與税。
其他 税
無 德國資本轉讓税(卡皮塔爾弗爾斯圖爾)、增值税(烏姆薩茲斯圖爾)、印花税(斯坦普爾蓋爾) 收購、持有或轉讓本公司股份時徵收或類似的税款。除非股東有效地選擇增值税,否則不徵收增值税。財富税淨額(Vermögensteuer)目前在德國沒有徵收。
2013年1月22日,歐盟理事會批准了11個歐盟成員國(包括德國)財政部長在加強合作框架內開徵金融交易税(FTT)的決議。2013年2月14日,歐洲委員會接受了一項理事會指令的提議,該指令旨在加強金融交易税領域的合作。 該計劃的重點是對金融工具的買賣徵收0.1%的金融税(衍生品為0.01%)。
2016年10月,歐盟11個參與國中的10個成員國發表了一份聯合聲明,重申了引入FTT的意圖。然而, 目前沒有太多細節可用。因此,尚不清楚在股票徵税方面將在多大程度上遵循歐盟委員會上一段概述的提案的要素。FTT提案仍有待參與成員國之間的談判,並受到政治討論的影響。因此,它可能會在實施之前進行更改, 具體時間尚不清楚。歐盟委員會承諾在2024年1月1日之前提出一項建議,並已於2023年6月發佈了一份工作文件。然而,預計短期內不會就任何提議達成一致。其他歐盟成員國 可以決定參與。建議潛在的股票持有者尋求有關FTT的專業建議。
9. 分紅和支付代理
不適用 。
10. 專家發言
不適用 。
11. 展示的文檔
我們 受《交易法》的信息要求約束。因此,我們需要向美國證券交易委員會提交報告和其他信息,包括年度報告和Form 6-K報告。美國證券交易委員會維護着一個網站,其中包含有關發行人的報告和其他信息, 這些發行人和我們一樣,以電子方式在美國證券交易委員會備案。該網站網址為www.sec.gov。
12. 子公司信息
不適用 。
-150-
第 項11.關於市場風險的定量和定性披露
市場風險來自我們對貨幣匯率波動的風險敞口。我們在正常的業務過程中面臨此類市場風險,因為我們對美元的風險敞口擴大了未來可能來自美國的支出和收入。 目前,我們沒有任何匯率對衝安排。
我們 不從事涉及其他市場價格風險的活動。有關市場風險的更多信息,請參閲我們已審計財務報表內的附註D.12“風險” ,以及根據IFRS-IASB編制的附註“項目18.財務報表”。
第 項12.股權證券以外的證券説明
| A. | 債務證券 證券 |
不適用 。
| B. | 認股權證 和權利 |
不適用 。
| C. | 其他 證券 |
不適用 。
| D. | 美國存托股份 |
不適用 。
-151-
第 第二部分
第 項13.違約、拖欠股息和拖欠
| A. | 缺省值 |
沒有 需要報告的事項。
| B. | 欠款 和拖欠 |
沒有 需要報告的事項。
第 項14.對擔保持有人的權利和收益的使用作出實質性修改
| A. | 材料 文書修改 |
不適用 。
| B. | 材料 權利修改 |
不適用 。
| C. | 退出 或資產替代 |
不適用 。
| D. | 更改 受託人或付款代理人 |
不適用 。
| E. | 使用收益的 |
不適用 。
-152-
第 項15.控制和程序
| A. | 披露 控制和程序 |
截至2023年12月31日,在包括首席執行官和首席財務官在內的管理層(包括首席執行官和首席財務官)的監督和參與下,我們對我們的披露控制和程序的設計和運營的有效性進行了評估 (如《交易法》第13a-15(E)條所定義)。任何披露控制和程序制度的有效性都有固有的侷限性,包括人為錯誤的可能性以及規避或推翻這些錯誤的可能性。即使有效,披露控制和程序也只能為實現其控制目標提供合理的保證。
基於這樣的評估,我們的首席執行官和首席財務官得出結論,我們的披露控制和程序是有效的 ,以提供合理的保證,我們必須在根據交易法提交或提交的報告中披露的信息:(1)在美國證券交易委員會規則和表格指定的時間段內記錄、處理、彙總和報告 和(2)累積並傳達給我們的管理層,以便及時做出有關所需披露的決定。
| B. | 管理層關於財務報告內部控制的年度報告 |
我們的管理層負責建立和維護對財務報告的充分內部控制,這一術語在《交易法》規則13a-15(F)中有定義。在包括首席執行官和首席財務官在內的管理層的監督和參與下,我們根據以下標準對財務報告內部控制的有效性進行了評估:內部控制--綜合框架特雷德韋委員會贊助組織委員會通過(2013)。根據這項評估,我們的管理層得出結論,我們對財務報告的內部控制自2023年12月31日起有效。
| C. | 註冊會計師事務所認證報告 |
獨立註冊會計師事務所安永會計師事務所發佈了一份關於截至2023年12月31日本公司財務報告內部控制有效性的認證報告,該報告就此發表了無保留意見 ,如本文所述。見“獨立註冊會計師事務所報告”第 F-2頁。
| D. | 財務報告內部控制變更 |
在本年度報告所述期間,我們對財務報告的內部控制發生了某些變化,這與在美國製造用於商業銷售的GOHIBIC(Viloblimab) 之後對庫存管理和估值實施的內部控制有關,這些變化對我們的財務報告內部控制產生了重大影響。
第 項16.已保留
第 項16A。審計委員會財務專家
董事會已確定Anthony Gibney先生、Nicolas Fulpius先生、Mark Kuebler先生和Richard Brudnick先生均為審計委員會財務專家,該術語由美國證券交易委員會定義,就美國證券交易委員會和納斯達克有關審計委員會獨立性的規則而言,所有四人都是獨立的。
第 16B項。道德準則
我們 通過了適用於我們所有員工、高級管理人員和董事的道德準則,並在我們網站www.inflarx.com的投資者關係部分公佈了我們的道德準則全文 。我們打算在我們的網站或公開文件中披露未來對我們的道德準則的修訂,或 對此類準則的任何豁免。我們網站上的信息不會以引用方式併入本年度報告,您不應將我們網站上包含的信息視為本年度報告的一部分。
-153-
第 項16C。首席會計師費用及服務
| A. | 審核 費 |
審計委員會已採納一項政策,要求我們的獨立註冊公共 會計師事務所為我們提供的所有服務均需事先批准。我們獨立註冊的公共會計師事務所提供的所有與審計相關的服務都經過審計委員會的事先批准,並且與維持審計師的獨立性是一致的。
以下列出了獨立註冊公共會計師事務所 或其關聯公司在過去兩年每年為提供審計和其他專業服務而按合併方式開具的(或預期開具的)費用總額。
2023年的審計費用為1,009,952歐元,與審計服務有關。這些服務由我們的主要會計師安永 GmbH & Co. KG Wirtschaftsprüfungsgesellschaft(前Ernst & Young GmbH Wirtschaftsprüfungsgesellschaft)提供, 與我們的年度審計、季度審查和公司註冊報表審查有關。
2022年的審計費用為988,541歐元,與審計服務有關。這些服務由我們的主要會計師 安永有限公司(安永有限公司)(前安永有限公司)提供, 我們的年度審計、季度審查和公司註冊報表審查。
| B. | 審計相關 費 |
沒有。
| C. | 税 費 |
沒有。
| D. | 全部 其他費用 |
沒有。
| E. | 審核 委員會的批准前政策和程序 |
審計委員會負責任命、更換、補償、評估和監督獨立 審計員的工作。作為此職責的一部分,審計委員會根據審計委員會的預批准政策,對 獨立審計師執行的所有審計和非審計服務進行預批准,以確保這些服務不會損害審計師對公司的獨立性。
| F. | 審核 由非主要會計師完成的工作,如超過50% |
不適用 。
第 項16D。豁免審計委員會遵守上市標準
不適用 。
-154-
項目 16E。發行人及附屬公司購買股權 購買者
沒有。
第 16F項。更改註冊人的認證會計師
不適用 。
第 項16G。公司治理
有關我們的公司治理實踐與納斯達克上市的美國公司所要求的公司治理實踐之間的重大差異的説明,請參閲“第6項.董事、高級管理人員和員工-3.董事會實踐-公司治理實踐 .”
第 16H項。煤礦安全信息披露
不適用 。
項目 16i.關於妨礙檢查的外國司法管轄區的披露
不適用 。
項目 16J。內幕交易政策
不適用 。
第 項16K。網絡安全風險管理和戰略
我們的董事會負責審查與公司財務報告相關的公司網絡安全風險管理和控制系統,包括公司的網絡安全戰略。我們的董事會已視情況將網絡安全風險管理的定期 監督授權給審計委員會,該委員會向我們的董事會報告。
我們的IT部門負責有針對性地定期監控網絡安全風險。他們獨立和持續地監測網絡安全風險和對策,以防禦此類威脅,並在發生網絡安全威脅或網絡安全事件時, 通知執行管理層、我們的審計委員會和我們的董事會。除了執行管理層與主要由各部門負責人組成的個人風險所有者之間的定期會議外,還酌情對內部和外部風險進行全面的網絡安全風險分析 。
IT部門確定和評估的網絡安全風險包括在總體風險目錄中。已識別的網絡安全風險在總體風險目錄中進行記錄、描述、記錄和評估。更改也相應地記錄在案。根據風險評估的結果,根據網絡安全風險的優先順序,應採取具體行動應對風險,並在適當的情況下采取必要的應對措施。為了能夠快速、靈活地應對網絡安全風險,風險管理被整合到現有的流程和報告渠道中。我們的風險管理計劃將網絡安全風險與其他 公司風險一起考慮,我們的企業風險專家諮詢公司主題專家以收集必要的信息,以識別網絡安全風險並評估其性質和嚴重性,以及確定緩解措施並評估這些緩解措施對剩餘風險的影響。我們可能會不時聘請第三方進行風險評估。
-155-
我們持續監控的主要網絡安全風險包括導致中央IT系統不可用的威脅和潛在事件、關鍵業務數據丟失、數據被盜、知識產權被盜、欺詐、敲詐勒索、對員工和患者的傷害、違反隱私法以及其他訴訟和法律風險,以及對我們聲譽的風險。這些網絡安全威脅的實現可能會對公司產生重大影響,也有可能對公司產生重大影響,包括公司的業務戰略、運營結果或財務狀況。例如,如果中央IT系統不可用,將導致公司的任何臨牀開發活動中斷或延遲。丟失關鍵業務數據(如臨牀研究報告的結果)和基本信息(如表格、清單和文件)可能會在事後毀掉數年的臨牀開發工作和相應的成本。如果涉及關鍵、機密或專有業務數據,數據盜竊可能會在競爭中造成損失 。知識產權盜竊可能危及我們為股東創造收入的能力。違反 隱私法可能會使我們的員工和患者面臨額外的網絡安全和個人風險,這可能會導致訴訟 併為公司帶來更多成本。網絡安全風險也可能導致我們公司的聲譽受到影響。
管理層、我們的IT部門和員工是有效的網絡安全風險管理的基礎。本公司實施了 IT安全指南,其中規定了安全處理個人數據的措施、為設備和軟件設置合理安全的密碼、處理移動IT設備以及正確使用互聯網和電子郵件通信軟件。 本公司還實施了訪問和使用公司用户帳户(包括電子郵件)的雙因素身份驗證。所有員工以及執行管理層的所有成員都定期接受IT安全培訓。
雖然我們不認為我們的業務戰略、運營結果或財務狀況受到任何網絡安全事件 的實質性不利影響,但我們在“第3項C風險因素-一般風險因素-IT系統中的網絡事件或其他故障 可能導致信息失竊、數據損壞和業務運營嚴重中斷”中描述未來事件是否以及如何對我們的業務戰略、運營結果或財務狀況產生實質性影響。此外, 雖然我們有網絡安全事件的保險覆蓋範圍,但不能保證我們將能夠維持我們的保險覆蓋範圍 或者它是否足以支付與一個或多個網絡安全事件相關的費用。見“項目3C.風險因素--一般風險因素--我們可能無法維持足夠的保險,以應對潛在的訴訟或其他風險。”
-156-
第 第三部分
項目 17.財務報表
我們 迴應了第18項,而不是此項。
項目 18.財務報表
財務報表作為本年度報告的一部分提交,參見本年度報告F—1至F—35頁。
物品 19.展品
| 展品編號: | 描述 | |
| 1.1 | InflaRx N.V.公司章程日期為2021年8月25日(英語翻譯)(通過引用附件1.1納入本文。2023年3月22日向SEC提交的截至2022年12月31日的年度20—F表格年度報告)。 | |
| 2.1 | 註冊權協議(通過引用附件4.2納入本協議,該附件2017年11月9日提交給SEC的F—1表格(文件號333—220962)的生效後修正案)。 | |
| 2.2+ | 高級契約表格(通過參考2023年6月30日向SEC提交的公司F—3表格註冊聲明(文件編號333—273058)的附件4.2納入本文)。 | |
| 2.3+ | 附屬契約的表格(通過參考2023年6月30日向SEC提交的公司F—3表格(文件號333—273058)的註冊聲明的附件4.3納入本文)。 | |
