
美國 美國
證券交易委員會
華盛頓特區,20549
表格
(標記 一)
| 根據1934年《證券交易法》第13或15(D)節提交的年度報告 |
對於
截止的財政年度
或
| 根據1934年《證券交易法》第13或15(D)條提交的過渡報告 |
對於 從過去到現在、從現在到現在的過渡期
佣金
文件編號:
(註冊人的確切名稱與其章程中規定的名稱相同)
(州或其他司法管轄區 公司(br}或組織) |
(I.R.S.僱主 標識 編號) |
| (主要執行辦公室地址 ) | (郵政編碼) |
註冊人的電話號碼,包括區號:
根據該法第12(B)條登記的證券:
| 每個班級的標題 | 交易 個符號 | 註冊的每個交易所的名稱 | ||
| 這個 |
根據該法第12(G)條登記的證券:無
如果登記人是《證券法》第405條規定的知名的經驗豐富的發行人,則應進行檢查。
是
如果註冊人不需要根據該法第13條或第15(D)條提交報告,請用勾號表示
。是的☐
用複選標記表示註冊人(1)是否在過去12個月內(或註冊人被要求提交此類報告的較短期限內)提交了1934年《證券交易法》第13或15(D)節要求提交的所有報告,以及(2)
在過去90天內是否符合此類提交要求。
用複選標記表示註冊人是否在過去12個月內(或在註冊人被要求提交此類文件的較短時間內)以電子方式提交了根據S-T規則(本章232.405節)第405條要求提交的每個交互數據文件。
用複選標記表示註冊人是大型加速申報公司、加速申報公司、非加速申報公司、較小的申報公司或新興成長型公司。請參閲《交易法》第12b-2條規則中“大型加速申報公司”、“加速申報公司”、“較小申報公司”和“新興成長型公司”的定義。
| 大型加速文件服務器 | ☐ | 加速文件管理器 | ☐ |
| ☒ | 規模較小的報告公司 | ||
| 新興成長型公司 |
如果 是一家新興成長型公司,請用複選標記表示註冊人是否已選擇不使用延長的過渡期來遵守根據《交易法》第13(A)節提供的任何新的或修訂的財務會計準則。☐
用複選標記表示註冊人是否提交了一份報告,並證明其管理層根據《薩班斯-奧克斯利法案》(《美國法典》第15編第7262(B)節)第404(B)條對其財務報告內部控制的有效性進行了評估
編制或發佈其審計報告的註冊會計師事務所。
如果證券是根據該法第12(B)條登記的,請用複選標記表示備案文件中包括的註冊人的財務報表是否反映了對以前發佈的財務報表的錯誤更正。
如果註冊人是空殼公司(如該法第12b-2條所界定),則用勾號表示
。是的☐
註冊人的非關聯公司持有的有投票權和無投票權普通股的總市值為#美元。
表明 截至2023年12月11日,註冊人的每一類普通股的流通股數量:
| 班級 | 股份數量: | |
| 普通股,面值0.0001美元 |
通過引用併入的文檔
目錄表
| 第 部分I | 1. | 業務 | 4 |
| 1A. | 風險因素 | 34 | |
| 1B. | 未解決的員工意見 | 75 | |
| 2. | 屬性 | 75 | |
| 3. | 法律訴訟 | 75 | |
| 4. | 煤礦安全信息披露 | 75 | |
| 第 第二部分 | 5. | 註冊人普通股市場、相關股東事項與發行人購買股權證券 | 76 |
| 7. | 管理層對財務狀況和經營成果的探討與分析 | 77 | |
| 7A. | 關於市場風險的定量和定性披露 | 88 | |
| 8. | 財務報表和補充數據 | 89 | |
| 9. | 會計與財務信息披露的變更與分歧 | 114 | |
| 9A. | 控制和程序 | 114 | |
| 9B. | 其他信息 | 114 | |
| 第 第三部分 | 10. | 董事、高管與公司治理 | 115 |
| 11. | 高管薪酬 | 119 | |
| 12. | 某些實益擁有人的擔保所有權以及管理層和相關股東的事項 | 123 | |
| 13. | 某些關係和相關交易,以及董事的獨立性 | 125 | |
| 14. | 首席會計費及服務 | 126 | |
| 第四部分 | 15. | 展示、財務報表明細表 | 127 |
| 16. | 表格10-K摘要 | 127 |
| 2 |
有關前瞻性陳述的特別説明
本報告包含1995年《私人證券訴訟改革法》所指的前瞻性陳述。前瞻性 陳述包括所有非歷史事實的陳述。在某些情況下,您可以使用術語 來識別前瞻性陳述,例如“可能”、“將”、“應該”、“可能”、“將”、“預期”、“ ”、“計劃”、“預期”、“相信”、“估計”、“項目”、“預測”、“潛在”或這些術語的負面含義,以及旨在識別前瞻性陳述的類似表述和類似術語。這些陳述反映了公司對未來事件的當前看法,是基於假設和受風險和不確定因素影響的,這些風險和不確定因素包括本10-K年度報告中第I部分第1A項“風險因素”中所述的風險和不確定因素。鑑於這些不確定性,您不應過度依賴這些前瞻性陳述。這些前瞻性 陳述僅代表公司截至本10-K年度報告之日的估計和假設,除法律規定的 外,公司沒有義務因本年度報告以10-K形式發佈之後的新信息、未來事件或其他原因而公開更新或審查任何前瞻性陳述。您應閲讀此Form 10-K年度報告和此Form 10-K年度報告中引用並作為附件存檔的文件,並瞭解公司未來的實際結果可能與公司預期的大不相同。本公司通過這些警告性聲明對其所有 前瞻性聲明進行限定。此類陳述可能包括但不限於以下有關 的陳述:
● 我們缺乏經營歷史和經營虧損歷史;
● 我們對大量額外資本的需求以及我們滿足資本需求的能力;
● 我們完成產品所需臨牀試驗並獲得FDA或不同 司法管轄區其他監管機構批准的能力;
● 我們維持或保護我們的專利和其他知識產權的有效性的能力;
● 我們留住關鍵執行成員的能力;
● 我們內部開發新發明和知識產權的能力;
● 對現行法律的解釋和未來法律的通過;
● 投資者接受我們的商業模式;
● 我們對費用和資本要求的估計的準確性;以及
● 我們充分支持增長的能力。
| 3 |
第 部分I
第 項1.業務
概述
Sonnet BioTherapeutics Holdings,Inc.,是一家臨牀階段的腫瘤生物技術公司,擁有一個專有平臺,用於創新單功能或雙功能生物藥物。被稱為FHAB™(全人血白蛋白結合),該技術利用 一種全人單鏈抗體片段,其結合人血清白蛋白(HSA)並在其上“搭便車”以轉運至 靶組織。我們設計了FHAB構建體以改善腫瘤中的藥物積累,以及延長體內 活性的持續時間。FHAB發育候選物在哺乳動物細胞培養物中產生,其能夠實現糖基化和 類似於天然細胞因子的生物結構 體內.我們相信我們的FHAB技術於二零二一年六月獲得 一項美國專利,是我們生物製藥平臺的顯著特色,非常適合未來在一系列人類疾病領域進行藥物開發 ,包括腫瘤學、自身免疫性、致病性、炎症和血液學病症。
我們目前的內部流水線開發活動專注於細胞因子,這是一類細胞信號肽,在其他重要功能中,它是有效的免疫調節劑。在獨立和協同作用下,特定的細胞因子已經顯示出調節免疫細胞的激活和成熟的能力,這些免疫細胞對抗癌症和病原體。然而,細胞因子本身並不會優先在特定的組織中積聚,很快就會從體內排出。通過細胞因子治療達到治療效果的傳統方法通常需要使用高劑量和頻繁劑量。這可能會導致治療效果降低,並伴隨着潛在的全身毒性,這對這類藥物的治療應用構成了挑戰。
十四行詩 構建了一個高效的研發平臺,其中包括外包供應商網絡,以幫助補救費用並改進執行時間 。大多數供應商都是戰略合作伙伴,為公司提供優先地位,並通過談判獲得成本。此方法的主要 優勢包括對項目的優化直接投資,費用可根據項目數量快速調整 。Sonnet平臺的成本優勢始於供應商網絡選擇過程,CMC是最初藥物開發步驟中最昂貴的組件之一。十四行詩已經選擇了CMC在印度的戰略合作伙伴,並通過談判將成本大大低於類似的美國或歐洲供應商所產生的費用。十四行詩正在澳大利亞進行該公司正在進行的三項臨牀試驗中的兩項,通過澳大利亞政府的研發税收抵免計劃,相對於美國的試驗,該試驗的成本大幅降低。十四行詩還與來自美國、英國、德國和瑞士的頂級研發供應商協調印度和澳大利亞的執行工作,目標是將該公司運營的 費用基礎設施的大部分用於其藥物開發管道。
管道
我們有一系列治療化合物,主要集中在高度未得到滿足的醫療需求的腫瘤學適應症上。
| ● | 我們的領先專利資產SON-1010是白介素12(IL-12)的全人類單鏈版本,與FHAB 結構,為此,我們正在進行實體腫瘤的臨牀開發。十四行詩已經完成了根據當前良好實驗室規範(CGLP)進行的非人類靈長類(NHP)毒性研究 ,併成功地生產了該藥物產品的液體和凍幹 臨牀使用。2022年3月,FDA批准了Sonnet的研究新藥(IND)申請 SON-1010。這使得該公司能夠在2022年第二個日曆季度啟動一項針對實體腫瘤腫瘤學患者的美國臨牀試驗(SB101)。2021年9月,公司成立了一家全資擁有的澳大利亞子公司SonnetBio Pty Ltd(“子公司”), 用於進行某些臨牀試驗。十四行詩獲得批准,並在2022年第三季度在澳大利亞健康志願者中啟動了一項關於SON-1010的臨牀研究(SB102)。SB101和SB102研究的臨時安全性和耐受性數據於2023年4月報告。2023年1月,Sonnet宣佈與羅氏達成合作協議,使用atezolizumab(Tecentriq)對SON-1010進行臨牀評估®)。這兩家公司已經簽訂了主臨牀試驗和供應協議(MCSA)以及輔助質量和安全協議,以研究SON-1010和阿替唑單抗在鉑類耐藥卵巢癌(PROC)患者環境中的安全性和有效性。此外,兩家公司將分別提供SON-1010和阿特唑珠單抗,用於1b期/2a期聯合安全性、劑量遞增和療效研究(SB221)。該試驗包括第1部分中修改的3+3劑量遞增設計,以確定具有固定劑量的SON-1010的最大耐受劑量(MTD)。PROC的臨牀益處將在一個擴展組中得到確認,以確定推薦的第二階段劑量(RP2D)。這項研究的第二部分隨後將通過隨機比較研究SON-1010單一療法或將其與阿替唑單抗和 PROC的護理標準(SOC)聯合使用,以顯示概念驗證(POC)。作為該公司正在進行的 成本削減評估的一部分,已暫停所有SON-1010的抗病毒開發。 |
| 4 |
| ● | 公司於2020年4月獲得了白介素6(IL-6)完全人類版本的全球開發權。我們將此候選對象稱為SON-080,因為它的目標適應症是化療誘導的周圍神經病變(CIPN)和糖尿病周圍神經病變(DPN)。Sonnet的CIPN 1b/2a階段臨牀試驗SB221正在澳大利亞進行。SB211研究的第一部分的登記即將完成,這將使DSMB在2024年第一個日曆季度完成對初步安全性數據的審查。根據公司於2021年5月與新加坡新生命治療有限公司(“新生命”)簽訂的許可協議,十四行詩和新生命將共同負責開發DPN中的SON-080 ,目的是在分析CIPN安全數據後評估一項前美國試點療效研究。 | |
| ● | SON-1210(IL12-FHAB-IL15),我們的先導雙功能化合物,將FH用單鏈IL-12和完全的人白介素15(IL-15)構建AB。這種化合物正在開發用於實體腫瘤的適應症,包括結直腸癌。2023年2月,該公司宣佈在NHP中成功完成了兩項啟用IND的SON-1210毒理學研究。十四行詩已準備好 啟動SON-1210的監管授權流程,等待任何合作活動的結果。 |
在我們的發現渠道中,我們正在調查:
| ● | SON-1410(IL18-FHAB-IL12),白介素18(IL-18)和IL-12的雙功能組合,用於治療黑色素瘤和腎癌。CELL 系列開發和工藝開發正在進行中,早期實驗藥物供應適合於配方和分析方法開發活動。經過2023年的一些開發延遲後,活動將持續到2024年,有可能產生一種適合於非臨牀研究和後續人體研究的藥物。 |
| ● | SON-3015(抗IL6-FHAB-抗轉化生長因子β)是一種抗白介素6和抗腫瘤生長因子β(β)的雙功能組合物,用於治療腫瘤和骨轉移。已生產出早期雙功能藥物,並已儲存以供將來使用體內老鼠研究。出於削減開支的目的,十四行詩選擇擱置SON-3015開發計劃。 |
在我們的治療化合物的開發和商業化方面,我們面臨着許多挑戰和不確定性,包括我們的FHAB技術。請參閲本招股説明書中其他部分包含的“風險因素”,以及通過引用併入本招股説明書的文件中標題為“風險因素”的章節。
戰略
我們的目標是快速推進我們的產品線,並利用我們的治療FHAB平臺成為生物藥物發現、開發和商業化的領導者。自成立以來,Sonnet一直專注於快速發展的管道候選人向 診所,同時也致力於建立與合適的合作伙伴的合作。隨着合作伙伴關係對話的發展,Sonnet打算 優先考慮具有最大戰略利益的資產的費用分配。為此,Sonnet在2023財年減少了運營費用,並打算談判一項許可協議,以幫助為未來的管道擴張提供資金。作為2022年10月宣佈的項目早期階段的一個例子,楊森對我們的三種管道化合物SON-1010、SON-1210和SON-1410及其細胞治療產品的評估仍在進行中。
FHAB 計劃進展:SON-1010已進入1b/2a期臨牀開發,以確定最大耐受劑量(MTD)並評估鉑類耐藥卵巢癌(PROC)的臨牀獲益。關於我們的第一個雙功能候選藥物SON-1210,已經成功完成了兩項在NHP中進行的IND啟動 毒理學研究,公司準備啟動監管授權程序, 等待任何合作活動的結果。
| 5 |
將 SON-080推進到下一階段的臨牀開發:SON-080是一種完全人用的低劑量IL-6,正在研究其用於化療誘導的 周圍神經病變(CIPN)。IL-6已在癌症患者的I期和II期臨牀試驗中成功進行研究,我們於2022年下半年在CIPN患者中開展了 一項初步療效1b/2a期研究。
製造 平臺:Sonnet化合物使用行業標準哺乳動物細胞(中國卵巢(CHO))宿主細胞系生產, 允許使用最先進的生產工藝和技術進行快速放大和商業生產。哺乳動物 細胞培養系統使糖基化和類似的生物結構的天然細胞因子 體內,這降低了免疫原性的 機會。用於臨牀應用的細胞因子的製造,即它們的生產和純化,提出了獨特的 技術挑戰。為此,Sonnet開發了一種專有的連續強化灌注生產工藝,包括 用於高效下游加工的專有配體以及穩定的凍乾製劑,我們正在為這些生產和下游工藝開發步驟尋求知識產權保護。
監管 策略:我們相信,Sonnet的候選藥物與現有療法有顯著差異,代表了生物製藥藥物開發的潛在突破。我們將努力尋求監管機構的突破性治療指定, 這可能會加快臨牀開發時間表。
管道 許可機會:我們正在尋求與領先的生物製藥公司合作的機會,以開發和 商業化我們的管道資產。
FHAB 技術擴展:Sonnet正在探索FHAB技術與有意擴大其 治療部署的外部合作伙伴進行了許可,我們相信這可能會導致該平臺在其他領域的應用,例如疫苗、抗體藥物 結合物,以及作為嵌合抗原受體(CAR)T細胞技術的補充 體內.一旦獲得支持性數據, 將提交臨時專利,以確保FHAB在這些領域
FHAB科技
我們的 專有FHAB技術被設計用於解決生物製藥 藥物開發的現有方法的幾個重要缺點。我們設計了FHAB結構域作為一種即插即用的模塊化構建體,用於創新新的化學實體, 易於重新配置用於不同的治療有效載荷。與所有生物藥物一樣,給藥的劑量水平和頻率 是關鍵變量,通常會對開發過程造成障礙。注射後,大分子治療劑,包括 肽、蛋白質、融合蛋白、抗體等,必須保持完整,並且能夠到達體內的指定靶點 ,而不超過特定的毒性閾值。最後,它們還必須使用具有商業吸引力的手段來生產。
十四行詩的平臺技術旨在將HSA作為一種治療性的穿梭分子。人血清白蛋白自然存在於血液中,是血漿中的主要蛋白質。白蛋白是發炎、高代謝組織的能量來源,包括腫瘤。由於對營養的需求旺盛,癌細胞過度表達白蛋白結合蛋白,如“富含半胱氨酸的酸性分泌蛋白”(SPARC)和gp60(白蛋白糖蛋白)。
根據2012年7月23日的發現合作協議和2019年5月7日的發現合作協議修正案(統稱為“合作協議”),XOMA(US)LLC(“XOMA”)向Sonnet授予非獨家、不可轉讓的 許可證和/或使用與發現、優化和開發 抗體和相關蛋白質有關的某些材料、技術和信息的權利,並據此開發和商業化產品(每個產品為“產品”)。合作協議包括使用全人噬菌體文庫的許可證,該文庫旨在產生完整的人類單鏈抗體 可變片段(ScFv),該片段包含完整的人類重鏈和輕鏈,用於為特定的 功能尋找生物序列。應用嚴格的標準,十四行詩將數百萬單鏈抗體結合到人血清白蛋白中以生成十四行詩的FHAB,它與人血清白蛋白(HSA)結合,是一種具有三個主要功能結構域的球狀蛋白。已知白蛋白結構域1和3參與了與FcRN的結合。這使得十四行詩能夠選擇和表徵特定於域2的單鏈抗體結合劑,這是十四行詩F的一個基本方面HAB站臺。
| 6 |
十四行詩 有義務在實現與產品相關的某些開發和審批里程碑時,按產品向XOMA支付總計375萬美元的或有里程碑付款。在這一點上,下一個預計為75萬美元的臨牀開發里程碑 預計將是開始註冊一種產品(即,Son-1010)在第二階段試驗中。十四行詩還同意為十四行詩銷售的產品的淨銷售額向XOMA支付較低的個位數版税。每種產品的版税應按國家/地區支付 ,直至(I)首次商業銷售(定義見合作協議)後十二(12)年和(Ii)合作協議涵蓋的已發佈專利的最後一項有效主張的最後到期日期兩者中較晚者為止。此外,十四行詩有權通過向XOMA支付指定金額來降低逐個產品的版税費率。 合作協議可由任何一方因故終止,幷包含慣常的賠償條款。
十四行詩的FHAB已顯示出跨物種(人、小鼠和食蟹猴)與血清白蛋白的高結合親和力,幾乎沒有免疫原性,並保留了新生兒FcRN介導的白蛋白循環延長血清半衰期的好處。與單抗不同,這種結合不會引起抗體依賴的細胞毒性(ADCC)或補體依賴的細胞毒性(CDC)。《F》HAB結構通過離子疏水機制物理結合血清白蛋白(圖1),我們認為這比依賴化學、共價結合的技術提供了明顯的優勢。共價鍵一旦斷裂,就不能改革,而十四行詩的FHAB能夠在動態平衡中與白蛋白結合、解除結合和重新結合。白蛋白還與白蛋白受體gp60和SPARC,F結合HAB利用先天的生物機制將靶向傳遞到腫瘤微環境並在腫瘤微環境中積累治療有效載荷。
臨牀前放射性標記研究已經驗證了F的腫瘤靶向性HAB結構,與沒有F的相同結構相比,腫瘤中有蓄積 HAB,不出所料,在肝臟、腎臟和其他器官中是一過性的。HAB在引流的淋巴結內也有可測量的蓄積。這些發現對任何單(ILX-F)的治療應用具有重要意義HAB)或雙功能(ILX-FH展示了增強的腫瘤靶向性和蓄積性,以及提高療效的潛力。
十四行詩F的另一個獨特優勢HAB是它的連接體設計(圖1),用於連接一個或兩個大分子 治療有效載荷,用於單功能或雙功能活性。我們的G4S(甘氨酸、絲氨酸)多肽連接體是靈活的,同時足夠長 以防止空間位阻,並可以採用棒狀結構以增強對緊密組織基質的滲透。除了保持治療功能區域之間的距離外,十四行詩連接體完全是人類的,在連接體結構上是非免疫原性的,包括在有效載荷結合區域。在雙功能結構中,可以操縱治療有效載荷的方向以改善潛在的治療效果。
| 7 |
作為 最後一個關鍵設計組件,FHAB在哺乳動物細胞培養物中產生,特別是中國人卵巢(CHO)細胞, 其能夠進行糖基化以降低或潛在消除免疫原性。使用CHO,我們已經產生了幾種不同的具有各種低分子量治療性蛋白質(例如,重組細胞因子,如IL-12、IL-15、IL-18、抗IL-6和抗TGF β)。重組治療性蛋白質,包括細胞因子,已經顯示出巨大的治療潛力,但可能缺乏組織 特異性,這可能導致毒性。由於它們的小尺寸(HAB衍生的化合物 已經顯示出顯著更長的血清半衰期,改善的組織積累,並且當 與它們各自的裸重組細胞因子相比時具有顯著的腫瘤減少活性。
總之,我們的FHAB技術支持模塊化的多功能支架,可定製以產生廣泛的多靶向 治療候選物。相對於現有的白蛋白結合技術,FHAB的區別在於具有線性、 棒狀形狀,其設計用於更好的靶組織滲透,是一種全人類設計,以降低免疫原性、哺乳動物糖基化、 和FcRn結合,從而延長血清半衰期。重要的是,FHAB衍生的治療劑具有靶向遞送 至腫瘤和淋巴組織、降低毒性和更寬治療窗的潛力,具有利用定製的單 或雙功能作用機制的額外益處。
F的擴展 應用HAB技術:
免疫療法: 我們相信,HAB平臺可以創新靶向特定組織的生物藥物,同時還可以延長治療半衰期。如FHAB構建體被設計為能夠同時部署兩種協同免疫治療化合物, 我們設想了一條通往先前未開發的免疫治療進步的道路。
藥物結合:使用FHAB技術,可將各種藥物化合物連接到FHAB支架組合 超出了我們的第一波細胞因子管道,這為無數疾病領域的發展提供了機會。
疫苗: 疫苗開發人員正在尋求通過將疫苗與天然載體(如白蛋白)結合來提高疫苗效率。我們相信 FH具有模塊化支架結構的AB平臺可以成為將疫苗遞送至淋巴結的有效載體, 改善滲透和呈遞,並延長半衰期。
CAR T細胞療法:CAR T細胞療法涉及遺傳修飾患者自身的T細胞以識別癌細胞,從而更有效地靶向和殺死腫瘤。我們相信利用白細胞介素的靶向Sonnet構建體可以全身共同施用 以增強CAR T細胞功效。
管道 概述
下表總結了我們已披露特定目標適應症的管道計劃的相關信息:

| 8 |
SON-1010
IL-12是一種循環細胞因子,已被證明對先天免疫和獲得性免疫有多種影響。這些免疫功能在攻擊癌細胞和病原體方面至關重要。IL-12是一種由樹突狀細胞、單核細胞和巨噬細胞產生的異源二聚體細胞因子,也稱為抗原提呈細胞(APC)。IL-12可誘導T細胞和自然殺傷細胞(NK細胞)分泌幹擾素-ɣ,促進活化的T細胞和NK細胞的擴增和存活,增強細胞毒T細胞的殺傷活性,支持Th1輔助效應細胞的分化,增強抗體依賴的細胞毒作用。IL-12也被證明具有刺激作用體外培養癌症患者外周血淋巴細胞的抗腫瘤活性體內黑色素瘤、結腸癌、乳腺癌和肉瘤小鼠模型的抗腫瘤活性。
小鼠的臨牀前研究
最初,Son-1010(mIL12-F)的小鼠版本H與不含F的重組mIL-12相比,臨牀前顯示出更大的腫瘤生長抑制HAB(裸露/獨立IL-12)在黑色素瘤小鼠模型中的作用。圖2,來自這項小鼠黑色素瘤研究,顯示了使用mIL12-F後腫瘤減少率增加了30到50倍HAB與獨立的MIL-12相比。
此外, 在同一型號中,mIL12-FHAB在腫瘤中積累的濃度較高,並在血清、脾和腫瘤中滯留 明顯長於未加F的mIL-12HAB,可能使給藥頻率更低,劑量更低。
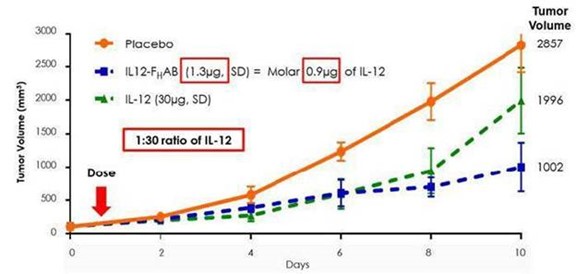
圖2:IL-12(0.9微克)的摩爾當量為IL-12-FHAB(1.3微克)和它們在體外具有相似的生物活性;而在體內,IL12-FHAB的效力大約是IL-12的30倍(在第10天,1.3微克IL-12-FHAB>IL-12 30微克)。
在另一項使用B16F10腫瘤模型mIL12-F的臨牀前研究中H與重組小鼠IL-12相比,AB顯示出更好的劑量反應,以及更長的生存時間(圖3和圖4)。這項研究的結果表明mIL12-FHAB 在縮小腫瘤體積和延長生存期方面可能比單獨使用mIL-12有更大的效果。
| 9 |

圖 3:對腫瘤體積的分析顯示,兩者的腫瘤均呈劑量依賴性減少mIL-12和mIL12-FHAB處理的 只小鼠,與載體對照組相比。IL12-FH與等摩爾劑量相比,AB處理的小鼠的腫瘤體積在統計上顯著減少,mIL-12處理的小鼠。結果表明,IL-12的抗腫瘤活性可能隨着血清半衰期的延長而增強HAB連鎖。
在圖4中,進行了Kaplan-Meier分析以比較用mIL12-F或mIL12-F處理的動物的存活率H這些數據説明瞭腫瘤生長減少(圖3)和生存期延長(圖4)之間的相關性。 在這項研究中,用mIL12-F治療的動物腫瘤生長較慢H與裸露mIL-12治療觀察到的更快的腫瘤生長相比,AB與更長的生存時間相關。最低劑量mIL12-F的存活率HAB(3微克) 相當於最高劑量的mIL-12(30微克)。所有劑量的mIL12-FH在第14天和17.5天,AB的存活率比賦形劑提高了50%。

圖 4:Kaplan-Meier對小鼠B16F腫瘤存活率的評估顯示,使用IL12-F可提高存活率HAB治療。 劑量為10微克和20微克的獨立mIL-12在第2天和第4天的存活率分別比賦形劑對照組(10天)高出50%。所有劑量的IL12-FHAB在第14天和17.5天的存活率分別為50%。IL12-F在最低劑量下的存活率HAB 相當於最高劑量的獨立IL-12
| 10 |
SON-1010的非人靈長類研究
我們 已完成體外培養親和力和結合動力學的藥理學研究表明,SON-1010對倉鼠、大鼠、食蟹猴和人的血清白蛋白具有物種交叉反應。結果表明,SON-1010對食蟹猴和人具有種屬特異性,為進一步的臨牀前毒理工作提供了物種選擇的指導。人源化小鼠模型(SCID) 旨在評估PK/PD和劑量反應的研究已經完成。這項工作為我們在一項非人類靈長類(NHP)研究中決定給藥提供了依據。
2021年2月,我們宣佈成功完成了SON-1010的NHP非GLP重複劑量毒理學研究,其中的數據被用於為cGLP毒性研究的設計提供信息,為IND提交做準備。非GLP研究的目標是評估SON-1010在幾個劑量水平重複給藥方案中的毒性,併為進一步的安全性和毒性研究的設計收集關鍵數據。研究包括靜脈(IV)和SC兩種給藥途徑,總共兩次注射,相隔14天。在這項研究中使用的最高劑量率大於患者暴露於患者的預期臨牀水平的50倍。研究結果包括:
| ● | 在檢查的兩個劑量水平下,靜脈和SC給藥途徑重複給藥是耐受的。正如通常使用IL-12所觀察到的那樣,白血球計數下降,肝酶(ALT和AST)升高。這些都是短暫的影響,在第二次服藥後7天內恢復到基線水平。 | |
| ● | SON-1010在生理觀察、體重、病理、細胞因子和免疫表型等方面發生了相關變化,所有這些變化都與以前在單劑研究中觀察到的靶向效應一致。 | |
| ● | 在第一次注射SON-1010後,幹擾素-γ水平顯著升高,而第二次注射後,幹擾素-γ水平降低。這一趨勢遵循了發表的關於IL-12在人類和NHP中的其他研究的數據。在研究中測試的所有劑量水平中,明顯沒有細胞因子失衡的跡象,或包括腫瘤壞死因子-α、IL-1β和IL-6在內的促炎細胞因子的不受控制的增加。 | |
| ● | 藥代動力學分析表明,通過SC注射SON-1010的動物的平均血清半衰期約為40小時。這與該研究先前進行的劑量遞增階段的數據是一致的,該階段的數據表明,與裸露的重組人IL-12 13-19小時的半衰期相比,半衰期有了顯著的改善。 | |
| ● | 這些結果建立在與B16F10黑色素瘤小鼠模型研究的基礎上,其中小鼠版本的SON-1010顯示,與小鼠IL-12相比,達到類似治療效果所需的劑量減少了20倍。綜上所述,我們認為,通過Sonnet的FHAB技術,觀察到的延長的半衰期、改善的治療窗口和減少的劑量需求代表了SON-1010作為潛在的免疫腫瘤治療的關鍵優勢。 |
2021年5月,我們宣佈成功完成了在NHP中對SON-1010進行的cGLP重複劑量研究。該研究的目的是評估SON-1010在NHP中的毒性,採用三種不同劑量水平的皮下(SC)重複給藥方案與未治療的對照組進行比較,並評估任何不良結果的潛在可逆性。研究結果包括:
| ● | 重複服用NHP後未觀察到的不良事件水平(NOAEL)是預期等量人類臨牀劑量的50倍以上,沒有細胞因子釋放綜合徵的證據。 | |
| ● | 血清樣本的藥代動力學(PK)分析證實了IL12-F的增強特徵HAB高於重組人IL-12,在NHP中的半衰期約為40小時。 | |
| ● | 在給予IL12-F後,觀察到與抗腫瘤機制相關的關鍵多效性細胞因子幹擾素-γ顯著增加H阿布。 | |
| ● | SON-1010在臨牀觀察、體重、臨牀病理、細胞因子和免疫表型方面發生了相關變化,所有這些都與以前在非人類靈長類動物中觀察到的靶向效應一致。 | |
| ● | 到第38天,所有研究對象恢復到基線(研究前)實驗室數值。 | |
| ● | 在檢查的所有劑量水平下,重複給藥都是耐受的。 |
| 11 |
生物分佈研究
在2023年9月,我們宣佈完成了兩個獨立的體內概念驗證(POC)研究顯示白細胞介素F的生物分佈HAB分子到腫瘤微環境(TME),使用在放射性標記生物製劑和 方面具有專業知識的實驗室體內生物分佈分析。這些實驗室採用了不同的放射性標記方法(99mTC或89Zr) 用於mIL-12和mIL12-FHAB,有或沒有多組氨酸標籤(His-Tag)。這兩項研究是使用B16F10小鼠黑色素瘤模型完成的,以測量不同時間點放射性標記產物的累積和腫瘤體積的抑制。H與mIL-12相比,AB的腫瘤積累量明顯更高,在較長時間點的平均水平是mIL-12的2.5-4.7倍,並增加了滯留。與正常小鼠相比,腫瘤中發現了蓄積,正如預期的那樣,肝、腎和其他器官中的蓄積是暫時的。重要的是,放射性標記的mIL12-FHAB還顯示可測量的蓄積在引流的淋巴結中。總體而言,這些發現對任何單(ILX-F)的治療應用具有重要意義HAB)或雙功能(ILX-FH展示了增強腫瘤靶向性和聚集性的分子,以及可能導致各種候選藥物的療效提高的可能性。
製造業 發展
除了藥物產品展示(液體或凍幹)外,表達SON-1010的主細胞庫的製造工作、配方開發和工藝開發活動都已完成。多個cGMP藥物產品批次已成功生產 併為正在進行的臨牀試驗提供庫存。
診所裏的SON-1010
我們於2022年4月啟動了首個人類(FIH)1期試驗(SB101),以評估患有晚期實體瘤和鉑耐藥卵巢癌(PROC)的成年患者的最大耐受劑量(MTD),並於2023年4月在AACR上公佈了該研究的初步數據。更多的PROC患者將被納入這項研究的擴展部分,以確認推薦的第二階段劑量(RP2D)。在SB101的前五個隊列中的15名患者中,有9名患者在第一次隨訪掃描時病情穩定,其中4名患者在進入研究階段時已經進展。經過4個月的隨訪,14名患者中有5名在第二次掃描時保持穩定,表明SON-1010在36%的患者中臨牀受益。第一位接受治療的患者患有侵襲性子宮內膜肉瘤,腫瘤明顯縮小,腹水一度完全消退,臨牀和放射學表現穩定了一年多。前3個隊列中的劑量是每4周進行一次,但現在新的隊列中每3周進行一次,以提高更高劑量的安全性。
我們 於2022年7月在澳大利亞健康志願者中啟動了一項單次遞增劑量(SAD)1期臨牀研究(SB102),以仔細研究PK和PD,為可能的聯合研究做準備。SB102研究的數據是在2023年第一個日曆季度 報告的。在SC給藥後,使用經過驗證的電化學發光分析(Meso Scale,LLC(MSD)),可以看到SON-1010在血清中的典型劑量相關增加。藥物濃度在約11小時內達到峯值,幾何平均最大濃度(C最大值50、100和150 ng/kg劑量組分別為29、68和125 pg/ml)(圖5)。平均消除半衰期(T½) 150 ng/kg劑量的SON-1010為112小時,而rh IL-12 SC為12小時。
| 12 |

圖5:在給藥後頻繁地評估SON-1010水平,然後在SB102研究的其餘部分指示的日期進行評估。插圖 更詳細地顯示了第一週的相同數據。
在細胞因子PD反應中,幹擾素-ɣ的升高最為明顯,且呈劑量依賴性、可控性和持續時間。在所有活性藥物受試者中,SON-1010誘導的幹擾素-ɣ在24~48小時達到高峯,2周後恢復到基線水平(圖6)。幹擾素-ɣ基因工程C最大值在50、100或150 ng/kg的SON-1010後的AUC分別為398、384和666 pg/mL,而在50、100或150 ng/kg的SON-1010後的AUC分別為398、384和666 pg/mL,0-48h為6050、10200和14600 h*pg/mL。用線性迴歸法預測IFN-γ C最大值在較高的 劑量下,這仍然在安全範圍內。以劑量依賴性方式誘導低量的IL-10,這也可能是對IFN-γ增加的響應。給藥後IL-6、IL-8和TNF-α出現小幅一過性升高 ,但IL-1β、IL-2或IL-4未觀察到一致模式,且無細胞因子釋放綜合徵(CRS)證據。安全性 與之前報告的一致;不良事件通常為輕度/中度、一過性, 均可耐受。

圖 6:在PD給藥後頻繁評估細胞因子水平,然後在SB 102研究剩餘時間的指定日期進行隨訪。
SON-1010在迄今為止測試的所有劑量下都是安全和可耐受的。不良事件通常為輕度/中度和一過性, 無因安全性原因而終止研究。此外,後續劑量的不良反應數量較少,強度較低。 幾何半衰期(T½SON-1010在SB 101中的作用時間為113小時,而在先前的研究中觀察到SC給予重組hIL-12的作用時間為12小時。我們兩項研究之間PK曲線的比較表明,SON-1010可能靶向 腫瘤,因為FHAB是為了做。與健康志願者一樣,癌症患者每次給藥後的細胞因子分析 顯示,IFN-γ的誘導作用類似,在24至48小時達到峯值,2至4周後恢復至基線水平 。與對IFN-γ的反應預期一致,每次給藥後觀察到IL-10小幅增加。在這些 劑量下,IL-1β、IL-6、IL-8或TNF-α的信號極小 或無信號,且無任何跡象表明可能發生細胞因子釋放綜合徵(CRS)。
| 13 |
SON-080治療化療引起的周圍神經病變
通過 我們的管道擴展工作,我們已經確定IL-6是一種細胞因子,當作為獨立的 分子遞送時具有重要的生物學特性。我們的主要臨牀階段資產SON-080是IL-6的天然人類版本,也是在中國人卵巢(CHO)細胞中生產的。在患有血小板減少症的癌症患者 和健康志願者中進行的I期和II期臨牀試驗中研究了先前版本的重組IL-6。Sonnet的類似版本將推進到化療誘導的周圍神經病(CIPN)的下一個發展階段,CIPN是癌症中使用化療藥物治療的常見副作用。CIPN是一種使人衰弱的病症,其表現為四肢的疼痛、麻木和刺痛。據報道,在接受特定癌症 治療方案的患者中,有多達70%的患者發生了這種情況,這是患者過早放棄化療的主要原因。在設計用於複製CIPN臨牀症狀的動物實驗中,重組IL-6呈現出改善疾病的特徵,包括修復受損神經的潛力。
Based on the preclinical work, we believe that SON-080 can potentially regenerate damaged nerves, thereby addressing not only the pain-related symptoms, but also the profound discomfort and motor disability CIPN patients often experience. In the nervous system, IL-6 has exhibited neurotrophic-like properties, inducing anti-apoptotic gene expression, protecting neurons from toxic injuries, and promoting nerve regeneration and remyelination. IL-6 has demonstrated the potential to elicit nerve regrowth and to re-establish both normal nerve function (Figure 7) and sensations (Figure 6) in various preclinical models of CIPN induced by cisplatin, taxol, or vincristine. Activity from treatment with SON-080 was also observed in preclinical models of type 2 diabetic neuropathy, outlining the potential for benefit in DPN, and other diseases affecting the nervous system or other organs. This broad activity suggests that the SON-080 mechanism of action might not be restricted to a given class of chemotherapeutic drugs and could elicit a universal neuroprotective-neurorestorative response. Additionally, preclinical data point to the potential of SON-080 to elicit both preventive and curative activity in neuropathies (Figure 8). This introduces the possibility of treating cancer survivors who still suffer from neuropathies, a population representing between 10% and 60% of the 14 million cancer survivors in the US.

