目錄表
已實現,以促進PAX-101用於某些神經學適應症的加速開發計劃,包括ASD、ME/CFS和LCS。根據我們在2021年3月與FDA舉行的IND前會議,以及部分基於我們從衞生部、馬拉維共和國和Lwala醫院(烏幹達索洛蒂)獲得的關於使用蘇拉明治療東非HAT患者的獨家許可數據的分析,我們相信我們已經制定了一項強大的發展戰略,我們計劃在尋求批准PAX-101用於治療東非HAT時採用該戰略。根據我們之前與FDA的互動,包括我們與FDA的Pre-IND和Type B會議,我們進一步認為,如果東非HAT獲得批准,我們可能會獲得FDA的優先審查憑證(PRV),我們可能會將其貨幣化,為我們未來的臨牀計劃提供資金。我們預計將需要對PAX-101進行進一步的臨牀研究,以治療ASD、FXS、FXTAS和ME/CFS,並需要類似的臨牀開發才能使PAX-102達到商業階段。2020年11月,FDA批准PAX-101為治療東非HAT的孤兒藥物。然而,不能保證我們將獲得FDA對用於治療東非HAT的PAX-101的批准,即使PAX-101獲得FDA的批准,也不能保證我們將獲得PRV。有關PRV過程以及我們如何從中受益的更多信息,請參閲本年度報告中Form 10-K的部分。“第1項。商業-政府監管 - 優先審查代金券計劃。”
開發管道
下表總結了我們目前的候選產品和適應症渠道。
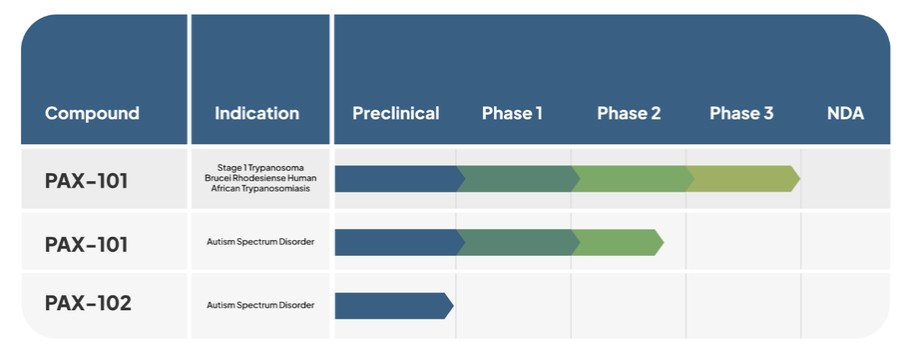
PAX-101(靜脈注射蘇拉明)
我們正在開發的主要候選產品是PAX-101,這是一種蘇拉明的靜脈製劑,我們正在開發用於多種適應症的PAX-101,包括東非HAT、ASD和ME/CFS。
我們正在開發PAX-101的最先進的適應症是用於“第一階段”的治療。布氏錐蟲(東非)HAT,HAT臨牀病程的階段,在外周循環中發現寄生蟲,但尚未滲透到中樞神經系統。我們擁有在全球範圍內獨家授權使用蘇拉明治療1期東非HAT的患者級別數據的權利,我們打算利用這些數據來證明PAX-101的安全性和有效性。我們已經就PAX-101的實施和開發與FDA舉行了三次正式會議。根據這些對話,並滿足FDA的要求,證明瞭實質性的有效性,我們進行了新的前瞻性臨牀試驗。2023年7月,我們完成了對2000年至2020年期間接受蘇拉明治療的東非HAT患者的回顧數據的分析和展示,我們獲得了獨家許可。除了這些回顧性數據,我們還將完成臨牀前和臨牀安全性研究,以支持提交PAX-101‘S東非HAT適應症的NDA。我們預計這項工作將在未來10個月內完成,打算在2024年下半年提交保密協議。此外,我們正在完成藥物物質和藥物產品專有供應鏈的開發,這將成為我們提交保密協議的基礎。如果不建立這一供應鏈,我們將無法提交東非HAT標誌的保密協議。有關我們建立供應鏈的預期時間表的其他信息,請參閲“製造”。2020年11月,FDA批准PAX-101為治療東非HAT的孤兒藥物。預計PAX-101如果被FDA批准用於東非HAT適應症,將有資格獲得新的化學實體獨家經營權(向該公司提供關於任何含有蘇拉明的產品在美國的獨家營銷權,最長可達七年),此外還有孤兒藥物獨家經營權,
4