| 2.4 | 根據《1934年證券交易法》第12條註冊的每種適用類別證券的權利説明(通過引用截至2022年12月31日止年度的表格2.4納入本報告)。 | |
| 4.3† | 2015年12月28日,InflaRx GmbH與北京德豐格雷生物技術有限公司簽訂的共同開發協議,由日期為2015年12月28日的附錄1補充(通過引用本公司於2017年11月7日向SEC提交的F—1表格註冊聲明(文件編號333—220962)的附件10.3納入本文件)。 | |
| 4.4 | 附錄2,日期為2022年11月9日,由InflaRx GmbH和北京Defengrei Biotechnology Co. Ltd.簽署(通過引用2023年3月22日向SEC提交的截至2022年12月31日的年度表格20—G年度報告的附件4.4納入本文)。 | |
| 4.5 | 附錄3,日期為2022年12月21日,由InflaRx GmbH和Staidson(Beijing)BioPharmaceuticals Co.簽署,有限公司,InflaRx GmbH與Staidson(Beijing)BioPharmaceuticals Co.於2015年12月28日簽訂的共同開發協議,北京德豐瑞生物技術有限公司(作為北京德豐瑞生物技術股份有限公司(BDB)的繼任者)(通過引用本公司於2022年12月21日提交給美國證券交易委員會的當前6—K表報告的附件10.1納入本文)。 | |
| 4.6 | InflaRx N.V.與Staidson Hong Kong Investment Company Limited於2022年12月21日簽署的購股協議(通過參考本公司於2022年12月21日提交給美國證券交易委員會的表格6—K的當前報告的附件10.2納入本協議)。 | |
| 4.7 | 董事和執行官的賠償協議表格(通過參考2017年10月13日向SEC提交的公司F—1表格(文件編號333—220962)的註冊聲明的附件10.4納入本協議)。 | |
| 4.8 | 2023年4月11日,InflaRx N.V.與Raymond James & Associates,Inc.簽訂了承保協議,作為附表1中指定的承銷商的代表(通過引用本報告表6—K,日期為2023年4月13日) | |
| 4.9 | InflaRx長期激勵計劃(通過參考2017年11月17日向SEC提交的公司S—8表格(文件號333—221656)的註冊聲明的附件99納入本文)。 | |
| 4.10 | InflaRx長期激勵計劃的修正案(通過引用2020年7月30日向SEC提交的公司S—8表格(文件號333—240185)的註冊聲明的附件99.2納入本文)。 | |
| 8.1* | 子公司名單。 | |
| 12.1* | 根據2002年《薩班斯-奧克斯利法案》第302條進行的認證。 | |
| 12.2* | 根據2002年《薩班斯-奧克斯利法案》第302條進行的認證。 | |
| 13.1* | 根據2002年《薩班斯-奧克斯利法案》第906條通過的《美國法典》第18編第1350條的認證。 | |
| 13.2* | 根據2002年《薩班斯-奧克斯利法案》第906條通過的《美國法典》第18編第1350條的認證。 | |
| 15.1* | 經安永股份有限公司同意。 | |
| 97.1* | 追回政策。 | |
| 101 | 以下是我們截至2023年12月31日的年度表格20—F年度報告中的材料,格式為內聯XBRL(可擴展 業務報告語言):(i)合併財務報表和(ii)合併財務報表附註, 標記為文本塊和細節。 | |
| 104 | 封面 頁面交互式數據文件(格式為內聯XBRL,見附件101)。 |
| * | 隨函存檔。 |
| + | 之前已提交 。 |
| † | 機密 對展覽部分給予的待遇。機密材料遺漏並存檔 分別與SEC。 |
-157-
簽名
註冊人特此證明,其符合表格20—F提交的所有要求,並已正式促使並授權 以下簽名人代表其簽署表格20—F的年度報告。
| InflaRx N.V. | |||
| 發信人: | /s/ Niels Riedemann | ||
| 姓名: | 尼爾斯 裏德曼 | ||
| 標題: | 主管 執行官兼董事 | ||
日期: 2024年3月21日
| 發信人: | /s/ 託馬斯·塔普肯 | ||
| 姓名: | Thomas 塔普肯 | ||
| 標題: | 首席財務官 | ||
日期: 2024年3月21日
-158-
合併財務報表索引
| 合併財務報表索引 | F-1 | |
| 獨立註冊會計師事務所報告(PCAOB ID: | F-2 | |
| 獨立註冊會計師事務所(PCAOB ID:1251) | F-5 | |
| 截至2023年、2022年及2021年12月31日止年度的綜合經營報表及全面虧損 | F-6 | |
| 截至2023年12月31日和2022年12月31日的合併財務狀況報表 | F-7 | |
| 截至2023年、2022年及2021年12月31日止年度合併股東權益變動表 | F-8 | |
| 截至2023年、2022年及2021年12月31日止年度合併股東權益變動表 | F-9 | |
| 2023年、2022年和2021年12月31日終了年度合併現金流量表 | F-10 |
F-1
獨立註冊會計師事務所報告{br
致 InflaRx N.V.的股東和董事會。
對財務報表的意見
我們 已審計隨附的InflaRx N.V.及其子公司的綜合財務狀況表(本公司)截至2023年及2022年12月31日,截至2023年12月31日止三年各年的相關合並經營報表及全面虧損、股東權益變動及現金流量,及相關附註(統稱為“綜合財務報表”)。我們認為,綜合財務報表在所有重大方面 公允列報了貴公司於2023年及2022年12月31日的財務狀況,以及截至2023年12月31日止期間三年各年的經營業績和現金流量,符合國際會計準則理事會頒佈的國際財務報告準則 。
我們 還根據上市公司會計監督委員會的標準進行了審計(美國)(PCAOB),公司 截至2023年12月31日的財務報告內部控制,基於特雷德韋委員會贊助組織委員會發布的內部控制—綜合框架 中確立的標準(2013年框架)和我們日期為2024年3月20日的報告發表了無保留意見。
徵求意見的依據
這些 財務報表由公司管理層負責。我們的責任是根據我們的審計對公司的財務報表發表意見。我們是一家在PCAOB註冊的公共會計師事務所,根據美國聯邦證券法以及美國證券交易委員會和PCAOB的適用規則和條例,我們必須與公司保持獨立。
我們 按照PCAOB的標準進行審計。這些標準要求我們計劃和執行審計,以獲得關於財務報表是否沒有重大錯報的合理保證,無論是由於錯誤還是舞弊。我們的審計 包括執行程序以評估財務報表重大錯誤陳述的風險,無論是由於錯誤還是欺詐, 並執行應對這些風險的程序。這些程序包括在測試的基礎上審查關於財務報表中的金額和披露的證據。我們的審計還包括評估管理層使用的會計原則和作出的重大估計,以及評估財務報表的整體列報。我們相信,我們的審計為我們的觀點提供了合理的基礎。
重大審計事項
以下傳達的 關鍵審計事項是指已傳達 或要求傳達給審計委員會的財務報表本期審計中產生的事項,並且:(1)與對財務報表具有重要意義的賬目或披露有關,以及(2)涉及我們特別具有挑戰性、主觀或複雜的判斷。傳達關鍵審計事項 不會以任何方式改變吾等對綜合財務報表整體的意見,且吾等不會透過傳達 以下重要審計事項,對關鍵審計事項或與之相關的賬目或披露 提供單獨意見。
F-2
確認 臨牀試驗和合同製造費用
| 事件描述 | 作為 在合併財務報表附註B.3中討論,公司確認研究 和開發(R & D)費用,其中包括臨牀試驗和合同費用 製造,由合同研究機構和合同製造機構負責 (統稱為“臨牀供應商”)。全部臨牀試驗和合同生產 截至2023年12月31日止年度確認的費用為3180萬歐元, 研發項目的相關預付款和應計負債為370萬歐元 截至2023年12月31日,分別為440萬歐元。
公司確定臨牀試驗和合同製造費用涉及估計完工時間, 由此,從臨牀供應商那裏為單個項目活動提供的服務的程度 由管理層評估和評估。雖然公司估計的臨牀試驗和合同製造費用 主要基於從其臨牀供應商處收到的與每項研究相關的信息,公司可能需要進行估計 根據管理層判斷產生的費用。這些活動的付款基於個別安排的條款, 這與成本的模式不同。
審核 由於管理層的判斷和主觀性,臨牀試驗和合同製造費用具有挑戰性 評估臨牀試驗進展和合同生產費用,相對於發生的成本,以估計 相關的應計負債和研發項目預付款,以及對數據完整性和準確性的評估 用於估計。 |
如何 我們在審計中解決了這個問題
|
我們 瞭解、評估設計並測試 與公司估計臨牀供應商成本相關的控制措施 試驗和合同製造費用。例如,我們測試了對管理層的控制 審查用於確定臨牀數量的估計完工時間 試製和合同製造費用以及對預付款和應計費用的相關影響 研發項目的負債。
至 評估臨牀試驗和合同製造費用的核算,我們的審計程序包括(除其他外)測試 通過評估 通過與公司的研發項目經理討論臨牀試驗活動,負責監督這些活動 並審查臨牀供應商向研發項目經理提供的進度報告、發票和其他通信。 我們檢查了公司的臨牀供應商合同、修訂案和待決變更單,以評估關鍵財務 合同條款與確認金額一致。我們還對完工時間的波動進行了分析審查 在整個期間內按項目分列。我們比較了臨牀供應商收到的發票和支付給臨牀供應商的現金 在年底之前和之後收到的臨牀供應商評估的信貸備忘錄。 |
F-3
成品存貨可變現淨值
| 事件描述 | 截至2023年12月31日,該公司的未完成產品淨庫存餘額為1,060萬歐元,所有這些都與其嚴重的新冠肺炎治療Gohibic有關。如合併財務報表附註 B.2、B.3和D.5所述,為了以成本或可變現淨值中較低的價格對庫存(包括未完成產品)進行估值,本公司審查其庫存 是否有超額或過時,主要使用對預期未來銷售額的估計,這對重要的輸入和假設非常敏感,例如預期的醫療需求和預期的市場滲透率。
審計 管理層對未完成產品庫存可變現淨值的估計,這部分是基於對預期未來銷售額的估計,由於公司需要考慮的銷售歷史有限,這是一項複雜且具有高度判斷性的工作。此外,這些估計在一定程度上依賴於管理層對公司無法控制的未來事件的假設,例如繼續 在美國的緊急使用授權和在歐盟的營銷授權。 |
我們如何在審計中處理該問題 |
為了測試管理層對未完成產品庫存可實現淨值的估計(部分基於對預期未來銷售的估計),我們執行了審計程序,其中包括將在制定醫療需求假設 時使用的投入與從美國政府新冠肺炎數據和該公司針對Gohibic的臨牀試驗結果 中觀察到的數據點進行比較。我們還比較了在制定市場滲透率假設時使用的投入,以及對製藥行業可實現的市場份額的研究。我們對醫療需求和市場滲透率假設進行了敏感性分析。我們評估了 管理層將未完成產品庫存與預期未來銷售額的估計進行的比較,其中包括考慮適用的庫存到期日。此外,我們通過參考與相關監管機構的通信和管理層對 公司為達到授權所需條件而採取的行動的詢問,評估了管理層評估 美國緊急使用授權仍然有效以及在歐盟授予營銷授權的可能性的合理性。我們還測試了公司預計未來銷售額所依據的計算 的文書準確性。 |
/s/ 安永有限公司
我們 自2020年起擔任本公司的審計師。
德國慕尼黑
2024年3月20日
F-4
獨立註冊會計師事務所報告{br
致 InflaRx N.V.的股東和董事會。
關於財務報告內部控制的意見
截至2023年12月31日,我們 根據Treadway委員會贊助組織委員會發布的內部控制綜合框架(2013年框架)(COSO標準)中確立的標準 審計了InflaRx N.V.及其子公司對財務報告的內部控制。我們認為,InflaRx N.V.及其子公司(本公司)在所有重大方面根據COSO標準對截至2023年12月31日的財務報告保持了有效的內部控制。
我們 還根據上市公司會計監督委員會的標準進行了審計(美國)(PCAOB),本公司截至2023年和2022年12月31日的綜合財務狀況表,相關的綜合經營報表和綜合虧損, 截至2023年12月31日止三年各年股東權益和現金流量的變動,以及相關附註和我們日期為3月20日的報告,2024對此發表無保留意見。
徵求意見的依據
公司管理層負責維持對財務報告的有效內部控制,並負責對隨附的管理層《財務報告內部控制年度報告》 財務報告內部控制的有效性進行評估 。我們的責任是根據我們的審計,就公司對財務報告的內部控制 發表意見。我們是一家在PCAOB註冊的公共會計師事務所,根據美國聯邦證券法以及美國證券交易委員會 和PCAOB的適用規則和條例,我們必須 獨立於本公司。