圖 7:在組織學(IENFD)或生理學(SNCV)水平上測量的IL-6對紫杉醇或順鉑誘導的大鼠神經病變的活性 .
| 14 |

圖 8:數據顯示預防和治療活性增強正常敏感性的恢復(在此,在順鉑誘導的周圍神經病變中使用對熱 刺激的行為反應)。
IL-6已在超過200名患有化療誘導的血小板減少症的癌症患者的I期和II期研究中進行了研究。試驗入組者 接受的SC劑量範圍為0.25 - 32 µg/kg,每日一次或每週三次。在這些試驗中,實體瘤癌症 存在於超過75%的治療患者中,IL-6的累積劑量平均在8000 μg範圍內(122 - 54880 μg), 平均治療持續時間等於28天。其中一項試驗涵蓋6個化療週期,IL-6治療期延長至203天。在這些試驗中均未觀察到癌症或神經病加重。
SON-080的MTD是在四項研究中通過使用已建立的共同毒性標準通過連續的IL-6劑量組的隊列劑量遞增來確定的。每天給藥時,每日注射SC後的MTD被確定為3至8μg/kg; 當每週給藥3次時,MTD估計>10μg/kg。在這些研究中,確定治療限制劑量的臨牀上最相關的毒性是流感樣症狀和神經皮質毒性,表現為嗜睡、躁動、困惑、幻覺、 和定向障礙。我們預計使用的SON-080劑量比之前的IL-6 MTD少50倍,並預計未來會有更良性的不良事件發生。
這些 數據構成了我們在CIPN進行的臨牀試驗的基礎,根據臨牀前研究的支持,劑量預計將大大低於MTD。作為比較,我們的目標劑量將提供的累積劑量是類似劑量期間達到的平均累積劑量的25倍。我們還相信,SON-080在治療包括DPN在內的其他神經疾病以及其他神經系統疾病方面具有巨大的潛力,我們目前正在評估這些機會的未來發展路徑。十四行詩於2022年7月在CIPN使用SON-080啟動了前美國階段1b/2a試點規模的療效研究。數據安全監控委員會(DSMB)計劃 在第1部分註冊完成後審查初步安全調查結果,預計在2024年第一個日曆季度。
治療糖尿病周圍神經病變的SON-080
除了我們與SON-080的CIPN計劃外,我們的DPN計劃可能會根據我們計劃與SON-080進行的CIPN研究收集的數據, 探索IL-6在糖尿病周圍神經病變(DPN)中的臨牀應用。目前有50%-80%的糖尿病患者被診斷為DPN。根據世界衞生組織(WHO)的預測,到2030年,糖尿病患病率估計將超過3.5億人。神經病變是進行性的,並在糖尿病的連續過程中發展。這種情況包括無明顯原因的頑固性疼痛,以及與疼痛無關的症狀,如失去平衡、感覺缺乏和自主神經功能障礙等。這些赤字損害了生活質量,並導致預期壽命縮短。糖尿病足潰瘍是與糖尿病醫療護理相關的主要成本,也與DPN的發展直接相關。
| 15 |
儘管病情嚴重,但目前的治療只針對DPN的疼痛部分,而沒有解決疾病進展和與疼痛無關的 症狀。此外,目前用於止痛的少數藥物(即欣百達、Lyrica、大麻素、阿片類藥物)僅部分有效,且與主要副作用相關,這通常會推遲它們進入患者的護理。因此,DPN仍然是一種具有很大商業市場潛力的未得到滿足的醫療需求。
長期以來,鍛鍊一直被世衞組織和護理人員視為治療和潛在預防糖尿病的有效手段,幾項試點研究已提供證據支持其在改善糖尿病營養方面的作用。然而,大多數糖尿病患者身體上無法進行鍛鍊。眾所周知,定期鍛鍊可以改善糖尿病相關指標,如糖化血紅蛋白和血糖穩態,改善心率變異性,並刺激神經功能和血流的恢復。最近的證據表明,IL-6在運動過程中被釋放,並介導了體力活動的一些有益效果。十四行詩已經完成了DPN動物模型的臨牀前工作,在該動物模型中,外源性給予IL-6在表皮神經密度、神經功能、血流和對疼痛或幹擾刺激的反應方面顯示出恢復活性。在這種情況下,SON-080可能成為未來治療DPN的關鍵疾病修正療法。
在試管中有關少突膠質細胞或器官培養的數據表明,IL-6可能通過雪旺細胞或少突膠質細胞誘導髓鞘基因的表達(圖9)。

圖9:髓鞘鹼性蛋白(MBP)、蛋白脂蛋白(PLP)及其剪接變異體表達(B)評估少突膠質細胞的存活(A)和分化。
Valerio 等人,Mol Cell Neurosci 21(2002)602-615。
Pizzi 等人,Mol Cell Neurosci 25(2004)301-311。
IL-6的神經保護活性已被用多種範式進行評估,包括興奮性毒性。除了保護神經元,IL-6還可能促進軸突再生和功能性突觸的恢復(圖10)。
| 16 |

圖1011:海馬體切片軸突再生活性(A),損傷切片中生長相關蛋白43(GAP43)表達增加,而正常切片(NL)中未見表達(B)。受抑制的(A)興奮性突觸後電位(EPSP)的軸突再生活性(C)和功能恢復(D).Hakkoum等,J Neurochem 100(2007)747-757.
DPN臨牀前模型中IL-6的活性已經由三個獨立的實驗室進行了評估。這項工作表明,IL-6在神經病變中以劑量依賴的方式顯示出 陽性活性,也可能有助於在神經病變建立後(即糖尿病和繼發性神經病變誘導後4周)恢復正常的生理參數。在運動(圖11A)和感覺(圖11B)神經功能(傳導速度)上觀察到有益的活動,並通過測量熱(圖11C)和觸覺(圖11D)在行為上觀察到有益的活動。除了先前觀察到的對髓鞘和軸突的直接影響體外培養,IL-6也被觀察到在恢復神經中的微血管血流方面具有活性體內(圖11e),這是糖尿病神經病變的主要驅動因素。在神經病的發展過程中接受IL-6預防性治療的動物的神經的組織學分析表明,IL-6對髓鞘具有保護活性,並可能在保護神經纖維完整性、神經傳導速度和感覺方面發揮作用。
| 17 |

圖11:IL-6對鏈脲佐菌素誘導的糖尿病神經病變大鼠的根治治療。卡梅倫等人,Exp Neurol 207(2007)23-29。
除了腫瘤學的適應症,15項試驗性研究共167名受試者,包括27名2型糖尿病患者,由不隸屬於十四行詩的獨立學術團體進行,以評估IL-6在運動和代謝中的作用。同行評議的結果表明,低劑量的IL-6模擬了運動的幾個有益方面,包括表達抗炎分子,增加脂肪代謝,減少胰島素分泌,以及激活肌肉中的STAT3信號通路。
我們相信這些數據為IL-6在DPN中的臨牀開發提供了強有力的支持。通過其作用機制和潛在的疾病改善活性,低劑量的IL-6可能為糖尿病患者的神經病變症狀和心臟自主神經病變(CAN)提供治療方案。我們打算使用從我們對SON-080進行的CIPN研究中收集的數據來為我們的決策提供信息,以確定SON-080在DPN中可能的下一步開發步驟。根據公司於2021年5月與新加坡新生命簽訂的許可協議,我們和新生命將共同負責在DPN開發SON-080,目標是在2023年下半年啟動一項前美國試點 療效研究。
SON-080: 新生命治療協議
2021年5月,我們宣佈簽署一項許可協議,具體內容如下(“新生活協議”),該協議導致我們的IL-6(SON-080)資產被授予新加坡新生活。許可的領土包括新加坡、馬來西亞、印度尼西亞、泰國、菲律賓、柬埔寨、文萊、越南、緬甸和老撾人民民主共和國這10個東盟國家。在2021年6月和7月, 我們修改了新生命協議,使Sonnet BioTreateutics,CH,SA(而不是Sonnet BioTreateutics,Inc.)新生命協議(第一修正案)的締約方和我們還分別使Sonnet BioTreeutics,Inc.成為新生命協議(第二修正案)下的履約擔保人。除了Sonnet在2020年8月簽署意向書時收到的最初500,000美元之外,Sonnet在簽署新生活協議時還收到了另一筆不可退還的預付款500,000美元。根據新生活協議的條款,十四行詩可以在實現早期商業銷售里程碑後30天內獲得100萬美元的遞延許可費、總計高達1900萬美元的里程碑付款和商業銷售12%至30%不等的分級特許權使用費。十四行詩 和New Life打算在決定是否繼續進行DPN適應症的臨牀研究之前,對CIPN研究的安全性數據進行評估。
| 18 |
SON-1210
SON-1210, 我們的主要雙功能構建體,將IL-12和IL-15結合到FH阿布。這些細胞因子是基於協同生物活性而選擇的。IL-15通過其特異性受體IL15Rα發揮作用,該受體表達於抗原提呈樹突狀細胞、單核細胞和巨噬細胞。除了上述IL-12的潛在抗腫瘤特性外,我們認為IL-15還可以潛在地增加以下互補活性:
| ● | 誘導T、自然殺傷(NK)和B細胞分化和增殖 | |
| ● | 增強CD8的細胞殺傷活性+T細胞 | |
| ● | 誘導長效CD8+ 記憶性T細胞在數月/數年內加強對癌症的免疫監視 | |
| ● | B細胞刺激分化和免疫球蛋白合成 | |
| ● | 誘導樹突狀細胞成熟 | |
| ● | Up調節IL-12β1受體表達 |
我們 已經對SON-1210(mIL12-F)的小鼠版本進行了一些臨牀前研究HAB-hIL15)。與對照組相比,注射所示劑量一次的小鼠抑制了B16F10黑色素瘤模型中的腫瘤生長(圖12)。白介素12和白介素15的聯合應用順式使用FHAB顯示出超出mIL12-F抑瘤作用的協同活性HAb (圖13)。總體而言,IL-12和IL-15的相互生物學活性表明:
| ● | IL-12:增加IL-15Rα受體、幹擾素-ɣ、NK/T細胞、TH1(腫瘤殺傷),降低Treg | |
| ● | IL-15:增加IL-12β-1受體、NK細胞、CD8記憶力,減少細胞凋亡 |

圖 12:這些數據顯示增強了腫瘤生長的抑制作用MIL12-FHAB-hIL15與同時給藥相比, 裸體mIL-12和hIL-15在黑色素瘤小鼠模型中的作用。

圖 13:IL-12和IL-15在順式使用FH與IL12-F相比,AB顯示出協同活性,導致腫瘤體積縮小H在黑色素瘤小鼠模型中單獨使用AB。
2023年2月,該公司宣佈在NHP中成功完成了兩項啟用IND的SON-1210毒理學研究。SON-1210的NHP非GLP 劑量遞增研究於2022年9月完成,GLP重複劑量NHP研究於2022年第四個日曆季完成。原料藥的cGMP生產完成,並於2023年初生產了凍乾製劑,以支持FIH的臨牀研究。最初的毒性材料支持非GLP研究,而GLP研究正在對與第一階段臨牀研究相同批次的GMP藥物進行 。SON-1210的監管授權流程定於 開始,等待任何合作活動的結果。
| 19 |
發現 資產:SON-1410(IL18-FHAB-IL12)和SON-3015(抗IL6-FHAB-抗轉化生長因子β)
2021年8月,我們在完成了對小鼠黑色素瘤模型的比較研究後,宣佈選擇了一種新的開發候選者。 該候選者代表了Sonnet的第二個雙功能化合物,將IL-12和IL18與該公司的FHAB 平臺。SON-1410的目標適應症將是黑色素瘤和腎癌。
IL18-FH與安慰劑相比,AB-IL12在一項小鼠黑色素瘤研究中顯示出統計上顯著的腫瘤體積縮小,以及劑量反應。 數據顯示:
| 化合物 | 第0天,單劑 腫瘤 @100 mm3 | 第8天腫瘤體積(mm3 +/-掃描電子顯微鏡),N=8 | 第8天腫瘤縮小8% | |||||
| 安慰劑 | 北美 | 1747 +/- 301 | - | |||||
| IL18-FHAB-IL12 | 1 µg | 918 +/- 130 | 47 | % | ||||
| IL18-FHAB-IL12 | 5 µg | 619 +/- 141 | 65 | % | ||||
還進行了單獨的小鼠研究,比較了所選的IL18-F版本HAB-IL12和另外兩個候選者,GMCSF-FHAB-IL18和GMCSF-FHAB-IL12。比較數據顯示,使用IL18-F後,腫瘤體積顯著縮小,幹擾素-γ水平和免疫細胞反應(NK、NKT、Th1和細胞毒性CD8T細胞)顯著增加HAB-IL12與GMCSF-F的比較HAB-IL12或GMCSF-FHAB-IL18。SON-1410(IL18-F)的臨牀前開發仍在繼續HAB-IL12),用於GMP應用的細胞系開發正在進行中。經過2023年的一些延遲之後,過程開發活動將持續到2024年,有可能產生一種適合NHP的GLP非臨牀研究和隨後的人體研究的藥物。
轉化生長因子-β/IL-6生物學效應是預測腫瘤總生存期的重要指標,聯合應用SON-3015靶向抑制IL-6和轉化生長因子β信號通路有望成為治療腫瘤和骨轉移的有效策略。轉化生長因子β從降解的骨中釋放出來,促進IL-6的產生,導致骨轉移的惡性循環。FcRN在骨環境中的高表達會導致雙重構建的抗IL6-F在骨中蓄積HAB-抗轉化生長因子β,從而潛在地抑制或阻斷骨轉移。 十四行詩已選擇擱置SON-3015開發計劃,以降低費用。
在我們的治療化合物的開發和商業化方面,我們面臨着許多挑戰和不確定性,包括我們的FHAB技術。請參閲本招股説明書中其他部分包含的“風險因素”,以及通過引用併入本招股説明書的文件中標題為“風險因素”的章節。
競爭
製藥和生物技術行業的特點是技術快速進步、競爭激烈以及對專有產品的高度重視。雖然我們相信我們的技術、開發經驗和科學知識為我們提供了競爭優勢,但我們面臨着來自許多不同來源的潛在競爭,包括大型製藥和生物技術公司、學術機構、政府機構和其他進行研究、尋求專利保護併為癌症免疫療法的研究、開發、製造和商業化建立合作安排的公共和私人研究組織。我們成功開發和商業化的任何候選產品都將與未來可能推出的新免疫療法競爭。
我們 在製藥、生物技術和其他開發免疫腫瘤治療的相關市場領域展開競爭。還有許多其他公司已經商業化和/或正在開發癌症免疫腫瘤學治療方法,包括大型製藥公司和生物技術公司,如安進、阿斯利康/醫學免疫公司、百時美施貴寶、默克、諾華、輝瑞和羅氏/基因泰克。
| 20 |
我們面臨着來自制藥和生物技術公司的激烈競爭,這些公司的目標是在癌症環境中使用特定的細胞因子或其他大分子作為免疫調節療法。這些通常包括單特異性或雙特異性抗體、融合蛋白、抗體藥物結合物和靶向疫苗。
對於我們的主要候選產品SON-080,我們知道還有其他公司正在開發治療CIPN的產品,包括但不限於Aphios Corporation、Asahi Kasei Corporation、MundiPharma EDO和Regenacy PharmPharmticals,Inc.;然而,我們相信我們是 唯一研究將疾病修改細胞因子用於適應症的公司。
對於我們的第一個FH除了AB派生的候選方案SON-1010,我們知道還有其他競爭對手的IL-12計劃,包括但不限於Celsion Corporation、Eli Lilly、Inovio PharmPharmticals,Inc.、Intrexon Corporation、Codiak Biosciences、Xolio Treeutics、Werewolf Treeutics、蜻蜓Treeutics和OncoSec Medical正在開發的計劃。我們相信我們的FH集成的IL-12是以腫瘤為靶點的,具有增強的PK配置文件,使其有別於競爭對手。
關於我們早期的流水線FHAB產品候選產品SON-1210、SON-2014和SON-3105,我們不知道有任何其他 競爭公司在這些特定的雙功能計劃上工作。
與我們相比,我們正在競爭或未來可能競爭的許多公司在研發、製造、臨牀前測試、進行臨牀試驗、獲得監管批准和營銷批准藥物方面擁有更多的財務資源和專業知識。製藥、生物技術和診斷行業的合併和收購可能會導致更多的資源集中在我們數量較少的競爭對手中。規模較小或處於早期階段的公司也可能成為重要的競爭對手,特別是通過與大公司和成熟公司的合作安排。這些競爭對手 還在招聘和留住合格的科學和管理人員、建立臨牀試驗場地、為我們的臨牀試驗招募受試者以及獲取與我們的計劃相輔相成或必要的技術方面與我們競爭。
如果我們的競爭對手開發和商業化比我們或我們的合作者 可能開發的任何產品更安全、更有效、副作用更少或更少、更方便或更便宜的產品,我們 可能會看到我們的商業機會減少或消失。我們的競爭對手也可能比我們更快地獲得FDA或外國監管機構對其產品的批准 ,這可能會導致我們的競爭對手在我們或我們的合作者能夠進入市場之前建立強大的市場地位 。如果獲得批准,影響我們所有候選產品成功的關鍵競爭因素可能是它們的療效、安全性、便利性、價格、配套診斷的有效性(如果需要)、生物相似或仿製藥競爭水平以及 政府和其他第三方付款人是否可以報銷。
製造業
我們依賴合同開發和生產組織(CDMO)根據FDA的 當前良好生產規範(CGMP)生產我們的候選藥物,以用於我們的臨牀試驗。生物藥品的生產受到廣泛的cGMP法規的約束,這些法規規定了各種程序和文件要求,並管理記錄保存、生產工藝和控制、人員和質量控制的所有領域。我們的流水線分子使用標準的工業中國倉鼠卵巢(CHO)平臺,使用現成的原材料進行常見的生化工程。
為了滿足我們對臨牀用品的預期需求,以通過監管審批和商業製造來支持我們的活動,我們目前與之合作的CDMO之一已經擴大了生產規模,並正在美國 建設一個cGMP製造基地,將於2024年第三季度投入使用。CDMO的前景很強勁,而且有多個潛在的代工來源。 我們尚未與替代供應商接洽,因為我們目前的CDMO能夠規模化生產,並繼續成功地生產 十四行詩的流水線。我們與CDMO的關係由在藥品開發和製造方面擁有豐富經驗的內部人員管理。
| 21 |
許可證 和其他商業安排
楊森 製藥公司(強生)
2022年10月,十四行詩宣佈與強生旗下的揚森製藥公司之一的揚森生物技術公司(Janssen Biotech,Inc.)達成合作協議,其中體外培養和體內藥效SON-1010(IL12-FHAB)、SON-1210(IL12-FHAB-IL15) 和SON-1410(IL18-FHAB-IL12)將與某些Janssen專利細胞治療資產一起進行評估。協議 由強生創新促成。根據協議條款,十四行詩應提供三種參考化合物 ,以供面對面使用在……裏面體外培養和體內功效研究。如果成功並受協議條款的約束, Janssen可以行使其選擇權,然後Sonnet可以尋求許可證和/或擴大合作。
羅氏
2023年1月,Sonnet宣佈與羅氏就SON-1010與泰唑珠單抗(Tecentriq®)的臨牀評估達成合作協議。 兩家公司已簽訂主臨牀試驗和供應協議,以及輔助質量和安全 協議,以研究SON-1010和阿替唑單抗在鉑耐藥卵巢癌(“PROC”)患者環境中的安全性和有效性。此外,兩家公司將分別提供SON-1010和阿特唑珠單抗,用於1b期/2a期安全性、劑量遞增和療效研究(SB221)。
新生活
於2021年5月,本公司與新生命治療私人有限公司(“新生命”)訂立許可協議(“新生命協議”)。根據新生命協議,本公司授予新生命於馬來西亞、新加坡、印度尼西亞、泰國、菲律賓、越南、文萊、緬甸、老撾人民民主共和國及柬埔寨(專屬地區)開發及商業化含有特定重組人IL-6、SON-080(“化合物”)(該等 製劑,“產品”)以預防、治療或緩解人類糖尿病周圍神經病變的藥物製劑(“DPN領域”)的獨家許可(連同再許可權利)。新生命可行使一項選擇權,以擴大(1)獨家許可的範圍以包括預防、治療或緩解人類化療引起的周圍神經病(“CIPN領域”),該選項是非獨家的 ,將於2021年12月31日到期;和/或(2)許可證的地區範圍包括人民Republic of China、 香港和/或印度,該選項是獨家的,也將於2021年12月31日到期。如果行使這些選項,CIPN油田和領土擴展的條款 將由雙方協商。2021年6月和7月,我們修改了新生命協議 ,將Sonnet BioTreateutics CH SA(而不是Sonnet BioTreateutics,Inc.)新生命協議(第一修正案)的當事人 和我們還分別使Sonnet BioTreateutics,Inc.成為新生命協議(第二修正案)下的履約擔保人。
該公司將保留在世界任何地方生產化合物和產品的所有權利。本公司與新生活將訂立 後續供應協議,根據該協議,本公司將按雙方協商的條款,在專屬地區的DPN油田(及CIPN油田,如適用)向新生活供應產品以供開發及商業化。公司 還將協助轉讓有助於新生活 從許可證中獲益的某些臨牀前和臨牀開發技術。
New Life將承擔並負責在專屬區域內進行臨牀研究和額外的非臨牀研究,以及DPN領域(和CIPN領域,如果適用)產品的其他開發和監管活動以及產品商業化的費用 。
New Life於2020年8月向公司支付了50萬美元不可退還的預付現金款項,並於2021年6月就執行New Life協議向公司支付了50萬美元不可退還的預付現金 。新生命還有義務在滿足某些里程碑時額外支付100萬美元的不可退還的延期許可費,以及根據某些開發和商業化里程碑的實現 可能向公司支付的高達1,900萬美元的額外里程碑付款。此外,在特許權使用費期限內(定義如下),新生命有義務 根據產品在專屬地區的年淨銷售額向公司支付12%至30%不等的分級兩位數特許權使用費。 “特許權使用費條款”是指在專屬地區內按產品和國家/地區的基礎上,從此類產品在專屬地區內首次商業銷售之日(視某些條件而定)起至 起至新生命停止在DPN領域(或CIPN領域)進行商業化為止。(如適用)。
| 22 |
新生命協議將按產品逐個國家/地區繼續有效,並將於最後到期國家/地區的最後到期產品的特許權使用費期限屆滿時終止,但須受以下條件限制:(I)各方的提前解約權 ,包括因另一方重大違約或破產或破產而提前終止的權利,以及(Ii)本公司的回購權和新生命的 回退權(定義如下)。
此外,New Life授予本公司回購本公司授予New Life的權利的獨家選擇權,而公司 授予New Life權利按待商定的條款在專屬地區的一個或多個國家或地區回購有關DPN現場和/或CIPN現場(如適用)的產品的權利,該等選擇權將於適用產品的第三階段試驗開始時終止。
Xoma
十四行詩 (作為Oncobiologics,Inc.(“Oncobiologics”)的利益繼承人,在Oncobiologics於2015年4月6日將某些資產剝離到Soncobiologics 並同時按比例向OncoBioics的股東分配其在Sonnet的所有股份後, 與XOMA(US)LLC(“XOMA”)是2012年7月23日的發現合作協議和2019年5月7日的發現 合作協議修正案(統稱為“合作協議”)的一方),根據該協議,XOMA向Sonnet 授予了一份非排他性的合作協議不可轉讓的許可證和/或使用與發現、優化和開發抗體及相關蛋白質相關的某些材料、技術和相關信息的權利,並據此開發和商業化產品(每個產品)。 十四行詩有義務在實現與產品相關的某些開發和批准里程碑時,按產品向XOMA支付總計375萬美元的或有里程碑付款。在這一點上,下一個預計為75萬美元的臨牀開發里程碑 預計將是開始註冊一種產品(即,Son-1010)在第二階段試驗中。十四行詩還同意為十四行詩銷售的產品的淨銷售額向XOMA支付較低的個位數版税。每種產品的版税應按國家/地區支付 ,直至(I)第一次商業銷售(如合作協議中所定義)之後的指定時間段、 和(Ii)合作協議涵蓋的已頒發專利的最後一項有效索賠到期之日兩者中較晚的一個為止。 此外,十四行詩有權通過向XOMA支付指定的金額來降低逐個產品的版税費率。 合作協議可由任何一方以原因終止,幷包含慣例賠償條款。
阿瑞斯
2015年8月28日,現為Sonnet全資子公司的救濟與默克KGaA(“ARES”)的全資子公司Ares Trading簽署了一份許可協議(“ARES許可協議”) 。根據ARES許可協議的條款,ARES已向本公司授予可再許可的、獨家的、全球範圍內的、有版税負擔的專有專利許可,以研究、開發、使用和商業化使用阿特克沙金阿爾法(“Atex akin”)的產品(每個,一個“產品”),阿特克沙金是一種治療周圍神經疾病和血管併發症的低劑量人類IL-6製劑。ARES許可協議中包括三項專利,保護使用Atex akin治療i)糖尿病神經病變、ii)化療引起的周圍神經病變和iii)血管併發症。
根據ARES許可協議,我們將根據公司銷售的產品的淨銷售額向ARES支付個位數的高額版税。版税按產品和國家/地區支付,直至(I)在該國家/地區進行第一次商業銷售(如ARES許可協議中所定義)後的指定時間段,以及(Ii)該產品在該國家/地區的有效索賠所涵蓋的最後日期 。如果產品不在國家/地區的有效索賠範圍內,或者在該產品在該國家/地區首次商業銷售之日起十二(12)週年之前,該有效索賠已過期或失效,則版税税率將減少50%(50%)。我們還將向ARES支付再許可費用,該費用是從再許可 事件(“再許可收據”)收到的收益的百分比,使用浮動比例表(在發生再許可事件的臨牀開發的後期階段,該百分比會下降),從較低的兩位數開始,然後減少到較高的個位數。為方便起見,公司可隨時終止ARES許可協議,或在任何一方違反協議時由另一方終止。許可證 協議包含慣例的賠償條款。
| 23 |
《Ares許可協議》已於2021年11月1日修訂,以澄清其中包含的與再許可相關的部分條款和條件的適用範圍。尤其是:
| ● | 十四行詩 現在被授權在未經ARES事先書面同意的情況下向第三方授予再許可,前提是任何此類再許可的財務狀況 應反映十四行詩善意確定的公平市場價值。 | |
| ● | 由於 十四行詩從再許可收據中向ARE支付報酬的初始條件尚不清楚,因此澄清了ARES許可協議 ,規定如果在第一階段臨牀試驗完成之前或之後簽署相關的再許可 協議,則十四行詩必須向ARES支付所有再許可收據的一定比例(而不是僅在最初的ARES許可協議中設定的在第一階段臨牀試驗完成後簽署相關的 再許可協議的情況下)。 | |
| ● | 雙方同意,上述澄清僅適用於未來的再許可協議,並適用於新生命協議可能產生的特許權使用費 (但不適用於里程碑付款)。 |
知識產權
在我們的專利組合方面,我們擁有四項已頒發的專利(美國、日本、新西蘭和俄羅斯),我們還提交了針對許多融合蛋白的專利申請 ,其中包括完全人類白蛋白結合結構域(FHAB)。如果獲得批准,由此產生的專利將在2038年至2041年之間到期,但在某些情況下可能會延長專利期限。專利申請 申請包括:
● 與WO/2018年/151868相對應的國家備案文件-此申請針對的是完全人類的白蛋白結合結構域(FHAB) 融合蛋白,“包括與scFv‘s(例如,抗轉化生長因子β、PD-L1、腫瘤壞死因子、IL-1、IL-6、IL-8等)、與細胞因子的融合蛋白(例如,IL-2-FHAB,IL-12-FHAB,IL-15-FHAB,IL-7-FHAb、 等。)以及兩種細胞因子的組合,如IL-12-FH單抗-IL-15、GM-CSF-FHAB-IL-18和IL-18-FHAB-IL-12; 以及使用該F的處理方法HAB融合蛋白。一項專利於2021年6月8日在美國頒發,名稱為美國專利號11,028,166。一項專利於2022年12月23日在日本授予,名稱為日本專利7200138號。一項專利於2022年12月21日在俄羅斯頒發,專利號為2786444。一項專利於2023年10月3日在新西蘭頒發,新西蘭專利號為 756674。美國專利號11,028,166目前預計將於2039年3月26日到期,而日本專利7200138號、俄羅斯專利2786444號和新西蘭專利756674號預計將於2038年2月20日到期。澳大利亞、巴西、加拿大、中國、歐洲、香港和印度也在等待申請。繼續申請和分區申請分別在美國和日本懸而未決。
● 美國專利第11,028,166號和PCT專利申請(PCT/US 2018/00085)最初收到的申請申請日期為2018年2月20日,這是美國臨時專利申請美國62/459,975和美國62/459,981都要求優先利益的美國臨時專利申請提交日期 一週年之後的四天。對於美國專利和PCT專利申請,批准了將優先權恢復到美國臨時專利申請US 62/459,975和US 62/459,981的申請。隨後,PCT專利申請在澳大利亞、巴西、加拿大、中國、歐洲、印度、日本、新西蘭和俄羅斯提交了國家階段專利申請。然而,由於這些司法管轄區的專利法不同,到目前為止,澳大利亞、歐洲、印度、日本、新西蘭和俄羅斯只接受了美國62/459,975和美國62/459,981的優先權權利要求。
● 美國針對抗IL-6-F的臨時申請HAB融合蛋白,包括抗IL-6-FHAB,抗IL-6-FH抗轉化生長因子β和抗IL-6-FHAB-抗-IL8融合蛋白;以及使用這種融合蛋白的治療方法於2021年9月22日重新提交US 63/245,702。然而,很大程度上由於科學上的挑戰,在申請臨時專利後的一年內沒有獲得支持數據,因此,該專利被放棄。
| 24 |
●美國臨時申請涉及抗原/白蛋白結合結構域結合物,以及使用這種結合物的治療方法於2021年5月11日重新提交US 63/187,278。沒有生成支持臨時專利權利要求的數據,因此,該專利 被放棄。
●美國臨時申請針對使用白介素6治療年齡相關性虛弱的方法,於2021年6月4日提交,申請號為63/197,097,並於2022年6月3日轉換為PCT專利。
● 美國針對基於抗體的藥物結合物的臨時申請於2021年12月7日提交,申請號為第63/286,996。由於一年時間內支持數據不足,此臨時 專利被放棄。
●於2022年5月27日作為申請號63/346,368提交的針對IL-12-白蛋白結合結構域融合蛋白製劑及其使用方法的美國臨時申請。這項臨時專利於2023年5月26日轉化為PCT申請(PCT/US2023/067566)。
●於2023年3月14日提交了針對低劑量IL-6製劑及其使用方法的美國臨時申請,作為申請編號63/490,202。
●於2022年9月30日提交了針對低劑量IL-6製劑及其使用方法的美國臨時申請,作為申請編號63/377,971。這項臨時專利於2023年9月29日轉化為PCT申請(PCT/US2023/075593)。
●美國臨時申請針對使用重組IL-12白蛋白結合結構域融合蛋白治療癌症的方法,於2022年11月2日提交,申請號為63/421,846。該臨時專利於2023年11月1日轉換為PCT申請(PCT/US2023/078366) 。
關於我們的商標組合,我們獲得了世界知識產權局(WIPO)對Sonnet BioTreateutics和F的國際註冊批准HAB標記,每個標記的生效日期為9月1日。17、2020年。此外,這兩個商標均由歐盟知識產權局(EUIPO)公佈,生效日期分別為2020年11月30日和2020年12月6日。2021年,美國專利商標局發佈了這兩個商標的補貼通知,表明這兩個申請都已成功完成了反對期,並已成熟到提交了可接受的使用聲明進行註冊。為此,USPTO發佈了Sonnet BioTreateutics和F的每一種使用聲明的津貼通知HAB申請和Sonnet BioTreeutics標誌已經獲得了註冊號為6,790,475的註冊證書。
● 瑞士商標局於9月9日授予Sonnet BioTreeutics和FHAB商標保護。分別於2021年10月14日和2021年10月26日 ,受國際商標註冊號保護。1558330和1558471。
● 加拿大知識產權局於2022年6月8日授予Sonnet BioTreateutics商標保護權,受國際商標註冊號1558330保護,而FHAB商標受國際商標註冊號15584471保護,反對期從2022年11月16日開始,為期18個月。
● 除了瑞士和加拿大,Sonnet BioTreeutics商標還在澳大利亞、歐盟、日本、墨西哥、韓國和英國獲得保護,每個國家都有一個註冊號。的有效註冊日期為1558330年9月17日,續訂日期為9月17日。17年,2030年。同樣,FHAB商標在澳大利亞、中國、歐盟、日本、墨西哥、韓國和英國獲得了保護,每個案例都有一個註冊號。1558471,授予保護日期為9月1日。2020年7月17日,續訂日期為9月9日17年,2030年。
● 儘管十四行詩生物治療商標最初在中國被駁回,原因是針對某些競爭對手 公司的潛在非使用索賠,但我們律師事務所非常有信心,由於最初的42類駁回被成功取消,同一商標的兩個新商標申請 也可以在2021年註冊和/或發佈;然而,我們要到2025年才能針對這些商標啟動 非使用註銷申請,這是預計這些未決的42類申請 可能在中國註冊的時間框架。
| 25 |
員工
截至2023年9月30日,我們有12名全職員工。我們的員工沒有工會代表,也沒有集體談判協議的覆蓋範圍,我們相信我們與員工的關係很好。此外,我們還利用獨立承包商和 其他第三方協助其業務的各個方面。
政府 法規
藥品(包括生物產品)的研究、開發、測試、製造、質量控制、批准、包裝、儲存、記錄保存、標籤、廣告、促銷、分銷、營銷、批准後監控和報告以及進出口均受到美國聯邦、州和地方政府當局以及其他國家和司法管轄區的廣泛監管。一些司法管轄區還對藥品的定價進行監管。在美國及其他國家和司法管轄區獲得營銷批准的流程,以及隨後對適用法規和法規及其他監管機構的遵守,都需要花費大量的時間和財力。
美國的生物製品許可證和監管
在美國,生物製品或生物製品受《公共衞生服務法》(PHSA)和《聯邦食品、藥品和化粧品法》(FDCA)及其實施條例的監管。在產品開發過程中的任何時間未能遵守適用的要求 可能會使申請人在進行研究、監管審查和批准、 和/或行政或司法處罰方面受到延誤。這些制裁可能包括但不限於FDA拒絕允許申請者繼續進行臨牀試驗、拒絕批准未決申請、吊銷或吊銷執照、撤回批准、 產品召回、產品扣押、暫停生產或分銷、禁令、罰款、調查以及民事和刑事處罰。生物製品候選產品必須獲得FDA頒發的生物許可證,才能在美國合法上市。
FDA在美國獲得生物許可證所需的 流程通常涉及以下內容:
● 完成廣泛的非臨牀或臨牀前實驗室試驗和臨牀前動物試驗,以及根據適用法規對實驗動物的人道使用和配方研究的適用要求,包括良好的實驗室實踐、或GLP;
● 在啟動任何人體臨牀試驗之前,向美國食品和藥物管理局提交正在研究的新藥或IND申請。必須在此類審判開始之前獲得繼續進行的許可;
● 進行充分且控制良好的人體臨牀試驗,以根據FDA的法規(通常稱為良好臨牀實踐)或GCP以及保護人體研究對象及其健康信息的任何附加要求,為每個建議的適應症確定候選產品的安全性、效力和純度,以確定建議的生物製品用於其預期用途的安全性和有效性。FDA還可能在我們的臨牀試驗之前或期間的任何時間,出於安全考慮或不符合規定的原因,對生物製品候選產品 實施臨牀擱置。如果FDA強制實施臨牀擱置,試驗 不能在沒有FDA授權的情況下重新開始,然後只能根據FDA授權的條款進行;
● 為請求營銷一個或多個建議適應症的生物製品準備並向食品和藥物管理局提交生物許可證申請,包括提交有關該產品在臨牀開發和建議標籤方面的製造和成分的詳細信息。
●由食品和藥物管理局審查部門確定的食品和藥物管理局顧問委員會對產品的審查;
● 圓滿完成FDA對生產產品或其組件的一個或多個製造設施(包括第三方)的一次或多次檢查,以評估符合當前良好製造規範或cGMP要求的情況,並確保設施、方法和控制足以保持產品的特性、強度、質量和純度;
| 26 |
●滿意地完成了一項或多項臨牀研究場地的FDA審核,以確保符合GCP,以及支持BLA的臨牀數據的完整性;
● 支付使用費,並確保FDA批准BLA和新生物製品的許可;
● 遵守任何審批後要求,包括實施風險評估和緩解戰略的潛在要求, 或REMS,以及FDA要求的任何審批後研究。
非臨牀研究和探索性新藥應用
每個候選產品在進行人體測試之前都必須經過非臨牀測試。這些測試包括產品化學、配方和穩定性的實驗室評估,以及評估活性和毒性潛力的動物研究,並且必須遵守適用的法規進行。非臨牀試驗的結果與生產信息和分析數據一起作為IND申請的一部分提交給FDA。IND在FDA收到後30天自動生效,除非在此之前FDA對擬議的臨牀試驗的產品或進行提出擔憂或問題,包括擔心人類研究對象將暴露在不合理的健康風險中。在這種情況下,IND贊助商和FDA必須在臨牀試驗開始之前解決FDA的任何懸而未決的問題。
提交IND可能導致FDA不允許試驗開始,或不允許試驗按發起人最初在IND中指定的條款進行。如果FDA提出擔憂或問題,它可以選擇在 臨牀試驗之前或期間的任何時間對生物製品候選產品實施臨牀擱置,原因是安全問題或不符合規定。如果FDA強制臨牀擱置,試驗不得在沒有FDA授權的情況下重新開始 ,並且只有在FDA授權的條款下才能重新開始。
人類 支持血乳酸的臨牀試驗
臨牀 試驗涉及根據GCP要求,在合格首席研究人員的監督下,將研究產品候選給健康志願者或要接受治療的疾病患者 。作為IND的一部分,必須向FDA提交每項臨牀試驗的方案和任何後續方案修改。希望在美國境外進行臨牀試驗的贊助商可以(但不需要)獲得FDA的授權,根據IND進行臨牀試驗。如果外國臨牀試驗不是在IND下進行的,贊助商可以將臨牀試驗的數據提交給FDA以支持BLA,只要臨牀試驗設計良好並符合GCP,包括由獨立的道德 委員會審查和批准,並且FDA能夠通過現場檢查(如有必要)驗證研究數據。
