美國
美國證券交易委員會
華盛頓特區,20549
表格10-K
| |
(馬克·奧內爾) | |
☒ | 根據1934年頒佈的《證券交易法》第13或15(D)節提交的年度報告 |
截至本財政年度止2023年12月31日 |
或 |
☐ | 根據1934年《證券交易法》第13或15(D)節提交的過渡報告 |
委員會文件編號:000-29889
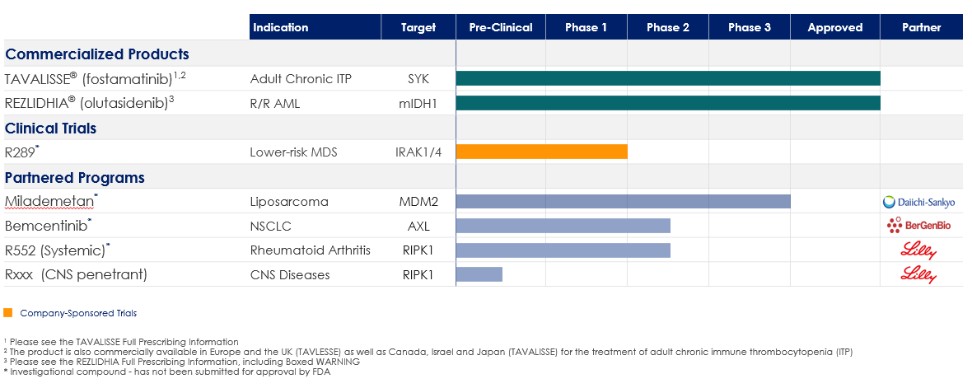
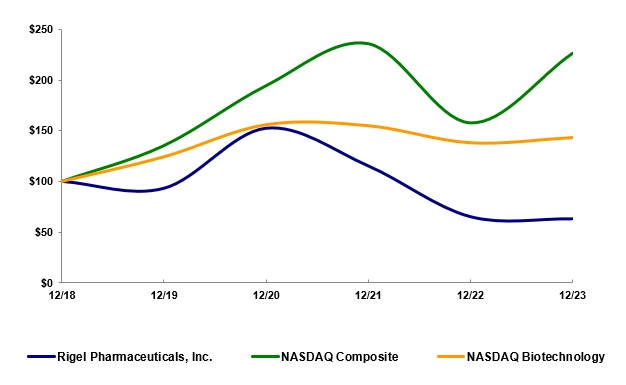
Rigel製藥公司。
(註冊人的確切姓名載於其章程)
| |
特拉華州(述明或其他司法管轄權
公司或組織) | 94-3248524(美國國税局僱主
識別號碼) |
門户大道611號,900號套房,
南舊金山, 加利福尼亞(主要執行辦公室地址) | 94080(郵政編碼) |
(650) 624-1100
(註冊人的電話號碼,包括區號)
根據該法第12(B)節登記的證券:
| | |
每節課的題目: | 交易代碼 | 在其註冊的每個交易所的名稱: |
普通股,每股票面價值.001美元 | 右上角 | 這個納斯達克股市有限責任公司 |
根據該法第12(G)節登記的證券:無
用複選標記表示註冊人是否為證券法規則第405條所定義的知名經驗豐富的發行人。☐ 不是 ☒
如果註冊人不需要根據該法第13節或第15(D)節提交報告,請用複選標記表示是。☐ 不是 ☒
用複選標記表示註冊人(1)是否在過去12個月內(或註冊人被要求提交此類報告的較短期限內)提交了1934年《證券交易法》第13或15(D)節要求提交的所有報告,以及(2)在過去90天內是否符合此類提交要求。是 ☒不是的。☐
用複選標記表示註冊人是否已在過去12個月內(或在註冊人被要求提交此類文件的較短時間內)以電子方式提交了根據S-T規則第405條(本章232.405節)要求提交的每個交互數據文件。是 ☒不是的。☐
用複選標記表示註冊人是大型加速申報公司、加速申報公司、非加速申報公司、較小的報告公司或新興成長型公司。請參閲《交易法》第12b-2條規則中“大型加速申報公司”、“加速申報公司”、“較小申報公司”和“新興成長型公司”的定義。
| | | |
大型加速文件服務器☐ | 加速文件管理器 ☒ | 非加速文件服務器☐ | 規模較小的報告公司☐ |
| | | 新興成長型公司☐ |
如果是一家新興的成長型公司,用複選標記表示註冊人是否已選擇不使用延長的過渡期來遵守根據《交易所法》第13(A)節提供的任何新的或修訂的財務會計準則。☐
用複選標記表示註冊人是否提交了一份報告,證明其管理層根據《薩班斯-奧克斯利法案》(《美國聯邦法典》第15編,第7262(B)節)第404(B)條對其財務報告的內部控制的有效性進行了評估,該評估是由編制或發佈其審計報告的註冊會計師事務所進行的。☒
如果證券是根據該法第12(B)條登記的,應用複選標記表示登記人的財務報表是否反映了對以前發佈的財務報表的錯誤更正。☐
用複選標記表示這些錯誤更正中是否有任何重述需要對註冊人的任何執行人員在相關恢復期間根據第240.10D-1(B)條收到的基於激勵的補償進行恢復分析。☐
用複選標記表示註冊人是否是空殼公司(如該法規則第12b-2條所定義)。☐不是的。☒
根據登記人的普通股在2023年6月30日,也就是登記人最近完成的第二財季的最後一個工作日在納斯達克全球精選上報告的收盤價,登記人的非關聯公司持有的普通股的大約總市值為$222.7百萬美元。每位高管、董事和關聯公司持有的註冊人已發行普通股的股票已被排除在外。就本計算而言,對附屬公司地位的確定不一定是對其他目的的決定性確定。
截至2024年2月28日,有175,377,812註冊人已發行普通股的股份。
以引用方式併入的文件
本10-K年度報告第III部分第10、11、12、13和14項引用了註冊人2024年股東年會的最終委託書中的信息,該最終委託書將於本10-K年度報告所涵蓋的財政年度結束後120天內按照第14A條提交給美國證券交易委員會(SEC)。.