我們 根據PCAOB的標準進行了審計。這些標準要求我們計劃和執行審計,以獲得對財務報告是否在所有重要方面都保持有效內部控制的合理保證。
我們的 審計包括瞭解財務報告的內部控制、評估存在重大缺陷的風險、根據評估的風險測試和評估內部控制的設計和操作有效性,以及執行我們認為在此情況下必要的其他 程序。我們相信,我們的審計為我們的觀點提供了合理的基礎。
財務報告內部控制的定義和侷限性
公司對財務報告的內部控制是一個過程,其目的是根據公認會計 原則,為財務報告的可靠性提供合理保證 ,併為外部目的編制財務報表。公司對財務報告的內部控制包括那些政策和程序:(1)與 維護記錄有關,這些記錄以合理的詳細程度、準確和公平地反映 公司資產的交易和處置;(2)提供合理的保證,即交易是必要的,以允許根據公認會計原則編制財務報表,並且公司的收入和支出僅根據公司管理層和董事的授權進行;以及(3)提供合理的保證以防止或及時發現可能對財務報表產生重大影響的未經授權的公司資產的獲取、使用或處置。
由於其固有的侷限性,財務報告的內部控制可能無法防止或發現錯誤陳述。此外,對未來期間進行任何有效性評估的預測可能會因為條件的變化而出現控制不足的風險, 或政策或程序的遵守程度可能惡化。
/s/
2024年3月20日
F-5
InflaRx N.V.和子公司
截至2023年、2022年及2021年12月31日止年度的綜合 經營及全面虧損表
| 注意事項 | 2023 | 2022 | 2021 | ||||||||||||
| (in歐元,份額數據除外) | |||||||||||||||
| 收入 | C.1. | ||||||||||||||
| 銷售成本 | C.2. | ( | ) | ||||||||||||
| 毛利 | ( | ) | — | — | |||||||||||
| 銷售和市場營銷費用 | C.3. | ( | ) | ||||||||||||
| 研發費用 | C.4. | ( | ) | ( | ) | ( | ) | ||||||||
| 一般和行政費用 | C.5. | ( | ) | ( | ) | ( | ) | ||||||||
| 其他收入 | C.6. | ||||||||||||||
| 其他費用 | ( | ) | ( | ) | ( | ) | |||||||||
| 運營結果 | ( | ) | ( | ) | ( | ) | |||||||||
| 財政收入 | C.8. | ||||||||||||||
| 財務費用 | C.8. | ( | ) | ( | ) | ( | ) | ||||||||
| 換匯結果 | C.8. | ( | ) | ||||||||||||
| 其他財務結果 | C.8. | ( | ) | ( | ) | ||||||||||
| 所得税 | — | — | — | ||||||||||||
| 當期虧損 | ( | ) | ( | ) | ( | ) | |||||||||
| 於其後期間可能重新分類至損益之其他全面收益(虧損): | |||||||||||||||
| 外幣換算匯兑差額 | |||||||||||||||
| 合計 綜合損失 | ( | ) | ( | ) | ( | ) | |||||||||
| 本集團的財務狀況。 | C.9. | ||||||||||||||
| 加權平均流通股數 | |||||||||||||||
| ( | ) | ( | ) | ( | ) | ||||||||||
| 本期虧損 | ( | ) | ( | ) | ( | ) | |||||||||
附註是這些合併財務報表的組成部分。
F-6
InflaRx N.V.和子公司
截至2023年和2022年12月31日的合併 財務狀況表
| 注意事項 | 2023年12月31日 | 12月31日, 2022 | |||||||||
| 資產 | (歐元) | ||||||||||
| 非流動資產 | |||||||||||
| 財產和設備 | D.1. | ||||||||||
| 使用權資產 | D.2. | ||||||||||
| 無形資產 | D.3. | ||||||||||
| 其他資產 | D.6. | ||||||||||
| 金融資產 | D.8. | ||||||||||
| 非流動資產總額 | |||||||||||
| 流動資產 | |||||||||||
| 盤存 | D.5. | ||||||||||
| 流動其他資產 | D.6. | ||||||||||
| 應收税金 | D.7. | ||||||||||
| 來自政府補助金的金融資產 | D.8. | ||||||||||
| 其他金融資產 | D.8. | ||||||||||
| 現金和現金等價物 | D.9. | ||||||||||
| 流動資產總額 | |||||||||||
| 總資產 | |||||||||||
| 權益和負債 | |||||||||||
| 權益 | D.10. | ||||||||||
| 已發行資本 | D.10.(a)。 | ||||||||||
| 股票溢價 | D.10.(a)。 | ||||||||||
| 其他資本儲備 | D.10.(a)。 | ||||||||||
| 累計赤字 | D.10.(a)。 | ( | ) | ( | ) | ||||||
| 股本的其他組成部分 | D.10.(a)。 | ||||||||||
| 總股本 | |||||||||||
| 非流動負債 | |||||||||||
| 租賃負債 | E.2. | ||||||||||
| 其他負債 | |||||||||||
| 非流動負債總額 | |||||||||||
| 流動負債 | |||||||||||
| 貿易和其他應付款 | D.11. | ||||||||||
| 來自政府贈款的負債 | |||||||||||
| 租賃負債 | E.2. | ||||||||||
| 員工福利 | |||||||||||
| 其他負債 | D.11. | ||||||||||
| 流動負債總額 | |||||||||||
| 總負債 | |||||||||||
| 權益和負債總額 | |||||||||||
附註是這些合併財務報表的組成部分。
F-7
InflaRx N.V.和子公司
截至2023年、2022年和2021年12月31日止年度的綜合股東權益變動表
| 注意事項 | 股票 傑出的 | 已發佈 資本 | 分享 補價 | ||||||||||||
| (歐元) | |||||||||||||||
| 截至2021年1月1日的餘額 | |||||||||||||||
| 當期虧損 | — | — | |||||||||||||
| 外幣換算匯兑差額 | — | — | |||||||||||||
| 全面損失總額 | — | — | |||||||||||||
| 普通股的發行 | |||||||||||||||
| 交易成本 | — | ( | ) | ||||||||||||
| 權益結算的股份支付 | C.10. | — | |||||||||||||
| 已行使的購股權 | |||||||||||||||
| 截至2021年12月31日的餘額 | |||||||||||||||
| 當期虧損 | — | — | |||||||||||||
| 外幣換算匯兑差額 | — | — | |||||||||||||
| 全面損失總額 | — | — | |||||||||||||
| 普通股的發行 | D.10.(a)。 | ||||||||||||||
| 交易成本 | — | ( | ) | ||||||||||||
| 權益結算的股份支付 | C.10. | ||||||||||||||
| 已行使的購股權 | |||||||||||||||
| 截至2022年12月31日的餘額 | |||||||||||||||
| 當期虧損 | — | — | |||||||||||||
| 外幣換算匯兑差額 | — | — | |||||||||||||
| 全面損失總額 | — | — | |||||||||||||
| 普通股的發行 | D.10.(a)。 | ||||||||||||||
| 交易成本 | — | — | ( | ) | |||||||||||
| 權益結算的股份支付 | C.10. | ||||||||||||||
| 已行使的購股權 | |||||||||||||||
| 截至2023年12月31日的餘額 | |||||||||||||||
附註是這些合併財務報表的組成部分。
F-8
InflaRx N.V.和子公司
截至2023年、2022年和2021年12月31日止年度的綜合股東權益變動表
| 注意事項 | 其他 資本儲備 | 累計 赤字 | 其他 組件 股權 | 總計 股權 | |||||||||||||||
| (歐元) | |||||||||||||||||||
| 截至2021年1月1日的餘額 | ( | ) | ( | ) | |||||||||||||||
| 當期虧損 | — | ( | ) | ( | ) | ||||||||||||||
| 外幣換算匯兑差額 | — | ||||||||||||||||||
| 全面虧損總額 | — | ( | ) | ( | ) | ||||||||||||||
| 普通股的發行 | D.10. | ||||||||||||||||||
| 交易成本 | — | — | — | ( | ) | ||||||||||||||
| 權益結算的股份支付 | C.10. | ||||||||||||||||||
| 已行使的購股權 | — | — | — | ||||||||||||||||
| 截至2021年12月31日的餘額 | ( | ) | |||||||||||||||||
| 當期虧損 | — | ( | ) | — | ( | ) | |||||||||||||
| 外幣換算匯兑差額 | — | — | |||||||||||||||||
| 全面損失總額 | — | ( | ) | ( | ) | ||||||||||||||
| 普通股的發行 | D.10. | — | — | — | |||||||||||||||
| 交易成本 | — | — | — | ( | ) | ||||||||||||||
| 權益結算的股份支付 | C.10. | ||||||||||||||||||
| 已行使的購股權 | — | — | — | ||||||||||||||||
| 截至2022年12月31日的餘額 | ( | ) | |||||||||||||||||
| 當期虧損 | — | ( | ) | — | ( | ) | |||||||||||||
| 外幣換算匯兑差額 | — | — | |||||||||||||||||
| 全面損失總額 | — | ( | ) | ( | ) | ||||||||||||||
| 普通股的發行 | — | — | — | ||||||||||||||||
| 交易成本 | — | — | — | ( | ) | ||||||||||||||
| 權益結算的股份支付 | C.10. | — | — | ||||||||||||||||
| 已行使的購股權 | C.10. | — | — | — | |||||||||||||||
| 截至2023年12月31日的餘額 | ( | ) | |||||||||||||||||
附註是這些合併財務報表的組成部分。
F-9
InflaRx N.V.和子公司
截至2023年、2022年和2021年12月31日止年度的合併現金流量表
| 注意事項 | 2023 | 2022 | 2021 | ||||||||||||
| (歐元) | |||||||||||||||
| 經營活動 | |||||||||||||||
| 當期虧損 | ( | ) | ( | ) | ( | ) | |||||||||
| 對以下各項進行調整: | |||||||||||||||
| 不動產和設備、使用權資產和無形資產的折舊和攤銷 | |||||||||||||||
| 財務淨收入 | C.8. | ( | ) | ( | ) | ( | ) | ||||||||
| 基於股份的支付費用 | C.10. | ||||||||||||||