此外, 每項臨牀試驗都必須由機構審查委員會或IRB進行審查和批准,審查委員會或IRB在將進行臨牀試驗的每個機構集中或單獨進行,對於在美國境外進行的試驗,則必須由上文提到的獨立倫理委員會進行審查和批准。IRB將考慮臨牀試驗設計、患者知情同意、倫理因素和人類受試者的安全等因素。IRB的運作必須符合FDA的規定。FDA、IRB或臨牀試驗贊助商可隨時出於各種原因暫停或中止臨牀試驗,包括髮現臨牀試驗未按照FDA要求進行,或受試者或患者面臨不可接受的健康風險。臨牀檢測還必須滿足廣泛的GCP規則和知情同意的要求。此外,一些臨牀試驗由臨牀試驗贊助商組織的獨立的合格專家小組監督,稱為數據安全監測委員會或委員會。 該小組可根據對研究中某些數據的訪問,建議按計劃繼續研究、改變研究方法或在指定檢查點停止研究。
臨牀試驗通常分三個連續階段進行,這些階段可能重疊,也可能合併。批准後可能需要進行額外的研究。
● 第一階段:該生物製品首先被引入健康的人體志願者中,並進行安全性測試。在一些嚴重或危及生命的疾病的產品的情況下,特別是當產品本身的毒性可能太高而無法合乎道德地給健康志願者使用時,最初的人體測試通常在患者身上進行,例如癌症患者。
| 27 |
● 第二階段:在有限的患者羣體中對候選生物製品進行評估,以確定可能的不良反應和安全風險,初步評估該產品對特定目標疾病的療效,並確定劑量耐受性、最佳劑量和劑量計劃。
● 第三階段:在擴大的患者羣體和地理分散的臨牀研究地點進行臨牀試驗,以進一步評估劑量、臨牀療效、效力和安全性。這些試驗旨在確定產品的總體風險/收益比率,併為產品標籤提供充分的依據。
● 第四階段:批准後臨牀試驗或第四階段臨牀試驗可在初步上市批准後進行。它們為預期治療適應症患者的治療提供了 額外經驗,特別是長期安全跟蹤。 如果FDA批准了一種產品,而一家公司正在進行不需要批准的臨牀試驗,公司可以 使用這些臨牀試驗的數據來滿足任何第四階段臨牀試驗的全部或部分要求,或請求更改 產品標籤。如果未能在進行所需的4期臨牀試驗方面進行盡職調查,可能會導致撤回對產品的批准。
符合cGMP要求
在批准BLA之前,FDA通常會檢查生產該產品的工廠,以確保製造工藝和工廠完全符合cGMP要求,並與所需規格保持一致。製造商和其他涉及產品製造和分銷的企業也必須向FDA和某些州的機構進行登記。國內和國外的製造企業必須在首次參與生產過程時向FDA登記並提供附加信息。由未註冊的工廠製造或從其進口的任何產品均被視為FDCA下的錯誤品牌。機構可能會受到政府當局的定期突擊檢查。製造商 可能需要提供有關其工廠的記錄。
審查和批准BLA
產品候選開發、非臨牀測試和臨牀試驗的結果作為BLA的一部分提交給FDA,以申請該產品的銷售許可證 。BLA必須包含有關產品製造和組成的廣泛而詳細的信息,以及建議的標籤和用户費用的支付。FDA在提交申請後有60天的時間進行初步審查,以確定BLA是否足以接受申請。一旦提交的申請被接受,FDA就開始進行深入的審查。FDA有12個月的時間完成對標準申請的初步審查(如果是優先審查,則有6個月的時間)並對申請人做出迴應。FDA並不總是達到其目標日期,審查過程可能會因FDA要求提供更多信息或澄清而顯著延長 。如果FDA要求,或者如果申請人在目標日期之前的最後三個月內提供了關於已在提交中提供的信息的其他信息或澄清,則審查過程和目標日期可以延長三個月。
根據FDA對申請的評估和相關信息,FDA可簽發批准信、拒絕函或完整的回覆信。批准函授權該產品的商業營銷,並提供特定適應症的具體處方信息 。根據PHSA,如果FDA確定產品是安全、純淨和有效的,並且將生產該產品的設施符合旨在確保其繼續安全、純淨和有效的標準,則FDA可以批准BLA。如果申請未獲批准,FDA可能會發出完整的回覆信,其中將包含為確保申請獲得最終批准而必須滿足的條件,並在可能的情況下列出贊助商為獲得申請批准而可能採取的建議行動。收到完整回覆信的贊助商可向FDA提交代表對FDA確定的問題的完整回覆的信息。根據《處方藥使用費法案》(PDUFA),此類重新提交根據申請人在回覆行動信函時提交的信息,被分為 1類或2類。根據FDA根據PDUFA同意的目標和政策,FDA有兩個月的時間審查1類重新提交的申請,有6個月的時間審查2類重新提交的申請。 FDA不會批准申請,直到完整的回覆信中確定的問題得到解決。如果FDA確定該機構或產品不符合該機構的要求,則會發出一封否認信。
| 28 |
FDA還可以將申請提交給諮詢委員會進行審查、評估和不具約束力的建議,以確定是否應批准申請 。特別是,FDA可能會將新的生物製品或提出安全性或有效性難題的生物製品的申請提交給諮詢委員會。
如果FDA批准一種新產品,FDA可以限制其批准的適應症使用,並要求在產品標籤中包括禁忌症、警告或預防措施。此外,FDA可能會要求進行批准後研究,包括4期臨牀試驗,以進一步評估批准後該產品的安全性。FDA還可能要求測試和監控計劃在產品商業化後對其進行監控,或者施加其他條件,包括分銷限制或其他風險管理機制,包括REMS,以幫助確保產品的益處大於潛在風險。FDA可能會根據上市後研究或監測計劃的結果,阻止或限制產品的進一步營銷。經批准後,對經批准的產品的許多類型的更改,如增加新的適應症、製造更改和額外的標籤聲明,都要接受進一步的測試要求 和FDA的審查和批准。
快速跟蹤、突破性治療和優先審查指定
FDA有權指定某些產品進行快速審查,如果這些產品旨在滿足在治療嚴重或危及生命的疾病或狀況時未得到滿足的醫療需求。這些計劃被稱為(I)快速通道指定、(Ii)突破性治療指定和(Iii)優先審查指定。
● 快速審查:如果一種產品打算(單獨或與一種或多種其他產品聯合使用)用於治療嚴重或危及生命的疾病或狀況,並且它顯示出解決此類疾病或狀況未得到滿足的醫療需求的潛力,則FDA可指定該產品進行快速軌道審查。贊助商可能會與FDA有更多的互動,FDA可能會在申請完成之前對Fast Track產品申請的部分進行審查。贊助商還必須提供提交剩餘信息的時間表,並且必須得到FDA的批准,並且贊助商必須支付適用的使用費。但是,FDA審查快速通道申請的時間段目標直到申請的最後部分提交後才開始。 FDA可能會撤回快速通道指定。
● 突破性療法:產品可被指定為突破性療法,如果該產品旨在單獨或與一個或多個其他產品聯合用於治療嚴重或危及生命的疾病或狀況,且初步臨牀證據表明該產品可能在一個或多個臨牀重要終點(如臨牀開發早期觀察到的重大治療效果)方面顯示出顯著改善,則該產品可能被指定為突破性療法,並有資格獲得快速審查。FDA可能會在突破性療法方面採取某些行動。
● 優先審查:如果一種產品治療嚴重情況,FDA可以指定該產品進行優先審查,如果獲得批准,與其他可用的治療方法相比, 將在安全性或有效性方面提供顯著改善。這項評估是由FDA根據具體情況進行的。優先指定旨在將整體注意力和資源引導到對此類申請的評估上,並將FDA對營銷申請採取行動的目標從10個月縮短至6個月。
加速了 審批途徑
FDA可以加速批准一種嚴重或危及生命的疾病的產品,該產品為患者提供了比現有治療更有意義的治療優勢 ,其基礎是確定該產品對替代終點有影響,而該替代終點很有可能預測臨牀益處。如果產品對中間臨牀終點有影響,且可早於對不可逆發病率或死亡率或IMM的影響進行測量,並且考慮到疾病的嚴重性、稀有性或流行率以及可用或缺乏替代治療, 合理地有可能預測對IMM或其他臨牀益處的影響,FDA也可批准加速批准此類疾病。獲得加速批准的產品必須符合與獲得傳統批准的產品相同的安全和有效性法定 標準。
就加速審批而言,替代終點是一個標記,如實驗室測量、放射圖像、體徵或其他被認為可預測臨牀益處但本身不是臨牀益處的衡量標準。代理終端 通常比臨牀終端更容易或更快速地進行測量。中間臨牀終點是對治療效果的測量,它被認為合理地可能預測產品的臨牀益處,例如對IMM的效果。加速審批路徑最常用於病程較長且需要延長時間來衡量產品的預期臨牀益處的環境中,即使對代用或中間臨牀終點的影響發生得很快。因此,加速批准已被廣泛用於開發和批准用於治療各種癌症的產品,其中治療的目標通常是提高存活率或降低發病率,典型病程的持續時間需要漫長的 ,有時還需要大型試驗來證明臨牀或生存益處。
| 29 |
加速審批途徑通常取決於贊助商同意以勤奮的方式在批准後進行額外的驗證性研究,以驗證和描述產品的臨牀益處。因此,在此基礎上批准的候選產品 必須遵守嚴格的上市後合規性要求,包括完成4期或批准後臨牀試驗以確認對臨牀終點的影響。如果不進行所需的批准後研究,或在上市後研究期間未能確認臨牀益處,將允許FDA加速將該產品從市場上召回。所有根據加速法規批准的候選產品的促銷材料 都必須經過FDA的事先審查。
審批後條例
即使獲得監管部門的批准,上市產品也必須遵守聯邦、州和外國法律法規的持續全面要求,包括不良事件報告、記錄保存、營銷和cGMP合規性方面的要求和限制。藥物批准後報告的不良事件可能會導致對上市產品的使用施加額外限制,或要求進行額外的上市後研究或臨牀試驗。
要基本遵守適用的聯邦、州和地方法律法規,需要花費大量的時間和財力。FDA對生物製品的嚴格和廣泛的監管在獲得批准後仍在繼續,特別是在cGMP要求方面。生物製品製造商和其他涉及經批准的生物製品的製造和分銷的實體必須向FDA和某些州機構登記其機構,並接受FDA和某些州機構的定期突擊檢查,以確保其遵守cGMP要求和其他法律。我們將依賴,並預計將繼續依賴第三方來生產我們可能商業化的任何產品的臨牀和商業批量。我們產品的製造商 必須遵守cGMP法規中適用的要求,包括質量控制和質量保證 以及記錄和文檔的維護。其他適用於生物製品的審批後要求包括記錄保存要求、不良反應報告和報告最新的安全和療效信息。
如果發現 以前未知的問題,或未能遵守與批准產品的製造商或促銷相關的適用法規要求 ,可能會導致限制該產品的銷售或將該產品從市場上召回 以及重大的行政、民事或刑事制裁。
孤兒 藥品名稱
美國的孤兒 藥物指定旨在鼓勵贊助商開發針對罕見疾病或疾病的產品。 在美國,法律將罕見疾病或疾病定義為在美國影響少於200,000人的疾病或疾病,或在美國影響超過200,000人的疾病,並且無法合理預期 針對該疾病或疾病的產品的開發和提供成本將從產品在美國的銷售中收回 。
孤兒藥 認定使公司有資格獲得税收抵免和市場獨佔權,期限為產品 上市批准之日起七年(如果獲得FDA批准)。可在提交產品上市批准申請之前的任何時間提出指定為孤兒藥的申請。根據可接受的申請,FDA孤兒藥開發辦公室 或OOPD可將產品指定為孤兒藥。然後,該產品必須像任何其他 產品一樣通過審核和批准流程。孤兒藥指定可以根據疾病發生率的變化而撤銷。
| 30 |
申辦者可以申請將以前未經批准的產品指定為孤兒藥,或將已上市產品指定為新的孤兒藥適應症。 此外,如果申辦者能夠提出合理的假設,證明其 產品在臨牀上可能優於第一種藥物,則與已批准的孤兒藥相同的產品的申辦者可以為用於相同罕見疾病或病症的後續產品尋求並獲得孤兒藥 認定。針對同一罕見疾病或病症的同一產品 ,可能有多個申辦者獲得孤兒藥認定,但每個尋求孤兒藥認定的申辦者必須提交完整的認定申請。
專營期從FDA批准上市申請之日開始,僅適用於該產品已被指定的 適應症。FDA可以批准同一產品的第二次申請用於不同的用途,或第二次申請該產品的臨牀更優版本用於相同的用途。然而,FDA不能在市場獨佔期內批准由另一製造商生產的相同產品用於相同的適應症,除非徵得贊助商的同意或 贊助商無法提供足夠的數量。
兒科 研究
根據《兒科研究公平法》,某些審批申請必須包括通常基於臨牀研究數據的對受試藥物在相關兒科人羣中的安全性和有效性的評估。FDA可以應公司的要求或FDA的倡議,免除或推遲兒科評估的要求。FDA可能會認定,風險評估和緩解策略對於確保新產品的益處大於風險是必要的。REM可能包括各種元素, 從藥物指南或患者包裝插入到對誰可以開出或分配藥物的限制,這取決於FDA認為安全使用藥物所必需的 。贊助商需要在與FDA的第二階段會議結束後向其IND 提交初步兒科研究計劃
歐盟藥品審批條例和程序
為了在美國以外銷售任何產品,公司還必須遵守其他國家/地區和司法管轄區的眾多法規要求。無論產品是否獲得FDA批准,申請人都需要獲得 可比外國監管機構的必要批准,然後才能在這些國家或司法管轄區啟動該產品的臨牀試驗或營銷。
臨牀試驗批准
根據目前適用的臨牀試驗指令2001/20/EC和關於GCP的指令2005/28/EC,歐盟已通過成員國的國家立法實施了臨牀試驗審批制度。根據這一制度,申請人必須獲得將在其中進行臨牀試驗的歐盟成員國的主管國家當局的批准,如果要在多個成員國進行臨牀試驗,則必須在多個成員國獲得批准。此外,只有在獨立的倫理委員會發表了贊成的意見後,申請者才可以在特定的研究地點開始臨牀試驗。臨牀試驗申請或CTA必須附有一份研究用藥品檔案,其中包含2001/20/EC號指令和2005/28/EC號指令以及成員國的相應國家法律規定的支持信息,並在適用的指南文件中進一步詳細説明。
2014年4月,歐盟通過了新的臨牀試驗條例(EU)第536/2014號,該條例將取代目前的臨牀試驗指令2001/20/EC。預計新的臨牀試驗條例將於2019年或2020年適用。它將徹底改革歐盟目前的臨牀試驗審批制度。具體地説,這項將在所有成員國直接適用的新規定旨在簡化歐盟臨牀試驗的審批程序。例如,新的 臨牀試驗條例規定了簡化的申請程序,使用單一入口點和嚴格定義的截止日期進行臨牀試驗申請的評估。
營銷 授權
要根據歐盟監管體系獲得產品的營銷授權,申請者必須提交MAA,該MAA由歐盟監管機構管理的中央程序或歐盟成員國主管當局管理的程序之一(分散程序、國家程序或互認程序)提交。只有在歐盟設立的申請者才能獲得營銷授權。申請人必須證明符合EMA批准的涵蓋兒科人羣所有子集的兒科調查計劃(PIP)中包括的所有措施,除非EMA已批准特定產品的豁免、類別豁免或推遲PIP中包括的一項或多項措施。
| 31 |
集中化程序規定由歐盟委員會授予對所有歐洲聯盟成員國均有效的單一營銷授權。對於特定產品是強制性的,包括通過某些生物技術工藝生產的藥品、被指定為孤兒藥品的產品、高級治療產品以及含有用於治療某些疾病的新活性物質的產品,包括用於治療癌症的產品。對於含有用於治療其他疾病的新活性物質的產品,以及高度創新的產品或符合患者利益的集中流程, 集中流程可能是可選的。
根據 集中程序,EMA設立的人用藥品委員會(CHMP)負責 對產品進行評估,以確定其風險/獲益特徵。在集中程序下, MAA評估的最長時限為210天,不包括申請人在迴應CHMP問題時提供額外信息或書面或口頭解釋的時鐘停止。在特殊情況下,當 藥品從公共衞生的角度,特別是從治療 創新的角度來看具有重大意義時,CHMP可以批准加速評價。
授權和續訂的期限
原則上, 上市許可的有效期為5年,5年後可根據EMA或授權成員國的主管機構對風險受益平衡的重新評估 進行更新。一旦更新,上市許可 無限期有效,除非歐盟委員會或主管機構基於與 藥物警戒相關的合理理由決定再進行一次為期五年的更新。在授權失效後 三年內,未將藥品 投放歐盟市場(在集中程序的情況下)或授權成員國市場的任何授權失效。
上市許可後的監管 要求
獲得 批准後,上市許可證持有人必須遵守適用於藥品生產、 上市、推廣和銷售的一系列要求。其中包括遵守歐盟嚴格的藥物警戒 或安全性報告規則,根據這些規則,可以實施許可後研究和額外的監測義務。此外, 授權產品的生產(必須獲得單獨的生產商許可證)也必須嚴格 遵守EMA的GMP要求和歐盟其他監管機構的類似要求,這些要求 規定了藥品生產、加工和包裝中使用的方法、設施和控制措施,以確保其安全性和特性。 最後,授權產品的營銷和推廣,包括行業贊助的針對藥物處方者和/或公眾的繼續醫學教育和廣告 ,在歐盟受到經修訂的指令2001/83 EC 的嚴格監管。
孤兒 藥品名稱和排他性
Regulation (EC) No. 141/2000 and Regulation (EC) No. 847/2000 provide that a product can be designated as an orphan drug by the European Commission if its sponsor can establish: that the product is intended for the diagnosis, prevention or treatment of (1) a life-threatening or chronically debilitating condition affecting not more than five in ten thousand persons in the European Union when the application is made, or (2) a life-threatening, seriously debilitating or serious and chronic condition in the European Union and that without incentives it is unlikely that the marketing of the drug in the European Union would generate sufficient return to justify the necessary investment. For either of these conditions, the applicant must demonstrate that there exists no satisfactory method of diagnosis, prevention, or treatment of the condition in question that has been authorized in the European Union or, if such method exists, the drug has to be of significant benefit compared to products available for the condition. An orphan drug designation provides benefits such as fee reductions, regulatory assistance and the possibility to apply for a centralized European Union marketing authorization. Marketing authorization for an orphan drug leads to a ten-year period of market exclusivity. The market exclusivity period may however be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria for orphan drug designation.
美國的組合 產品
某些 產品,即組合產品,可能由通常由不同類型的監管機構監管的成分組成,通常由FDA的不同中心監管。組合產品可以是(I)由兩個或兩個以上受監管的 成分組成的產品,這些成分以物理、化學或其他方式組合或混合,作為一個單一實體生產;(Ii)兩個或兩個以上單獨的產品,包裝在一個單獨的包裝中或作為一個單元,由藥品和器械產品、器械和生物製品或生物和藥品產品組成;(3)單獨包裝的藥品、器械或生物製品,根據其研究計劃或擬議的標籤僅擬與經批准的單獨指定的藥品、器械或生物製品一起使用,如果兩者都需要達到預期的用途、適應症或效果,並且在擬議的產品獲得批准後,需要更改 核準產品的標籤,例如,以反映預期用途、劑型、濃度、給藥途徑的變化、 或劑量的重大變化;或(Iv)單獨包裝的任何研究用藥物、裝置或生物製品,根據 其建議的標籤僅用於另一單獨指定的研究用藥、裝置或生物製品,而這兩種藥物、裝置或生物製品都需要 以達到預期的用途、適應症或效果。FDA負責指定具有主要管轄權的中心或牽頭中心對組合產品進行審查,這一決定基於組合產品的 “主要行動模式”。贊助商可以通過向組合藥品辦公室提交指定請求來請求管轄權確定。
| 32 |
與強安蒂克利爾合併並收購Relipment
本10-K表格年度報告由Sonnet BioTreateutics Holdings,Inc.(“Sonnet Holdings”、“WE”、“Us”、 “Our”或“Company”)提交,前身為強安蒂克利爾控股公司。在2020年3月31日之前,該公司一直在國內和國際擁有、經營和特許經營快速休閒餐飲概念。如先前披露,本公司於2020年4月1日完成與十四行詩生物治療公司(“十四行詩”)的合併交易,而十四行詩成為本公司的全資附屬公司(“合併”)。2020年4月1日,與合併有關,公司更名為“Sonnet BioTreateutics Holdings,Inc.”。十四行詩於2015年4月6日註冊成立為新澤西州公司。
合併被本公司視為反向合併,並根據美國公認會計原則(“美國公認會計原則”)作為反向資本重組入賬。就會計目的而言,十四行詩被視為已收購該公司。
在合併後及合併前,本公司將與本公司餐飲業務有關的所有資產及負債轉讓給新成立的本公司全資附屬公司amergent Hotel Group,Inc.(“amergent”)。該股息連同上述對公司餐飲業務的貢獻和轉讓,稱為“分拆”。在剝離之前,阿美金特沒有從事任何業務或運營。
作為分拆和合並的結果,自2020年4月1日以來,公司一直通過十四行詩及其直接和間接子公司運營 ,公司的持續業務是十四行詩業務。
此外,於2020年4月1日,與合併有關及於合併前,十四行詩已完成向救濟治療控股公司(“救濟 控股”)收購阿特沙金阿爾法(低劑量白介素6,IL-6,現為“SON-080”的低劑量製劑)的全球開發權,收購救濟控股的全資附屬公司救濟治療公司(“救濟”),以換取在合併中向救濟控股發行合共2,460股公司普通股的十四網普通股。
公司 和現有信息
根據特拉華州的法律,該公司成立於1999年10月21日,原名為Tulvine Systems,Inc.。2005年4月25日,Tulvine Systems,Inc.成立了全資子公司Chonancleer Holdings,Inc.,2005年5月2日,Tulvine Systems,Inc.與Chancleer Holdings,Inc.合併並更名為Chancleer Holdings,Inc.。2020年4月1日,根據本公司、Sonnet和Biosub Inc.之間日期為2019年10月10日的合併協議和計劃的條款,本公司完成了與Sonnet BioTreateutics,Inc.(以下簡稱Sonnet)的業務合併。本公司的全資附屬公司(“合併附屬公司”)(“合併協議”),根據該協議,合併附屬公司與十四行詩合併並併入十四行詩,而十四行詩作為本公司的全資附屬公司(“合併”)繼續存在(“合併”)。根據合併協議的條款,本公司於緊接合並前向十四行詩股東發行普通股 ,匯率為每股已發行十四行詩普通股換0.106572股已發行普通股 。與合併相關的是,該公司將其名稱從“強安蒂克利爾控股公司”改為“Sonnet BioTreateutics Holdings,Inc.”,公司經營的業務變成了Sonnet開展的業務。
我們的主要執行辦公室位於新澤西州普林斯頓08540號第102室俯瞰中心100號。我們的電話號碼是(609) 375-2227,公司網址是https://www.sonnetbio.com/.我們將網站地址包含在本年度報告的表格 10-K中,僅作為非活動文本參考,並不打算將其作為指向我們網站的活動鏈接。網站上的信息未以參考方式併入本10-K表格年度報告中。
本 年度報告Form 10-K、季度報告Form 10-Q、當前報告Form 8-K和對這些報告的所有修訂,以及我們提交給美國證券交易委員會(“美國證券交易委員會”)的其他文件,在以電子方式提交給美國證券交易委員會或以電子方式提交給美國證券交易委員會後,可在合理可行的範圍內儘快通過我們網站的投資者部分免費獲取。公眾可以獲得我們向美國證券交易委員會備案的文件,網址為Www.sec.gov。
| 33 |
第 1a項。風險因素。
投資我們的普通股涉及很高的風險,包括您的全部投資損失的風險。您應仔細 考慮以下描述的風險和不確定性,以及本報告和我們提交給美國證券交易委員會的其他報告中包含的其他信息。下面列出的風險並不是我們面臨的唯一風險。可能存在其他風險和不確定因素,這些風險和不確定性也可能對我們的業務、運營和財務狀況產生不利影響。如果實際發生以下任何風險,我們的業務、財務狀況和/或運營都可能受到影響。在這種情況下,我們普通股的價值可能會下降,您可能會損失所有 或您為我們普通股支付的很大一部分錢。
風險因素摘要
| ● | 我們 有重大運營虧損的歷史,預計在可預見的未來將出現重大且不斷增加的虧損, 我們可能永遠不會實現或保持盈利。 | |
| ● | 我們在運營中的反覆虧損令人對我們作為持續經營企業的持續經營能力產生了極大的懷疑。 | |
| ● | 我們 將需要大量額外資金,如果我們無法在需要時籌集資金,我們可能會被迫推遲、減少 或取消我們的產品發現和開發計劃或商業化努力。 | |
| ● | 我們 在很大程度上依賴於我們內部開發計劃的成功,我們的候選產品可能無法 成功完成臨牀試驗、獲得監管批准或成功商業化。 | |
| ● | 我們 處於開發工作的非常早期階段,我們的候選產品代表着一種新的藥物類別,可能會 受到更嚴格的監管審查,直到它們被確立為治療方式。 | |
| ● | 即使我們完成了必要的臨牀前研究和臨牀試驗,營銷審批流程也非常昂貴、耗時 且不確定,可能會阻止我們或任何合作伙伴獲得部分或全部候選產品商業化的審批。 因此,我們無法預測我們或任何合作伙伴將在何時、是否以及在哪些地區獲得將候選產品商業化的營銷批准。 | |
| ● | 我們面臨着激烈的競爭,如果我們的競爭對手開發和銷售比我們開發的候選產品更有效、更安全或更便宜的產品 ,我們的商業機會將受到負面影響。 | |
| ● | 任何當前或未來候選產品的商業成功將取決於醫生、患者、付款人和醫學界其他人對市場的接受程度。 | |
| ● | 對於 某些候選產品,我們可能依賴開發和商業化合作夥伴來開發和進行臨牀試驗 ,獲得監管部門對候選產品的批准,如果獲得批准,則營銷和銷售候選產品。如果此類合作者未能按預期執行 ,我們從此類候選產品中獲得未來收入的潛力將顯著降低,我們的業務將受到損害。 | |
| ● | 我們 將依靠包括獨立臨牀研究人員和CRO在內的第三方來進行和贊助我們候選產品的一些臨牀試驗 。第三方未能履行其在候選產品臨牀開發方面的義務 候選產品可能會推遲或削弱我們獲得監管部門批准候選產品的能力。 | |
| ● | 如果我們無法為我們的產品和候選產品獲得並維護專利和其他知識產權保護,或者 如果獲得的專利和其他知識產權保護的範圍不夠廣泛,我們的競爭對手可能會開發 與我們相似或相同的產品並將其商業化,我們成功將我們的產品和候選產品商業化的能力可能會受到不利影響 。 | |
| ● | 我們 希望擴大我們的組織,因此,我們在管理我們的增長時可能會遇到困難,這可能會中斷我們的 運營。 | |
| ● | 我們 預計在可預見的未來不會支付現金股息,因此投資者不應預期他們的 投資將獲得現金股息。 |
| 34 |
與我們的財務狀況和額外資本需求相關的風險
我們 有重大運營虧損的歷史,預計在可預見的未來將出現重大且不斷增加的虧損, 我們可能永遠不會實現或保持盈利。
我們 預計短期內不會產生為我們的運營融資所必需的收入或盈利能力。截至2023年9月30日和2022年9月30日,我們的淨虧損分別為1,880萬美元和2,970萬美元。截至2023年9月30日,我們的累計赤字為1.102億美元。
到目前為止,我們還沒有將任何產品商業化,也沒有從產品銷售中獲得任何收入,如果沒有實現足夠的產品銷售收入,我們未來可能永遠不會實現盈利。我們將幾乎所有的財政資源和努力都投入到研究和開發上,包括臨牀前研究和臨牀試驗。我們的淨虧損可能會因季度和年度而大幅波動 。淨虧損和負現金流已經並將繼續對我們的股東(赤字)權益和營運資本產生不利影響。
我們 預計,如果我們:
● 繼續針對我們的主要候選產品SON-080和其他候選產品進行開發和臨牀試驗;
● 為任何未來的候選產品啟動並繼續研究、臨牀前和臨牀開發工作;
● 尋求發現和開發更多候選產品,並進一步擴大我們的臨牀產品線;
● 為成功完成臨牀試驗的任何候選產品尋求營銷和監管批准;
● 需要為臨牀開發和潛在的商業化生產更大數量的候選產品;
● 維護、擴大和保護我們的知識產權組合;
● 擴展我們的研發基礎設施,包括招聘和保留更多人員,如臨牀、質量控制和科學人員;
● 在未來建立銷售、營銷、分銷和其他商業基礎設施,以便將我們獲得 市場批准的產品商業化(如果有);
● 增加運營、財務和管理信息系統和人員,包括支持我們的產品開發和商業化的人員,並幫助我們履行上市公司的義務;以及
● 添加設備和物理基礎設施以支持我們的研發。
我們實現盈利並保持盈利的能力取決於我們授權產品和創造收入的能力。產品收入的產生 將取決於我們是否有能力為我們的一個或多個候選產品獲得市場批准,併成功將其商業化。
成功的 商業化將需要實現關鍵里程碑,包括完成我們的候選產品的臨牀試驗,獲得這些候選產品的營銷批准,製造、營銷和銷售我們或任何合作伙伴可以獲得營銷批准的產品,滿足任何上市後要求,並從私人保險 或政府付款人那裏獲得我們產品的報銷。由於與這些活動相關的不確定性和風險,我們無法準確預測收入的時間和金額,以及我們是否或何時可能實現盈利。我們和任何合作者可能永遠不會在這些活動中成功 ,即使我們或任何合作者成功了,我們也可能永遠不會產生足夠大的收入來實現盈利。即使我們確實實現了盈利,我們也可能無法維持或提高季度或年度盈利能力。
我們的 如果不能盈利並保持盈利,將壓低我們普通股的市場價格,並可能削弱我們籌集資金、擴大業務、使產品多樣化或繼續運營的能力。如果我們繼續蒙受損失,投資者可能無法從他們的投資中獲得任何回報,並可能失去他們的全部投資。
| 35 |
我們有限的運營歷史可能會使您很難評估我們業務到目前為止的成功程度,也難以評估我們未來的生存能力。
我們的業務於2015年開始運營。到目前為止,我們的業務僅限於為我們的公司提供資金和人員,開發我們的 技術,為我們的候選產品進行臨牀前研究和早期臨牀試驗,並尋求戰略合作 來推進我們的候選產品。我們尚未證明有能力成功進行後期臨牀試驗、獲得 上市批准、製造商業規模的產品或安排第三方代表我們這樣做,或進行成功的產品商業化所需的銷售和營銷活動。因此,您應該根據公司在開發早期階段經常遇到的成本、不確定性、延誤和困難來考慮我們的前景,特別是像我們這樣的臨牀階段的生物製藥公司。如果我們擁有更長的運營歷史或成功開發和商業化醫藥產品的歷史,您對我們未來成功或生存能力的任何預測都可能不像 那樣準確。
我們 在實現我們的業務目標時可能會遇到不可預見的費用、困難、複雜情況、延誤和其他已知或未知因素。 我們最終需要從一家專注於發展的公司過渡到一家能夠支持商業活動的公司。 我們在這種過渡中可能不會成功。
我們 預計,由於各種因素,我們的財務狀況和經營業績將繼續在每個季度和每年大幅波動 ,其中許多因素是我們無法控制的。因此,您不應依賴任何季度或年度業績作為未來運營業績的指標。
我們在運營中的反覆虧損令人對我們作為持續經營企業的持續經營能力產生了極大的懷疑。
我們 自成立以來一直在運營活動中產生經常性虧損和負現金流,我們預計在可預見的未來,主要由於我們潛在產品的研發成本,運營活動將產生虧損和負現金流 。截至2023年9月30日,我們擁有230萬美元的現金,股東赤字為20萬美元。我們相信,我們在2023年9月30日的現金,加上我們在2023年10月24日公開發行普通股和認股權證所獲得的410萬美元淨收益,將為我們預計到2024年3月的運營提供資金。我們還預計將從澳大利亞的研發税收激勵計劃獲得80萬美元的現金淨退款(請參閲綜合財務報表的附註2),並且最近初步批准了我們的申請,即通過技術營業税證書轉讓計劃出售最多480萬美元的新澤西州淨營業虧損 ,但須執行此類銷售(請參閲綜合財務報表的附註10)。我們將需要大量額外資金來為我們的運營提供資金。這些因素使人對我們作為一家持續經營的公司繼續經營的能力產生很大懷疑。隨附的綜合財務報表 乃以持續經營為基礎編制,考慮正常業務過程中的資產變現及負債清償情況 。合併財務報表不包括這種不確定性的結果 可能導致的任何調整。
我們 未來將通過股權或債務融資、合作伙伴關係、合作或其他來源需要更多資金,以開展我們計劃的開發活動。如果在需要時無法獲得額外資金,我們可能需要推遲或縮減運營 ,直到收到此類資金。各種內部和外部因素將影響我們的候選產品是否以及何時獲得市場營銷和成功商業化的批准 。我們候選產品的監管批准和市場接受度、開發和商業化這些候選產品的時間長度 以及這些候選產品的開發和商業化成本和/或它們在審批過程的任何階段失敗都將對我們的財務狀況和未來運營產生重大影響。
自成立以來,運營 主要包括組織我們、獲得融資、通過進行研究和開發來開發技術以及進行臨牀前研究。我們面臨着與其產品正在開發中的公司相關的風險。這些 風險包括需要額外資金來完成其研發、實現其研發目標、 保護其知識產權、招聘和留住技術人員以及對關鍵管理層成員的依賴。
我們作為持續經營企業的持續經營能力取決於我們籌集額外股本或債務資本或剝離非核心資產的能力 以籌集額外現金。如果我們無法籌集足夠的額外資金,我們可能需要採取成本削減措施 ,包括推遲或停止某些臨牀活動。
未來任何融資的來源、時間和可用性將主要取決於市場狀況,更具體地説,取決於我們臨牀開發計劃的進展。在需要的時候,可能根本就沒有資金,或者是在我們可以接受的條件下。由於缺乏必要的資金,我們可能需要推遲、縮減或取消部分或全部計劃中的臨牀試驗。除其他因素外,這些 因素使人對我們作為一家持續經營企業的持續經營能力產生了極大的懷疑。
| 36 |
我們 將需要大量額外資金,如果我們無法在需要時籌集資金,我們可能會被迫推遲、減少或取消我們的產品發現和開發計劃或商業化努力。
開發藥物產品,包括進行臨牀前研究和臨牀試驗,是一個非常耗時、昂貴和不確定的過程,需要數年時間才能完成。例如,在截至2023年9月30日和2022年9月30日的年度中,我們分別使用了2,130萬美元和2,780萬美元的淨現金用於我們的經營活動,基本上所有這些活動都與研發活動有關。我們 預計與我們正在進行的活動相關的費用將增加,特別是當我們啟動新的臨牀試驗,為我們當前的候選產品或任何未來的候選產品 啟動新的研究和臨牀前開發工作並尋求營銷批准 。此外,如果我們的任何候選產品獲得營銷批准,我們可能會產生與產品銷售、營銷、製造和分銷相關的鉅額商業化費用 ,因為此類銷售、營銷、製造和分銷不是合作伙伴的責任。此外,由於合併,我們將繼續產生與上市公司運營相關的鉅額成本。因此,我們將需要獲得與我們持續運營相關的大量額外資金。如果我們無法在需要時或在有吸引力的條件下籌集資金,我們可能會被迫推遲、減少或取消我們的研發計劃或任何未來的商業化努力。
我們 將需要花費大量資金來推進我們正在開發的候選產品以及我們可能尋求開發的其他候選產品的開發。此外,雖然我們可能會為我們的 候選產品的未來開發尋找一個或多個合作伙伴,但我們可能無法以合適的 條款及時或根本無法與我們的任何候選產品合作。無論如何,我們現有的現金將不足以為我們計劃進行的所有工作提供資金,也不足以為我們的任何候選產品的開發完成提供資金。因此,我們將被要求通過公開或私募股權發行、債務融資、合作和許可安排或其他來源獲得更多 資金。我們沒有任何承諾的外部資金來源。我們可能無法按可接受的條款或全部獲得足夠的額外融資。我們未能在需要時籌集資金,將對我們的財務狀況和我們實施業務戰略的能力產生負面影響。
我們的 估計可能被證明是錯誤的,我們可以比目前預期的更快地使用可用的資本資源。此外,不斷變化的情況,其中一些情況可能超出我們的控制,可能會導致我們消耗資本的速度大大快於我們目前的預期,我們 可能需要比計劃更早地尋求額外資金。我們未來的資金需求,無論是短期還是長期,將取決於許多因素,包括:
● 我們當前和未來的候選產品的臨牀試驗的範圍、進度、時間、成本和結果,以及研究和臨牀前開發工作;
● 我們訂立任何合作、許可或其他安排的能力、條款和時間安排;
● 我們為我們的管道確定一個或多個未來候選產品的能力;
● 我們追求的未來候選產品的數量及其開發要求;
● 尋求監管批准的結果、時間和成本;
● 任何獲得營銷批准的候選產品的商業化活動的成本,只要此類成本 不是任何合作者的責任,包括建立產品銷售、營銷、分銷 和製造能力的成本和時間;
● 從我們當前和未來候選產品的商業銷售中獲得的營銷批准、收入(如果有);
● 隨着我們擴大研發和建立商業基礎設施,我們的員工人數增長和相關成本;
● 準備、提交和起訴專利申請、維護和保護我們的知識產權(包括 執行和辯護知識產權相關索賠)的成本;以及
● 作為上市公司運營的成本。
| 37 |
籌集 額外資本可能會稀釋我們現有股東的權益,限制我們的運營或導致我們放棄寶貴的權利。
We may seek additional capital through a combination of public and private equity offerings, debt financings, strategic partnerships and alliances, licensing arrangements or monetization transactions. To the extent that we raise additional capital through the sale of equity, convertible debt securities or other equity-based derivative securities, your ownership interest will be diluted and the terms may include liquidation or other preferences that adversely affect your rights as a shareholder. Any indebtedness we incur would result in increased fixed payment obligations and could involve restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. Furthermore, the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our common stock to decline and existing shareholders may not agree with our financing plans or the terms of such financings. If we raise additional funds through strategic partnerships and alliances, licensing arrangements or monetization transactions with third parties, we may have to relinquish valuable rights to our technologies, or our product candidates, or grant licenses on terms unfavorable to us. Adequate additional financing may not be available to us on acceptable terms, or at all. If we are unable to raise additional funds when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
與我們候選產品的發現、開發和監管批准相關的風險
冠狀病毒COVID-19大流行或任何其他傳染病的廣泛爆發可能對我們的 業務、財務狀況及經營業績造成重大不利影響。
我們 面臨與流行病或傳染病爆發相關的風險,例如,最近在世界各地爆發的 高傳染性和致病性冠狀病毒COVID-19。此類傳染病的爆發可能導致廣泛的健康 危機,從而對許多國家的一般商業活動以及經濟和金融市場產生不利影響。
於 2019年12月,一種新型冠狀病毒株COVID-19據報道在中國武漢出現,並於2020年3月11日被世界衞生組織宣佈為大流行。COVID-19對我們臨牀前和臨牀試驗運營的影響程度將 取決於未來的發展,這些發展具有高度不確定性,無法有信心地預測,例如爆發的持續時間和地理範圍、COVID-19的嚴重程度以及遏制和治療COVID-19的行動的有效性。
世界各地許多 國家對旅行和大型集會實施禁令和限制,以減緩COVID-19的傳播,並關閉了非必要的業務,儘管這些限制目前在大多數情況下已經失效。此類事件可能導致 業務、供應和藥品生產中斷,並導致運營減少,其中任何一項都可能對我們的業務、 財務狀況和運營結果產生重大影響。
這種 大流行或爆發可能導致難以確保臨牀試驗中心位置、CRO和/或試驗監查員以及支持試驗的其他關鍵 供應商和顧問的安全。此外,在臨牀試驗地點附近爆發或感知到爆發 可能會影響我們招募患者的能力。這些情況或其他與COVID-19相關的情況可能導致我們的臨牀 試驗計劃延遲,並可能增加預期成本,所有這些都可能對我們的業務及其財務狀況產生重大不利影響。
特別是,我們的管道產品(SON-1010除外)的生產因COVID-19相關供應鏈問題而延遲,特別是原材料(包括培養基、樹脂和分析試劑盒)的供應,加上國際運輸延遲。然而,COVID-19目前並未影響我們的業務。
| 38 |
雖然 新冠肺炎或任何其他傳染病的廣泛爆發可能帶來的潛在經濟影響及其持續時間可能難以評估或預測 但大流行可能導致全球金融市場嚴重混亂, 降低我們獲得資本的能力,這可能在未來對我們的流動性產生負面影響。此外,新冠肺炎或任何其他傳染性疾病傳播導致的經濟衰退或 市場回調可能會對我們的業務和 我們普通股的價值產生重大影響。
疫情還可能影響我們的員工和與我們合作的各方開展我們的非臨牀、臨牀和藥物生產活動的能力。我們依賴或將來可能依賴臨牀站點、研究人員和其他研究人員、顧問、獨立承包商、合同研究組織和其他第三方服務提供商來幫助我們管理、監控 以及以其他方式執行我們的非臨牀研究和臨牀試驗。我們還依賴或將來可能依賴顧問、獨立承包商、合同製造組織和其他第三方服務提供商來協助我們管理、監控和以其他方式執行我們的原料藥生產、配方和藥品製造活動。大流行將影響任何這些外部人員、組織或公司將足夠的時間和資源投入到我們的計劃或為我們執行工作的旅行的能力。
新冠肺炎爆發或任何其他傳染病的廣泛爆發對當前或未來臨牀研究的進行可能產生的負面影響包括延遲從監管機構獲得反饋、啟動新的臨牀研究以及招募 受試者參加正在登記的研究。潛在的負面影響還包括無法在研究地點進行研究訪問, 安全性和有效性數據收集不完整,正在進行的研究中受試者輟學率較高,研究數據進入數據庫的地點延遲,由於對研究地點的物理訪問限制,研究數據的監控延遲,站點對查詢的響應延遲,數據庫鎖定延遲,數據分析延遲,頂級數據的時間延遲,以及完成研究報告的延遲 。新的或惡化的新冠肺炎(或任何其他傳染性疾病)中斷或限制可能會 對我們的非臨牀研究、臨牀試驗和藥物生產活動產生進一步的負面影響。
我們 在很大程度上依賴於我們內部開發計劃的成功,我們的候選產品可能無法 成功完成臨牀試驗、獲得監管批准或成功商業化。
我們未來的成功將在很大程度上取決於我們內部開發計劃和我們渠道計劃候選產品的成功。
我們成功地將我們的渠道和其他候選產品商業化的能力將取決於我們的能力,其中包括:
● 成功完成臨牀前研究和臨牀試驗;
● 獲得美國食品和藥物管理局、歐洲藥品管理局和其他類似監管機構的監管批准;
● 建立並維護與第三方的合作,以開發和/或商業化我們的候選產品,或者 建立並維護強大的開發、銷售、分銷和營銷能力,足以開發產品並啟動任何經批准的產品的商業銷售 ;
● 從政府醫療保健系統和保險公司等付款人那裏獲得保險和足夠的補償,並實現具有商業吸引力的定價水平;
● 確保醫生、醫療保健付款人、患者和醫學界接受我們的產品候選;
● 通過驗證的流程,在經過監管機構(包括食品和藥物管理局)檢查和批准的製造設施中生產足夠數量的候選產品,以實現成功的商業化;
● 在臨牀試驗和商業化導致費用增加的情況下管理我們的支出;以及
● 為任何經批准的產品和候選產品獲取並強制執行足夠的知識產權。
| 39 |
在製藥業大量正在開發的藥物中,只有一小部分藥物 向FDA提交了新藥申請(NDA)或生物製品許可申請(BLA),更少的藥物獲得了商業化批准。此外, 即使我們確實獲得了營銷我們的候選產品的監管批准,任何此類批准也可能會受到我們可能營銷該產品的指定用途或患者羣體的限制。因此,即使我們能夠獲得必要的資金以繼續為我們的開發計劃提供資金,我們也不能向您保證我們的候選產品將成功開發或商業化。 如果我們無法開發或獲得監管部門的批准,或者如果我們的候選產品無法成功商業化, 我們可能無法產生足夠的收入來繼續我們的業務。
我們 處於開發工作的非常早期階段,我們的候選產品代表着一種新的藥物類別,可能會 受到更嚴格的監管審查,直到它們被確立為一種治療方式。
我們的 候選流水線產品代表了一種新的治療方式,包括使用完全人類白蛋白結合結構域來提供 治療產品。我們的候選產品可能無法在患者身上展示我們認為它們 可能具有的任何或全部藥理益處。我們還沒有、也可能永遠不會成功地證明這些或任何其他候選產品的有效性和安全性 在臨牀試驗中或之後獲得市場批准。
監管機構對我們的候選產品沒有經驗,可能要求提供超出我們 在開發計劃中所包含的安全性和有效性的證據。在這種情況下,我們候選產品的開發可能比 預期的成本或時間更高,我們的候選產品可能被證明是不可行的。
如果我們的開發努力不成功,我們可能無法推進候選產品的開發、將 產品商業化、籌集資金、擴大業務或繼續運營。
我們的候選產品和任何合作伙伴的產品都將需要經過耗時且昂貴的臨牀前和臨牀試驗, 其結果不可預測,而且失敗的風險很高。如果我們或他們的候選產品的臨牀前或臨牀試驗未能令人滿意地向FDA、EMA和任何其他類似的監管機構證明安全性和有效性, 可能會產生額外的成本或延遲完成,這些候選產品的開發或他們的開發可能會被放棄 。
美國的FDA、歐盟和歐洲經濟區的EMA以及其他司法管轄區的其他類似監管機構必須批准新的候選產品,然後才能在這些地區進行營銷、推廣或銷售。我們之前沒有 向FDA提交過IND或BLA,也沒有向類似的外國監管機構提交過類似的藥品審批文件。我們必須向這些監管機構提供來自臨牀前研究和臨牀試驗的數據,以證明我們的候選產品對於特定適應症是安全有效的,然後才能被批准用於商業銷售。 我們不能確定我們的候選產品的臨牀試驗是否會成功,或者我們的任何候選產品將 獲得FDA、EMA或任何其他類似監管機構的批准。
臨牀前研究和臨牀試驗是漫長、昂貴和不可預測的過程,可能會受到廣泛延誤的影響。我們不能保證 任何臨牀試驗將按計劃進行或按計劃完成(如果有的話)。完成候選產品商業化所需的臨牀前研究和臨牀試驗可能需要數年時間並需要大量的 支出,而延遲或失敗在本質上是不可預測的,並且在任何階段都可能發生。除了我們預期的試驗和測試之外,我們還可能需要對我們的候選產品進行額外的臨牀試驗或其他測試 ,這可能會導致我們產生額外的計劃外成本 或導致臨牀開發的延遲。此外,我們可能需要重新設計或以其他方式修改與正在進行或計劃中的臨牀試驗有關的計劃,而更改臨牀試驗的設計可能既昂貴又耗時。在一個或多個試驗中出現不利的 結果將是我們的候選產品和我們的重大挫折。一個或多個試驗中的不利結果 可能需要我們推遲、縮小或取消一個或多個產品開發計劃,這可能會對我們的業務、財務狀況、運營結果和未來增長前景產生實質性的不利影響。
許多導致或導致臨牀試驗延遲開始或完成的因素也可能最終導致我們的候選產品被拒絕上市批准。FDA、EMA或任何其他類似的監管機構可能不同意我們的臨牀試驗設計和我們對臨牀試驗數據的解釋,或者即使在審查了 並對我們的臨牀試驗設計進行了評論後,也可能更改審批要求。
| 40 |
在我們候選產品的臨牀試驗中,我們面臨許多風險,包括以下風險:
● 對於相同的適應症,候選產品無效或劣於現有批准的產品;
● 候選產品導致或與不可接受的毒性或具有不可接受的副作用相關;
● 患者可能死亡或遭受不良反應,其原因可能與正在測試的候選產品有關,也可能與之無關;
● 該結果可能不能證實早期試驗的陽性結果;
● 結果可能不符合食品和藥物管理局、食品藥品監督管理局或其他相關監管機構所要求的統計顯著性水平,以確定我們的候選產品的安全性和有效性,以供繼續試驗或上市批准;以及
● 我們的協作者可能無法或不願履行其合同。
此外, 出於規劃目的,我們有時會估計完成各種科學、臨牀、法規和其他產品開發目標的時間。 這些里程碑可能包括我們對開始或完成科學研究、臨牀試驗、提交監管文件或商業化目標的期望。我們可能會不時地公開宣佈其中一些里程碑的預期時間,例如完成正在進行的臨牀試驗、啟動其他臨牀計劃、 收到市場批准或產品的商業發佈。其中許多里程碑的實現可能超出了我們的控制範圍。所有這些里程碑都基於各種假設,這可能會導致里程碑的實現時間與我們的估計大不相同。如果我們未能在預期的時間範圍內實現里程碑,我們的候選產品的商業化可能會被推遲,我們可能沒有資格獲得某些合同付款,這可能會對我們的業務、財務狀況、運營結果和未來增長前景產生實質性的不利影響 。
我們 可能會發現很難將患者納入我們的臨牀試驗,這可能會推遲或阻止我們繼續進行候選產品的臨牀試驗 。
確定 並使患者有資格參與我們候選產品的臨牀試驗對我們的成功至關重要。我們臨牀試驗的時間取決於我們招募患者參與的能力以及所需的隨訪期的完成情況。由於與新治療方法相關的不良事件的負面宣傳、類似患者羣體的競爭性臨牀試驗、當前治療方法的存在或其他原因,患者可能 不願參與我們的臨牀試驗。對於我們可能針對一個或多個可能是罕見疾病的候選產品的某些跡象,登記風險 會增加,這可能會限制我們計劃的臨牀試驗中可能登記的患者池。招募患者、進行 試驗和獲得我們候選產品的監管批准的時間表可能會延遲,這可能會導致成本增加、延遲推進我們的候選產品、延遲測試候選產品的有效性或完全終止臨牀試驗。
我們 可能無法識別、招募和招募足夠數量的患者或具有所需或所需特徵的患者, 無法及時完成我們的臨牀試驗。例如,由於我們最初針對的適應症的性質,具有晚期疾病進展的患者可能不適合使用我們的候選產品進行治療,並且可能沒有資格參加我們的臨牀試驗。因此,我們的目標疾病患者的早期診斷對我們的成功至關重要。患者 登記和試驗完成受以下因素影響:
● 患者羣體的規模和識別受試者的流程;
● 試驗方案設計;
| 41 |
● 資格和排除標準;
● 到目前為止,正在研究的候選產品的安全概況;
● 被研究的產品候選產品的感知風險和收益;
● 我們治療疾病的方法的風險和益處;
● 競爭療法和臨牀試驗的可用性;
● 正在調查的疾病的嚴重程度;
● 入組時受試者疾病的進展程度;
● 潛在受試者的臨牀試驗地點的鄰近性和可用性;
● 獲得和保持受試者知情同意的能力;
● 入組受試者在完成試驗前退出的風險;
● 醫生的患者轉診實踐;以及
● 在治療期間和治療後充分監測受試者的能力。
此外,SON-080中試規模可行性研究的臨牀開發目前計劃在美國境外進行。我們 在任何外國成功啟動、入組和完成臨牀試驗的能力受制於 在外國開展業務所特有的眾多風險,包括:
● 難以建立或管理與學術合作伙伴或合同研究組織或CRO和醫生的關係;
● 開展臨牀試驗的不同標準;
● 在一些國家,缺乏具有足夠監管專業知識的既定小組來審查與我們的新 方法相關的協議;
● 我們無法找到合格的當地顧問、醫生和合作夥伴;以及
● 遵守各種外國法律、醫療標準和監管要求的潛在負擔,包括藥品和生物技術產品和治療的監管 。
如果 我們難以招募足夠數量的患者來按計劃開展臨牀試驗,我們可能需要延遲、限制或 終止正在進行或計劃進行的臨牀試驗,任何此類措施都將對我們的業務、財務狀況、 經營業績和前景產生不利影響。
臨牀前研究和早期臨牀試驗的結果可能不能預測未來臨牀試驗的結果。
臨牀前研究和早期臨牀試驗的 結果可能無法預測後期臨牀試驗的成功,臨牀試驗的中期結果 也不一定能預測已完成臨牀試驗結果的成功。製藥 和生物技術行業的許多公司在早期 開發取得積極成果後,在後期臨牀試驗中遭遇重大挫折,我們也可能面臨類似的挫折。例如,SON-080的IIa期試驗將在美國境外進行,並且 在我們或 我們的合作者進行的後期臨牀試驗中,這些發現可能不會在全球臨牀試驗中心的未來試驗中重現。臨牀試驗的設計可以確定其結果是否支持產品的批准,並且臨牀試驗設計中的缺陷可能在臨牀試驗充分推進之前不會變得明顯。我們可能無法設計和執行 臨牀試驗以支持上市批准。
| 42 |
臨牀前 和臨牀數據通常容易受到不同解釋和分析的影響。許多公司認為其候選產品 在臨牀前研究和臨牀試驗中表現令人滿意,但未能獲得候選產品的上市批准。即使我們或任何合作者認為我們候選產品的臨牀試驗結果值得上市 批准,FDA或類似的外國監管機構可能不同意,並且可能不會授予我們候選產品的上市批准。
在 某些情況下,由於許多因素,包括方案中規定的試驗程序的變化、 患者人羣的規模和類型的差異、給藥方案和其他臨牀試驗方案的變化和依從性以及 臨牀試驗參與者的脱落率,同一候選產品 的不同臨牀試驗之間的安全性或有效性結果可能存在顯著差異。如果我們的候選產品的臨牀試驗未能收到積極的結果,我們最先進的候選產品的開發時間軸 和監管批准以及商業化前景,以及相應地,我們的業務 和財務前景將受到負面影響。
我們的 當前或未來候選產品在單獨使用或與 其他獲批產品或研究性新藥聯合使用時,可能會導致不良副作用或具有其他特性,從而可能停止其臨牀開發、阻止其上市批准、限制 其商業潛力或導致重大負面後果。
可能會發生不良 或臨牀上無法管理的副作用,導致我們或監管機構中斷、延遲或停止臨牀試驗 ,並可能導致FDA或類似的外國監管機構對標籤進行更嚴格的限制或延遲或拒絕上市批准 。我們的試驗結果可能會揭示出副作用或意外特徵的高和不可接受的嚴重程度和患病率。
如果我們的候選產品在開發過程中出現不可接受的副作用,我們、FDA或類似的外國監管機構、機構審查委員會或IRBs或我們進行研究的機構的獨立道德委員會,或者 數據安全監控委員會或DSMB可以暫停或終止我們的臨牀試驗,或者FDA或類似的外國監管機構可以命令我們停止臨牀試驗或拒絕批准我們的產品候選產品的任何或所有目標適應症。與治療相關的副作用也可能影響患者招募或受試者完成試驗的能力,或導致潛在的產品責任索賠。此外,治療醫務人員可能無法正確識別或管理這些副作用。 我們可能需要對使用我們的候選產品的醫務人員進行培訓,以瞭解我們的臨牀試驗以及我們的任何候選產品商業化後的副作用概況。在識別或管理我們的候選產品的潛在副作用方面培訓不足 可能會導致患者受傷或死亡。這些情況中的任何一種都可能阻止我們實現或保持 市場對受影響候選產品的接受程度,並可能嚴重損害我們的業務、財務狀況和前景。
此外,我們候選產品的臨牀試驗是在精心定義的患者組中進行的,這些患者已同意進入臨牀 試驗。因此,我們的臨牀試驗可能會顯示候選產品的明顯正面效果大於實際正面效果(如果有的話),或者無法識別不良副作用。如果在 候選產品批准後,我們或其他人發現該產品的效果不如之前認為的那樣有效,或者造成了以前未確定的不良副作用 ,則可能會發生以下任何後果:
● 監管部門可以撤銷對該產品的批准或扣押該產品;
● 我們或任何合作者可能需要召回產品,或被要求更改產品的給藥方式,或進行額外的 臨牀試驗;
● 可對特定產品的營銷或製造工藝施加額外限制;
| 43 |
● 我們可能會受到罰款、禁令或施加民事或刑事處罰;
● 監管機構可能要求添加標籤聲明,如方框警告或禁忌症;
● 我們或任何合作者可能被要求創建一份藥物指南,概述以前未確定的副作用的風險 以分發給患者;
● 我們或任何合作者可能會被起訴,並對給患者造成的傷害承擔責任;
● 產品可能變得不那麼有競爭力;以及
● 我們的聲譽可能會受到影響。
如果我們當前或未來的任何候選產品在臨牀試驗中不能證明安全性和有效性,或者沒有獲得市場批准,我們將無法產生收入,我們的業務將受到損害。這些事件中的任何一個都可能損害我們的業務和運營, 並可能對我們普通股的價格產生負面影響。
我們 在確定或發現其他候選產品方面可能不會成功。
儘管 除了我們目前正在開發的候選產品外,我們打算探索其他治療機會,但由於多種原因,我們可能無法確定用於臨牀開發的其他候選產品。例如,我們的研究方法可能無法成功識別潛在的候選產品,或者我們發現的產品可能具有有害的副作用或其他特徵,使其無法銷售或不太可能獲得監管部門的批准。在商業銷售之前,其他候選產品將需要額外、耗時的 開發工作,包括臨牀前研究、臨牀試驗和FDA和/或適用的外國監管機構的批准。所有候選產品都容易出現藥品開發中固有的失敗風險 。如果我們不能發現和開發更多潛在的候選產品,我們可能無法發展我們的業務,我們的運營結果可能會受到實質性的損害。
我們 可能會花費有限的資源來追求特定的候選產品或指示,而無法利用可能更有利可圖或成功可能性更大的候選產品或指示 。
由於 我們的財務和管理資源有限,因此我們打算專注於為 我們認為最有可能成功的特定適應症開發候選產品,無論是在市場批准方面還是在商業化方面。因此,我們 可能會放棄或推遲尋求其他候選產品或可能被證明具有更大商業潛力的其他指示 。
我們的資源分配決策可能會導致我們無法利用可行的商業產品或有利可圖的市場機會。我們在當前和未來研發計劃以及特定適應症候選產品上的支出可能不會產生任何商業上可行的候選產品。如果我們不能準確評估特定候選產品的商業潛力或目標市場, 在保留候選產品的獨家開發和商業化權利對我們更有利的情況下,我們可能會通過協作、許可或其他版税安排放棄對該候選產品的寶貴權利。
| 44 |
我們 面臨潛在的產品責任,如果針對我們的索賠成功,我們可能會招致大量責任和成本。如果 使用我們的候選產品傷害了患者,或者即使這種傷害與我們的候選產品無關,也被認為傷害了患者, 我們的監管批准可能會被撤銷或以其他方式受到負面影響,我們可能會受到代價高昂且具有破壞性的產品責任索賠 。
在臨牀試驗中使用我們的候選產品以及銷售我們獲得市場批准的任何產品,都會使我們面臨產品責任索賠的風險。患者、醫療保健提供者、製藥公司或銷售或以其他方式接觸我們產品的其他人可能會向我們提出產品責任索賠。我們的候選產品可能會引發 不良事件。如果我們不能成功地抗辯產品責任索賠,我們可能會招致大量的責任和成本。此外,無論優點或最終結果如何,產品責任索賠可能會導致:
| ● | 我們的商業聲譽受到損害; | |
| ● | 臨牀試驗參與者的退出; | |
| ● | 給予患者或其他索賠人鉅額的金錢獎勵; | |
| ● | 因相關訴訟產生的費用 ; | |
| ● | 將管理層的注意力從我們的主要業務上分散; | |
| ● | 無法將我們的候選產品商業化;以及 | |
| ● | 減少了對我們的候選產品的需求(如果獲得批准用於商業銷售)。 |
鑑於我們目前的臨牀計劃,我們 打算購買產品責任保險;但是,我們可能無法以合理的成本或足夠的金額維持 保險範圍,以保護我們免受因責任造成的損失。我們打算在每次商業化新產品時擴大我們的保險範圍;但是,我們可能無法以商業上合理的條款或足夠的金額獲得產品責任保險。有時,在基於 具有意想不到的不良影響的藥物或治療的集體訴訟中,會做出大額判決。成功的產品責任索賠或針對我們提出的一系列索賠 可能會導致我們的股票價格下跌,如果判決超出我們的保險範圍,可能會對我們的運營和業務業績產生不利影響 。
患有我們的某些候選產品所針對的疾病的患者 通常已經處於嚴重的 和晚期疾病,並且具有已知和未知的重大預先存在的和潛在威脅生命的健康風險。 在治療過程中,患者可能會因可能與我們的候選產品相關的原因而遭受包括死亡在內的不良事件。 此類事件可能會使我們面臨代價高昂的訴訟,要求我們向受傷的患者支付鉅額費用,延誤、產生負面影響或終止我們獲得或保持監管部門批准以營銷我們產品的機會,或要求我們暫停或放棄我們的商業化努力。即使在我們不認為不良事件與我們的產品相關的情況下,對該情況的 調查也可能很耗時或不確定。這些調查可能會中斷我們的銷售工作,推遲我們的監管審批流程,或者影響和限制我們的產品候選接收或維護的監管審批類型。由於這些因素,產品責任索賠即使成功辯護,也可能對我們的業務、財務狀況或運營結果產生重大不利影響。
我們 可以向FDA和其他類似的監管機構為我們的候選產品尋求認證,這些認證旨在提供更快的開發過程或更快的監管途徑等好處,但不能保證我們會成功 獲得此類認證。此外,即使我們的一個或多個候選產品獲得此類指定,我們也可能無法 實現此類指定的預期好處。
FDA和其他類似的監管機構為候選產品提供了特定的名稱,旨在鼓勵 針對重大未得到滿足的醫療需求的情況進行藥物產品的研發。這些指定可能會 帶來好處,例如與監管機構的額外互動、可能加速的監管途徑和優先審查。我們不能保證我們的任何其他候選產品都能成功獲得這樣的認證。此外,雖然此類指定可以加快開發或審批流程,但它們通常不會更改審批標準。 即使我們獲得了一個或多個候選產品的此類指定,也不能保證我們將實現預期的 好處。
| 45 |
例如,我們可能會為我們的一個或多個候選產品尋求突破性治療認證。突破性療法被定義為:如果初步臨牀證據表明,突破性療法在一個或多個臨牀重要終點(例如在臨牀開發早期觀察到的實質性治療效果)較現有療法有顯著改善,則該療法旨在單獨或與一種或多種其他療法相結合來治療嚴重或危及生命的疾病或狀況。對於已被指定為突破性療法的療法,FDA和試驗贊助商之間的互動和溝通可以幫助 確定臨牀開發的最有效途徑,同時將無效對照方案的患者數量降至最低。 被FDA指定為突破性療法的療法也有資格獲得加速批准。指定為突破性療法 是FDA的自由裁量權。因此,即使我們認為我們的候選產品之一符合指定為突破性療法的標準,FDA也可能不同意,而是決定不進行此類指定。在任何情況下,與根據FDA傳統程序考慮批准的療法相比,收到針對候選產品的突破性治療指定 可能不會導致更快的開發過程、審查或批准,也不能確保FDA最終批准。此外,即使我們的一個或多個候選產品符合突破性療法的條件,FDA也可能在以後決定這些候選產品不再 符合資格條件。
我們 可能還會為我們的一些候選產品尋求快速通道認證。如果一種療法旨在治療一種嚴重或危及生命的疾病,並且該療法顯示出解決這種疾病未得到滿足的醫療需求的潛力,則治療贊助商可以申請 快速通道認證。FDA擁有廣泛的自由裁量權來決定是否授予該稱號,因此,即使我們認為特定的候選產品有資格獲得該稱號,也不能保證FDA會決定授予該稱號。即使我們確實獲得了快速通道認證,與傳統的FDA程序相比,我們可能不會體驗到更快的開發流程、審查或批准, 而且獲得快速通道認證並不能保證FDA的最終批准。如果FDA認為我們的臨牀開發計劃的數據不再支持該指定,它可能會撤回該指定 。
我們 可能會為我們的一個或多個候選產品尋求優先審查指定,但我們可能不會收到這樣的指定,即使我們這樣做了,這樣的指定也可能不會帶來更快的監管審查或審批過程。
如果FDA確定一種候選產品提供了治療嚴重疾病的方法,並且如果獲得批准,該產品將在安全性或有效性方面提供 顯著改善,FDA可以指定該候選產品進行優先審查。指定優先審查 意味着FDA審查申請的目標是六個月,而不是標準的十個月審查期。 我們可能會要求對我們的候選產品進行優先審查。FDA在是否向候選產品授予優先審查地位方面擁有廣泛的自由裁量權,因此,即使我們認為特定候選產品有資格獲得此類資格或地位, 特別是如果該候選產品已獲得突破療法指定,FDA也可能決定不授予該資格。此外,與FDA的傳統程序相比,優先審查指定並不一定會加快開發,也不一定會加快監管審查或審批過程,也不一定會帶來任何批准方面的優勢。獲得FDA的優先審查並不保證在六個月的審查週期內或根本不能獲得批准。
在一個司法管轄區獲得並保持我們當前和未來候選產品的營銷批准,並不意味着我們將成功 在其他司法管轄區獲得當前和未來候選產品的營銷批准。
在一個司法管轄區獲得並維護我們當前和未來候選產品的營銷批准並不能保證我們將能夠在任何其他司法管轄區獲得或保持營銷批准,而在一個司法管轄區未能或延遲獲得營銷批准可能會對其他司法管轄區的營銷審批流程產生負面影響。例如,即使FDA批准銷售候選產品,外國司法管轄區的可比監管機構也必須批准候選產品在這些國家/地區的製造、營銷和推廣。審批程序因司法管轄區而異,可能涉及與美國不同或大於美國的要求 和行政審查期限,包括額外的臨牀前研究 或臨牀試驗,因為在一個司法管轄區進行的臨牀研究可能不會被其他司法管轄區的監管機構接受。 在美國以外的許多司法管轄區,候選產品必須先獲得報銷批准,然後才能在該司法管轄區 銷售。在某些情況下,我們打算為我們的產品收取的價格也需要得到批准。我們沒有在國際市場獲得報銷或定價審批的經驗。
| 46 |
獲得市場批准並遵守監管要求可能會給我們帶來重大延誤、困難和成本,而 可能會推遲或阻止我們的產品在美國以外的某些國家/地區推出。如果我們不遵守國際市場的監管要求和/或獲得相應的營銷批准,我們的目標市場將會減少 ,我們充分發揮候選產品市場潛力的能力將受到損害。
與我們的候選產品商業化相關的風險以及其他合規問題
即使我們完成了必要的臨牀前研究和臨牀試驗,營銷審批流程也是昂貴、耗時且不確定的,可能會阻止我們或任何合作者獲得將部分或全部候選產品商業化的批准。 因此,我們無法預測我們或任何合作者將在何時、是否以及在哪些地區獲得將候選產品商業化的營銷審批。
在美國和國外獲得營銷批准的過程漫長、昂貴且不確定。如果最終獲得批准,可能需要 年,而且可能會根據各種因素而變化很大,這些因素包括所涉及的候選產品的類型、複雜性和新穎性。要獲得上市批准,需要針對每個治療適應症向監管機構提交廣泛的臨牀前和臨牀 數據和支持信息,以確定候選產品的安全性和有效性。要獲得市場批准,還需要向監管機構提交有關產品製造過程的信息,並由監管機構檢查製造設施。FDA或其他監管機構可能會認定我們的候選產品不安全有效、僅有中等效果或有不良或意外的副作用、毒性或其他使我們無法獲得上市批准或阻止或限制商業使用的特徵。我們 最終獲得的任何營銷批准都可能受到限制,或受到限制或批准後的承諾,從而使批准的產品在商業上無法 生存。
此外,開發期間市場審批政策的更改、附加法規、法規或指南的制定或頒佈的更改,或針對每個提交的產品申請的監管審查的更改,都可能導致申請審批的延遲 或拒絕。監管機構在審批過程中擁有相當大的自由裁量權,可能拒絕接受任何 申請,或可能決定我們的數據不足以獲得批准,需要進行額外的臨牀前、臨牀或其他研究。 對從臨牀前和臨牀測試中獲得的數據的不同解釋可能會推遲、限制或阻止候選產品的上市批准。在適當的監管機構審核並批准候選產品之前,我們不能將產品商業化。即使我們的候選產品在臨牀試驗中表現出安全性和有效性,監管機構也可能無法及時完成其審查流程,或者我們可能無法獲得監管部門的批准。如果FDA諮詢委員會或其他監管機構建議不批准或限制批准,可能會導致額外的延遲。此外,在產品開發、臨牀試驗和審查過程中,我們可能會因未來立法或行政行動中的額外政府監管或監管機構政策的變化而出現延誤或拒絕。我們最終 獲得的任何營銷批准都可能受到限制,或受到限制或批准後的承諾,從而使批准的產品在商業上不可行。
此外,我們臨牀試驗的首席研究員可能會不時擔任我們的科學顧問或顧問,並獲得與此類服務相關的報酬 。在某些情況下,我們可能需要向FDA或其他監管機構報告其中一些關係。FDA或其他監管機構可能會得出結論,認為我們與主要調查員之間的財務關係造成了利益衝突或以其他方式影響了對研究的解釋。因此,FDA或其他監管機構可能會質疑在適用的臨牀試驗現場生成的數據的完整性,臨牀試驗本身的效用可能會受到威脅。這可能會導致FDA或其他監管機構延遲批准或拒絕我們的上市申請,並可能最終導致我們的一個或多個候選產品的上市審批被拒絕。
此外,監管機構可能不會批准我們的候選產品成功商業化所必需或需要的標籤聲明 。例如,監管機構可能批准的候選產品的適應症比要求的更少或更少,也可能根據上市後研究的表現給予批准。監管機構可能會批准適用於患者人數較少、不同藥物配方或不同製造工藝的候選產品,而不是我們正在尋求的產品。如果我們無法 獲得必要的監管批准,或者監管批准比我們預期的更有限,我們的業務、前景、財務狀況 和運營結果可能會受到影響。
任何延遲獲得或未能獲得所需批准的 都可能對我們從特定候選產品獲得收入的能力產生負面影響,這可能會對我們的財務狀況造成重大損害,並對我們的普通股價格產生不利影響。
| 47 |
我們 目前沒有與我們的候選產品相關的營銷、銷售或分銷基礎設施。如果我們不能通過自己或通過與營銷合作伙伴的合作來發展我們的銷售、營銷和分銷能力,我們將不會成功地將我們的候選產品商業化。
我們 目前沒有營銷、銷售或分銷能力,在我們組織內的銷售或營銷經驗有限。 如果我們的一個或多個候選產品獲得批准,我們打算建立一個具有技術專業知識和支持分銷能力的銷售和營銷組織,以將該候選產品商業化,或者將此功能外包給第三方 。建立我們自己的銷售和營銷能力並與 第三方達成協議來執行這些服務都存在風險。
招聘和培訓內部商業組織既昂貴又耗時,可能會推遲任何產品發佈。這些 部分或全部費用可能會在我們的任何候選產品獲得批准之前發生。如果我們招聘銷售團隊並建立營銷能力的 候選產品的商業發佈因任何原因而推遲或沒有發生,我們將過早地 或不必要地產生這些商業化費用。如果我們不能留住或重新定位我們的銷售和營銷人員,成本可能會很高,而且我們的投資將會損失。此外,我們可能無法在美國或在我們打算瞄準的醫療市場中擁有足夠規模或具有足夠專業知識的其他目標市場招聘銷售人員。
可能會阻礙我們將候選產品商業化的因素 包括:
● 無法招聘、培訓和留住足夠數量的有效銷售和營銷人員;
● 銷售人員無法接觸醫生或説服足夠數量的醫生開出我們可能開發的任何未來產品的處方 ;
● 銷售人員提供的補充治療不足,這可能使我們相對於擁有更廣泛產品線的公司 處於競爭劣勢;以及
● 與創建獨立的銷售和營銷組織相關的意外成本和費用。
如果我們與第三方達成協議來執行銷售、營銷和分銷服務,我們的產品收入或從這些收入流中為我們帶來的盈利能力可能會低於我們營銷和銷售我們自己開發的任何候選產品的情況。 此外,我們可能無法成功地與第三方達成協議來銷售和營銷我們的候選產品,或者 可能無法以對我們有利的條款這樣做。我們可能對這些第三方几乎沒有控制權,他們中的任何一方都可能 無法投入必要的資源和注意力來有效地銷售和營銷我們的候選產品。如果我們不能成功地建立銷售 和營銷能力,無論是我們自己還是與第三方合作,我們都可能無法成功地將我們的候選產品商業化。
如果獲得批准,我們開發的任何當前或未來候選產品的 市場機會可能僅限於那些 沒有資格接受現有療法或之前的療法失敗的患者,因此可能很小。
癌症療法有時被描述為一線、二線或三線,FDA通常最初只批准三線使用的新療法。當癌症被發現得足夠早時,一線治療,通常是化療、激素治療、手術、放射治療、免疫治療或這些療法的組合,有時足以治癒癌症或延長生命,而不需要治癒。當先前的治療無效時,對患者實施二線和三線治療。我們最初可能會尋求SON-080和 我們開發的任何其他候選產品的批准,作為對接受過一種或多種先前治療的患者的治療。隨後,對於那些被證明具有足夠益處的產品(如果有的話),我們預計可能會尋求批准作為一線療法,但 不能保證我們開發的候選產品即使獲得批准也會被批准用於一線療法,在獲得任何此類批准之前,我們可能必須進行額外的臨牀試驗。
| 48 |
我們所針對的癌症患者數量可能會低於預期。此外,如果獲得批准,我們當前計劃或未來候選產品的潛在可尋址患者數量可能會受到限制。即使我們為任何候選產品獲得了相當大的市場份額,如果獲得批准,如果潛在目標人羣較少,我們可能永遠無法實現盈利 如果沒有獲得額外適應症的營銷批准,包括用作一線或二線治療。
即使我們獲得了候選產品的上市批准,我們也將受到持續的監管義務和持續的監管 審查,這可能會導致大量額外費用,如果我們未能遵守監管要求 或遇到意外的產品問題(如果獲得批准),我們可能會受到處罰。