| 淨匯差 | |||||||||||||||
| 以下內容中的更改: | |||||||||||||||
| 來自政府補助金的金融資產 | D.8. | ( | ) | ||||||||||||
| 其他資產 | ( | ) | ( | ) | |||||||||||
| 員工福利 | ( | ) | ( | ) | |||||||||||
| 其他負債 | |||||||||||||||
| 收到的政府補助金的負債 | D.8. | ( | ) | ( | ) | ||||||||||
| 貿易和其他應付款 | D.11. | ( | ) | ||||||||||||
| 盤存 | D.5. | ( | ) | ||||||||||||
| 收到的利息 | |||||||||||||||
| 支付的利息 | ( | ) | ( | ) | ( | ) | |||||||||
| 用於經營活動的現金淨額 | ( | ) | ( | ) | ( | ) | |||||||||
| 投資活動 | |||||||||||||||
| 購買無形資產及不動產和設備 | ( | ) | ( | ) | ( | ) | |||||||||
| 購買流動和非流動金融資產 | ( | ) | ( | ) | ( | ) | |||||||||
| 流動金融資產到期所得款項 | |||||||||||||||
| 投資活動產生/(用於)現金淨額 | ( | ) | ( | ) | |||||||||||
| 融資活動 | |||||||||||||||
| 發行普通股所得款項 | D.10. | ||||||||||||||
| 發行普通股的交易成本 | D.10. | ( | ) | ( | ) | ( | ) | ||||||||
| 行使購股權所得款項 | C.10 | ||||||||||||||
| 償還租賃債務 | ( | ) | ( | ) | ( | ) | |||||||||
| 融資活動的現金淨額 | |||||||||||||||
| 現金和現金等價物淨減少 | ( | ) | ( | ) | ( | ) | |||||||||
| 匯率變動對現金及現金等價物的影響 | ( | ) | |||||||||||||
| 期初現金及現金等價物 | |||||||||||||||
| 期末現金及現金等價物 | D.9. | ||||||||||||||
附註是這些合併財務報表的組成部分。
F-10
InflaRx N.V.和子公司
a. 綜合財務報表附註
1.企業信息
InflaRx N.V.及其附屬公司(統稱“本集團”)截至2023年12月31日止年度的 綜合財務報表已根據董事會於2024年3月20日的決議案獲授權刊發。InflaRx N.V.(“公司”) 是一家荷蘭上市公司,有限責任公司(Naamloze Vennootschap),其公司總部位於荷蘭阿姆斯特丹, 並在荷蘭商會商業登記處註冊,CCI編號為68904312。 公司的註冊辦事處位於德國耶拿07745的Winzerlaer Straße 2。自2017年11月10日起,公司的 普通股已在納斯達克全球精選市場上市,代碼為“IFRX”。
公司及其子公司,統稱為一個生物技術集團,通過應用其專有的 抗C5a和抗C5aR技術,發現、開發和商業化補體激活 因子(稱為C5a及其受體C5aR)的高效和特異性抑制劑,開創了抗炎療法的先河。
子公司 是指本集團對其擁有控制權的所有實體。當本集團因參與該實體而承受或有權享有可變回報 且可能通過其指導該實體活動的權力影響該等回報時,本集團即控制該實體。 附屬公司自本集團取得控制權之日起全面綜合入賬。自控制 停止之日起,將其取消合併。本集團採用收購會計法將業務合併入賬。公司間交易、結餘 和集團公司間交易的未實現收益予以抵銷。未實現虧損亦會對銷,除非交易 提供已轉讓資產減值的證據。
本集團於2023年12月31日的附屬公司載列如下。
| 業務所在地/國家/地區 | 功能性 | 所有權
權益 由公司持有 | |||||||||||||
| 名字 | 成立為法團 | 貨幣 | 2023 | 2022 | 主要活動 | ||||||||||
| InflRx GmbH | % | % | |||||||||||||
| InflRx製藥公司 | % | % | |||||||||||||
B. 材料核算政策
1. 準備依據
本集團的綜合財務報表乃根據國際會計準則委員會(以下簡稱“IFRS”)發佈的國際財務報告準則編制。
The consolidated financial statements have been prepared on a historical cost basis. These consolidated financial statements of the Group comprise the Company and its wholly owned subsidiaries, InflaRx GmbH and InflaRx Pharmaceuticals, Inc. The consolidated financial statements are presented in Euro (€). The presentation currency of the Group is the Euro, as the functional currency of the largest operating company, InflaRx GmbH, continues to be the Euro. Effective January 1, 2023, the functional currency of InflaRx N.V. changed from the U.S. dollar to the Euro due to a change in the Company’s operational function and, in turn, a change in the primary currency of its underlying transactions. This change in functional currency has been accounted for prospectively. The functional currency of InflaRx Pharmaceuticals, Inc. is the U.S. dollar ($), as most of their income and expenses occurred in U.S. dollars in 2023. All financial information presented in Euro has been rounded to the nearest Euro, unless stated otherwise.
F-11
2. 重大會計政策摘要
本 節描述編制該等綜合財務報表時採納的重大會計政策。除另有説明外,此等政策 已貫徹應用於所有呈列年度。
(a) 本集團採納之新訂及經修訂準則
以下修訂自2023年1月1日起已採納,且對本集團的綜合財務報表並無重大影響 :
| ● | IFRS 第17號保險合同 | |
| ● | 修正案 國際會計準則第8號會計政策、會計估計變更和錯誤:會計定義 估計 | |
| ● | 修正案 根據國際會計準則第12號,與單一交易產生的資產和負債有關的遞延税項 | |
| ● | 修正案 《國際會計準則第1號》和《國際財務報告準則第2號》—會計政策披露 |
自2023年第二季度開始,以下IFRS準則的會計 政策首次應用,因為之前未確認 這些IFRS準則範圍內的交易。
| ● | IAS 2號存貨 |
根據國際會計準則第2號,存貨按其成本或其可變現淨值的較低數額列報。成本包括直接材料成本 和(如適用)直接人工成本以及在將存貨運到其當前 位置和狀態時發生的間接費用。成本乃採用加權平均成本法計算。可變現淨值指估計售價減去所有估計完工成本以及市場營銷、銷售和分銷成本。按可變現淨值確認存貨 包括減記被視為超額或過時的存貨。
| ● | IFRS 15號客户合約收益 |
目前,公司僅使用分銷商向最終客户銷售其產品(例如,醫院)。最終客户(如醫院) 已被確定為這些銷售安排中的客户。因此,從分銷商收到的付款不是合同 負債,而是在"其他應計負債"中確認的"其他負債"。因此,當通過向客户轉讓承諾貨物或服務履行履約義務時,即當客户獲得該資產的控制權時, 確認收入,並考慮估計退貨負債和預期回扣或現金 折扣進行計量。
(b) 新標準尚未採用
以下已頒佈的準則將在未來期間被採納,並正在評估對本集團綜合財務報表的潛在影響(如有):
| ● | 修正案 國際財務報告準則第16號租賃:售後租賃及回租租賃 | |
| ● | 修正案 國際會計準則第1號財務報表列報:負債分類為流動或 有契約的非流動和非流動負債 |
| ● | 修正案 國際會計準則第21號外匯匯率變動的影響:缺乏匯率 |
(c) 當期和非當期分類
本集團根據流動/非流動分類在財務狀況表中呈列資產和負債。
流動 資產包括作為正常運營週期(假設運營週期為12個月)一部分出售、消耗或變現的資產,或現金和現金等價物,除非在報告期後至少12個月內被限制交換或用於結算負債。所有其他資產分類為非流動資產。
流動 負債,如貿易應付款項、租賃負債或期限最長為12個月的僱員福利,以及應付運營成本或社會保障費用的款項,是公司正常運營週期中使用的營運資金的一部分。即使該等經營項目於報告期後超過12個月到期結算,該等經營項目仍被分類為流動負債。所有其他 負債分類為非流動。
F-12
(d) 外幣交易
外幣交易 最初使用 交易日期的即期匯率換算為相應的功能貨幣。不以功能貨幣計值的貨幣項目隨後使用期末適用的匯率進行換算 。所產生之貨幣收益及虧損直接於損益確認。
在 合併時,以歐元(公司的列報貨幣)以外的貨幣計算的業務資產和負債 按報告日期的匯率換算為歐元,其業務報表則按報告期內的 月平均匯率換算。綜合換算產生的匯兑差額於 “其他全面收益”(OCI)確認。在出售海外業務時,與該特定海外業務有關的其他全面收益部分重新分類至損益。其他全面收益在綜合財務狀況表中披露為“權益的其他組成部分”。
(e) 政府和類似機構的贈款
集團從政府機構和類似機構獲得資助,用於積極參與特定的研究和開發項目。 當有合理保證將收到補助金,並符合所有補助金條件時,確認補助金。如果 補助金是在發生符合條件的費用或購買資產之前或在滿足所有補助金條件之前收到的,則 此類金額記錄為其他負債中的負債。如果基金報銷費用,則負債在相應費用發生的期間(或者,對於在滿足所有補助條件之前發生的費用,在達到所有補助條件將得到滿足的合理保證期間)攤銷為其他 營業收入。如果資金償還已購買的 資產,則在記錄合格 資產時,負債將相應金額從資產的賬面值中扣除。根據補助條款,補助人一般有權在完成政府資助的項目後最多 五年內審計集團提交的合格費用。
於
二零二一年十月,本集團宣佈其收到最高達歐元的補助金,
(f) 現金流量表附註、現金及現金等價物
合併現金流量表已使用間接法編制經營活動現金流量表。綜合現金流量表中披露的現金 包括現金及現金等價物。現金包括庫存現金和活期存款 。現金等價物是指短期銀行存款,可隨時轉換為已知數額的現金,且 原到期日為三個月或更短時間內不存在價值變動的重大風險。已付和已收利息包括在 經營活動現金中。
(g) 研發費用
研究和開發費用包括第三方服務、工資和薪金、材料成本、知識產權相關費用、 相關設備和無形資產的折舊和攤銷以及管理費用。研發費用主要包括 公司臨牀藥品的臨牀試驗和生產費用;此外,臨牀前活動 以及基礎研究活動產生的費用。
如果符合IAS 38的標準,則開發 費用必須資本化。在所列期間,沒有將開發費用資本化,因為 管理層評估認為,並非符合國際會計準則第38號的所有確認標準。這種評估是由於藥物開發中的普遍不確定性 和監管要求的不可預測性。因此,研發支出在發生時計入費用 。
F-13
(h) 員工福利
(i) 短期僱員福利
工資和薪金及現金獎金的負債 按債務清償時預計支付的金額計量。負債 在合併財務狀況表中作為員工福利列示。如果本集團因僱員過去提供的服務而負有支付該金額的當前法律或推定義務,且該義務 能夠可靠地估計,則確認該責任。
(2) 基於股份的支付交易
授予員工的以股權結算的股份支付安排的授予日公允價值通常被確認為獎勵歸屬期間的支出,並相應增加股本。確認為費用的金額將進行調整,以反映 預計將滿足相關服務條件的獎勵數量,包括估計的沒收金額,以便最終確認的金額基於在歸屬日期滿足相關服務條件的獎勵數量。對於立即歸屬的基於股份的支付獎勵,基於股份的支付的授予日公允價值被計量以反映該等條件,並且 預期結果與實際結果之間的差異不確認收益或損失。
(i) 租約安排
該集團租賃各種物業、實驗室和辦公設備以及汽車。租賃合同通常是以固定的期限
(I) 使用權資產
集團於租賃開始日(即標的資產可供使用的日期)確認使用權資產。 使用權資產按成本減去任何累計折舊和減值損失計量,並根據租賃負債的任何重新計量進行調整 。使用權資產成本包括已確認的租賃負債額、已產生的初始直接成本、 以及在生效日期或之前支付的租賃付款減去收到的任何租賃獎勵。除非本集團合理地確定 將於租賃期結束時取得租賃資產的所有權,否則已確認使用權資產將按其估計使用年限及租賃期中較短的一項按直線 基準進行折舊。於2023年12月31日,本公司使用權資產的剩餘使用年限為3至41個月。使用權資產應計提減值。
(Ii) 租賃負債
於租賃開始日期 ,本集團確認按租賃付款現值計量的租賃負債將於租賃期內支付 。租賃付款包括固定付款(包括實質固定付款)減去任何應收租賃獎勵、取決於指數或費率的可變租賃付款以及根據剩餘價值擔保預期支付的金額。租賃付款 還包括本集團合理確定將行使的購買選擇權的行使價,以及支付終止租賃的罰金 (如果租期反映本集團行使終止選擇權)。不依賴於指數或費率的可變租賃付款在觸發付款的事件或條件發生的期間被確認為費用。
在計算租賃付款現值時,本集團採用租賃開始日的遞增借款利率,因為租賃中隱含的利率無法輕易確定。生效日期後,租賃負債額增加以反映利息的增加,並減少所支付的租賃付款。此外,如果租賃負債的賬面價值發生變動、租賃期限的變化、實質固定租賃付款的變化或購買相關資產的評估發生變化,租賃負債的賬面價值將被重新計量。
F-14
(3) 短期租賃和低價值資產租賃
本集團將短期租約確認豁免適用於其短期設備租約(即租期自開始日期起計為12個月或以下且不含購買選擇權的租約)。它還將低價值資產的租賃 確認豁免適用於被視為低價值的辦公設備租賃。短期租賃和低價值資產租賃的租賃付款 在租賃期內按直線原則確認為費用。
(四)確定合同租賃期限
於開始日期 後,如本集團控制範圍內發生重大事件或情況變化,並影響其行使續期選擇權的能力,本集團將重新評估租賃期。
集團進一步決定租期為租約的不可撤銷期限,以及在合理確定將行使租約的情況下延長租約的選擇權所涵蓋的任何期限,或如果合理地確定不行使租約的選擇權所涵蓋的任何期限。
目前也導致使用權資產資本化的租賃不包括任何續訂選擇權。對於具有潛在續訂選擇權的未來租賃 合同,公司在評估是否合理確定行使 續訂選擇權時應用判斷。在這樣做時,管理層將考慮為執行 續約創造經濟激勵的所有相關因素。
(j) 利息收入
利息 收入來自計息金融資產,包括現金等價物。現金及現金等價物、使用實際利率法計算的按攤餘成本計量的金融資產的利息收入在綜合經營報表 和全面虧損中確認為融資收入的一部分。
(k) 無形資產
無形
資產主要包括採購的IT軟件。無形資產初步按收購成本計量,包括準備資產投入其擬定用途的任何直接應佔成本減累計攤銷及累計減值虧損(如有)。攤銷
在資產可供使用時開始,並且攤銷是使用直線法計算的,以便在估計
使用壽命內分配成本。無形資產之可使用年期於各報告日期審閲。軟件攤銷
(l) 財產和設備
實驗室 及辦公室設備按歷史成本減累計折舊及累計減值虧損(如有)列賬。歷史 成本包括直接歸屬於購置項目的支出。
所有 維修和保養在發生的財政期間內在損益中確認,因為它們不 構成單獨的資產。
實驗室和辦公室設備的折舊 採用直線法計算,以在其估計使用壽命內分配其成本, 如下所示:
| ● | 實驗室設備: | |
| ● | 辦公設備: |
資產的剩餘價值和使用壽命在每個報告期結束時進行審查,並在適當情況下進行調整。
出售收益和虧損是通過比較收益和賬面金額來確定的,並在綜合經營報表和全面虧損的“其他收入” 或“其他費用”中確認。
F-15
(m) 資產減值
於每個報告日期,本集團會評估是否有資產可能減值的跡象。如有任何減值跡象 或需要進行年度減值測試,本集團估計資產的可收回金額。資產的可收回金額 是資產的公允價值減去處置成本及其使用價值後的較高者。它是針對單個資產確定的, 除非該資產不產生基本上獨立於其他資產或資產組的現金流入,在這種情況下,它在現金產生單位的水平上確定。如果一項資產的賬面金額超過其可收回金額,則該資產將減值並減記至其可收回金額。在評估使用價值時,估計的未來現金流量使用税前貼現率折現至其現值,該貼現率反映了當前市場對貨幣時間價值和資產特有風險的評估 。
若自上次確認減值虧損以來,用以釐定資產可收回金額的估計數字發生變動,則先前確認的任何減值虧損將予以撥回。沖銷不得超過在攤銷或折舊後確定的賬面金額 如果該資產在以前的期間沒有確認減值損失的話。沖銷金額 在當期損益中確認。
在2021年、2022年或2023年沒有減值或減值逆轉。
(n) 金融資產和負債(金融工具)
(一)定義
金融工具是指產生一個實體的金融資產和另一個實體的金融負債或權益工具的任何合同。本集團的金融資產包括主要報價的固定利息債務證券。金融負債 包括貿易和其他應付款(包括研發項目的應計負債)。
(二)確認和解除確認的標準、初步衡量
於 中,一般購買或出售金融資產於結算日確認,即本集團呈交或收到櫃枱表現(通常為現金)之日。本集團最初按其公允價值加交易成本計量金融資產。
本集團於按公允價值扣除直接應佔交易成本後,初步確認非衍生金融負債。當合同義務被解除、註銷或到期時,本集團將不再確認金融負債。
(三)後續測量方法
考慮到 本集團管理金融資產的業務模式,目標是持有該等金融資產以收取合約現金流量及其合約現金流特徵,即僅就未償還本金支付本金及利息,本集團按固定利率將報價的債務證券分類,其後採用實際利息法(EIR)按攤銷成本計量。金融資產也應計提減值準備。
本集團的財務負債按其後按攤銷成本計量分類,而攤銷成本是根據收購的任何折讓或溢價以及作為企業內部回報率組成部分的費用或成本計算而得。
按計量類別分列的綜合財務狀況表中的賬面金額分析在‘3.8金融資產和金融負債’項下披露。
F-16
(四)實現收支的標準
利息 收入按相關的實際利率計提。負債的利息支出(如果有)也是根據實際利率計提的。
當所有重大風險和回報轉移完畢後,將全額確認處置金融工具的收益和損失。 在風險和回報部分轉移的情況下,將區分控制權仍然屬於公司還是 轉移。
減值 金融資產的損失在損益中確認。本集團確認所持金融資產的預期信貸損失準備(ECL),見附註‘C.8。淨財務結果‘。
ECL 基於根據合同到期的合同現金流與本集團 預期收到的所有現金流之間的差額,按原始實際利率的近似值貼現。ECL通常分兩個階段確認。 對於自初始確認以來信用風險沒有顯著增加的信用風險敞口,提供ECL是為了應對因未來12個月內可能發生的違約事件而造成的信用損失(12個月ECL)。對於自初始確認以來信用風險顯著增加的信用風險敞口 ,無論違約的時間如何(終身ECL),都需要為風險敞口剩餘壽命內預期的信用損失 計提損失準備金。對於具有較高信用評級且自初始確認以來信用風險沒有顯著增加的固定利率報價債務證券, 集團使用信用機構發佈的CDS定價信息(即信用違約互換價值)來確定信用違約風險敞口,並確認12個月的ECL。
(o) 公允價值計量
本集團並不按公允價值計量任何金融資產或負債。所有金融工具的賬面價值均接近其公允價值,但披露公允價值的報價債務證券除外(見附註‘D.8)。財務 資產和財務負債‘)。
在計量資產或負債的公允價值時,本集團將盡可能使用可觀察到的市場數據。公允價值根據估值技術中使用的投入在公允價值層次結構中劃分為不同的級別,具體如下:
| ● | 第1級,相同資產或負債在活躍市場上的報價。 |
| ● | 第2級,第1級中可觀察到的報價以外的輸入, 直接(作為價格)或間接(從價格派生)。 |
| ● | 第 3級,不基於可觀測市場數據的工具的投入(不可觀測投入)。 |
如果 用於計量資產或負債的公允價值的投入屬於公允價值層次結構的不同級別,則 公允價值計量整體歸類於公允價值層次結構中與對整個計量重要的最低水平投入相同的級別。
在發生變動的報告期結束時, 集團將確認公允價值層級之間的轉移。
(p) 所得税
所得税 税包括當期税和遞延税。本期及遞延税項於損益中確認,但與直接於權益或其他全面虧損中確認的項目有關者除外。
(一)當期所得税
當期所得税資產和負債按預期向税務機關收回或支付的金額計量。預期 本年度應課税收入或虧損的應付或應收税款,是根據報告日期頒佈或實質頒佈的税率以及對往年應付税款的任何調整而計算的。
於所列期間內,本集團並無產生所得税開支。銀行代扣代繳、匯入税務機關的税款在年度納税申報後退還。
(二)遞延所得税
遞延税項是就財務報告用途的資產及負債賬面值與税務用途的金額之間的暫時性差異而確認的。如果導致資產和負債初步確認的交易不是業務合併交易,且 不影響會計或税項損益,則不會就與資產和負債相關的暫時性差異確認遞延税金。
遞延税項是根據截至報告日期已頒佈或實質頒佈的法律,按暫時性差額轉回時預期適用的税率計量。
由税項虧損結轉產生的遞延税項資產只有在本集團有足夠的應課税暫時性差異 或有令人信服的證據證明有足夠的未來應課税利潤可用於抵銷未使用的税項虧損時才予以確認。 於2023年、2023年及2022年12月31日,根據管理層的判斷,不可能有應課税利潤可用於可動用未使用税項虧損的 ;因此,並無遞延税項資產於綜合財務報表 確認。
F-17
3.重大會計判斷、估計和假設
根據《國際財務報告準則》編制合併財務報表需要管理層作出判斷、估計和假設,以影響會計政策的應用以及資產、負債、收入和費用的報告金額。實際結果 可能與這些估計值不同。
估計 和基本假設將持續進行審查。會計估計的修訂在 估計被修訂的期間以及任何受影響的未來期間確認。在編制該等綜合財務報表時,管理層在應用本集團會計政策時作出的關鍵判斷 涉及以下方面:
(a) 基於股份支付的會計核算
此外, 公司必須估計將在未來期間歸屬的權益工具的數量,因為獎勵可能會在歸屬之前被沒收 ,因為獲獎者未能滿足履約條件,包括由於終止僱用。沒收率 的假設是根據歷史信息定期作出的,並進行調整以反映未來的預期。對沒收率 的修訂可能導致當期和前期估計數變動的累積影響在變動期間確認。
(b) 第三方研發臨牀試驗和合同製造費用的計量
在 計量報告期內的研發費用時,公司估計要確認的費用和要產生的負債 ,前提是尚未收到公司的合同研究組織(“CRO”)和合同製造組織(“CDMO”)的發票且超過任何預付款。CRO為項目服務開具發票的時間遵循 合同計費時間表,可以在報告期前幾個月或之後幾個月進行。此估算涉及確定 完成時間,據此,由內部研發項目經理評估和估算,並由控制部門審查 CRO和CDMO承包的單個項目活動的服務程度。此完工時間 用於衡量截至報告日期已完成的未開票項目活動的金額,以及因此確認的相關 研發費用和負債。
完工時間估計值基於當時可用的最佳信息。但是,將來可能會 提供額外信息,管理層可能會在這些未來時期調整估計數。在這種情況下,當實際活動水平變得更加確定時,公司可能需要 記錄未來期間對研發費用的調整。 公司將由此產生的費用增加或減少視為估計的變化,並在確定的期間內反映研究和開發費用的變化 。
公司應計歐元
F-18
(c) 存貨的變現能力
存貨 按成本和可變現淨值兩者中較低者計價。可變現淨值包括估計銷售所得款項減去直至銷售時的必要 預期成本。為確定可變現淨值,在每個報告日期,本公司主要使用預期未來銷售額模型和使用具有重大估計不確定性的假設 (如預期醫療需求和預期市場滲透率)來估計超額 和過時存貨。
此外, 這些估計部分依賴於管理層對公司控制之外的未來事件的假設,例如 繼續在美國進行緊急使用授權和在歐盟授予上市授權。在 作出這些假設時,管理層通過考慮與相關監管機構的通信以及公司為達到授權所需條件而採取的行動 ,評估這些授權仍然有效或被授予(如適用)的可能性。
此外, 考慮了原材料、未加工產品和成品的可能替代用途。
管理層 定期評估市場和銷售趨勢、市場條件、疾病流行率、競爭格局和監管環境 ,以完善過剩和過時庫存的估計。基於這些評估,並考慮到製造提前期, 管理層根據庫存老化和上述標準做出運營決策,訂購庫存。
截至2023年12月31日止年度的庫存
減記金額為歐元
預計未來需求模型中包含的假設 可能需要在未來期間進行修訂,這可能導致超額和過時庫存估計 以及庫存減記發生變化。
C.綜合經營報表和全面虧損
1.