對於任何當前或未來的候選產品,我們收到的任何 營銷批准可能會受到產品上市的批准指示用途或批准條件的限制,或者包含可能代價高昂的上市後 測試和監控要求,以監控候選產品的安全性和有效性。FDA還可能要求將風險評估和緩解策略或REMS作為批准任何候選產品的條件,其中可能包括對藥物指南、醫生溝通計劃或確保安全使用的其他要素的要求,例如受限分配方法、患者登記 和其他風險最小化工具。如果FDA或類似的外國監管機構批准候選產品,候選產品的製造、標籤、包裝、分銷、不良事件報告、儲存、廣告、促銷、進出口和記錄 將受到廣泛和持續的監管要求。這些要求包括禁止推廣批准的產品用於產品批准的標籤中未包括的用途,提交安全和其他上市後信息和報告,註冊,以及對於我們在批准後進行的任何臨牀試驗,繼續遵守當前的良好製造實踐(CGMP)和良好臨牀實踐(GCP)。後來發現任何已批准的候選對象存在以前未知的問題,包括意外嚴重性或頻率的不良事件,或我們的第三方製造商或製造流程,或未能遵守監管要求,可能會導致以下情況,其中包括:
● 限制產品的標籤、分銷、營銷或製造,將產品從市場上召回,或 產品召回;
● 無標題和警告信,或暫停臨牀試驗;
● 食品和藥物管理局拒絕批准未決申請或對我們提交的已批准申請的補充,或暫停或吊銷許可證批准 ;
● 進行上市後研究或臨牀試驗的要求;
● 對第三方付款人承保的限制;
● 罰款、返還或返還利潤或收入;
● 暫停或撤回上市審批;
● 產品被扣押或扣留,或拒絕允許產品進口或出口;以及
● 禁止令或施加民事或刑事處罰。
FDA和其他監管機構的政策可能會改變,可能會頒佈額外的政府法規,以阻止、限制或推遲產品的上市審批。我們無法預測美國或國外未來的立法或行政行動可能產生的政府監管的可能性、性質或程度。如果我們緩慢或無法使 適應現有要求的變化或採用新的要求或政策,或者如果我們無法保持合規性, 我們可能會失去我們可能獲得的任何營銷批准,我們可能無法實現或保持盈利。
| 49 |
我們 面臨激烈的競爭,如果我們的競爭對手開發和銷售比我們開發的候選產品更有效、更安全或更便宜的產品,我們的商業機會將受到負面影響。
生命科學行業競爭激烈。我們目前正在開發的療法如果獲得批准,將與其他產品和目前存在的、正在開發的或未來將開發的療法 競爭,其中一些我們目前可能還不知道。
我們在美國和國際上都有競爭對手,包括大型跨國製藥公司、老牌生物技術公司、專業製藥公司、大學和其他研究機構。與我們相比,我們的許多競爭對手擁有更多的財務、製造、營銷、產品開發、技術和人力資源。大型製藥公司,尤其是大型製藥公司,在臨牀測試、獲得市場批准、招募患者和製造藥品 產品方面擁有豐富的經驗。這些公司的研究和營銷能力也比我們強得多,可能還擁有已獲批准或處於開發後期階段的產品,以及在我們的目標市場與領先公司和研究機構的合作安排。老牌製藥公司還可能大舉投資,以加快新化合物的發現和開發,或者授權使用可能使我們開發的候選產品過時的新化合物。製藥和生物技術行業的合併和收購 可能導致更多的資源集中在我們數量較少的競爭對手 中。由於所有這些因素,我們的競爭對手可能會在我們之前成功獲得專利保護和/或營銷批准,或者在我們之前發現、開發和商業化我們領域的產品。
有大量的公司開發或營銷癌症治療方法,包括許多主要的製藥和生物技術公司。 這些治療方法既包括傳統化療等小分子藥物產品,也包括新型免疫療法。例如,許多跨國公司以及大型生物技術公司,包括Astellas Pharma Inc.、Seattle Genetics、 Inc.、阿斯利康和葛蘭素史克,正在為我們的流水線項目探索的目標開發項目。
如果我們的競爭對手開發和商業化比我們可能開發的任何產品更安全、更有效、影響更少或更不嚴重、更方便、更廣泛的標籤、更有效的營銷、得到報銷或更便宜的產品,我們的商業機會可能會減少或消失。我們的競爭對手也可能比我們更快地獲得FDA、EMA或其他產品的市場批准 ,這可能會使我們的競爭對手在我們能夠進入市場之前建立強大的市場地位。即使我們開發的候選產品獲得了市場批准,它們的定價也可能比競爭產品高出顯著的 溢價(如果到那時已獲得批准),從而導致競爭力下降。
規模較小的 和其他初創公司也可能被證明是重要的競爭對手。這些第三方在招聘和留住合格的科學和管理人員、建立臨牀試驗場地和臨牀試驗患者註冊方面以及在獲取與我們的計劃互補或必要的技術方面與我們展開競爭。此外,生物製藥行業的特點是技術變化迅速。如果我們不能保持在技術變革的前沿,我們就可能無法有效地競爭。 技術進步或競爭對手開發的產品可能會使我們的候選產品過時、競爭力下降或不經濟。
任何當前或未來候選產品的商業成功將取決於醫生、患者、付款人和醫學界其他人對市場的接受程度。
我們 從未將產品商業化,即使我們為我們的候選產品獲得任何監管批准,我們候選產品的商業成功也將在一定程度上取決於醫療界、患者和付款人是否接受我們的候選產品是有效、 安全和經濟高效的。我們向市場推出的任何產品都可能得不到醫生、患者、付款人和醫學界其他人的認可。醫生通常不願將患者從現有的治療方法中轉換出來,即使是在新的、可能更有效或更方便的治療方法進入市場時也是如此。此外,患者通常適應他們目前正在接受的治療 ,除非醫生建議他們更換產品,或者由於現有治療缺乏 報銷而要求他們更換治療,否則患者不想更換。
| 50 |
如果這些候選產品獲準用於商業銷售,其市場接受度將取決於許多因素,包括:
| ● | 與替代療法相比的潛在療效和潛在優勢; |
| ● | 任何副作用的頻率和嚴重程度,包括產品批准的標籤中包含的任何限制或警告; |
| ● | 對我們的候選產品進行管理的後續要求導致的任何副作用的頻率和嚴重程度; |
| ● | 相對方便和容易管理; |
| ● | 目標患者羣體嘗試新療法的意願以及醫生開出這些療法的意願; |
| ● | 市場營銷和分銷支持的實力以及競爭產品進入市場的時機; |
| ● | 宣傳我們的產品或競爭對手的產品和治療方法;以及 |
| ● | 充足的第三方保險覆蓋範圍和充足的報銷。 |
即使 如果候選產品在臨牀前研究和臨牀試驗中表現出良好的療效和安全性,如果該產品被批准用於商業銷售,市場接受程度也要等到該產品推出後才能知道。我們努力讓醫療界和付款人瞭解我們的候選產品的好處,這可能需要大量資源,而且可能永遠不會成功。這種教育市場的努力可能需要比我們的競爭對手銷售的傳統技術所需的更多的資源,特別是 由於我們的十四行詩接近。如果這些產品沒有達到足夠的接受度,我們可能不會產生顯著的產品收入,也可能無法盈利。
如果我們的候選產品的市場機會比我們認為的要小,我們的產品收入可能會受到不利影響 ,我們的業務可能會受到影響。
我們 目前將我們的研究和產品開發重點放在腫瘤適應症的治療和我們的產品FHAB候選對象 旨在針對實體腫瘤。我們對患有這些疾病的人數以及有可能從我們的候選產品治療中受益的這些疾病患者的子集的瞭解是基於估計的。這些估計可能被證明是不正確的,新的研究可能會降低這些疾病的估計發病率或流行率。患者識別 工作也會影響解決患者羣體的能力。如果在患者識別方面的努力不成功或效果不如預期 ,我們可能無法解決我們正在尋找的整個機會。
新批准產品的保險覆蓋範圍和報銷狀態不確定。如果我們的任何候選產品未能獲得或保持足夠的承保範圍和報銷,如果獲得批准,可能會限制我們營銷這些產品的能力,並降低我們創造收入的能力 。
我們 預計,當我們的候選產品獲得市場批准時,他們的成本將會很高。政府和私人付款人的可獲得性和報銷範圍是大多數患者能夠負擔得起昂貴治療費用的關鍵。我們候選產品的銷售將在很大程度上取決於我們的候選產品在國內和國外的銷售情況 我們候選產品的費用將在多大程度上由私人付款人支付,如私人健康保險公司、健康維護、管理式醫療、藥房福利和類似的醫療保健管理組織,或由政府醫療保健計劃(如Medicare和Medicaid)報銷。我們可能無法 提供足夠的數據來獲得承保和報銷方面的認可。如果無法獲得報銷或僅限量獲得報銷,我們可能無法成功地將我們的候選產品商業化,即使獲得批准。即使提供了保險 ,批准的報銷金額也可能不足以讓我們建立或保持足夠的定價來實現足夠的投資回報。
| 51 |
與新批准產品的保險覆蓋範圍和報銷相關的重大不確定性。在美國,有關新藥承保和報銷的主要決定通常由醫療保險和醫療補助服務中心(CMS)做出,CMS是美國衞生與公眾服務部的一個機構,由CMS決定新藥是否在Medicare下承保和報銷,以及在多大程度上報銷。私人支付者傾向於在很大程度上遵循CMS。很難預測CMS將就像我們這樣的新產品的承保範圍和報銷做出什麼決定,因為這些新產品沒有既定的 實踐和先例。第三方付款人的承保範圍和報銷可能取決於許多因素, 包括第三方付款人對產品使用的確定:(1)其醫療計劃下的承保福利;(2)安全、 有效且具有醫療必要性;(3)適用於特定患者;(4)成本效益;以及(5)既不是試驗性的,也不是試驗性的。 在美國,第三方付款人之間沒有統一的產品承保和報銷政策。因此,保險範圍確定過程通常是一個耗時且昂貴的過程,需要我們為每個付款人分別提供使用我們產品的科學和臨牀支持 ,但不能保證保險範圍和足夠的報銷將始終如一地應用 或首先獲得。即使我們獲得了特定產品的保險,由此產生的報銷付款率也可能不足以 使我們實現或維持盈利能力,或者可能需要患者認為不可接受的高額自付費用。第三方付款人 可以將承保範圍限制到已批准清單(也稱為處方)上的特定藥品,該清單可能不包括特定適應症的所有已批准 藥物。
此外, 第三方付款人可能無法支付或提供足夠的補償,用於使用 候選產品後所需的長期後續評估。患者不太可能使用我們的候選產品,除非提供了覆蓋範圍,並且報銷足以 覆蓋我們候選產品的大部分成本。由於我們的候選產品的商品成本可能高於 傳統療法,並且可能需要長期隨訪評估,因此承保範圍和報銷率可能不足以 使我們實現盈利的風險可能更大。新 批准產品的保險範圍和報銷存在重大不確定性。目前很難預測第三方付款人將如何決定我們候選產品的承保範圍和報銷 。
此外, 美國和國外的政府和第三方付款人越來越多地限制或降低醫療保健成本,可能會導致 此類組織限制新批准產品的承保範圍和報銷水平,因此,他們可能無法為我們的候選產品承保 或提供足夠的付款。 美國對特殊藥品定價實踐的立法和執法興趣越來越大。具體而言,最近美國國會進行了幾次調查,並 提出了聯邦和州立法,旨在提高藥品定價的透明度,降低Medicare下處方藥的成本,審查定價與製造商患者計劃之間的關係,以及改革政府計劃藥品報銷 方法。由於 醫療保健管理的趨勢、健康維護組織的影響力日益增強、成本控制計劃和其他 立法變化,我們預計在銷售任何候選產品時都會遇到定價壓力。
Outside the United States, certain countries, including a number of member states of the European Union, set prices and reimbursement for pharmaceutical products, or medicinal products, as they are commonly referred to in the European Union. These countries have broad discretion in setting prices and we cannot be sure that such prices and reimbursement will be acceptable to us or our collaborators. If the regulatory authorities in these jurisdictions set prices or reimbursement levels that are not commercially attractive for us or our collaborators, our revenues from sales by us or our collaborators, and the potential profitability of our drug products, in those countries would be negatively affected. An increasing number of countries are taking initiatives to attempt to reduce large budget deficits by focusing cost-cutting efforts on pharmaceuticals for their state-run health care systems. These international price control efforts have impacted all regions of the world, but have been most drastic in the European Union. Additionally, some countries require approval of the sale price of a product before it can be lawfully marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. To obtain reimbursement or pricing approval in some countries, we, or any collaborators, may be required to conduct a clinical trial that compares the cost-effectiveness of our product to other available therapies. As a result, we might obtain marketing approval for a product in a particular country, but then may experience delays in the reimbursement approval of our product or be subject to price regulations that would delay our commercial launch of the product, possibly for lengthy time periods, which could negatively impact the revenues we are able to generate from the sale of the product in that particular country.
| 52 |
Moreover, efforts by governments and payors, in the United States and abroad, to cap or reduce healthcare costs may cause such organizations to limit both coverage and level of reimbursement for new products approved and, as a result, they may not cover or provide adequate reimbursement for our product candidates. There has been increasing legislative and enforcement interest in the United States with respect to specialty drug practices. Specifically, there have been several recent U.S. Congressional inquiries and proposed federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs. We expect to experience pricing pressures in connection with the sale of any of our product candidates, due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be harmed.
如果 FDA或類似的外國監管機構批准了我們任何獲得上市 批准的候選產品的仿製藥版本,或者此類機構在批准 此類產品的仿製藥版本之前未授予此類產品適當的數據獨佔期,則此類產品的銷售可能會受到不利影響。
在美國,製造商可以通過提交簡化的生物許可證申請或ABLAS來尋求FDA根據BLA批准的生物相似版本的批准。為了支持ABLA,生物相似製造商通常必須證明其產品與原始生物產品相似。生物相似產品推向市場的成本可能低於原始生物產品 ,生產生物相似產品的公司有時能夠以更低的價格提供這些產品。因此,在推出生物相似產品後,原始生物產品的銷售額可能會有很大一部分被生物相似產品搶走,原始生物產品的價格可能會降低。
在原始生物的任何適用的非專利專有期到期之前,FDA不得接受或批准生物相似產品的ABLA進行審查或批准。公共衞生服務(PHS)法案為根據BLA批准的生物提供了12年的非專利專有期。
我們的產品可能面臨來自生物相似版本產品的競爭 可能會對我們未來的收入、盈利能力和現金流產生負面影響,並極大地限制我們從這些候選產品中獲得投資回報的能力。
我們 可能直接或間接受到聯邦和州醫療欺詐和濫用法律、虛假聲明法律、健康信息隱私和安全法律以及其他醫療法律和法規的約束。如果我們不能遵守或沒有完全遵守這些法律, 我們可能面臨鉅額處罰。
如果 我們的任何候選產品獲得了FDA的批准,並開始在美國將這些產品商業化,我們的運營 將直接或間接通過我們的處方者、客户和購買者遵守各種聯邦和州欺詐和濫用法律法規,包括但不限於聯邦醫療保健計劃反回扣法規、 聯邦民事和刑事虛假索賠法案以及醫生支付陽光法案和法規。這些法律將影響我們提議的銷售、營銷和教育計劃等。此外,我們可能受到聯邦政府和我們開展業務所在州的患者隱私法的約束。將影響我們運營的法律包括但不限於:
| ● | 《反回扣條例》,除其他事項外,禁止個人或實體直接或間接、公開或隱蔽地以現金或實物形式直接或間接、公開或隱蔽地索要、收受、提供或支付任何報酬(包括任何回扣、賄賂或回扣),以引誘或回報個人推薦,或購買、租賃、訂購、安排或推薦根據聯邦醫療保健計劃可全部或部分支付的任何商品、設施、物品或服務。例如聯邦醫療保險和醫療補助計劃。“報酬”一詞被廣泛解釋為包括任何有價值的東西。個人或實體不需要實際瞭解《反回扣法規》或違反該法規的特定意圖即可實施 違規。此外,政府可以斷言,根據聯邦虛假索賠法案或聯邦民事罰款的目的,包括因違反反回扣法規而產生的物品或服務的索賠構成虛假或欺詐性索賠。 反回扣法規已被解釋為適用於藥品製造商與 處方者、購買者和處方經理之間的安排。有一些法定例外和監管避風港可以保護一些常見活動免受起訴; |
| 53 |
| ● | 聯邦民事和刑事虛假申報法和民事金錢懲罰法,包括FCA,對個人或實體處以刑事和民事處罰,原因包括:故意向聯邦政府提交或導致向聯邦政府提交虛假或欺詐性的付款索賠;故意對向聯邦政府支付或轉移金錢或財產的虛假或欺詐性索賠或義務作出、使用或導致做出或使用記錄材料的虛假陳述 。製造商 可以根據FCA承擔責任,即使他們沒有直接向政府付款人提交索賠,但如果他們被認為“導致” 提交虛假或欺詐性索賠。FCA還允許充當“告密者”的個人代表聯邦政府提起訴訟,指控違反FCA的行為,並分享任何金錢追回; |
| ● | 《議定書》的 受益人誘因條款,除其他事項外,禁止提供或給予報酬,其中包括但不限於免費或低於公平市場價值的任何物品或服務的轉讓(有限的例外情況), 個人知道或應該知道可能影響受益人選擇可由聯邦或州政府計劃報銷的物品或服務的特定供應商; |
| ● | 1996年聯邦健康保險攜帶和責任法案,或HIPAA,創建了新的聯邦刑法,禁止 任何人故意和故意執行或試圖執行詐騙任何醫療福利計劃的計劃,或 通過虛假或欺詐性的藉口、陳述或承諾,獲得任何醫療福利計劃擁有或託管或控制的任何金錢或財產,而無論付款人(例如,公共或私人),並故意和故意偽造,以任何詭計或手段隱瞞或掩蓋重大事實,或作出任何重大虛假、虛構或欺詐性的陳述或陳述,涉及與醫療事宜有關的醫療福利、項目或服務的交付或付款 ;與《反回扣法規》類似,個人或實體不需要實際瞭解法規或違反法規的具體意圖即可實施違規; |
| ● | HIPAA,經2009年《衞生信息技術促進經濟和臨牀健康法》及其各自的實施條例修訂的《HIPAA》, 對某些醫療保健提供者、健康計劃和醫療信息交換所(稱為覆蓋實體)及其各自的商業夥伴、代表其提供服務的個人和實體提出要求,涉及使用或披露涉及個人可識別健康信息的隱私、安全和傳輸的個人可識別健康信息 ; |
| ● | 經《醫療保健和教育協調法》修訂的《患者保護和平價醫療法案》(或統稱為ACA)規定的美國聯邦透明度要求,包括通常稱為《醫生支付陽光法案》的條款,該條款要求適用的藥品、設備、生物製品和醫療用品製造商可根據Medicare付款,醫療補助或兒童健康保險計劃(某些例外情況除外)每年向CMS報告與向醫生(定義為包括醫生、牙醫、視光師、足科醫生和脊椎按摩師) 和教學醫院,以及上述醫生及其直系親屬持有的所有權和投資權益。從2022年開始,適用的製造商還將被要求報告向醫生助理、護士從業者、臨牀護士專家、註冊護士麻醉師和註冊護士助產士支付和轉移價值的信息; |
| ● | 聯邦政府價格報告法,要求我們準確、及時地計算並向政府項目報告複雜的定價指標 ;以及 |
| ● | 聯邦 消費者保護和不正當競爭法,對市場活動和可能損害消費者的活動進行廣泛監管 。 |
此外, 我們受制於上述每個醫療保健法律法規的州和國外等價物,其中包括一些可能範圍更廣且可能適用於任何付款人的 。美國許多州已經通過了類似於《反回扣法規和虛假索賠法案》的法律,可能適用於我們的商業實踐,包括但不限於研究、分銷、銷售或營銷安排以及涉及由非政府付款人(包括私人保險公司)報銷的醫療項目或服務的索賠。 此外,一些州還通過了法律,要求製藥公司遵守2003年4月總監察長辦公室 藥品製造商合規計劃指南和/或美國藥品研究和製造商與醫療保健專業人員互動的 守則。幾個州還實施了其他營銷限制,或要求製藥公司向州政府進行營銷或價格披露。對於需要遵守這些州的要求存在歧義 ,如果我們不遵守適用的州法律要求,我們可能會受到處罰。最後,還有管理健康信息隱私和安全的國家法律和外國法律,其中許多法律在很大程度上彼此不同,而且HIPAA往往不會先發制人,從而使合規工作複雜化。
| 54 |
由於這些法律的廣度,以及可用的法定例外和監管安全港的狹窄,我們的一些業務活動可能會受到一項或多項此類法律的挑戰。執法部門越來越多地 專注於執行欺詐和濫用法律,我們的一些做法可能會受到這些法律的挑戰。確保我們當前和未來與第三方的業務安排以及我們的業務總體上符合適用的醫療法律法規的努力 將涉及大量成本。如果我們的業務,包括我們與醫生和其他醫療保健提供者的安排,其中一些獲得股票期權作為所提供服務的補償,被發現違反了適用於我們的任何此類法律或任何其他政府法規,我們可能會受到懲罰,包括但不限於行政、民事和刑事處罰、損害賠償、罰款、返還、合同損害、聲譽損害、利潤減少和未來收入, 我們業務的削減或重組,被排除在聯邦和州醫療保健計劃(如Medicare 和Medicaid)的參與之外,如果我們受到公司誠信協議或類似的 協議的約束,以解決有關不遵守這些法律的指控和個人監禁,則會有額外的報告要求和/或監督,其中任何一項都可能對我們的業務運營能力和財務業績產生不利影響。任何違反這些法律的行為,即使成功辯護, 也可能導致製藥商招致鉅額法律費用,並轉移管理層對業務運營的注意力 。禁止或限制銷售或撤回未來上市的產品可能會以不利的方式對業務產生重大影響。
醫療保健 立法改革措施和對國家預算社會保障制度的限制可能會對我們的業務和運營結果產生實質性的不利影響 。
付款方,無論是國內還是國外,或政府或私人,都在開發越來越複雜的方法來控制醫療成本 這些方法並不總是專門適用於我們正在開發的那些新技術。在美國和某些外國司法管轄區,醫療保健系統的立法和監管發生了許多變化, 可能會影響我們銷售產品盈利的能力。尤其是在美國,ACA於2010年頒佈,除其他事項外,它使生物產品受到低成本生物仿製藥的潛在競爭;解決了一種新的方法,通過該方法,對於吸入、輸液、滴注、植入或注射的藥物,計算製造商在Medicaid藥品回扣計劃下所欠的回扣;增加了大多數製造商在Medicaid藥品回扣計劃下所欠的最低Medicaid回扣;將Medicaid 藥品回扣計劃擴大到使用在Medicaid管理的護理組織中登記的個人的處方;要求製造商 對某些品牌處方藥徵收新的年費和税;併為增加聯邦政府比較有效性研究的項目提供激勵。
自頒佈以來,ACA的某些方面一直受到司法和國會的挑戰,以及 本屆政府最近為廢除或取代ACA的某些方面所做的努力。此外,自2017年1月以來,總裁·特朗普簽署了兩項行政命令和其他指令,旨在推遲ACA某些條款的實施,或以其他方式規避ACA授權的一些醫療保險要求。此外,CMS最近發佈了一項最終規則,從2020年開始,將給予各州更大的靈活性,在為個人和小團體市場的保險公司設定基準方面,這可能會放寬ACA對通過此類市場銷售的保險計劃所要求的基本健康福利。
同時, 國會審議了將廢除和取代全部或部分ACA的立法。雖然國會還沒有通過全面的廢止立法,但兩項影響ACA下某些税收實施的法案已經簽署成為法律。2017年減税和就業法案(TCJA)包括一項條款,從2019年1月1日起廢除ACA對未能在一年的全部或部分時間內維持合格醫療保險的個人實施的基於税收的分擔責任付款, 通常被稱為“個人強制醫保”。此外,2018年1月22日,總裁·特朗普簽署了一項關於2018財年撥款的持續決議 ,推遲了ACA規定的某些費用的實施,包括對某些高成本僱主贊助的保險計劃徵收所謂的“凱迪拉克”税 ,對某些高成本僱主贊助的保險計劃徵收年費,根據市場份額向某些醫療保險提供者徵收年費,以及對非豁免的醫療器械徵收醫療器械税 。此外,2018年兩黨預算法案(BBA)等修訂了ACA,自2019年1月1日起生效, 將參與Medicare D部分的製藥製造商所欠的銷售點折扣從50%提高到70%,並縮小大多數Medicare藥物計劃的覆蓋缺口,通常被稱為“甜甜圈洞”。最近, 在2018年7月,CMS發佈了一項最終規則,允許根據ACA風險調整計劃對某些ACA合格健康計劃和 健康保險發行商進行進一步的收款和付款,以迴應聯邦地區法院關於CMS確定此風險調整的方法的訴訟結果。國會還可以考慮額外的立法,以廢除或取代ACA的其他要素。因此,ACA的全部影響、任何廢除或取代它的內容的法律,以及圍繞我們業務的任何廢除或取代立法的政治不確定性仍不清楚。
| 55 |
此外,自ACA頒佈以來,美國還提出並通過了其他立法修改。2011年8月,除其他事項外,《2011年預算控制法案》制定了國會削減開支的措施。赤字削減聯合特別委員會的任務是建議2013年至2021年至少削減1.5萬億美元的赤字,但該委員會無法 達到所需的目標,從而觸發了立法對幾個政府計劃的自動削減。這包括在2013年4月生效的每個財年向提供商支付的醫療保險總金額減少2%,並且由於隨後的立法修訂,包括BBA,除非採取額外的國會行動,否則將一直有效到2027年。2013年1月,2012年《美國納税人救濟法》簽署成為法律,除其他事項外,進一步減少了對包括醫院和癌症治療中心在內的幾家醫療服務提供者的醫療保險支付,並將政府 追回向醫療服務提供者多付款項的訴訟時效從三年延長至五年。
此外, 政府最近加強了對藥品製造商為其上市產品定價的方式的審查,這導致了幾次國會調查,並提出並頒佈了聯邦和州立法,旨在提高產品定價的透明度,審查定價與製造商患者計劃之間的關係,並改革 政府計劃藥品報銷方法。例如,本屆政府發佈了一份《藍圖》 ,以降低藥品價格和降低藥品的自付成本,其中包含增加製造商競爭的額外建議, 增加某些聯邦醫療保健計劃的談判力,激勵製造商降低其產品的標價 ,並減少消費者支付的藥品的自付成本。例如,2018年11月,CMS發佈了一項建議規則,以徵求 意見,其中將為D部分下的Medicare處方藥計劃提供更大的定價透明度和更大的 靈活性,以談判六個受保護的處方類別中的藥物的折扣,並在某些情況下排除這些藥物,並允許Medicare Advantage計劃使用特定的藥物管理工具,如醫生管理的藥物的階梯療法。儘管其中許多措施和其他擬議措施需要通過額外的立法授權才能生效,但國會和特朗普政府都表示將繼續尋求新的立法和/或行政措施來控制 藥品成本。
外國、聯邦和州各級已經並可能繼續提出立法和監管建議,旨在 擴大醫療保健的可獲得性,並控制或降低醫療保健成本。我們無法預測未來可能採用的計劃。這些政府和其他付款人繼續努力控制或降低醫療保健成本和/或實施價格管制,可能會對以下方面產生不利影響:
● 對我們候選產品的需求,如果我們獲得監管部門的批准;
● 我們有能力為我們的產品設定一個我們認為是公平的價格;
● 我們創造收入、實現或保持盈利的能力;
● 我們須繳交的税項水平;及
● 資金的可得性。
任何拒絕承保或減少聯邦醫療保險或其他政府計劃的報銷都可能導致類似的拒絕或減少私人支付者的付款 ,這可能會對我們未來的盈利能力產生不利影響。
| 56 |
我們 受修訂後的美國1977年《反海外腐敗法》或《反海外腐敗法》和其他反腐敗法律以及出口管制法律、進口和海關法、貿易和經濟制裁法律以及其他管理我們業務的法律的約束。
我們的運營受到反腐敗法律的約束,包括《反海外腐敗法》、美國《反海外腐敗法》第18章第201節中包含的美國國內賄賂法規、《美國旅行法》以及適用於我們開展業務的國家/地區的其他反腐敗法律。《反賄賂法》、《反海外腐敗法》和其他 法律一般禁止我們以及我們的員工和中介機構直接或間接授權、承諾、提供或提供不正當或被禁止的款項或任何其他有價值的東西給政府官員或其他人員,以獲得或保留業務或 獲得其他業務優勢。根據《反賄賂法》,我們還可能因未能阻止與我們有關聯的人實施賄賂犯罪而承擔責任。我們和我們的商業合作伙伴在多個司法管轄區開展業務,這些司法管轄區可能存在違反《反賄賂法》或《反海外腐敗法》的風險,我們參與與第三方的合作和關係,這些第三方的腐敗或非法活動 可能使我們根據《反賄賂法》、《反海外腐敗法》或當地反腐敗法承擔責任,即使我們沒有明確授權 或實際瞭解此類活動。此外,我們無法預測未來監管要求的性質、範圍或效果 我們的國際業務可能受制於這些監管要求,也無法預測現有法律可能被管理或解釋的方式。
我們 還受制於管理我們國際業務的其他法律和法規,包括由英國和美國政府以及歐盟當局管理的法規,包括適用的出口管制法規、對某些國家和人員的經濟制裁和禁運、反洗錢法、進口和海關要求以及貨幣兑換法規,統稱為貿易管制法。
不能保證我們將完全有效地確保我們遵守所有適用的反腐敗法律,包括《反賄賂法》、《反海外腐敗法》或其他法律要求,包括貿易控制法。如果我們不遵守《反賄賂法》、《反海外腐敗法》和其他反腐敗法或貿易控制法,我們可能會受到刑事和民事處罰、返還和其他 制裁和補救措施,以及法律費用,這可能會對我們的業務、財務狀況、運營結果和流動性產生不利影響。同樣,對英國、美國或其他當局可能違反《反賄賂法》、《反海外腐敗法》、其他反腐敗法律或貿易管制法律的任何調查也可能對我們的聲譽、我們的業務、經營結果和財務狀況產生不利影響。
如果 我們未能遵守環境、健康和安全法律法規,我們可能會受到罰款或處罰,或產生 成本,這可能會對我們的業務成功產生重大不利影響。
我們 受眾多環境、健康和安全法律法規的約束,包括管理實驗室程序以及危險材料和廢物的處理、使用、儲存、處理和處置的法律法規。我們的業務涉及使用危險和易燃材料,包括化學品和生物材料。我們的業務還會產生危險廢物產品。我們通常與第三方簽訂處理這些材料和廢物的合同。我們無法消除這些材料造成污染或傷害的風險。如果我們使用危險材料造成污染或傷害,我們可能要對由此產生的任何損害承擔責任,任何責任都可能超出我們的資源範圍。我們還可能產生與民事或刑事罰款相關的鉅額成本 和處罰。此外,環境法律和法規復雜,變化頻繁,並有變得更加嚴格的趨勢。 我們無法預測此類變化的影響,也無法確定我們未來的合規性。此外,為了遵守當前或未來的環境、健康和安全法律法規,我們可能會產生鉅額成本 。這些當前或未來的法律法規 可能會損害我們的研究、開發或生產努力。不遵守這些法律法規還可能導致鉅額罰款、處罰或其他制裁。
雖然 我們維持工人補償保險,以支付因使用危險材料或其他與工作有關的傷害而對員工造成的傷害所產生的成本和開支,但該保險可能無法為潛在的 責任提供足夠的保險。此外,為了遵守當前或未來的環境、健康和安全法律和法規,我們可能會產生鉅額成本。這些現行或未來的法律法規可能會損害我們的研究、開發或生產努力。不遵守這些法律法規還可能導致鉅額罰款、處罰或其他制裁或責任,這可能會對我們的業務、財務狀況、運營結果和前景產生重大不利影響。
| 57 |
與我們國際業務相關的風險
由於我們的子公司救濟位於美國境外,因此我們面臨與國際業務相關的經濟、政治、監管和其他風險 。
由於Relipment的總部設在瑞士,我們的業務在美國以外的地區開展業務會受到風險的影響。 我們的許多供應商和臨牀試驗關係都位於美國以外。因此,我們未來的業績可能會受到多種因素的影響,包括:
| ● | 經濟疲軟,包括通貨膨脹,或特別是非美國經濟體和市場的政治不穩定; |
| ● | 不同的 和不斷變化的產品審批法規要求; |
| ● | 不同的法域在確保、維持或獲得在此類法域運作的自由方面可能會出現不同的問題; |
| ● | 可能會減少對知識產權的保護; |
| ● | 在遵守多個司法管轄區不同、複雜和不斷變化的法律、法規和法院系統以及遵守各種外國法律、條約和法規方面遇到困難 ; |
| ● | 改變美國以外的法規和關税、關税和貿易壁壘; |
| ● | 英鎊、美元、歐元和貨幣管制的非美國貨幣匯率變化 ; |
| ● | 貿易 各國政府的保護措施、進出口許可要求或其他限制行動; |
| ● | 在某些非美國市場實行不同的報銷制度和價格管制; |
| ● | 税法變更帶來的負面後果; |
| ● | 遵守國外居住或旅行員工的税法、就業法、移民法和勞動法,包括在不同司法管轄區對根據我們的股票期權計劃或股權激勵計劃授予的期權的可變税收 待遇; |
| ● | 勞動力 勞工騷亂比美國更普遍的國家的不確定性; |
| ● | 訴訟 或因現任或前任員工或顧問單獨或作為集體訴訟的一部分而對我們提起的行政訴訟,包括對錯誤解僱、歧視、錯誤分類或其他違反勞動法的索賠或 其他被指控的行為; |
| ● | 與人員配置和管理國際業務有關的困難,包括不同的勞資關係; |
| ● | 生產 任何影響國外原材料供應或製造能力的事件造成的短缺; |
| ● | 業務 由於地緣政治行動(包括戰爭和恐怖主義)或自然災害(包括地震、颱風、洪水和火災)造成的中斷。 |
| 58 |
歐洲 數據收集受有關個人信息的使用、處理和跨境傳輸的限制性法規管轄。
歐盟信息系統中個人健康數據的收集和使用受數據保護指令的規定管轄,自2018年5月25日起,該指令已被GDPR取代。這些指令對個人數據涉及的個人的同意、向個人提供的信息、向主管國家數據保護機構通知數據處理義務以及個人數據的安全和保密提出了幾項要求。數據保護指令和GDPR還對將個人數據從歐盟轉移到美國實施了嚴格的規則。如果 未能遵守數據保護指令、GDPR以及歐盟成員國的相關國家數據保護法的要求,可能會受到罰款和其他行政處罰。雖然數據保護指令不適用於總部位於歐盟以外的組織,但GDPR已將其覆蓋範圍擴大到向歐盟居民提供商品或服務的任何企業,無論其位置如何 。這一擴展將納入歐盟成員國的任何潛在臨牀試驗活動。GDPR對個人數據的控制器和處理器施加了 嚴格的要求,包括對包括居住在歐盟的數據對象的健康和遺傳信息的“敏感信息” 的特殊保護。GDPR允許個人有機會反對 處理其個人信息,允許他們在某些情況下請求刪除個人信息,並且 為個人提供了明確的權利,在個人認為其權利受到侵犯的情況下尋求法律補救。此外,GDPR對將個人數據從歐盟轉移到美國或其他被認為沒有提供“充分”隱私保護的地區實施了嚴格的規則。如果不遵守GDPR和歐盟成員國的相關國家數據保護法的要求,可能會稍微偏離GDPR, 可能會被處以高達全球收入的4%或20,000,000歐元的罰款,以金額較大者為準。作為實施GDPR的結果,我們可能需要建立額外的機制,以確保遵守新的數據保護規則。