| 2023 | 2022 | 2021 | ||||||||||
| (歐元) | ||||||||||||
| 收入 | ||||||||||||
| 總計 | ||||||||||||
2023年6月,本集團開始在美國商業化GOHIBIC(vilobelimab)。關於商業化的開始, 本集團與Cencora Inc.的某些子公司簽訂了協議。("Cencora")(原名AmerisourceBergen Corp.)作為本集團的美國分銷商,並提供GOHIBIC(vilobelimab)供美國醫院客户訂購。Cencora 提供冷藏、冷鏈配送服務、庫存管理和二次標籤/包裝等服務。
2023年,公司自成立以來首次實現產品銷售收入。報告的收入為 最終客户(醫院)的銷售額。向分銷商的銷售並不構成對客户的履約義務的完成,因此 不會導致根據國際財務報告準則第15號確認本公司的收入。
2.
| 2023 | 2022 | 2021 | ||||||||||
| (歐元) | ||||||||||||
| 銷售成本 | ||||||||||||
| 總計 | ||||||||||||
截至2023年12月31日止12個月確認的銷售成本 與GOHIBIC(vilobelimab)在美國的收入 和存貨減記有關。
F-19
在這些期間銷售的產品的銷售成本 不包括材料成本,因為這些材料的相關成本 是在美國食品藥品監督管理局(以下簡稱"FDA")於2023年4月授予GOHIBIC(vilobelimab)緊急使用許可(以下簡稱"EUA") 之前發生的。這些材料在其發生期間被記錄為"研發費用" 。
截至2023年12月31日止十二個月的 銷售成本主要包括將在 預期銷售前到期的存貨減記。
3.
| 2023 | 2022 | 2021 | ||||||||||
| (歐元) | ||||||||||||
| 第三方費用 | ||||||||||||
| 員工福利支出 | ||||||||||||
| 其中以股權結算的股份支付費用 | ||||||||||||
| 律師費和諮詢費 | ||||||||||||
| 其他費用 | ||||||||||||
| 銷售和營銷費用總額 | ||||||||||||
截至2023年12月31日止十二個月,本集團發生了歐元
4.研發費用
| 2023 | 2022 | 2021 | ||||||||||
| (歐元) | ||||||||||||
| 第三方服務 | ||||||||||||
| 其中臨牀材料和相關製造服務 | ||||||||||||
| 其中臨牀前研究 | ||||||||||||
| 員工福利支出 | ||||||||||||
| 其中以股權結算的股份支付費用 | ||||||||||||
| 律師費和諮詢費 | ||||||||||||
| 其他費用 | ||||||||||||
| 總計 | ||||||||||||
5.一般及行政開支
| 2023 | 2022 | 2021 | ||||||||||
| (歐元) | ||||||||||||
| 員工福利支出 | ||||||||||||
| 其中以股權結算的股份支付費用 | ||||||||||||
| 律師費和諮詢費 | ||||||||||||
| 保險費 | ||||||||||||
| 折舊和攤銷費用 | ||||||||||||
| 非執行董事的薪酬支出 | ||||||||||||
| 其他費用 | ||||||||||||
| 總計 | ||||||||||||
F-20
6.其他收入
其他收入為歐元
| 2023 | 2022 | 2021 | ||||||||||
| (歐元) | ||||||||||||
| 政府補助金的其他收入 | ||||||||||||
| 其他收入 | ||||||||||||
| 總計 | ||||||||||||
7.僱員福利開支
| 2023 | 2022 | 2021 | ||||||||||
| (歐元) | ||||||||||||
| 工資和薪金 | ||||||||||||
| 社會保障繳款(僱主的份額) | ||||||||||||
| 以權益結算以股份為基礎的付款開支(見附註C. 10。以股份為基礎的付款) | ||||||||||||
| 其他 | ||||||||||||
| 總計 | ||||||||||||
截至2023年12月31日, 員工人數由2022年底的48名員工(相當於 至44. 3名全職員工)和2021年底的59名員工(相當於55. 9名全職員工)增加至66名(相當於62. 2名全職員工)。這些數字是截至12月31日的, 並不構成年度平均數字。
8.
| 2023 | 2022 | 2021 | ||||||||||
| (歐元) | ||||||||||||
| 利息收入 | ||||||||||||
| 利息支出 | ( | ) | ( | ) | ( | ) | ||||||
| 租賃負債利息 | ( | ) | ( | ) | ( | ) | ||||||
| 財務結果 | ||||||||||||
| 外匯收入 | ||||||||||||
| 外匯費用 | ( | ) | ( | ) | ( | ) | ||||||
| 換匯結果 | ( | ) | ||||||||||
| 其他財務結果 | ( | ) | ( | ) | ||||||||
| 淨財務業績 | ||||||||||||
淨
財務結果減少歐元
外幣 收入和支出來自以外幣計值的現金及現金等價物、有價證券及其他金融資產 和負債,按結算日的現行匯率換算。所有由此產生的換算 差異均在損益表中確認。這些收益和損失是由報告日期 匯率變動引起的,可能最終無法實現。
F-21
9.每股虧損
每股普通股虧損
的計算方法是將本期虧損除以本期已發行普通股加權平均數
。二零二三年財政年度之已發行普通股加權數目為
由於 公司處於虧損狀況,每股攤薄虧損與每股基本虧損相同,因為行使購股權(唯一已發行的攤薄工具)時將發行的股份的加權平均數 將產生反攤薄影響 。參見附註'C.10。未行使的購股權餘額
10.股份為基礎之付款
a)以股權結算的股份支付安排
| 2023選項 | 2023 WAEP * | 2022 選項 | 2022 WAEP * | |||||||||||||
| 截至1月1日的未償還款項 | € | € | ||||||||||||||
| 年內進行的運動 | ||||||||||||||||
| 截至12月31日的未償還款項 | € | € | ||||||||||||||
| 可於12月31日行使 | € | € | ||||||||||||||
| * |
2016年之前授出的所有購股權在年底尚未行使的行使價為歐元
根據
2016年購股權計劃(“2016年計劃”)的條款和條件,InflaRx GmbH向董事、高級管理人員和主要員工授予認購
InflaRx GmbH普通股的權利。
| 2023 選項 | 2023 WAEP * | 2022 選項 | 2022 WAEP * | |||||||||||||
| 截至1月1日的未償還款項 | $ | $ | ||||||||||||||
| 年內進行的運動 | ||||||||||||||||
| 截至12月31日的未償還款項 | $ | $ | ||||||||||||||
| 可於12月31日行使 | $ | $ | ||||||||||||||
| * |
根據
F-22
| 2023 選項 | 2023 WAEP * | 2022 選項 | 2022 WAEP * | |||||||||||||
| 截至1月1日的未償還款項 | $ | $ | ||||||||||||||
| 年內批出 | $ | $ | ||||||||||||||
| 在本年度內被沒收 | ( | ) | $ | ( | ) | $ | ||||||||||
| 年內進行的運動 | ( | ) | $ | ( | ) | $ | ||||||||||
| 截至12月31日的未償還款項 | $ | $ | ||||||||||||||
| 可於12月31日行使 | $ | $ | ||||||||||||||
| * |
截至2023年12月31日,2017年計劃項下尚未行使購股權的
加權平均剩餘合約年期為
於二零二三年授出的所有購股權均於一年後歸屬,惟於二零二三年七月七日授出的購股權部分歸屬於三年後除外。2023年之前授出的購股權 可在一年、兩年或三年內歸屬,視乎授出情況而定,其中1/2或1/3的購股權 分別在歸屬開始後的第一年結束後歸屬,其餘購股權其後按季度按相等比例歸屬。這些未歸屬購股權的歸屬 受歸屬時服務條件的約束,且不適用市場或表現條件 。
2023年授出購股權的
加權平均公允價值為美元
年內就行使購股權而發行的所有 股份已於2023年12月31日前登記於商業登記冊。
b) 根據二零一七年計劃授出的購股權的公允價值計量
根據二零一七年計劃授出之購股權之 公平值乃使用柏力克—舒爾斯估值模式釐定。由於本公司的普通股 在納斯達克全球精選市場上市,因此使用普通股在授出日期的收盤價。
| 授出購股權 | 選項 | 公允價值 選擇權 | 作為贈款
的外匯匯率 日期 | 公允價值 選擇權 | 股票價格
日期/ 鍛鍊 價格 | 預期 波動 | 預期壽命 (中點 | 無風險
費率 (內插, 美國 主權 條曲線) | |||||||||||||||||||||||||
| 2021 | |||||||||||||||||||||||||||||||||
| 1月4日 | $ | € | $ | % | |||||||||||||||||||||||||||||
| 1月4日 | $ | € | $ | % | |||||||||||||||||||||||||||||
| 7月2日 | $ | € | $ | % | |||||||||||||||||||||||||||||
| 7月2日 | $ | € | $ | % | |||||||||||||||||||||||||||||
第
個
| 授出購股權 | 選項 | 每股公允價值期權 | 截至授予日的外匯匯率 | 每股公允價值期權 | 授出日股價/行權價 | 預期波動率 | 預期壽命(基於中間值) | 無風險利率(內插,美國主權條帶曲線) | |||||||||||||||||||||||||||
| 2022 | |||||||||||||||||||||||||||||||||||
| 1月12日 | $ | € | $ | ||||||||||||||||||||||||||||||||
| 1月12日 | $ | € | $ | ||||||||||||||||||||||||||||||||
| 重新定價,4月13日 | $ | € | $ | ||||||||||||||||||||||||||||||||
| 11月21日 | $ | € | $ | ||||||||||||||||||||||||||||||||
F-23
第
個
| 授出購股權 | 數 | 公允價值 選擇權 | 外匯匯率 截至 | 公允價值 選擇權 | 分享 鍛鍊 | 預期 波動率 | 預期 (中點) | 無風險
費率 (內插, 美國 主權 條曲線) | |||||||||||||||||||||||||
| 2023 | |||||||||||||||||||||||||||||||||
| 1月24日 | $ | € | $ | % | |||||||||||||||||||||||||||||
| 1月24日 | $ | € | $ | % | |||||||||||||||||||||||||||||
| 5月31日 | $ | € | $ | % | |||||||||||||||||||||||||||||
| 7月7日 | $ | € | $ | % | |||||||||||||||||||||||||||||
| 7月7日 | $ | € | $ | % | |||||||||||||||||||||||||||||
| 7月19日 | $ | € | $ | % | |||||||||||||||||||||||||||||
| 九月18 | $ | € | $ | % | |||||||||||||||||||||||||||||
第
個
預期 股息為 以上所列的所有購股權。
股價 波動性是根據估值日期前過去五年 公司股價的年化月波動率計算的。
工具預期壽命的 結果範圍基於所考慮的情景中對期權持有人行為的預期 。
股息收益率不受2017年計劃中定義的反稀釋條款的影響。
D.綜合財務狀況表附註
1.