與我們依賴第三方相關的風險
對於 某些候選產品,我們可能依賴開發和商業化合作夥伴來開發和進行臨牀試驗, 獲得監管部門對候選產品的批准,如果獲得批准,則營銷和銷售候選產品。如果此類合作者的表現達不到預期, 我們從這些候選產品中獲得未來收入的潛力將顯著降低,我們的業務將受到損害。
對於 某些候選產品,我們依賴或將依賴我們的開發和商業合作伙伴來開發、進行候選產品的臨牀 試驗,並在獲得批准後將其商業化。
我們當前的合作以及我們未來參與的任何合作都會面臨許多風險,包括:
| ● | 協作者 在確定他們將應用於協作的工作和資源方面有很大的自由裁量權; |
| ● | 協作者 可能未按預期履行義務,或未能及時履行責任,或根本不履行責任; |
| ● | 合作者 不得繼續開發和商業化獲得監管批准的任何候選產品,或選擇不繼續 或基於臨牀前研究或臨牀試驗結果更新開發或商業化計劃,合作者 戰略重點或可用資金或外部因素,如轉移資源或產生競爭優先事項的收購; |
| ● | 合作者 可能會延遲臨牀前研究或臨牀試驗,為臨牀試驗提供的資金不足,停止臨牀前研究或臨牀 試驗或放棄候選產品,重複或進行新的臨牀試驗,或需要候選產品的新配方,以 臨牀試驗; |
| 59 |
| ● | 我們 可能無法訪問或可能被限制披露有關正在開發的候選產品的某些信息,或 在合作下商業化,因此可能無法向我們的股東告知此類 候選產品; |
| ● | 合作者 可以獨立開發或與第三方合作開發與我們的候選產品直接或間接競爭的產品 如果合作者認為競爭產品更有可能在 在經濟上比我們更具吸引力的條件; |
| ● | 該 合作可能不會導致候選產品的開發和/或臨牀前研究或臨牀試驗作為 合作可能不會成功; |
| ● | 產品 與合作者共同開發的候選產品可能會被我們的合作者視為與他們自己的候選產品或產品競爭, 這可能導致合作者停止我們的候選產品的商業化; |
| ● | a 擁有我們一個或多個獲得監管批准的候選產品的營銷和分銷權的合作者可以 不投入足夠的資源用於任何此類候選產品的營銷和分銷;以及 |
| ● | 合作者 可能無法正確維護或捍衞我們的知識產權,或可能以邀請 可能危及或使我們的知識產權或專有信息無效或使我們面臨潛在訴訟的訴訟。 |
此外,某些合作和商業化協議為我們的合作者提供了終止此類協議的權利, 這些權利可能受條件限制,也可能不受條件限制,如果行使這些權利,將對我們的產品開發工作產生不利影響, 並可能使我們難以吸引新的合作者。在這種情況下,我們可能需要限制此類候選產品或產品的開發和商業化的規模和範圍;我們可能需要 尋求額外融資,以資助進一步開發或確定替代戰略合作;我們從這些候選產品或產品的特許權使用費和里程碑付款產生未來收入的潛力將顯著降低,延遲或取消; 而且可能會對我們的業務和未來增長前景產生不利影響。我們收回有形和無形資產的權利 以及在合作終止後推進候選產品或產品所需的知識產權可能受到合同的限制 ,我們可能無法在終止後推進計劃。
如果與我們的開發和商業化合作者或許可人發生衝突,他們可能會出於自身利益行事,這可能會對我們公司的利益不利。
我們 未來可能會與我們的開發和商業化合作者或許可方發生分歧。由於以下一項或多項原因,我們與第三方的協作和許可安排可能會出現衝突:
| ● | 爭議 關於里程碑、特許權使用費和其他根據適用協議應支付的款項; |
| ● | 分歧 關於知識產權的歸屬或者許可範圍; |
| ● | 分歧 關於任何報告義務的範圍; |
| ● | 不情願 作為合作者,讓我們瞭解其開發和商業化活動的進展情況,或 允許公開披露這些活動;以及 |
| ● | 爭議 關於合作者或我們對我們的產品和候選產品的開發或商業化努力。 |
| 60 |
與我們的開發和商業化合作夥伴或許可方的衝突 可能會對我們的業務、財務狀況或運營結果和未來增長前景產生重大不利影響。
我們 將依賴第三方(包括獨立臨牀研究者和CRO)來開展和贊助我們候選產品的部分臨牀試驗 。第三方未能履行其在候選產品臨牀開發方面的義務可能會延遲或損害我們獲得候選產品監管批准的能力。
我們 將依賴並計劃繼續依賴第三方(包括獨立臨牀研究者、學術合作伙伴、 法規事務顧問和第三方CRO)來開展我們的臨牀前研究和臨牀試驗(包括在某些情況下 贊助此類臨牀試驗),並與監管機構合作,監測和管理我們正在進行的臨牀前和 臨牀項目的數據。鑑於我們認為我們的候選產品可能具有實用性的臨牀治療領域的廣度,我們打算 繼續依賴外部服務提供商,而不是建立內部監管專業知識。
在某些情況下,這些第三方中的任何一方都可以終止與我們的合同。我們可能無法達成替代的 安排或以商業上合理的條款這樣做。此外,當新的合同研究機構開始工作時,還有一個自然的過渡期。因此,可能會出現延遲,這可能會對我們滿足預期臨牀開發時間表的能力產生負面影響,並損害我們的業務、財務狀況和前景。
We remain responsible for ensuring that each of our preclinical studies and clinical trials is conducted in accordance with the applicable protocol and legal, regulatory and scientific standards, and our reliance on these third parties does not relieve us of our regulatory responsibilities. We and our third-party contractors and CROs are required to comply with GCP requirements, which are regulations and guidelines enforced by the FDA, the Competent Authorities of the Member States of the European Economic Area, or EEA, and comparable foreign regulatory authorities for all of our products in clinical development. Regulatory authorities enforce these GCP requirements through periodic inspections of trial sponsors, principal investigators and trial sites. If we fail to exercise adequate oversight over any of our academic partners or CROs or if we or any of our academic partners or CROs do not successfully carry out their contractual duties or obligations, fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements, or for any other reasons, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, the EMA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon a regulatory inspection of us, our academic partners or our CROs or other third parties performing services in connection with our clinical trials, such regulatory authority will determine that any of our clinical trials complies with GCP regulations. In addition, our clinical trials must be conducted with product produced under applicable CGMP regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process.
此外, 代表我們進行臨牀試驗的第三方不是我們的員工,除了根據我們與此類承包商的協議向我們提供的補救措施外,我們無法控制他們是否將足夠的時間、技能和資源投入到我們正在進行的開發項目中。 這些承包商還可能與其他商業實體有關係,包括我們的競爭對手,他們也可能為這些實體進行臨牀試驗或其他藥物開發活動,這可能會阻礙他們將適當的時間投入我們的臨牀項目的能力。 如果這些第三方,包括臨牀研究人員,沒有成功履行合同職責,如果我們無法在預期的最後期限內完成 或根據法規要求或我們規定的方案進行臨牀試驗,我們可能無法獲得或延遲獲得我們候選產品的上市批准。如果發生這種情況,我們將無法或可能會推遲將我們的候選產品成功商業化的努力。
| 61 |
In addition, with respect to investigator-sponsored trials that may be conducted, we would not control the design or conduct of these trials, and it is possible that the FDA or EMA will not view these investigator-sponsored trials as providing adequate support for future clinical trials or market approval, whether controlled by us or third parties, for any one or more reasons, including elements of the design or execution of the trials or safety concerns or other trial results. We expect that such arrangements will provide us certain information rights with respect to the investigator-sponsored trials, including access to and the ability to use and reference the data, including for our own regulatory submissions, resulting from the investigator-sponsored trials. However, we would not have control over the timing and reporting of the data from investigator-sponsored trials, nor would we own the data from the investigator- sponsored trials. If we are unable to confirm or replicate the results from the investigator-sponsored trials or if negative results are obtained, we would likely be further delayed or prevented from advancing further clinical development. Further, if investigators or institutions breach their obligations with respect to the clinical development of our product candidates, or if the data proves to be inadequate compared to the firsthand knowledge we might have gained had the investigator-sponsored trials been sponsored and conducted by us, then our ability to design and conduct any future clinical trials ourselves may be adversely affected. Additionally, the FDA or EMA may disagree with the sufficiency of our right of reference to the preclinical, manufacturing or clinical data generated by these investigator-sponsored trials, or our interpretation of preclinical, manufacturing or clinical data from these investigator-sponsored trials. If so, the FDA or EMA may require us to obtain and submit additional preclinical, manufacturing, or clinical data.
我們 打算依賴第三方生產候選產品,這增加了我們無法以可接受的成本獲得足夠數量的此類候選產品或產品或此類數量的風險,這可能會延遲、阻止或損害我們的開發 或商業化努力。
我們 不擁有或經營生產我們 在開發計劃中正在開發或評估的候選產品的臨牀或商業供應的生產設施。我們擁有藥品生產經驗的人員有限,並且缺乏 資源和能力來生產臨牀或商業規模的任何候選產品。我們依賴第三方 提供候選產品,我們的策略是將所有候選產品和產品的製造外包給第三方 。
為了對候選產品進行臨牀試驗,我們可能需要大量生產這些產品。我們的第三方製造商可能無法及時或以具有成本效益的方式或根本不能成功地提高我們的任何候選產品的生產能力。此外,在擴展活動期間和任何其他時間都可能出現質量問題。例如, 有關我們候選產品穩定性的持續數據可能會縮短我們候選產品的有效期,並導致臨牀試驗材料供應短缺,並可能導致臨牀試驗延遲。如果這些第三方製造商無法以足夠的質量和數量成功擴大我們候選產品的生產 ,則該候選產品的開發、測試和臨牀試驗可能會被推遲或不可行,該候選產品的監管批准或商業發佈可能會延遲或無法獲得, 這可能會嚴重損害我們的業務。
我們使用新的第三方製造商會增加生產延遲或候選產品供應不足的風險,因為 我們將製造技術轉讓給這些製造商,隨着他們獲得製造我們候選產品的經驗。即使 第三方製造商在製造我們的候選產品方面獲得了豐富的經驗,或者即使我們認為我們已經成功地優化了製造流程,也不能保證該製造商將及時或隨着時間的推移連續生產足夠數量的我們候選產品,或者根本不能保證。
如果我們需要更改第三方使用的製造流程,我們 可能會延遲。此外,如果我們更改批准的製造流程 ,如果FDA或類似的外國機構需要在新的製造流程可以使用之前對其進行審查,則我們可能會被推遲。
| 62 |
我們 採用外包模式生產我們的候選產品,並與獲得許可的藥品合同開發和製造組織簽訂了良好的製造規範或GMP。雖然我們已經聘請了幾家第三方供應商來提供臨牀用品和非臨牀用品以及填充劑服務,但我們目前沒有與第三方製造商簽訂任何長期商業用品的協議。未來,我們可能無法與第三方製造商就我們開發的任何候選產品的商業供應 達成協議,或者無法以可接受的條款這樣做。即使我們能夠與第三方製造商建立並維護 安排,依賴第三方製造商也會帶來風險,包括:
| ● | 依賴第三方進行製造流程開發、合規和質量保證; |
| ● | 由於第三方的能力和進度限制而造成的供應可用性限制 ; |
| ● | 由於我們無法控制的因素,第三方可能違反制造協議;以及 |
| ● | 在對我們來説代價高昂或不方便的情況下, 第三方可能終止或不續訂製造協議。 |
第三方 製造商可能無法遵守cGMP要求或美國以外的類似監管要求。我們未能遵守適用要求或第三方製造商未能遵守相關要求可能導致對我們實施制裁,包括罰款、禁令、民事處罰、延遲、暫停或撤回審批、吊銷許可證、扣押或召回候選產品或產品、運營限制和/或刑事起訴,其中任何一項都可能對我們候選產品的供應造成重大和不利的 影響。此外,我們打算開發的一些候選產品,包括SON-080,使用毒素 或其他只能在具有特定授權和許可的專門設施中生產的物質,並且無法 保證我們或我們的製造商能夠保持此類授權和許可。這些專業化要求還可能限制我們可以聘請的潛在製造商的數量,以生產我們的候選產品,並破壞過渡到替代製造商的任何努力 。
我們未來的候選產品和我們可能開發的任何產品都可能與其他候選產品和產品競爭生產設施。按照cGMP要求運營並有能力為我們生產產品的製造商數量有限。
如果受聘為我們的臨牀前測試和臨牀試驗提供任何材料或產品的第三方因任何原因而停止提供任何材料或產品,我們很可能會在確定和 確定替換供應商或製造商資格時延遲推進這些測試和試驗,並且我們可能無法以對我們有利的條款獲得替換供應。 此外,如果我們無法獲得足夠的候選產品或用於製造這些候選產品的物質,我們將更難開發候選產品並有效競爭。
我們目前和預期的未來對其他候選產品生產的依賴可能會對我們未來的利潤率以及我們開發候選產品和將任何及時獲得市場批准的產品進行商業化的能力產生不利影響。
我們對第三方的依賴要求我們共享我們的商業祕密,這增加了競爭對手發現這些祕密或我們的商業祕密被盜用或泄露的可能性。
由於 我們依賴第三方來生產候選產品,並且由於我們與各種組織和學術機構 合作開發候選產品,因此我們有時必須與他們分享商業祕密。我們在開始研究或披露專有信息之前, 通過與我們的合作者、顧問、員工和顧問簽訂保密協議,以及材料轉讓協議、合作研究協議、 諮詢協議或其他類似協議(如適用)來保護我們的專有技術。這些協議通常會限制第三方使用或披露我們的機密 信息(如商業機密)的權利。
| 63 |
儘管與第三方合作時採用了合同條款,但共享商業祕密和其他機密信息的需要 會增加此類商業祕密被我們的競爭對手知曉、被無意中併入其他人的技術中、或在違反這些協議的情況下被披露或使用的風險。鑑於我們的專有地位在一定程度上基於我們的專有技術和商業祕密,競爭對手發現我們的商業祕密或其他未經授權的使用或披露將損害我們的競爭地位,並可能對我們的業務產生實質性的不利影響。
此外,這些協議通常會限制我們的合作者、顧問、員工和顧問發佈可能與我們的商業祕密相關的數據。我們的學術合作者通常有權發佈數據,前提是提前通知我們 ,並且可能會將發佈推遲一段指定的時間,以保護我們因合作而產生的知識產權。在 其他情況下,發佈權由我們獨家控制,儘管在某些情況下我們可能與其他各方共享這些權利。 儘管我們努力保護我們的商業祕密,但我們的競爭對手可能會通過違反這些協議、獨立開發或發佈信息(包括我們的商業祕密)來發現我們的商業祕密,在發佈時我們沒有專有權或其他 受保護的權利。競爭對手發現我們的商業機密將損害我們的競爭地位 並對我們的業務產生不利影響。
與我們知識產權相關的風險
如果 我們無法為我們的產品和候選產品獲得並維護專利和其他知識產權保護,或者如果 獲得的專利和其他知識產權保護的範圍不夠廣泛,我們的競爭對手可能會開發 與我們相似或相同的產品並將其商業化,我們成功將我們的產品和候選產品商業化的能力可能會受到不利影響。
我們有效競爭的能力將在一定程度上取決於我們保持技術和製造流程的專有性質的能力 。我們依靠研究、製造和其他技術、專利、商業祕密、許可協議和合同條款來確立我們的知識產權並保護我們的產品和候選產品。然而,這些法律手段只能提供有限的保護,可能無法充分保護我們的權利。截至2023年12月11日,我們的知識產權組合包括19項未決專利申請和已頒發專利,包括在美國的1項已發佈專利,日本、俄羅斯和新西蘭各1項授權決定,以及5007專利系列中的9項PCT申請-此外,還有6項未決的臨時申請,涉及配方、製造工藝和使用方法。
在 某些情況下,在被認為合適的情況下,我們已經並打算繼續尋求通過在美國提交專利申請來保護我們的專有地位,在至少某些情況下,在美國以外的一個或多個國家/地區提交與當前和未來的產品以及對我們業務重要的候選產品有關的專利申請。然而,我們無法預測目前正在申請的專利 是否會作為專利頒發,或者任何由此產生的專利的權利要求是否會為我們提供競爭優勢,或者我們是否能夠在未來成功地申請與我們當前或未來的產品和候選產品相關的專利申請 。此外,專利申請和審批過程既昂貴又耗時。我們可能無法以合理的成本或及時提交和起訴所有必要或可取的專利申請。此外,我們或任何未來的合作伙伴、合作者或被許可人可能無法在獲得專利保護之前確定在開發和商業化活動中的發明的可申請專利的方面。因此,我們可能會錯過尋求額外專利保護的預期潛在機會 。專利申請的準備或提交過程中可能存在形式上的缺陷, 或將來可能會出現,例如,在適當的優先權權利要求、清單、權利要求範圍或專利期限調整請求方面。如果我們未能建立、維護或保護此類專利和其他知識產權,此類權利可能會減少或取消。如果我們的專利或專利申請在形式、準備、起訴或執行方面存在重大缺陷, 此類專利可能無效和/或無法強制執行,並且此類申請可能永遠不會產生有效的、可強制執行的專利。
即使我們的專利和專利申請不受質疑,但如果它們被頒發,我們的專利和專利申請可能不會為我們提供任何有意義的保護,或者阻止 競爭對手通過以非侵權方式開發類似或替代技術或療法來繞過我們的專利主張進行設計 。例如,第三方可能開發一種競爭性療法,提供與我們的一個或多個候選產品類似的益處 ,但這超出了我們的專利保護範圍。如果我們對候選產品持有或申請的專利和專利申請提供的專利保護不夠廣泛,不足以阻礙此類競爭,我們將候選產品成功商業化的能力可能會受到負面影響。
| 64 |
正如在“業務”標題下討論的那樣,我們的國際專利申請號為PCT/US2018/00085的PCT專利申請收到的申請提交日期為2018年2月20日,這是在美國臨時專利申請US 62/459,975和US 62/459,981提交日期一週年之後的四天,PCT專利申請要求因PCT接收辦公室的計算機問題而享有優先權。儘管PCT已將優先權恢復到美國臨時專利申請的申請日(美國62/459,975和美國62/459,981),但一些從本PCT專利申請中提交的國家階段專利申請的國家階段申請不接受這種恢復,包括加拿大和中國,恢復程序在巴西懸而未決 。在優先權未恢復的情況下,現有技術可能適用於這些專利申請,否則這些專利申請可能不適用於PCT/US2018/00085提交的其他專利申請。這可能會影響我們 在巴西、加拿大和中國提出的專利申請的範圍或廣度,或者可能導致無法在這些國家/地區獲得專利。
其他 方,其中許多人擁有更多的資源,並在競爭技術方面進行了大量投資,已經開發出 或可能開發出與我們的方法相關或競爭的技術,並且可能已經提交或可能提交專利申請 ,並且可能已經頒發或可能頒發專利,其權利要求與我們的專利申請重疊或衝突,或者通過要求 相同的成分,配方或方法,或要求保護可能主導我們專利地位的主題。此外,外國的 法律可能無法像美國法律那樣保護我們的權利。因此,我們 將來可能獲得的任何專利可能無法為我們提供充分和持續的專利保護,足以阻止其他人將與我們的產品和候選產品類似的 產品商業化。
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain. No consistent policy regarding the breadth of claims allowed in biotechnology and pharmaceutical patents has emerged to date in the United States or in many foreign jurisdictions. In addition, the determination of patent rights with respect to pharmaceutical compounds commonly involves complex legal and factual questions, which has in recent years been the subject of much litigation. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain. Our competitors may also seek approval to market their own products similar to or otherwise competitive with our products. Alternatively, our competitors may seek to market generic versions of any approved products by submitting ANDAs to the FDA in which they claim that our patents are invalid, unenforceable or not infringed. In these circumstances, we may need to defend or assert our patents, or both, including by filing lawsuits alleging patent infringement. In any of these types of proceedings, a court or other agency with jurisdiction may find our patents invalid or unenforceable, or that our competitors are competing in a non-infringing manner. Thus, even if we have valid and enforceable patents, these patents still may not provide protection against competing products or processes sufficient to achieve our business objectives.
將來,我們的一個或多個產品和候選產品可能會從第三方獲得許可。因此,在某些情況下,潛在專利保護的 可用性和範圍會根據我們的許可方或發明人先前的決定而受到限制,例如 關於何時提交專利申請或是否提交專利申請的決定。我們因任何原因未能獲得、維護、執行 或捍衞此類知識產權,可能會允許第三方,特別是其他已建立的、資金更充足的 競爭對手(具有已建立的開發、製造和分銷能力)生產競爭產品或影響我們開發、製造和營銷我們的產品和候選產品的能力, 即使這些產品和候選產品在商業上可行的基礎上獲得批准,如果有話, 這可能對我們的業務產生重大不利影響。
除專利保護外,我們預計還將嚴重依賴商業祕密、專有技術和其他難以 保護的非專利技術。儘管我們通過與我們的供應商、員工、顧問 和其他可能訪問專有信息的人簽訂保密協議來尋求此類保護,但我們無法確定這些協議不會被違反,是否有足夠的 補救措施來應對任何違約行為,或者我們的商業祕密,專有技術和其他非專利專有技術不會 被我們的競爭對手所知或獨立開發。如果我們未能成功保護我們的知識產權, 我們產品的銷售可能會受到影響,我們產生收入的能力可能會受到嚴重影響。
| 65 |
如果在法庭或行政 訴訟中受到質疑,則涵蓋我們的產品和候選產品的已頒發 專利可能被認定無效或不可執行。在法庭上我們可能無法保護我們的商業祕密。
如果 我們針對第三方發起法律訴訟以強制執行覆蓋我們的產品或候選產品的專利,如果 出現此類專利問題,被告可以反訴覆蓋我們的產品或候選產品的專利無效或不可強制執行。 在美國的專利訴訟中,被告聲稱無效或不可強制執行的反訴很常見。 質疑有效性的理由可能是據稱未能滿足幾項法定要求中的任何一項,包括缺乏新穎性、明顯、 書面描述或未啟用。不可執行性主張的理由可能是,與專利起訴有關的人在起訴期間向美國專利商標局隱瞞了有關專利可專利性的信息,或做出了誤導性的聲明。 第三方也可以向美國或國外的行政機構提出類似的索賠,即使在 訴訟的背景之外也是如此。此類機制包括重新審查、授權後審查、各方間審查以及在外國司法管轄區進行的同等程序。 在上述任何程序中做出不利裁決可能會導致我們的 專利被撤銷、取消或修改,從而不再涵蓋我們的產品或候選產品。在法律上斷言無效和不可強制執行之後的結果是不可預測的。例如,關於有效性問題,我們不能確定沒有使專利審查員和我們在起訴期間不知道的現有技術無效。如果被告或第三方勝訴,則我們可能至少部分甚至全部失去對我們的一個或多個產品和候選產品的專利保護。這種專利保護的喪失可能會對我們的業務產生實質性的不利影響。
此外,我們的商業祕密可能會被競爭對手知曉或獨立發現。競爭對手和其他第三方 可能購買我們的產品和候選產品,並試圖複製我們從我們的開發工作中獲得的部分或全部競爭優勢,故意侵犯、挪用或以其他方式侵犯我們的知識產權,圍繞我們 受保護的技術進行設計,或開發他們自己的不屬於我們知識產權的競爭技術。如果我們的任何商業祕密是由競爭對手或其他第三方合法獲取或獨立開發的,我們將無權 阻止他們或他們向其傳達信息的人使用該技術或信息與我們競爭。如果我們的商業祕密沒有得到充分保護或不足以提供相對於競爭對手的優勢,我們的競爭地位可能會受到不利的 影響,我們的業務也可能受到影響。此外,如果為保護我們的商業祕密而採取的措施被認為是不充分的,我們可能沒有足夠的追索權 針對第三方盜用我們的商業祕密。
我們 可能會受到質疑專利和其他知識產權的發明權或所有權的索賠。
我們 可能會受到前員工、合作者或其他第三方對我們擁有或將來可能擁有或許可的專利和知識產權的所有權權益的索賠。雖然我們的政策是要求 可能參與知識產權開發的員工和承包商執行將此類知識產權轉讓給我們的協議,但我們可能無法與實際開發我們視為自己的知識產權的每一方執行此類協議,或者 此類轉讓可能不是自動執行的或可能被違反。例如,由於參與開發我們產品或候選產品的員工、顧問或其他人的義務衝突,我們可能會受到所有權糾紛的影響。可能需要 針對任何質疑庫存或所有權的索賠進行辯護。如果我們或未能為任何此類索賠辯護,我們可能需要支付金錢損害賠償,並可能失去寶貴的知識產權,如知識產權的獨家所有權或使用權 ,這可能會對我們的業務、運營結果和財務狀況產生不利影響。
獲得和維持專利保護取決於遵守政府專利機構提出的各種程序、文件提交、費用支付和其他要求 ,如果不符合這些要求,我們的專利保護可能會減少或取消。
定期 專利和申請的維護費、續期費、年費和各種其他政府費用需要在專利和申請的有效期內分幾個階段向USPTO和美國以外的各種政府專利機構支付 。美國專利商標局和各種非政府專利機構要求在專利申請過程中和專利頒發後遵守一些程序、文件、費用支付和其他類似條款。在 情況下,不遵守規定可能會導致專利或專利申請被放棄或失效,從而導致相關司法管轄區的專利權部分或全部喪失。我們未來簽訂的一個或多個許可證的條款可能無法為我們提供 維護或起訴產品組合中的專利的能力,因此必須依賴第三方來維護或起訴。
| 66 |
如果我們沒有為我們的產品和候選產品獲得專利期延長和數據獨佔權,我們的業務可能會受到嚴重損害。
專利 的壽命有限。在美國,如果及時支付所有維護費,專利的自然有效期通常為自其在美國最早的非臨時申請日期起計 20年。可能有各種延期,但專利的有效期及其提供的保護是有限的。即使獲得了涵蓋我們候選產品的專利,一旦候選產品的專利有效期過期,我們可能會接受來自競爭產品的競爭。考慮到新產品候選產品的開發、測試和監管審查所需的時間,保護這些候選產品的專利可能會在這些候選產品商業化之前或之後不久 到期。因此,我們的專利組合可能無法為我們提供足夠的權利來排除其他公司將與我們相似或相同的產品商業化。
In the future, if we obtain an issued patent covering one of our present or future product candidates, depending upon the timing, duration and specifics of any FDA marketing approval of such product candidates, such patent may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, or Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent extension term of up to five years as compensation for patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent may be extended and only those claims covering the approved drug, a method for using it or a method for manufacturing it may be extended. A patent may only be extended once and only based on a single approved product. However, we may not be granted an extension because of, for example, failure to obtain a granted patent before approval of a product candidate, failure to exercise due diligence during the testing phase or regulatory review process, failure to apply within applicable deadlines, failure to apply prior to expiration of relevant patents or otherwise our failure to satisfy applicable requirements. A patent licensed to us by a third party may not be available for patent term extension. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain patent term extension or the term of any such extension is less than we request, our competitors may obtain approval of competing products following our patent expiration, and our revenue could be reduced, possibly materially.
美國和其他司法管轄區專利法的變化 可能會降低專利的價值,從而削弱我們保護產品和候選產品的能力。
Changes in either the patent laws or the interpretation of the patent laws in the United States or other jurisdictions could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. When implemented, the Leahy-Smith Act included several significant changes to U.S. patent law that impacted how patent rights could be prosecuted, enforced and defended. In particular, the Leahy-Smith Act also included provisions that switched the United States from a “first-to-invent” system to a “first-to-file” system, allowed third- party submission of prior art to the USPTO during patent prosecution and set forth additional procedures to attack the validity of a patent by the USPTO administered post grant proceedings. Under a first-to-file system, assuming the other requirements for patentability are met, the first inventor to file a patent application generally will be entitled to the patent on an invention regardless of whether another inventor had made the invention earlier. The USPTO developed new regulations and procedures governing the administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, only became effective on March 16, 2013. It remains unclear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business.
此外,公司在生物製劑和藥品開發和商業化中的專利地位尤其不確定。美國聯邦巡迴上訴法院和美國最高法院最近的裁決縮小了在某些情況下專利保護的範圍,並削弱了專利所有人在某些情況下的權利。這些事件的組合 已經對專利的有效性和可撤銷性(一旦獲得)產生了不確定性。根據美國國會、聯邦法院和USPTO未來的行動 ,管理專利的法律和法規可能會以不可預測的方式發生變化 ,這可能會對我們現有的專利組合以及我們未來保護和執行知識產權的能力 產生重大不利影響。
| 67 |
我們 無法向您保證,我們為一種或多種產品和候選產品尋求專利保護的努力不會受到上述決定、其他案件的裁決或USPTO發佈的指南或程序變更的負面影響。我們無法 完全預測法院在歷史和未來案件中的判決可能會對生命科學公司 未來獲得或執行與其產品相關的專利的能力產生什麼影響。這些決定、USPTO發佈的指南以及 其他案例中的裁決或USPTO指南或程序的變更可能會對我們現有的專利權以及 未來保護和執行我們知識產權的能力產生重大不利影響。
我們 可能無法在全球範圍內保護我們的知識產權。
在全球所有國家/地區對產品和候選產品申請、起訴、維護、辯護和強制執行專利的費用將高得令人望而卻步,而且我們在美國以外的一些國家/地區的知識產權可能沒有美國的知識產權那麼廣泛。對專利性的要求在某些國家可能有所不同,特別是在發展中國家; 因此,即使在我們確實尋求專利保護的國家,也不能保證任何專利都會發布 涵蓋我們產品的聲明。不能保證我們將根據未來的任何許可協議在美國境內或境外獲得或維護專利權。此外,一些國家的法律對知識產權的保護程度不如美國的聯邦法律和州法律。因此,我們可能無法阻止第三方在美國以外的所有國家/地區實施我們的發明,即使在我們尋求專利保護的司法管轄區,也無法阻止第三方在美國或其他司法管轄區銷售或進口使用我們的發明製造的產品。競爭對手可以在我們沒有申請和獲得專利保護的司法管轄區使用我們的技術來開發他們自己的產品,並進一步將其他侵權產品出口到我們擁有專利保護的地區,但執法力度不如美國。這些產品 可能與我們的產品和候選產品競爭,而我們的專利或其他知識產權可能無法有效或不足以阻止它們競爭。
許多公司在保護和捍衞外國司法管轄區的知識產權方面遇到了嚴重的問題。某些國家的法律制度,尤其是某些發展中國家的法律制度,不支持專利、商業祕密和其他知識產權保護的實施,特別是與生物技術和醫藥產品有關的保護,這可能使我們很難在總體上阻止侵犯我們的專利或營銷競爭對手產品侵犯我們的專有權 。例如,許多外國國家都有強制許可法,根據這些法律,專利所有者必須將許可授予第三方 。在外國司法管轄區執行我們的專利權的訴訟,即使獲得,也可能導致鉅額成本,並將我們的努力和注意力從我們業務的其他方面轉移,可能使我們的專利面臨被無效或狹義解釋的風險 ,我們的專利申請有可能無法發佈,並可能引發第三方對我們提出索賠。我們可能不會在我們發起的任何訴訟中獲勝,並且如果有損害賠償或其他補救措施,可能沒有商業意義。雖然我們打算 保護我們產品在主要市場的知識產權,但我們不能確保我們能夠在我們希望銷售產品的所有司法管轄區開展或保持類似的努力。因此,我們在全球範圍內執行知識產權的努力可能不足以從我們 開發的知識產權中獲得顯著的商業優勢。
如果我們因侵犯第三方知識產權而被起訴,此類訴訟可能代價高昂且耗時,並可能 阻止或推遲我們的候選產品的開發或商業化。
我們的商業成功在一定程度上取決於我們在不侵犯第三方知識產權和其他專有權利的情況下開發、製造、營銷和銷售我們的候選產品的能力。第三方可能擁有美國和非美國頒發的專利,以及與化合物、化合物製造方法和/或用於治療我們正在開發的候選產品的疾病適應症的使用方法相關的 未決專利申請。如果發現任何第三方專利或專利申請 涵蓋我們的候選產品或其使用或製造方法,我們和我們的合作者或分被許可人可能無法在未獲得許可的情況下按計劃自由生產或銷售我們的候選產品,而許可可能無法以商業合理的條款獲得, 或根本無法獲得。在這種情況下,我們還可能被要求對我們的合作者或分被許可人進行賠償。
| 68 |
在生物技術和製藥行業有大量的知識產權訴訟,我們可能成為與我們的產品知識產權有關的訴訟或其他對抗訴訟的一方,或受到威脅 候選產品,包括幹擾和美國專利商標局的授權後訴訟。可能存在與我們候選產品的組成、使用或製造有關的材料、配方、製造方法或處理方法的第三方專利或專利申請 。我們不能保證我們的任何專利搜索或分析,包括但不限於相關專利的識別、專利權利要求的範圍或相關專利的到期,都是完整或徹底的,我們也不能確定 我們已經識別了與我們的候選產品在任何司法管轄區商業化或必要的 在美國和國外的每一項專利和待決申請。由於專利申請可能需要數年時間才能發佈,因此可能存在當前正在處理的專利申請,這些申請可能會導致我們的候選產品被指控侵犯已頒發的專利。 此外,第三方可能會在未來獲得專利,並聲稱使用我們的技術侵犯了這些專利。因此, 第三方可能會根據現在存在或將來出現的知識產權向我們提出侵權索賠。知識產權訴訟的結果受到不確定因素的影響,這些不確定因素事先無法充分量化。製藥和生物技術行業產生了大量專利,包括我們在內的行業參與者可能並不總是清楚哪些專利涵蓋各種類型的產品或使用或製造方法。專利提供的保護範圍受到法院的解釋,解釋並不總是一致的。如果我們因專利侵權而被起訴,我們 需要證明我們的候選產品、產品或方法沒有侵犯相關專利的專利主張 ,或者專利主張無效或不可執行,而我們可能無法做到這一點。證明無效性是困難的。例如,在美國, 要證明無效性,需要出示清楚且令人信服的證據,以推翻已頒發專利所享有的有效性的推定。即使我們在這些訴訟中勝訴,我們也可能會產生鉅額成本,我們的管理層和科學人員的時間和注意力可能會被轉移到這些訴訟中,這可能會嚴重損害我們的業務 和經營業績。此外,我們可能沒有足夠的資源來圓滿完成這些行動。
如果 我們被發現侵犯了第三方的知識產權,我們可能會被迫停止開發、製造或商業化侵權候選產品或產品,包括法院命令。或者,我們可能需要獲得此類第三方的許可,才能使用侵權技術並繼續開發、製造或營銷侵權產品 候選產品或產品。
但是, 我們可能無法以商業合理的條款或根本無法獲得任何所需的許可證。即使我們能夠獲得許可證,它也可能是非排他性的,從而使我們的競爭對手能夠訪問授權給我們的相同技術;此外,它可能包含阻礙或破壞我們在商業市場上成功競爭的能力的條款。此外,如果我們被發現故意侵犯了一項專利,我們可能會被判承擔金錢損害賠償責任,包括三倍損害賠償和律師費。侵權發現可能會阻止我們將候選產品商業化或迫使我們停止部分業務 ,這可能會損害我們的業務。有關我們盜用第三方的機密信息或商業祕密的指控可能會對我們的業務產生類似的負面影響。
我們 可能會受到第三方的索賠,聲稱我們的員工或我們挪用了他們的知識產權,或者要求 擁有我們認為是我們自己的知識產權。
我們的許多現任和前任員工,包括我們的高級管理層,以前受僱於大學或其他生物技術 或製藥公司,包括一些可能是競爭對手或潛在競爭對手的公司。其中一些員工可能 受制於與此類先前工作相關的專有權利、保密和競業禁止協議或類似協議。 儘管我們盡力確保我們的員工在為我們工作時不使用他人的專有信息或專有技術,但我們 可能會受到指控,即我們或這些員工使用或披露了任何此類第三方的知識產權,包括商業祕密或其他專有 信息。可能有必要提起訴訟來對此類索賠進行辯護。如果我們未能為任何此類索賠辯護, 除了支付金錢損害賠償外,我們還可能失去寶貴的知識產權或人員,或遭受損害。此類知識產權 可以授予第三方,我們可能需要從該第三方獲得許可才能將我們的技術或產品商業化。這樣的許可可能不會以商業上合理的條款或根本不存在。即使我們成功地對此類索賠進行了辯護,訴訟也可能導致鉅額成本,並分散管理層的注意力。
| 69 |
此外,雖然我們通常要求可能參與知識產權開發的員工、顧問和承包商執行將此類知識產權轉讓給我們的協議,但我們可能無法成功地與 實際開發我們視為自己的知識產權的每一方執行此類協議,這可能會導致我們向或針對我們提出與此類知識產權所有權相關的索賠。如果我們不能起訴或為任何此類索賠辯護,除了支付金錢損害賠償外,我們還可能失去寶貴的知識產權。即使我們成功地起訴或抗辯此類索賠, 訴訟也可能導致鉅額成本,並分散我們高級管理人員和科學人員的注意力。
我們 可能會捲入保護或強制執行我們的專利和其他知識產權的訴訟,這可能是昂貴、耗時的 ,而且不會成功。
競爭對手 可能會侵犯我們的專利、商標、版權或其他知識產權。為了反擊侵權或未經授權的使用,我們可能會被要求提出侵權索賠,這可能既昂貴又耗時,並分散了我們的管理人員和科學人員的時間和注意力。此外,我們的專利可能會捲入發明權、優先權或有效性糾紛。對此類索賠進行反擊或 辯護可能既昂貴又耗時,而且我們的對手可能有能力投入比我們更多的 資源來起訴這些法律行動。我們對被認定的侵權者提出的任何索賠都可能促使這些當事人 對我們提出反索賠,聲稱我們侵犯了他們的專利,此外還聲稱我們的專利無效或不可強制執行,或兩者兼而有之。
在侵權訴訟中,法院可以裁定某項專利無效或不可強制執行,或以我們的專利不涵蓋相關技術為由,拒絕阻止另一方使用相關技術。因此,儘管我們做出了努力,但我們可能無法阻止第三方侵犯或挪用我們擁有或控制的知識產權。 任何訴訟程序中的不利結果可能會使我們擁有或授權的一個或多個專利面臨被無效或狹義解釋的風險。此外,由於與知識產權訴訟相關的大量發現, 在此類訴訟期間,我們的一些機密信息可能會因披露而泄露。
即使 如果裁決對我們有利,法院也可能決定不授予禁止進一步侵權活動的禁令,而只判給 金錢損害賠償,這可能是也可能不是足夠的補救措施。與知識產權索賠有關的訴訟或其他法律程序可能會導致我們產生鉅額費用,並可能分散我們人員的正常責任。此外, 可能會公佈聽證會、動議或其他臨時程序或事態發展的結果,如果證券分析師或投資者認為這些結果是負面的,可能會對我們普通股的價格產生重大不利影響。此類 訴訟或訴訟可能大幅增加我們的運營虧損,並減少可用於開發活動或任何未來銷售、營銷或分銷活動的資源。
我們 可能沒有足夠的財政或其他資源來充分開展此類訴訟或訴訟程序。我們的一些競爭對手可能 能夠比我們更有效地承擔此類訴訟或訴訟的費用,因為他們擁有更多的財力 以及更成熟和發展的知識產權組合。專利 訴訟或其他訴訟的發起和繼續所產生的不確定性可能會對我們在市場上的競爭能力產生重大不利影響。
如果 我們未能履行未來與第三方的任何知識產權許可義務,我們可能會失去對我們的業務非常重要的許可權 。
鑑於我們正在努力建立我們的候選產品渠道,我們可能會在未來簽訂許可協議。我們預計 此類許可協議將對我們施加各種盡職調查、里程碑付款、特許權使用費、保險和其他義務。如果我們未能遵守這些許可下的義務,我們的許可人可能有權終止這些許可協議,在這種情況下,我們可能無法銷售這些協議涵蓋的任何產品,或者我們的許可人可能會將許可轉換為非獨家許可,這可能會對根據許可協議開發的候選產品的價值產生負面影響。 終止這些許可協議或減少或取消我們的許可權利也可能導致我們不得不協商條款較差的新許可或恢復許可。
| 70 |
如果 我們的商標和商號沒有得到充分保護,那麼我們可能無法在我們感興趣的標記中建立名稱認知度 ,我們的業務可能會受到不利影響。
我們的商標或商號可能會受到質疑、侵犯、規避或宣佈為通用商標,或被認定為侵犯了其他商標。 我們的商標依賴於註冊和普通法保護。我們可能無法保護我們對這些商標和商品名稱的權利,或者可能會被迫停止使用這些名稱,我們需要這些名稱才能在我們感興趣的市場中獲得潛在合作伙伴或客户的名稱認可。在商標註冊過程中,我們可能會收到拒絕。儘管我們將有機會 迴應這些拒絕,但我們可能無法克服這些拒絕。此外,在美國專利商標局和許多外國司法管轄區的類似機構中,第三方有機會反對未決的商標申請並尋求註銷註冊商標。可能會對我們的商標提起反對或取消訴訟,我們的商標可能無法繼續存在。 如果我們不能根據我們的商標和商號建立名稱認可,我們可能無法有效競爭, 我們的業務可能會受到不利影響。
與員工事務和管理增長相關的風險
我們 只有有限的員工來管理和運營我們的業務。
截至2023年9月30日,我們約有12名全職員工。此外,我們利用獨立承包商和其他第三方 來協助我們業務的各個方面。我們對候選產品開發的關注要求我們優化現金使用 並以高效的方式管理和運營我們的業務。我們不能向您保證我們將能夠僱用或保留足夠的 人員水平來開發我們的候選產品或運營我們的業務,或實現我們本來會 尋求實現的所有目標。
網絡攻擊 或電信或信息技術系統中的其他故障可能導致信息被盜、數據損壞和嚴重的業務運營中斷。
我們 利用信息技術或IT、系統和網絡來處理、傳輸和存儲與我們的業務活動相關的電子信息。隨着數字技術的使用越來越多,網絡事件,包括蓄意攻擊和企圖未經授權進入計算機系統和網絡,也增加了頻率和複雜性。這些威脅對我們的系統和網絡的安全、數據的保密性以及可用性和完整性構成了風險。
我們未來的成功取決於我們能否留住關鍵員工、顧問和顧問,以及吸引、留住和激勵合格的 人員。
我們 高度依賴高管團隊的主要成員和關鍵員工,失去他們的服務可能會對我們目標的實現 產生不利影響。雖然我們已經與我們的某些高管簽訂了僱傭協議,但他們中的任何人都可以在任何時間 離開我們的工作崗位。我們不為這些個人的生命或我們任何其他員工的生命維護“關鍵人物”保險單。失去一名或多名現有員工的服務可能會阻礙我們實現研發和商業化目標。此外,更換高管或其他關鍵員工可能很困難,而且可能需要較長的時間,因為我們行業中擁有成功開發、獲得營銷批准和產品商業化所需的技能和經驗的人員數量有限。
為我們的業務招聘和留住其他合格的員工、顧問和顧問,包括科學和技術人員,也將是我們成功的關鍵。目前,我們的行業缺乏有技能的高管,這種情況很可能會持續下去。因此,對技能人才的競爭非常激烈,流失率可能很高。我們可能無法以可接受的條件吸引和留住人才 ,因為眾多製藥和生物技術公司都在爭奪擁有類似技能的人員 。此外,未能在臨牀前或臨牀試驗中取得成功,可能會使招聘和留住合格人員變得更具挑戰性。
此外,我們還依賴顧問和顧問,包括科學和臨牀顧問,來幫助我們制定我們的研究、開發和商業化戰略。我們的顧問和顧問可能受僱於其他實體,並可能根據與這些實體的諮詢或諮詢合同作出承諾,這可能會限制他們對我們的可用性。如果我們無法繼續吸引和留住高素質人才,我們開發和商業化候選產品的能力將受到限制。
| 71 |
無法招聘或失去任何高管、關鍵員工、顧問或顧問的服務可能會阻礙我們 研發和商業化目標的進展。
我們的 員工、獨立承包商、顧問、合作者和合同研究組織可能從事不當行為或其他 不當活動,包括不遵守監管標準和要求,這可能會給我們造成重大責任並損害我們的聲譽。
我們 面臨員工、獨立承包商、顧問、合作者和合同研究組織可能 從事欺詐行為或其他非法活動的風險。這些各方的不當行為可能包括故意、魯莽和/或疏忽 從事或向我們披露違反以下規定的未經授權的活動:(1)FDA的法規或類似的非美國監管機構的類似法規,包括要求向這些機構報告真實、完整和準確信息的法律, (2)製造標準,(3)聯邦和州醫療欺詐和濫用法律法規以及由類似的非美國監管機構制定並執行的類似法律法規,以及(4)要求準確報告財務信息或數據的法律。特別是,醫療保健行業的銷售、營銷和業務安排受到旨在防止欺詐、不當行為、回扣、自我交易、賄賂和其他濫用行為的廣泛法律法規的約束。這些法律法規限制或禁止廣泛的定價、折扣、營銷和促銷、銷售佣金、客户激勵計劃和其他業務安排 。員工或合作者的不當行為還可能涉及不當使用(包括交易)在臨牀試驗過程中獲得的信息,這可能會導致監管制裁併嚴重損害我們的聲譽。雖然我們有行為和商業道德準則,但並不總是能夠識別和阻止不當行為,我們為檢測和防止此類活動而採取的預防措施可能無法有效控制未知或未管理的風險或損失,或保護我們免受政府 調查或因未能遵守此類法律、標準或法規而引發的其他行動或訴訟。如果對我們提起任何此類訴訟,而我們未能成功為自己辯護或維護自己的權利,這些訴訟可能會對我們的業務和運營結果產生重大影響,包括施加民事、刑事和行政處罰, 損害賠償,罰款,可能被排除在聯邦醫療保險、醫療補助和其他聯邦醫療保健計劃之外,如果我們受到公司誠信協議或類似協議的約束,如果我們受到公司誠信協議或類似協議的約束,以解決有關違反這些法律的指控、監禁、合同損害、聲譽損害、利潤減少和未來收益,則可能會受到額外的報告要求和/或監督。以及 削減我們的業務,其中任何一項都可能對我們的業務運營能力和我們的運營結果產生重大不利影響 。
我們 希望擴大我們的組織,因此,我們在管理我們的增長時可能會遇到困難,這可能會中斷我們的運營。
我們 預計我們的員工數量和業務範圍將大幅增長,特別是在藥品製造、監管事務和銷售、營銷和分銷領域,並支持我們的上市公司運營。要管理這些增長活動,我們必須繼續實施和改進我們的管理、運營和財務系統,擴大我們的設施,並繼續招聘和培訓更多合格的人員。我們的管理層可能需要投入大量精力來管理這些增長活動。此外,我們的預期增長可能需要我們搬遷到我們歷史上所在地區以外的地理區域。例如,我們在新澤西州普林斯頓設有辦事處,我們的許多財務、管理和行政人員都在那裏工作。由於我們的財務資源有限,而且我們的管理團隊在管理具有如此預期增長的公司方面的經驗也有限 ,我們可能無法有效地管理我們業務的擴張或搬遷, 留住關鍵員工,或確定、招聘和培訓更多合格的人員。我們無法有效地管理我們業務的擴展或搬遷 可能會導致我們的基礎設施薄弱,導致操作錯誤、失去商機、員工流失以及剩餘員工的生產率下降。我們的預期增長還可能需要大量的資本支出 ,並可能會將財務資源從其他項目中轉移出來,例如開發更多候選產品。如果我們不能 有效管理我們的預期增長,我們的支出可能會比預期增加得更多,我們的創收能力可能會降低 ,我們可能無法實施我們的業務戰略,包括我們候選產品的成功商業化。
| 72 |
與我們的普通股相關的風險
我們普通股的市場價格可能會有很大波動。
我們普通股的市場價格可能波動較大,可能會受到以下因素的影響:
| ● | 本公司季度或年度經營業績的實際或預期波動; |
| ● | 財務或業務估計或預測的變化 ; |
| ● | 一般的市場狀況; |
| ● | 與我們類似的公司的經濟表現或市場估值的變化 ;以及 |
| ● | 美國或其他地方的一般經濟或政治狀況。 |
特別是,像我們這樣的生物技術公司的市場價格一直非常不穩定,原因包括但不限於 :
| ● | 任何延誤或未能對我們的產品進行臨牀試驗或獲得FDA和其他監管機構的批准; |
| ● | 有關公司知識產權的事態發展或糾紛; |
| ● | 此類公司或其競爭對手的技術創新; |
| ● | 類似公司的市場估值變化 ; |
| ● | 此類公司或其競爭對手宣佈重大合同、收購、戰略夥伴關係、合資企業、資本承諾、新技術或專利;以及 |
| ● | 未能完成重大交易或與供應商合作生產產品。 |
證券市場不時經歷與特定公司的經營業績無關的重大價格和成交量波動。這些市場波動也可能對我們普通股的市場價格產生實質性的不利影響。
我們 預計在可預見的未來不會支付現金股息,因此投資者不應預期他們的投資會有現金股息。
我們的董事會不打算在可預見的未來支付現金股息,而是打算保留任何和所有收益 為業務增長提供資金。到目前為止,我們沒有支付任何現金股息,也不能保證我們的普通股將永遠支付現金股息 。
作為一家上市公司,我們 會產生巨大的成本並投入大量的管理時間,我們預計這些成本 將會增加。
作為一家上市公司,我們承擔了大量的法律、會計和其他費用。例如,我們被要求遵守《薩班斯-奧克斯利法案》和《多德-弗蘭克華爾街改革和消費者保護法案》的某些 要求,以及美國證券交易委員會隨後實施的規則和條例 ,包括建立和維持有效的信息披露和財務控制,以及公司治理實踐中的變化 。我們預計,遵守這些要求將增加我們的法律和財務合規成本 ,並將使某些活動更加耗時和成本高昂。此外,我們預計我們的管理層和其他人員將需要 將注意力從運營和其他業務事務上轉移,以便將大量時間用於這些上市公司要求。 尤其是,我們預計將產生鉅額費用,並投入大量管理工作以確保遵守《薩班斯-奧克斯利法案》第404條的要求 。我們目前沒有內部審計職能,我們可能需要招聘或簽約聘請具有適當上市公司經驗和技術會計知識的額外會計和財務人員。
我們的內部控制的有效性可能存在限制,如果我們的控制系統不能防止錯誤或欺詐,可能會對我們的公司造成重大損害。
根據《薩班斯-奧克斯利法案》第404條,我們 必須由我們的管理層提交一份關於財務報告內部控制有效性的報告。這項評估將需要包括披露我們管理層在財務報告內部控制中發現的任何重大弱點。重大缺陷是指財務報告的內部控制存在缺陷或缺陷的組合,使得年度或中期合併財務報表的重大錯報有合理的可能性無法及時防止或發現。
| 73 |
有效的財務報告內部控制對於我們提供可靠和及時的財務報告是必要的,並與適當的 披露控制和程序一起旨在合理地發現和防止欺詐。任何未能實施所需的新的或改進的控制措施,或在實施過程中遇到的困難,都可能導致我們無法履行我們的報告義務。未發現的材料 我們對財務報告的內部控制存在弱點,可能導致財務報表重述,並要求我們產生補救費用 。
此外, 我們預計財務報告的披露控制或內部控制不會阻止所有錯誤和所有欺詐。控制系統,無論設計和操作有多好,都只能提供合理的保證,而不是絕對的保證,以確保控制系統的目標能夠實現。此外,控制系統的設計必須反映這樣一個事實,即存在資源限制,並且必須考慮控制相對於其成本的好處。由於所有控制系統的固有侷限性,任何對控制的評估都不能絕對保證所有控制問題和舞弊實例(如果有)都已被檢測到。如果我們的控制系統未能檢測或防止錯誤或欺詐,可能會對我們造成嚴重的負面影響。
如果發生上述任何情況,都可能對公眾對我們公司的看法產生負面影響,這可能會對我們的股價產生負面影響。
我們 可能無法及時完成我們對財務報告的內部控制的分析,或者這些內部控制 可能無法確定為有效,這可能會對投資者對我們公司的信心產生不利影響,從而影響我們的 普通股的價值。
我們 可能無法完成對財務報告內部控制的評估和測試。在評估和測試過程中 如果我們在財務報告的內部控制中發現一個或多個重大弱點,我們將無法斷言我們的內部控制有效。
如果我們無法斷言我們對財務報告的內部控制是有效的,或者如果適用,我們的獨立註冊會計師事務所無法對我們的內部控制的有效性發表意見,我們可能會失去投資者對我們財務報告的準確性和完整性的信心 ,這將導致我們的普通股價格下跌,我們可能 受到美國證券交易委員會的調查或處罰。我們還將被要求每季度披露在內部控制和程序方面所做的更改。
特拉華州法律的反收購條款可能會使收購合併後的公司變得更加困難,並可能阻止合併後的公司股東試圖更換或撤換合併後的公司管理層。
由於合併後的公司將在特拉華州註冊成立,因此它受特拉華州通用公司法第203節或DGCL的條款管轄,該條款禁止持有已發行合併公司投票權股票超過15%的股東合併 或與合併後的公司合併。儘管我們相信這些條款將通過要求潛在收購者與合併後的公司董事會進行談判,從而提供獲得更高出價的機會,但如果某些股東可能認為要約是有益的,這些條款甚至可以適用。此外,這些規定可能會使股東更難更換負責任命管理層成員的董事會成員,從而挫敗或阻止 合併後公司股東更換或撤換現有管理層的任何企圖。
董事 和高級船員責任有限。
由於 特拉華州法律允許,我們的章程限制了董事因違反董事的受託責任而承擔的金錢損害賠償責任,但在某些情況下的責任除外。由於我們的章程條款和特拉華州法律,股東可能只有有限的權利 向違反受託責任的董事追償。
| 74 |
一般風險因素
網絡攻擊 或電信或信息技術系統中的其他故障可能導致信息被盜、數據損壞和嚴重的業務運營中斷。
我們 利用信息技術或IT、系統和網絡來處理、傳輸和存儲與我們的業務活動相關的電子信息。隨着數字技術的使用越來越多,網絡事件,包括蓄意攻擊和企圖未經授權進入計算機系統和網絡,也增加了頻率和複雜性。這些威脅對我們的系統和網絡的安全、數據的保密性以及可用性和完整性構成了風險。
由於增發普通股、可轉換證券、認股權證或期權,我們的普通股可能會進一步稀釋。
過去,為了籌集資金,我們發行了普通股、可轉換證券(如可轉換票據)和權證。我們 還發行了普通股,作為對我們的員工、董事和某些供應商的服務補償和激勵性補償。 我們保留了普通股,以便在行使其中某些證券時發行,並可能在未來增加為這些目的保留的股份 。我們增發普通股、可轉換證券、期權和認股權證可能會影響我們股東的權利,可能會降低我們普通股的市場價格,或者可能導致對已發行認股權證的行權價格進行調整(導致這些證券可以行使更多的普通股 股票),或者可能迫使我們向某些股東發行額外的普通股。
有資格未來出售的股票 可能會對市場產生不利影響。
根據證券法頒佈的第144條規則,我們的某些股東可能不時有資格通過普通經紀交易在公開市場上出售其全部或部分普通股,但受某些限制。總體而言,根據第144條,前三個月為非關聯公司的股東可以在六個月後自由出售我們普通股的股票,但必須遵守當前的公開信息要求。關聯公司可以在六個月後根據規則144的數量、銷售方式、最新公開信息和通知要求出售我們普通股的股票。根據規則144出售我們的普通股,可能會對我們普通股的市場價格產生重大不利影響。
項目 1B。未解決的員工評論。
不適用 。
第 項2.屬性。
該公司依賴短期辦公使用合同來採購辦公和會議空間。
第3項:法律訴訟。
我們 目前沒有受到任何重大法律程序的影響。但是,我們可能會不時地成為我們正常業務過程中出現的各種法律程序的一方。
第 項4.礦山安全信息披露
不適用 。
| 75 |
第 第二部分
第 項5.註冊人普通股市場、相關股東事項和發行人購買股權證券。
市場信息
該公司的普通股在納斯達克資本市場上交易,代碼為“SONN”。
持有者
截至2023年12月11日,我們共有76名普通股持有者。記錄持有人的數量是根據轉讓代理的記錄確定的,不包括普通股的受益所有人,其股票以各種證券經紀人、交易商和註冊結算機構的名義持有。我們普通股的轉讓代理是證券轉讓公司,地址:達拉斯公園路北2901號,地址:380Suite380,Plano,TX 75093。
分紅
我們 從未宣佈或支付過普通股的現金股息。在可預見的未來,我們不打算宣佈或支付普通股的現金股息 ,但目前打算保留任何未來的收益,為我們業務的發展和增長提供資金。 普通股支付現金股息(如果有的話)將完全由我們的董事會酌情決定,並將取決於我們的收益、資本要求、財務狀況和其他相關因素。
第 項6.[已保留]
| 76 |
第 項7.管理層對財務狀況和經營成果的討論和分析
以下 管理層對財務狀況和運營結果的討論和分析(“MD&A”)旨在 幫助瞭解我們的財務狀況和我們在本報告所述期間的歷史運營結果。本MD&A應與本年度報告(Form 10-K)中包含的經審計的合併財務報表及其附註一併閲讀。本MD&A可能包含涉及風險和不確定因素的前瞻性陳述。有關前瞻性陳述的討論,請參閲上文“關於前瞻性陳述的特別説明”標題下的信息,該信息通過引用併入本文。
概述
十四行詩 BioTreateutics Holdings,Inc.(“十四行詩”,“我們”或“公司”), 是一家臨牀階段、專注於腫瘤學的生物技術公司,擁有創新單一或雙功能生物藥物的專利平臺。稱為FHAB™(完全人血白蛋白結合),該技術利用完全人單鏈 抗體片段,其結合人血白蛋白並在人血白蛋白上“搭便車”以運輸至靶組織。我們設計了 結構,以改善藥物在特定組織中的積累,以及延長體內活性的持續時間。FHAB 發育候選物在哺乳動物細胞培養物中產生,其能夠糖基化,從而降低免疫原性的風險。 我們相信我們的FHAB技術於二零二一年六月獲得美國專利,是我們生物製藥 平臺的顯著特色,非常適合未來在一系列人類疾病領域進行藥物開發,包括腫瘤學、自身免疫性、致病性、 炎症和血液學疾病。
我們 目前的內部管道開發活動主要集中在細胞因子上,這是一類細胞信號肽,除其他重要功能外,還可用作強效免疫調節劑。特異性細胞因子既可獨立發揮作用,也可協同發揮作用, 能夠調節對抗癌症和病原體的免疫細胞的激活和成熟。然而,因為它們不優先 在特定組織中積累並從體內快速消除,所以用 細胞因子療法實現治療效果的常規方法通常需要高劑量和頻繁劑量的施用。這可能導致治療效果降低,伴隨 全身毒性的可能性,這對這類藥物的治療應用提出了挑戰。
我們的 主要專有資產SON-1010是一種完全人源化的白細胞介素12(“IL-12”),與FHAB 構建體,我們正在實體瘤適應症中進行臨牀開發,包括卵巢癌、非小細胞肺癌 和頭頸癌。於2022年3月,FDA批准我們的SON-1010新藥研究(“IND”)申請。 這使我們能夠在 2022年第二季度在實體瘤患者中啟動美國臨牀試驗(SB 101)。於2021年9月,我們成立了一家全資擁有的澳大利亞附屬公司SonnetBio Pty Ltd(“附屬公司”),以進行若干臨牀試驗。我們獲得批准,並於2022年第三季度在健康 志願者中啟動了SON-1010的澳大利亞臨牀研究(SB 102)。SB 101和SB 102研究的中期安全性和耐受性數據於2023年4月報告 。
2023年1月,我們宣佈與羅氏達成合作協議,對SON-1010與atezolizumab(Tecentriq®)進行臨牀評價。 兩家公司已簽署了一份臨牀試驗和供應主協議(“MCSA”)以及輔助質量和安全性協議 ,以研究SON-1010和atezolizumab聯合治療鉑類耐藥卵巢癌(“PROC”)患者的安全性和有效性 。此外,兩家公司將分別提供SON-1010和atezolizumab,用於1b期/2a期聯合用藥的安全性、劑量遞增和療效研究(SB 221)。本2部分研究的第1部分於2023年6月由澳大利亞當地人類研究倫理委員會 根據CT-2023-CTN-01399-1批准,並已通知治療產品管理局。2023年8月,FDA接受了SB 221的IND。 該試驗包括第1部分的改良3+3劑量遞增設計,以確定SON-1010與 固定劑量atezolizumab的最大耐受劑量(MTD)。將在擴展組中確認PROC的臨牀獲益,以確定推薦的II期劑量 (RP 2D)。然後,研究的第2部分將在隨機比較中研究SON-1010單藥治療、其與atezolizumab聯合使用或PROC的標準治療 (SOC),以顯示概念驗證(POC)。
| 77 |
我們 於二零二零年四月透過收購Relief Therapeutics SA的流通股份,取得我們最先進的化合物SON-080(一種全人類版本的白細胞介素6(“IL-6”))的全球開發權。我們正在推進 SON-080在化療誘導的周圍神經病變(“CIPN”)和糖尿病周圍神經病變 (“DPN”)目標適應症中的應用。我們獲得批准,在CIPN中啟動SON-080的美國以外1b/2a期研究。SB 211化療誘導的周圍神經病變(CIPN)研究的第一 部分入組工作即將完成,這將使 DSMB能夠在2024年第一個日曆季度完成對初步安全性數據的審查。根據我們於二零二一年五月與新加坡的New Life Therapeutics Pte,Ltd.(“New Life”)訂立的許可協議,Sonnet及New Life將共同負責在DPN開發SON-080。目的是分析數據,並在評估CIPN安全性數據後考慮啟動 II期研究。
SON-1210(IL12-F)HAB-IL 15),我們的領先雙特異性構建體,結合FHAB與完全人IL-12和完全人白細胞介素 15(“IL-15”)。該化合物正在開發用於實體瘤適應症,包括結直腸癌。2023年2月,我們宣佈成功完成了兩項SON-1210在非人類靈長類動物中的IND毒理學研究。我們準備 啟動SON-1210的監管授權流程,等待任何合作活動的結果。
SON-1410(IL18-F)HAB-IL12)是白細胞介素18(“IL-18”)和IL-12治療實體瘤的雙特異性組合。細胞系開發和工藝開發正在進行中,早期實驗性藥物供應適合配方和分析方法開發活動。經過2023年的一些延遲之後,活動將持續到2024年,有可能產生一種適合臨牀前研究和後續人體研究的藥物。
我們 已完成SON-3015(抗IL6-F)的序列確認HAB-抗轉化生長因子β)。已生產出早期雙特效藥物 ,並正在儲存以備將來使用體內老鼠研究。出於降低費用的目的,我們已選擇擱置SON-3015開發計劃 。