| 屬性
和 設備 | 預付款 付款 | 總計 | ||||||||||
| 成本 | (歐元) | |||||||||||
| 2022年1月1日 | ||||||||||||
| 加法 | ||||||||||||
| 匯兑差異 | ||||||||||||
| 2022年12月31日 | ||||||||||||
| 加法 | ||||||||||||
| 處置 | ( | ) | ( | ) | ||||||||
| 匯兑差異 | ( | ) | ( | ) | ||||||||
| 2023年12月31日 | ||||||||||||
| 累計折舊 | ||||||||||||
| 2022年1月1日 | ( | ) | ( | ) | ||||||||
| 當年的折舊費用 | ( | ) | ( | ) | ||||||||
| 匯兑差異 | ( | ) | ( | ) | ||||||||
| 2022年12月31日 | ( | ) | ( | ) | ||||||||
| 當年的折舊費用 | ( | ) | ( | ) | ||||||||
| 處置 | ||||||||||||
| 匯兑差異 | ||||||||||||
| 2023年12月31日 | ( | ) | ( | ) | ||||||||
| 賬面淨值 | ||||||||||||
| 2022年12月31日 | ||||||||||||
| 2023年12月31日 | ||||||||||||
F-24
2.
| 建築物 | 汽車 | 總計 | ||||||||||
| 成本 | (歐元) | |||||||||||
| 2022年1月1日 | ||||||||||||
| 加法 | ||||||||||||
| 匯兑差異 | ||||||||||||
| 2022年12月31日 | ||||||||||||
| 加法 | ||||||||||||
| 匯兑差異 | ( | ) | ( | ) | ||||||||
| 2023年12月31日 | ||||||||||||
| 累計折舊 | ||||||||||||
| 2022年1月1日 | ( | ) | ( | ) | ( | ) | ||||||
| 當年的折舊費用 | ( | ) | ( | ) | ( | ) | ||||||
| 匯兑差異 | ( | ) | ( | ) | ||||||||
| 2022年12月31日 | ( | ) | ( | ) | ( | ) | ||||||
| 當年的折舊費用 | ( | ) | ( | ) | ( | ) | ||||||
| 匯兑差異 | ||||||||||||
| 2023年12月31日 | ( | ) | ( | ) | ( | ) | ||||||
| 賬面淨值 | ||||||||||||
| 2022年12月31日 | ||||||||||||
| 2023年12月31日 | ||||||||||||
3.
| 採購 IT軟件 | 預付款 支付 軟件 | 總計 | ||||||||||
| 成本 | (歐元) | |||||||||||
| 2022年1月1日 | ||||||||||||
| 加法 | ||||||||||||
| 匯兑差異 | ||||||||||||
| 2022年12月31日 | ||||||||||||
| 加法 | ||||||||||||
| 處置 | ( | ) | ( | ) | ||||||||
| 匯兑差異 | ( | ) | ( | ) | ||||||||
| 2023年12月31日 | ||||||||||||
| 累計攤銷 | ||||||||||||
| 2022年1月1日 | ( | ) | ( | ) | ||||||||
| 本年攤銷費用 * | ( | ) | ( | ) | ||||||||
| 匯兑差異 | ( | ) | ( | ) | ||||||||
| 2022年12月31日 | ( | ) | ( | ) | ||||||||
| 本年度攤銷費用 | ( | ) | ( | ) | ||||||||
| 處置 | ||||||||||||
| 匯兑差異 | ||||||||||||
| 2023年12月31日 | ( | ) | ( | ) | ||||||||
| 賬面淨值 | ||||||||||||
| 2022年12月31日 | ||||||||||||
| 2023年12月31日 | ||||||||||||
無形資產的攤銷
計入"研發費用"行項目(2023年:歐元
F-25
4.租契
租賃 義務包括根據主要與公司辦公空間租賃有關的不可撤銷租賃協議支付的款項。公司場地的租賃期限如下:2025年12月德國耶拿,2027年5月德國Martinsried,2026年4月美國密歇根州安阿伯。
| 租賃負債 | 2023 | 2022 | ||||||
| (歐元) | ||||||||
| 截至1月1日 | ||||||||
| 加法 | ||||||||
| 不再認識 | ( | ) | ( | ) | ||||
| 付款 | ( | ) | ( | ) | ||||
| 應計利息支出短期負債 | ( | ) | ||||||
| 外匯差價 | ( | ) | ||||||
| 截至12月31日 | ||||||||
| 2023 | 2022 | 2021 | ||||||||||
| (歐元) | ||||||||||||
| 使用權資產折舊費用(見附註E. 2.) | ||||||||||||
| 租賃負債利息支出 | ||||||||||||
| 租賃租金支出 | ||||||||||||
| 其中短期租賃(計入行政費用)。 | ||||||||||||
| 其中低價值資產租賃(計入行政費用)。 | ||||||||||||
| 在損益中確認的總金額 | ||||||||||||
本集團的租賃現金流出總額為歐元
5.
| 2023 | 2022 | 2021 | ||||||||||
| (歐元) | ||||||||||||
| 原材料和供應品 | ||||||||||||
| 未完成的產品 | ||||||||||||
| 成品 | ||||||||||||
| 總計 | ||||||||||||
公司的庫存包括與GOHIBIC(vilobelimab)相關的材料,這些材料主要是在2023年4月初在美國市場上根據EUA生產的
(參見附註A.2)。截至2023年12月31日止年度,存貨減記為歐元
F-26
| 6. |
| 2023年12月31日 | 2022年12月31日 | | ||||||
| (歐元) | ||||||||
| 非流動其他資產 | — | — | ||||||
| 預付費用 | ||||||||
| 總計 | ||||||||
| 流動其他資產 | ||||||||
| 研發項目預付款 | ||||||||
| 預付費用 | ||||||||
| 其他 | ||||||||
| 總計 | ||||||||
| 其他資產總額 | ||||||||
研發項目預付款 包括CRO和製造合同預付款。預付費用主要包括預付保險費用。
| 7. | 所得税所得税對賬 |
| InflaRx集團 | 2023 | 2022 | 2021 | |||||||||
| (歐元) | ||||||||||||
| 本期間虧損(除所得税前會計溢利) | ( | ) | ( | ) | ( | ) | ||||||
| 税率 | % | % | % | |||||||||
| 按税率計算的税收優惠 | ||||||||||||
| 未確認遞延税項資產的暫時性差異和税務虧損 | ( | ) | ( | ) | ( | ) | ||||||
| 不確認以股份為基礎的付款的税務影響 | ( | ) | ( | ) | ( | ) | ||||||
| 税務方面的不可扣除費用 | ( | ) | ( | ) | ( | ) | ||||||
| 因税率而產生的其他差異 | ( | ) | ||||||||||
| 所得税 | ||||||||||||
上述適用的税率代表德國和美國法定税率的加權平均值。在德國,InflaRx N.V.及其子公司InflaRx GmbH須繳納企業所得税(2023/2022/2021:
F-27
| a) | 税項虧損結轉 |
本集團的總税務損失結轉為
€
截至2023年12月31日,本集團擁有歐元
| ● | 截至2017年12月31日的InflRx GmbH的税收損失(歐元 |
| ● | 此外,集團仍有税項虧損結轉#美元。 |
截至2023年12月31日、2022年12月31日和2021年12月31日,沒有任何遞延 税項資產被確認為未使用税項損失的結轉。
| b) | 應收當期所得税 |
應收當期所得税包括因本集團就金融資產賺取的利息收入預扣所得税而提出的税務申索
(2023:歐元
| 8. | 金融資產和金融負債 |
| 金融資產和金融負債 | 12月31日, 2023 | 12月31日, 2022 | ||||||
| (歐元) | ||||||||
| 按攤銷成本計算的金融資產 | ||||||||
| 非流動金融資產 | ||||||||
| 來自政府補助金的金融資產 | ||||||||
| 其他流動金融資產 | ||||||||
| 按攤銷成本計算的財務負債 | ||||||||
| 來自政府贈款的負債 | ||||||||
| 貿易和其他應付款 | ||||||||
流動和非流動金融資產
的公允價值為歐元
截至2023年12月
31日持有的所有證券的到期日為1至17個月(2022年:1至16個月);它們的名義固定利息範圍為
截至2023年12月31日,來自政府補助金的金融資產和
負債總額為歐元, 由於2023年6月30日授予期結束(歐元
F-28
| 9. |
| 12月31日, 2023 | 12月31日, 2022 | |||||||
| (歐元) | ||||||||
| 短期存款 | — | — | ||||||
| 美元存款 | ||||||||
| 歐元存款 | ||||||||
| 總計 | ||||||||
| 銀行裏的現金 | ||||||||
| 持有美元現金 | ||||||||
| 歐元持有現金 | ||||||||
| 總計 | ||||||||
| 現金和現金等價物合計 | ||||||||
| 10. | 權益 |
| a) | 已發行資本 |
截至2023年12月31日,
本公司已發行股本分為
2020年7月8日,本公司向美國證券交易委員會(“SEC”)提交了關於本公司證券要約和出售的
F—3表格(2020—登記聲明)。該公司還向美國證券交易委員會提交了一份招股説明書補充,其中涉及一項上市計劃,該計劃規定出售最多美元,
截至2022年12月31日,公司已發行
通過2023年4月的承銷公開發行,本公司出售和發行了
關於於2022年12月21日修訂與Staidson(北京)生物製藥有限公司(“Staidson”)的共同開發協議,本公司與Staidson訂立股份購買協議,根據該協議,Staidson購買本公司普通股,總金額為
$
2023年,本公司共發行
F-29
| b) | 法定資本 |
根據
公司章程,
為了阻止收購出價,公司
股東大會批准了荷蘭法律規定的獨立基金會或保護性基金會的權利,根據認購期權協議行使
認購期權,在此基礎上,公司將向保護性基金會
發行優先股,
這些優先股將具有清算 和股息優先權優於公司普通股,並將按預先確定的比率累計現金股息。保護性的 基礎預計將要求公司註銷其優先股,一旦對公司及其利益相關者的感知威脅已經消除或充分減輕或消除 。本公司認為,認購期權並不代表基於第3級估值的重大 公允價值,因為優先股在使用上受到限制,可以被本公司註銷。
截至2023年12月31日止年度,本公司
支出歐元
| c) | 股本儲備的性質和用途 |
除已發行股本外,本公司 披露以下其他儲備:
| ● | 股票溢價記錄發行普通股時支付的金額超過面值歐元 |
| ● | 這個其他資本儲備包括髮行購股權所產生的開支。 |
| ● | 累計赤字包括以往報告期的損失。 |
股本的其他組成部分僅包括 從轉換成外幣的財務報表中獲得的貨幣儲備。
| 11. |
| 12月31日, 2023 | 12月31日, 2022 | |||||||
| (歐元) | ||||||||
| 研發項目應計負債 | ||||||||
| 商業活動應計負債 | ||||||||
| 應付帳款 | ||||||||
| 其他應計負債和應付款項 | ||||||||
| 貿易和其他應付款項總額 | ||||||||
研發項目的應計負債包括 截至報告日期尚未向公司開具發票的公司正在進行的項目的服務。
商業活動的應計負債 包括截至報告日期尚未向公司開具發票的商業製造合作伙伴提供的服務。
其他應計負債和應付款項包括
根據所有權分銷模式從我們的分銷合作伙伴處收到的GOHIBIC(vilobelimab)付款,收入將
在最終銷售和交付給醫院客户時確認。這些應計負債付款額為歐元
F-30
| 12. | 金融風險管理 |
| a) | 金融風險管理目標和政策 |
本集團的財務風險主要 由董事會於2022年11月3日批准並於2023年10月27日修訂的投資政策下的中央財資活動控制。這些財務活動識別、評估和管理符合集團經營 需要的財務風險。董事會提供全面風險管理政策,涵蓋特定領域,如外匯風險和 信貸風險。本公司不打算使用衍生金融工具,因為無法 可靠地預測本集團的未來風險(業務活動量、流動性需求、外匯風險)。
不採用套期保值,因為大多數業務 活動都打算以美元執行,並以公開發行中籌集的美元資金支付。以歐元以外的貨幣產生的成本的外匯風險 被視為不重要。
本集團的主要金融資產 包括具有高信貸評級的報價債務證券。除該等金融資產外,本集團擁有大量現金及現金等價物。 本集團的主要金融負債包括貿易及其他應付款項。該等金融資產、現金╱現金等價物及負債的主要目的是為本集團的發展活動提供資金。
| 暴露 | 量測 | 風險管理 | |
| 市場風險 | 實現自然對衝 在未來 | ||
| 信用風險 | 現金和現金等價物, 流動及非流動金融資產 |
信用 評級 |
銀行存款多樣化、投資準則 債務投資 |
| 流動性 | 滾動 現金流量預測 |
| b) | 市場風險 |
市場風險是指市場 價格變動的風險(例如,由於外匯匯率的原因)將影響集團的收入、支出或持有的金融工具的價值。市場風險管理的目標是識別、管理及控制市場風險,並在可接受的參數內。
Foreign exchange risk arises when commercial transactions or recognized assets or liabilities are denominated in a currency that is not an entity’s functional currency. The Group is exposed to transactional foreign currency risk to the extent that there is a mismatch between the currencies in which costs and purchases are denominated and the respective functional currencies of Group companies. The functional currencies of Group companies are primarily the Euro and U.S. dollars. The currencies in which these transactions and financial assets are primarily denominated are Euro and U.S. dollars. The Group is exposed to the exchange rate between the Euro and the U.S. dollars. Due to the Company’s various registered offerings of ordinary shares in U.S. dollars, the Group has significant cash and cash equivalents in U.S. dollars. Currently the Group does not hedge U.S. dollars but intends to achieve a natural hedge by contracting suppliers in U.S. dollars in the future. In 2023, the Group recognized significant foreign exchange gains and losses as the natural hedge is not yet achieved and the functional currency for InflaRx N.V. and InflaRx GmbH is Euro.
本集團主要面臨 美元兑歐元匯率變動的風險。損益對匯率變動的敏感度主要來自InflaRx N.V.和InflaRx GmbH以美元 計值的金融工具。
F-31
| 以美元計值的現金、現金等價物和金融資產,InflaRx N.V.和InflaRx GmbH | 12月31日, 2023 | 12月31日, 2022 * | ||||||
| (歐元) | ||||||||
| 流動及非流動金融資產(證券及應計利息) | ||||||||
| 現金和現金等價物 | ||||||||
| 面臨風險的資產總額 | ||||||||
| 報告日期歐元兑美元的匯率1/1.1050 | ||||||||
| * |
| 敏感性分析: | 轉換 費率 | 利潤/(虧損) | 攜載 金額 | |||||||||
| (歐元) | ||||||||||||
| 歐元兑美元走軟 | ||||||||||||
| 歐元兑美元走強 | ( | ) | ||||||||||
根據
過去三年的匯率波動,公司預計歐元對美元的匯率波動
| c) | 信用風險 |
信貸風險是指交易對手 不履行其義務導致公司財務損失的風險。本公司面臨的信貸風險主要來自其融資活動,包括銀行和金融機構存款、外匯交易和其他金融工具。
本公司根據本公司的投資政策管理銀行和金融機構餘額的信貸風險。 目前未用於資助研發或G & A活動的財務資源投資僅在 投資政策批准的信貸限額內與交易對手進行。對於歐元或美元債務證券的投資,需要BBB+至AAA的信用評級(標準普爾和惠譽 評級;或穆迪和DBRS的同等評級)。投資政策不允許以歐元或美元以外的貨幣計價的複雜金融產品以及其他投資 。交易對手信貸限額和投資 政策每年與公司審計委員會討論,並可在獲得公司審計委員會批准的情況下全年更新 。設置限額是為了最大限度地減少風險集中,從而減少交易對手可能無法支付的財務損失 。
交易對手信貸風險的最大風險
為€
| d) | 流動性風險 |
公司在每個季度預測中以及持續的基礎上監控其 資金短缺的風險。本公司在附註E“承諾”項下披露了其主要負債的到期日。審慎的流動性風險管理涉及維持充足的現金和有價證券,以及 有資金可供償還到期債務。本集團使用短期和 中期流動性計劃持續監控資金短缺風險。這考慮到所有活動的預期現金流量。管理團隊對預算進行定期 審查。
本公司有重大經營損失的歷史。管理層預計,公司在可預見的將來將產生重大且不斷增加的虧損;由於公司可能無法 在不久的將來實現或維持盈利能力,因此其依賴於出資或其他資金。
F-32
本集團從各種 登記發行中籌集了大量資金,估計這些資金將使本集團能夠為自2023年12月31日起至少 24個月的運營費用和資本支出需求提供資金。本集團預期將需要額外資金以繼續推進候選產品的開發。 如果獲得監管部門的批准,並且公司實施了將產品商業化的戰略,集團將需要 額外的資本。
2023年,由於BMBF協議(見
附註C.6)。公司收到€
| 流動性 | 12月31日, 2023 | 12月31日, 2022 | ||||||
| (歐元) | ||||||||
| 短期存款 | ||||||||
| 銀行裏的現金 | ||||||||
| 有價證券(流動和非流動) | ||||||||
| 其他(非流動部分) | ||||||||
| 其他(當前) | ||||||||
| 可動用資金總額 | ||||||||
| 13. | 資本管理 |
本集團的資本管理政策 是確保其保持流動性,以便為其經營活動、未來業務發展提供資金,並在到期時償還債務 。本集團主要透過權益管理其資本架構。本集團並無任何金融負債, 貿易及其他應付款項或租賃負債除外。
年內,資本管理的目標、政策 或流程均未發生任何變更。
| E. | 承付款 |
| 1. | 業務合同或服務 |
本集團在正常的 業務過程中與CRO和臨牀研究中心簽訂合同,以進行臨牀試驗,與專業顧問提供專家諮詢,以及與其他 供應商簽訂合同,提供臨牀供應品製造或其他服務。這些合同通常可提前30至180天通知終止。 除此最短期限外,這些合同還要求為已經提供的服務支付全額費用。
於2023年,本集團並無任何購買物業、廠房及設備或專利及商標的承諾 (分別 2022年)。
| 2. | 租賃義務 |
| 2023年資本化租賃到期日分析 | 合同 最低要求 租賃 義務 | 的效果 打折 | 租賃負債 | |||||||||
| (歐元) | ||||||||||||
| 一年內 | ||||||||||||
| 一年後但不超過五年 | ||||||||||||
| 五年多 | ||||||||||||
| 總計 | ||||||||||||
F-33
| 2022年資本化租賃到期日分析 | 合同 最低要求 租賃 義務 | 的效果 打折 | 租賃 負債 | |||||||||
| (歐元) | ||||||||||||
| 一年內 | ||||||||||||
| 一年後但不超過五年 | ||||||||||||
| 五年多 | ||||||||||||
| 總計 | ||||||||||||
| 2023年所有租賃債務到期日分析 | 總計 | 低值 租契 | 短期 租賃 | 大寫 租賃 | ||||||||||||
| 一年內 | ||||||||||||||||
| 一年後但不超過五年 | ||||||||||||||||
| 五年多 | ||||||||||||||||
| 總計 | ||||||||||||||||
| 2022年所有租賃責任到期日分析 | 總計 | 低值 租賃 | 短期 租賃 | 大寫 租賃 | ||||||||||||
| (歐元) | ||||||||||||||||
| 一年內 | ||||||||||||||||
| 一年後但不超過五年 | ||||||||||||||||
| 五年多 | ||||||||||||||||
| 總計 | ||||||||||||||||
預計未來租賃費用按截至2023年12月31日的匯率換算
,
本集團應用“租賃低價值資產”確認豁免。本集團亦就到期 少於12個月的租賃應用“短期租賃”豁免。
| F. | 其他信息 |
| 1. | 細分市場報告 |
該集團主要作為一家專注於研發的生物製藥公司運營,應用其專有的抗C5a和C5aR技術開發針對高醫療需求疾病的新型治療產品。然而,自2023年4月其用於治療重症COVID—19患者的主導產品vilobelimab獲得EUA以來,該公司也圍繞GOHIBIC(vilobelimab)在美國的銷售和營銷開展商業活動。 集團不受細分市場的支配。董事會為主要經營決策者。資源管理和向決策者報告 基於集團整體。
所有業務活動均在德國
和美國進行。收入數額為美元
| ● | 2023年12月31日:€ |
| ● | 2022年12月31日:€ |
所有非流動資產均不在公司註冊所在國家 (荷蘭)。
F-34
| 2. | 關聯方交易 |
| 行政人員和董事會薪酬 | 2023 | 2022 | 2021 | |||||||||
| (歐元) | ||||||||||||
| 執行管理 | ||||||||||||
| 短期僱員福利 | ||||||||||||
| 基於股份的支付 | ||||||||||||
| 小計 | ||||||||||||
| 非執行董事會成員 | ||||||||||||
| 短期僱員福利 | ||||||||||||
| 基於股份的支付 | ||||||||||||
| 小計 | ||||||||||||
| 全額補償 | ||||||||||||
執行管理層包括董事會執行董事 和本公司高級管理層成員。
上表披露了董事會和執行管理層根據合同商定的短期僱員福利。截至2023年12月31日,歐元
本集團行政管理層的薪酬 包括固定和可變部分以及以股份為基礎的薪酬獎勵。此外,高級管理人員還可獲得補充福利 和津貼。
公司與其董事和高級管理層簽訂了賠償協議 。賠償協議和公司的組織章程要求公司 在法律允許的最大範圍內賠償其董事。
公司現任和未來董事 (以及董事會指定的其他高級管理人員或僱員)享有InflaRx N.V.的《章程》 中的賠償條款。這些條款賦予索賠人向公司收回款項的權利,包括 但不限於訴訟費用,以及他們被命令支付的任何損害賠償,與履行職責時的作為或不作為有關。然而,對於被認為構成惡意、嚴重過失、故意魯莽和/或嚴重罪責的行為或不行為,則無權要求賠償。這些協議還規定, 除某些例外情況外,賠償相關費用,其中包括律師費、判決、罰款、 罰款和任何這些個人在任何訴訟或訴訟中所產生的和解金額。除此類賠償外, 公司還為其董事提供董事和高級管理人員責任保險。
| G. | 報告日期後的重大事項 |
沒有。
F-35