作為正在進行的成本削減評估的一部分,SON-1010的所有抗病毒開發已暫停。
自成立以來,我們 發生了經常性的運營虧損和負現金流。我們能否產生足以實現盈利的產品或許可收入 將在很大程度上取決於我們當前或未來的一個或多個候選產品的成功開發和最終商業化。截至2023年9月30日和2022年9月30日的年度,我們的淨虧損分別為1880萬美元和2970萬美元。截至2023年9月30日,我們擁有230萬美元的現金。我們預計,至少在未來幾年內,我們將繼續產生鉅額費用並 增加運營虧損。我們預計,與我們正在進行的活動相關的費用和資本需求將大幅增加 ,特別是當我們:
| ● | 為候選產品進行 個額外的臨牀試驗; | |
| ● | 繼續 發現和開發其他候選產品; | |
| ● | 獲取 或許可其他候選產品和技術; | |
| ● | 維護, 擴大和保護我們的知識產權組合; | |
| ● | 僱用 增加臨牀、科學和商業人員; |
| 78 |
| ● | 建立 商業製造來源和安全的供應鏈能力,足以提供 我們可能獲得監管批准的任何候選產品的商業數量; | |
| ● | 查找 成功完成臨牀試驗的候選產品的監管批准; | |
| ● | 建立 銷售、市場營銷和分銷基礎架構,用於將 我們可能會獲得監管批准;以及 | |
| ● | 添加 業務、財務和管理信息系統和人員,包括人事 以支持我們的產品開發和計劃的未來商業化工作,以及 以支持我們作為一家公開報道公司的運作。 |
我們 不會從產品銷售中獲得收入(如果有),除非我們獲得許可收入和/或成功完成臨牀 開發並獲得候選產品的監管批准。如果我們的任何候選產品 獲得監管部門的批准,但沒有建立商業化合作夥伴關係,我們預計將產生與開發我們的內部商業化能力相關的大量費用, 以支持產品銷售、營銷和分銷。我們將繼續承擔與作為上市公司運營 相關的重大成本。
因此,我們將需要大量的額外資金來支持我們的持續運營和實現我們的增長戰略。在 我們能夠從產品銷售中獲得可觀收入之前(如果有的話),我們希望通過出售股權、 債務融資或其他資本來源(可能包括與其他公司合作或其他戰略交易)為我們的運營提供資金。我們可能 無法籌集額外資金或在需要時以優惠條款訂立此類其他協議或安排,或根本無法訂立此類協議或安排。 如果我們未能在需要時籌集資金或簽訂此類協議,我們可能不得不大幅推遲、減少或取消 一個或多個候選產品的開發和商業化,或推遲我們尋求潛在的許可或收購。
由於 與產品開發相關的眾多風險和不確定性,我們無法預測 費用增加的時間或金額,也無法預測我們何時或是否能夠實現或保持盈利能力。即使我們能夠產生產品銷售,我們也可能 無法盈利。如果我們無法實現盈利,或無法在持續基礎上保持盈利能力,或無法籌集額外資本 或簽訂合作或許可協議,則我們可能無法繼續按計劃水平運營,並被迫 減少或終止運營。
自 2015年成立以來,我們投入了大量的精力和財務資源來組織和配備公司人員, 業務規劃,籌集資金,收購或發現候選產品並確保相關知識產權,以及 為候選產品進行發現,研究和開發活動。我們沒有任何批准銷售的產品, 也沒有從產品銷售中獲得任何收入。迄今為止,我們的運營資金主要來自出售普通股、 認股權證和發行可轉換債券的收益。
| 79 |
運營結果的組成部分
協作 收入
協作 收入目前來自於2021年5月與New Life簽訂的許可協議,該協議授予New Life 獨家許可權(具有分許可權)開發和商業化含有特定重組人IL-6 SON-080的藥物製劑(“化合物”)(此類製劑,“產品”)用於預防、治療或緩解人類糖尿病周圍神經病變(“DPN領域”)。我們確定了協議項下的以下義務 :(i)在專屬區域內開發、銷售、進口、使用和商業化產品的許可 (下稱“許可”);以及(ii)轉讓專有技術和臨牀開發及監管活動(下稱“研發活動”)。 我們確定,許可證和研發活動彼此並無區別,因此,將這些重大承諾 合併為一項履約義務。根據該協議,我們收到了總額為100萬美元的預付現金付款,這些款項已完全 分配給單一履約義務,並在研發服務的估計履約期內確認。
運營費用
研究和開發費用
研究 和開發費用主要包括與發現和開發我們的候選產品有關的成本。 我們按實際發生的研究和開發成本支出,此類成本包括:
| ● | 與銷售相關的 員工的開支,包括工資、基於股份的薪酬和相關福利 從事研究和開發工作; | |
| ● | 與我們候選產品的臨牀前和臨牀開發相關的費用 ,包括與第三方(如顧問和臨牀研究組織)達成的協議; | |
| ● | 生產用於我們的臨牀前研究和臨牀試驗的藥物產品的成本,包括與第三方(如顧問和合同製造組織)達成的協議; | |
| ● | 設施、 折舊和其他費用,包括租金和設施維護和保險的直接費用或分攤費用; | |
| ● | 與遵守法規要求有關的成本 ; | |
| ● | 根據第三方許可協議進行付款 。 |
我們 根據服務提供商提供的信息對完成特定任務的進度進行評估,從而確認外部開發成本。此流程包括審核未結合同和採購訂單,與其人員溝通以確定已代表我們執行的服務,以及在我們尚未收到發票或以其他方式通知實際成本的情況下,估算執行的服務級別和為該服務產生的相關成本。未來收到的用於研發活動的貨物或服務的預付款不可退還 記為預付費用。當貨物已交付或服務已完成時,此類金額將確認為費用。
我們的直接研發費用主要包括外部成本,例如支付給外部顧問、CRO、CMO和研究實驗室的費用,這些費用與臨牀前開發、流程開發、製造和臨牀開發活動有關。 我們的直接研發費用還包括根據第三方許可協議產生的費用。我們不會將員工成本和與發現工作、實驗室用品和設施相關的成本(包括折舊或其他間接成本)分配給特定的候選產品,因為這些成本部署在多個計劃中,因此沒有單獨分類。 我們主要使用內部資源進行研究和發現,以及管理臨牀前開發、流程開發、製造和臨牀開發活動。這些員工在多個計劃中工作,因此,我們不按候選產品跟蹤成本 。
| 80 |
我們 在可預見的未來將繼續產生研發費用,因為我們試圖推進我們的候選產品的開發 。我們的候選產品能否成功開發具有很大的不確定性。目前,由於與臨牀開發相關的許多風險和不確定性,包括以下方面的風險和不確定性,我們無法合理估計或 知道完成我們當前管道的剩餘開發或我們可能開發的任何未來候選產品所需的工作的性質、時間和成本:
| ● | 臨牀前和臨牀開發活動的時間和進度; | |
| ● | 我們決定開展的臨牀前和臨牀項目的數量和範圍; | |
| ● | 我們 有能力維持當前的研發計劃並建立新的計劃; | |
| ● | 通過研究性新藥使能研究建立適當的安全概況; | |
| ● | 成功的 患者登記,並啟動和完成臨牀試驗; | |
| ● | 成功完成臨牀試驗,其安全性、耐受性和療效狀況令FDA或任何類似的外國監管機構滿意; | |
| ● | 收到適用監管機構的監管批准; | |
| ● | 來自適用監管機構的任何上市批准的時間、接收和條款; | |
| ● | 我們 能夠建立新的許可或協作安排; | |
| ● | 與第三方製造商建立 協議,為我們的臨牀試驗和商業生產(如果我們的任何候選產品獲得批准)提供臨牀供應; | |
| ● | 開發並及時交付可用於我們的臨牀試驗和商業投放的臨牀級和商業級藥物配方; | |
| ● | 取得、維護、捍衞和執行專利權利要求和其他知識產權; | |
| ● | 啟動 候選產品的商業銷售(如果獲得批准),無論是單獨還是與其他合作 ; | |
| ● | 在批准後,保持候選產品的持續可接受的安全概況;以及 | |
| ● | 新冠肺炎對運營的潛在影響,可能會影響臨牀試驗的時間、原材料的可用性以及訪問和保護檢測設施的能力 。 |
| 81 |
對於我們的候選產品的開發,這些變量中的任何一個的結果發生變化都可能顯著改變與該候選產品的開發相關的成本和時間安排。我們的任何候選產品可能永遠無法獲得監管部門的批准 。
一般費用 和管理費用
一般費用和行政費用主要包括行政、財務和行政職能人員的薪金和相關費用,包括按股份計算的薪酬。一般和行政費用還包括直接和分攤的設施相關成本,如 以及法律、專利、諮詢、會計和審計服務的專業費用。
隨着我們增加員工人數以支持持續的研究活動和候選產品的開發,我們的 一般和管理費用未來將會增加。我們將繼續產生更多與上市公司相關的會計、審計、法律、監管、合規和董事 和高管保險成本,以及投資者和公關費用。
外匯損失
外匯損失包括以美元以外貨幣計價的交易的匯率變動。
運營結果
2023年和2022年9月30日終了年度比較
下表彙總了截至2023年9月30日和2022年9月30日的年度運營結果:
| 截至9月30日止年度, | ||||||||||||
| 2023 | 2022 | 變化 | ||||||||||
| 協作收入 | $ | 147,805 | $ | 349,943 | $ | (202,138 | ) | |||||
| 運營費用: | ||||||||||||
| 研發 | 11,814,690 | 21,444,019 | $ | (9,629,329 | ) | |||||||
| 一般和行政 | 7,125,732 | 8,575,283 | (1,449,551 | ) | ||||||||
| 總運營費用 | 18,940,422 | 30,019,302 | (11,078,880 | ) | ||||||||
| 運營虧損 | (18,8792,617 | ) | (29,669,359 | ) | 10,876,742 | |||||||
| 匯兑損失 | (40,077 | ) | (52,482 | ) | 12,405 | |||||||
| 淨虧損 | $ | (18,832,694 | ) | $ | (29,721,841 | ) | $ | 10,889,147 | ||||
協作 收入
我們 在截至2023年9月30日的年度內確認了與新生活協議相關的收入10萬美元,而截至2022年9月30日的年度內為30萬美元。減少20萬美元是由於延遲提供研發服務。
| 82 |
研究和開發費用
截至2023年9月30日止年度的研究和開發費用為1180萬美元,而截至2022年9月30日止年度為2140萬美元。減少960萬美元的主要原因是通過將產品開發活動 轉移到印度和澳大利亞等成本較低的地區,通過減少三級項目(如SON-3015)的支出(該項目已被擱置開發),以及暫停與SON-1010相關的抗病毒藥物開發,以及基於股票的薪酬 費用的減少。
一般費用 和管理費用
截至2023年9月30日止年度的一般及行政開支為710萬美元,而截至2022年9月30日止年度為860萬美元。150萬美元的減少主要與基於股份的薪酬、法律和業務開發費用的減少有關, 因為我們正在出於流動性目的管理費用,並正在加緊關注我們評估 具有最大近期潛力的研發項目。
流動性 與資本資源
我們 迄今為止主要通過出售普通股、認股權證和發行可轉換債券的收益為運營提供資金。如果我們認為需要此類融資計劃來推進我們的業務計劃,並且符合我們股東的最佳利益,我們可能會根據市場狀況或其他情況提供額外的證券供銷售。無法確定 將來是否會提供股權或債務融資,也無法確定其條款是否可以接受,目前無法 預測這些事項的結果。
我們 截至2023年和2022年9月30日止年度分別產生淨虧損1880萬美元和2970萬美元。我們預計 在未來12個月及以後將繼續產生大量運營費用和淨虧損。我們的淨虧損 可能會在季度和年度之間大幅波動,這取決於我們的研發研究和相關支出的階段和複雜性, 我們技術許可的額外付款(如果有),以及我們可能進入的任何當前或未來 合作下的付款。
我們 已評估是否存在對我們 繼續作為持續經營的能力產生重大疑問的情況或事件(總體考慮)。我們相信,我們在2023年9月30日的現金,以及 我們在2023年10月24日公開發行普通股和認股權證收到的410萬美元淨收益,將為我們預計到2024年3月的運營提供資金。我們還預計將從澳大利亞的研發税收激勵計劃中獲得 80萬美元的淨現金返還(見合併財務報表附註2),並於最近獲得初步批准,我們申請通過技術營業税證書轉讓計劃出售高達480萬美元的新澤西州淨經營虧損,視乎有關出售的執行情況而定(見綜合 財務報表附註10)。我們將需要大量 額外融資來為我們的運營提供資金。這些因素對我們繼續 持續經營的能力提出了重大質疑。
下表 總結了我們在每個所列期間的現金來源和使用情況:
| 截至九月三十日止年度, | ||||||||
| 2023 | 2022 | |||||||
| 用於經營活動的現金淨額 | $ | (21,341,842 | ) | $ | (27,773,528 | ) | ||
| 用於投資活動的現金淨額 | (443,250 | ) | (810,227 | ) | ||||
| 融資活動提供的現金淨額 | 21,006,472 | 4,014,567 | ||||||
| 現金淨減少 | $ | (778,620 | ) | $ | (24,569,188 | ) | ||
| 83 |
操作 活動
在截至2023年9月30日的 年度,我們在經營活動中使用了2130萬美元的現金,這主要歸因於我們的淨 虧損1880萬美元,預付費用和其他資產淨增加50萬美元,主要是由於研究和 開發活動的現金流出,應付賬款和應計費用淨減少240萬美元,主要是由於研究和開發費用減少,被收購的正在進行的研究和開發費用30萬美元和基於股票的薪酬費用20萬美元所抵消。
在截至2022年9月30日的 年度,我們在經營活動中使用了2780萬美元的現金,這主要歸因於我們的淨 虧損2970萬美元。這一金額被90萬美元的股份薪酬費用和100萬美元的收購過程中 研究和開發費用以及10萬美元的運營資產和負債淨減少所抵消。
投資 活動
在截至2023年9月30日的年度內,我們在投資活動中使用了40萬美元現金,用於購買收購的正在進行的研究和開發 。
在截至2022年9月30日的年度內,我們在投資活動中使用了80萬美元現金,用於購買收購的正在進行的研究和開發 。
為 活動提供資金
在截至2023年9月30日的年度內,融資活動提供的現金淨額為2,100萬美元,主要包括在市場融資機制下出售普通股以及通過包銷的公開發行和登記直接發行所得的淨收益。
在截至2022年9月30日的年度內,融資活動提供的現金淨額為400萬美元,主要包括出售優先股和認股權證的淨收益以及出售普通股的淨收益。
資金需求
我們 預計將繼續產生與我們正在進行的活動相關的鉅額費用,特別是在我們推進臨牀前活動和正在開發的候選產品的臨牀試驗的情況下。此外,我們預計將繼續產生與上市公司運營 相關的成本。我們的營運開支的時間和數額將主要視乎:
| ● | 我們當前或未來候選產品的 範圍、數量、啟動、進度、時間、成本、設計、持續時間、任何潛在延遲、 臨牀試驗和非臨牀研究的結果 ; | |
| ● | 我們為這些候選產品制定的 臨牀開發計劃; | |
| ● | 我們開發或可能獲得許可的候選產品和程序的數量和特徵; |
| 84 |
| ● | 監管審查、批准或其他行動的結果、時間和成本,以滿足FDA和類似的外國監管機構建立的監管要求,包括 FDA或類似的外國監管機構可能要求我們 對我們的候選產品進行比我們目前預期的更多的研究; | |
| ● | 我們 獲得候選產品上市批准的能力; | |
| ● | 申請、起訴、辯護和執行專利主張和涵蓋我們候選產品的其他知識產權的 成本; | |
| ● | 我們 維護、擴展和捍衞我們的知識產權組合範圍的能力,包括為知識產權糾紛辯護的成本,包括第三方對我們或我們的候選產品提起的專利侵權訴訟 ; | |
| ● | 與候選產品有關的商業規模外包製造活動的 成本和完成時間; | |
| ● | 我們 以優惠條款建立和維護許可、協作或類似安排的能力,以及我們是否以及在多大程度上根據任何新的許可、協作或類似安排保留開發或商業化責任 ; | |
| ● | 為我們選擇自行商業化產品的地區獲得監管批准的任何候選產品建立銷售、營銷和分銷能力的 成本 ; | |
| ● | 我們收購或投資的任何其他業務、產品或技術的成功; | |
| ● | 收購、許可或投資企業、候選產品和技術的成本; | |
| ● | 我們需要和有能力聘請更多的管理人員以及科學和醫療人員; | |
| ● | 在美國作為上市公司運營的 成本,包括為我們的業務實施額外的財務和報告系統以及其他內部系統和基礎設施的需要 ; | |
| ● | 市場接受我們的候選產品,只要任何產品被批准用於商業銷售; | |
| ● | 競爭的技術和市場發展的影響;以及 | |
| ● | 新冠肺炎疫情對我們的臨牀試驗和運營的潛在影響。 |
在 我們能夠產生可觀的產品收入之前,我們預計將通過股權發行、債務融資、合作、戰略聯盟以及與第三方的營銷、分銷或許可安排的組合來滿足我們的現金需求。 如果我們通過出售股權或可轉換債務證券籌集額外資本,我們的所有權權益可能會被大幅稀釋,此類證券的條款可能包括清算或其他對我們股東權利產生不利影響的優惠。債務融資和優先股權融資(如果可用)可能涉及包括限制性 契約的協議,這些契約限制我們採取特定行動的能力,例如招致額外債務、進行資本支出或宣佈 股息。如果我們通過與第三方合作、戰略聯盟或營銷、分銷或許可安排來籌集資金,我們可能不得不放棄對技術、未來收入流、研究計劃或候選產品的寶貴權利,或者 以可能對我們不利的條款授予許可證。如果我們無法在需要時通過股權或債務融資或其他安排籌集更多資金,我們可能需要推遲、減少或取消產品開發或未來的商業化努力, 出售資產,或授予開發和營銷我們原本更願意開發和營銷的候選產品的權利。
| 85 |
2023年2月提供
2023年2月10日,我們通過Chardan Capital Markets,LLC和EF Hutton,Benchmark Investments LLC的部門作為承銷商,完成了普通股和某些認股權證的公開發行,通過發行和出售530,222股普通股,向某些投資者發行和出售預資金權證,獲得1360萬美元的淨收益,購買101,090股普通股,以及隨附的普通權證,購買最多1,262,618股普通股(“2月發售”)。每股普通股和購買一股普通股的預融資權證與購買兩股普通股的普通權證一起出售。普通股和配套普通權證的公開發行價為每股23.76美元,每份預籌資權證和配套普通權證的公開發行價為23.7578美元。普通權證可立即以每股普通股23.76美元的價格行使,自發行之日起滿五年,並載有替代的無現金行使條款 ,根據該條款,在符合某些條件的情況下,可在普通股的無現金交易中以每股普通股0.5股的費率行使認股權證,否則可通過現金行使。預先出資的認股權證可在任何時間立即行使,直至全部行使為止,價格為每股普通股0.0022美元。此外,向承銷商發行了購買44,190股普通股的認股權證,作為對承銷商與此次發行相關服務的補償。這些普通股認股權證 的行使價為每股29.70美元,自發行之日起五年到期。
2023年6月產品
2023年6月30日,我們通過Chardan Capital Markets LLC作為配售代理完成了登記直接發售普通股(以及普通股等價物)和同時私募若干普通股認股權證,通過發行和出售166,363股我們的普通股以及向某些投資者發行購買60,909股普通股的預融資權證和隨附的認股權證以購買最多227,272股我們的普通股 (“六月發售”),獲得190萬美元的淨收益。每股普通股和購買一股普通股的預融資權證與購買一股普通股的普通權證一起出售。每股普通股及配套普通權證的公開發行價為9.90美元。普通股認股權證從2023年12月30日起以普通股每股14.8478美元的價格行使,自發行之日起三年半到期,幷包含替代的無現金行使條款。預先出資的認股權證可隨時立即行使,直至全部行使為止,價格為每股普通股0.0022美元。此外,向配售代理髮行了購買6,818股普通股的認股權證,作為對其與發行相關服務的補償。 這些普通股認股權證的行使價為每股14.8478美元,自發行之日起三年半到期。
| 86 |
2023年10月提供服務
2023年10月26日,我們通過Chardan Capital Markets、LLC和Ldenburg Thalmann&Co.Inc.作為承銷商完成了普通股和某些認股權證的公開發行,通過發行和出售1,306,250股普通股獲得淨收益410萬美元,並向某些投資者發行了購買1,537,500股普通股的預資金權證,以及 購買最多5,687,500股普通股的配套普通權證(“十月發售”)。每股普通股 和購買一股普通股的預付資金權證與購買兩股普通股的普通權證一起出售。每股普通股及附帶普通權證的公開發行價為1.6美元,每份預籌資權證及附帶普通權證的公開發行價為1.5999美元。普通權證可立即按普通股每股1.6美元的價格行使,自發行之日起滿五年,幷包含替代的無現金行使條款。 預先出資的權證可隨時立即行使,直至全部行使,普通股價格為每股0.0001美元。 此外,還向承銷商發行了購買85,312股普通股的權證,作為對承銷商提供的與發行相關的服務的補償。這些普通股認股權證的行權價為每股2.00美元,自發行之日起5年內到期。
在市場上發行普通股
我們 於2022年8月15日與BTIG,LLC(“BTIG”)訂立市場銷售協議(“2022年銷售協議”)。 根據2022年銷售協議,我們可不時透過BTIG作為銷售代理及/或委託人發售及出售我們普通股的股份 ,總髮行價最高可達2,500萬美元,但須受2022年銷售協議對我們可提供及出售的普通股 金額的若干限制所規限。由於適用於我們的發售限制,我們 根據2022年銷售協議 為出售我們普通股的股份提交了招股説明書補充資料,總髮行價高達780萬美元。在截至2023年9月30日的年度內,我們根據與BTIG的2022年銷售協議,出售了總計136,701股普通股,總收益為580萬美元,淨收益為540萬美元。截至2023年9月30日, 根據《2022年銷售協議》,已無登記股份待售。
合同義務和承諾
下表彙總了截至2023年9月30日我們的合同義務,以及此類義務預計將對我們未來的流動性和現金流產生的影響:
| 不到1年 | 1至3年 | 4至5年 | 5年以上 | 總計 | ||||||||||||||||
| 經營租賃(1) | $ | 93,614 | $ | 143,703 | $ | — | $ | — | $ | 237,317 | ||||||||||
| 總計 | $ | 93,614 | $ | 143,703 | $ | — | $ | — | $ | 237,317 | ||||||||||
(1) 反映了我們在新澤西州普林斯頓的辦公室租約規定的義務。
除了我們在上表中反映的具有付款承諾的合同外,我們還在 正常業務過程中與某些CRO、CMO和其他第三方就臨牀前研究和測試、臨牀 試驗和製造服務簽訂了其他合同。這些合同不包含任何最低採購承諾,可在事先通知後取消 ,因此不包括在上述合同義務和承諾表中。取消時應支付的款項僅包括所提供服務的付款和所發生的費用,包括對我們的服務提供商的不可取消義務,直至取消日期。
| 87 |
關鍵會計政策和估算
我們管理層對財務狀況和運營結果的討論和分析基於我們的合併財務報表, 該報表是根據美國公認會計準則編制的。在編制這些合併財務報表時,我們需要做出影響資產、負債和費用報告金額以及或有資產和負債在財務報表中的披露的估計和判斷。我們會持續評估我們的估計和判斷,包括與研發費用應計相關的估計和判斷。我們的估計基於歷史經驗、已知趨勢和事件以及各種被認為在當時情況下是合理的其他因素,這些因素的結果構成了對資產和負債的賬面價值作出判斷的基礎,而這些資產和負債的賬面價值從其他來源看起來並不是很明顯。在不同的假設或條件下,實際結果可能與這些 估計值不同。
雖然我們的主要會計政策在本10-K表格中其他地方包括的綜合財務報表附註中有更詳細的描述,但我們認為以下會計政策對編制綜合財務報表所使用的判斷和估計最為關鍵。
研究和開發費用
研究和開發費用包括與我們的生物製藥產品開發相關的所有直接和間接成本。這些 費用包括人員成本、諮詢費以及向第三方支付的研發和製造服務費用。 這些成本在發生時計入費用。
在每個報告期結束時,我們會根據合同中定義的進度衡量標準,將向第三方服務提供商支付的款項與相關項目完成的估計進度進行比較。我們在編制評估時考慮的因素包括: 服務提供商產生的成本、實現的里程碑以及與我們的服務提供商的努力相關的其他標準。隨着獲得更多信息,此類 估計值可能會發生變化。根據向第三方服務提供商付款的時間 以及我們估計所提供服務取得的進展,我們將記錄與這些成本相關的預付費用或應計負債。或有發展或監管里程碑付款在此類或有事項的相關解決方案 中確認。截至2023年9月30日,我們沒有對應計研究和開發費用的先前估計進行任何實質性調整。
最近 發佈了會計公告
最近發佈的可能影響我們財務狀況和經營結果的會計聲明的説明 在本表格10-K其他部分包括的合併財務報表的附註2中披露。
第 7A項。關於市場風險的定性和定量披露。
不適用 。
| 88 |
第 項財務報表和補充數據
十四行詩 生物治療控股公司
合併財務報表索引
| 頁面 | |
| 獨立註冊會計師事務所報告(畢馬威,LLP,費城,賓夕法尼亞州,審計師事務所ID: |
90 |
| 合併資產負債表 | 91 |
| 合併業務報表 | 92 |
| 合併股東權益變動表(虧損) | 93 |
| 合併現金流量表 | 94 |
| 合併財務報表附註 | 95 |
| 89 |
獨立註冊會計師事務所報告
致股東和董事會 Sonnet BioTreeutics Holdings,Inc.
關於合併財務報表的意見
我們已審計了所附的Sonnet BioTreateutics Holdings,Inc.及其子公司(本公司)截至2023年9月30日、2023年9月和2022年9月30日的合併資產負債表、截至2023年9月30日和2022年9月30日的相關合並經營報表、截至2023年9月30日的股東赤字變化和現金流量以及相關的 附註(統稱為合併財務報表)。我們認為,合併財務報表公平地反映了公司截至2023年9月30日、2023年9月和2022年9月30日的財務狀況,以及截至2023年9月30日和2022年9月30日的經營業績和現金流量。符合美國公認的會計原則。
正在進行 關注
隨附的綜合財務報表已於 編制時假設本公司將繼續作為持續經營企業。正如綜合財務報表附註1所述,本公司自成立以來已出現經常性虧損及營運現金流為負的情況,並將需要大量額外的 融資以繼續為其研發活動提供資金,這令人對其作為持續經營企業的能力產生重大懷疑。附註1也説明瞭管理層在這些事項上的計劃。合併財務報表 不包括可能因這種不確定性的結果而產生的任何調整。
徵求意見的依據
這些合併財務報表由公司管理層負責。我們的責任是根據我們的 審計對這些合併財務報表發表意見。我們是一家在美國上市公司會計監督委員會(PCAOB)註冊的公共會計師事務所,根據美國聯邦證券法以及美國證券交易委員會和PCAOB的適用規則和法規,我們必須與公司保持獨立。
我們按照PCAOB的標準進行審核。這些標準要求我們計劃和執行審計,以獲得關於合併財務報表是否沒有重大錯報的合理保證,無論是由於錯誤還是欺詐。本公司不需要,也沒有聘請 對其財務報告的內部控制進行審計。作為我們審計的一部分,我們需要了解財務報告的內部控制,但不是為了對公司財務報告內部控制的有效性發表意見。因此,我們不表達這樣的意見。
我們的審計包括執行評估合併財務報表重大錯報的風險的程序 ,無論是由於錯誤還是欺詐,以及執行應對這些風險的程序。這些程序包括在測試的基礎上審查關於合併財務報表中的金額和披露的證據。我們的審計還包括評估管理層使用的會計原則和作出的重大估計,以及評估合併財務報表的整體列報。我們相信,我們的審計為我們的觀點提供了合理的 基礎。
| 90 |
重大審計事項
下文所述的關鍵審計事項是指已向審計委員會傳達或要求傳達給審計委員會的、因本期對合並財務報表進行審計而產生的事項,並且:(1)涉及對綜合財務報表具有重大意義的賬目或披露,以及(2)涉及我們特別具有挑戰性的、主觀或複雜的判斷。關鍵審計事項的傳達不會以任何方式改變我們對綜合財務報表的整體意見,我們不會通過傳達下面的關鍵審計事項來提供對關鍵審計事項或與之相關的賬目或披露的單獨意見。
預付 研發費用和應計研發費用
如綜合財務報表附註2及附註3所述,研究及發展成本於產生時計提,包括應付第三方的研究、發展及製造服務費用。在每個報告期結束時,公司根據合同規定的進度衡量標準,將向第三方服務提供商支付的款項與相關項目完成的估計進度進行比較。公司在編制估算時考慮的因素包括服務提供商產生的成本、實現的里程碑以及與其服務提供商的努力相關的其他標準。根據向第三方服務提供商付款的時間和公司估計所提供的服務取得的進展,公司將記錄與這些成本相關的預付費用或應計負債 。截至2023年9月30日,該公司報告的預付費用和其他流動資產為170萬美元,其中一部分與這些成本有關,應計研發費用為90萬美元。
我們將評估第三方服務提供商的某些預付和應計研發費用確定為一項重要的審計事項。評估完成研發項目的估計進度,包括上述因素,尤其需要審計師的主觀判斷。
以下是我們為解決這一關鍵審計問題而執行的主要程序。為了評估公司對截至2023年9月30日發生的費用的估計,對於選定的預付和應計研發費用,我們(1)檢查了從第三方服務提供商收到的合同、發票和通信 中與項目狀態有關的條款;(2)向第三方服務提供商發送了確認書; 和(3)詢問了負責監測和跟蹤研發活動狀況的個人
/s/
自2015年以來,我們一直擔任本公司的審計師。
2023年12月14日
| 91 |
十四行詩 生物治療控股公司
合併資產負債表
| 9月30日, | ||||||||
| 2023 | 2022 | |||||||
| 資產 | ||||||||
| 流動資產: | ||||||||
| 現金和現金等價物 | $ | $ | ||||||
| 預付費用和其他流動資產 | ||||||||
| 激勵性應收税款 | ||||||||
| 流動資產總額 | ||||||||
| 財產和設備,淨額 | ||||||||
| 經營性租賃使用權資產 | ||||||||
| 遞延發售成本 | ||||||||
| 其他資產 | ||||||||
| 總資產 | $ | $ | ||||||
| 負債和股東赤字 | ||||||||
| 流動負債: | ||||||||
| 關聯方票據 | $ | $ | ||||||
| 應付帳款 | ||||||||
| 應計費用和其他流動負債 | ||||||||
| 經營租賃負債的當期部分 | ||||||||
| 遞延收入 | ||||||||
| 流動負債總額 | ||||||||
| 經營租賃負債,扣除當期部分 | ||||||||
| 總負債 | ||||||||
| 承付款和或有事項(附註4) | ||||||||
| 股東赤字: | ||||||||
| 普通股,$面值:授權股份;和分別於2023年9月30日及2022年9月30日發行及未償還 | ||||||||
| 額外實收資本 | ||||||||
| 累計赤字 | ( | ) | ( | ) | ||||
| 股東總虧損額 | ( | ) | ( | ) | ||||
| 總負債和股東赤字 | $ | $ | ||||||
見 合併財務報表附註
| 92 |
十四行詩 生物治療控股公司
合併的操作報表
| 截至9月30日止年度, | ||||||||
| 2023 | 2022 | |||||||
| 協作收入 | $ | $ | ||||||
| 運營費用: | ||||||||
| 研發 | ||||||||
| 一般和行政 | ||||||||
| 總運營費用 | ||||||||
| 運營虧損 | ( | ) | ( | ) | ||||
| 匯兑損失 | ( | ) | ( | ) | ||||
| 淨虧損 | $ | ( | ) | $ | ( | ) | ||
| 每股信息: | ||||||||
| 每股基本和稀釋後淨虧損 | $ | ) | $ | ) | ||||
| 加權平均流通股、基本股和稀釋股 | $ | $ | ||||||
見 合併財務報表附註
| 93 |
十四行詩 生物治療控股公司
合併 股東權益變動表(虧損)
| 優先股 | 普通股 | 額外實收 | 累計 | |||||||||||||||||||||||||
| 股票 | 金額 | 股票 | 金額 | 資本 | 赤字 | 總計 | ||||||||||||||||||||||
| 2021年10月1日的餘額 | $ | $ | $ | $ | ( | ) | $ | |||||||||||||||||||||
| 出售優先股和普通股認股權證,扣除發行成本 | ||||||||||||||||||||||||||||
| 將優先股轉換為普通股 | ( | ) | ( | ) | ||||||||||||||||||||||||
| 出售普通股,扣除發行成本 | ||||||||||||||||||||||||||||
| 限制性股票單位歸屬時發行普通股 | ||||||||||||||||||||||||||||
| 基於股份的薪酬 | — | — | ||||||||||||||||||||||||||
| 淨虧損 | — | — | ( | ) | ( | ) | ||||||||||||||||||||||
| 2022年9月30日的餘額 | ( | ) | ( | ) | ||||||||||||||||||||||||
| 出售普通股,扣除發行成本 | ||||||||||||||||||||||||||||
| 認股權證淨股份結算 | ( | ) | ||||||||||||||||||||||||||
| 限制性股票單位歸屬時發行普通股 | ( | ) | ||||||||||||||||||||||||||
| 認股權證的行使 | ||||||||||||||||||||||||||||
| 基於股份的薪酬 | — | — | ||||||||||||||||||||||||||
| 淨虧損 | — | — | ( | ) | ( | ) | ||||||||||||||||||||||
| 2023年9月30日的餘額 | $ | $ | $ | $ | ( | ) | $ | ( | ) | |||||||||||||||||||
見 合併財務報表附註
| 94 |
十四行詩 生物治療控股公司
合併的現金流量表
| 截至9月30日止年度, | ||||||||
| 2023 | 2022 | |||||||
| 經營活動的現金流: | ||||||||
| 淨虧損 | $ | ( | ) | $ | ( | ) | ||
| 對淨虧損與經營活動中使用的現金淨額進行的調整: | ||||||||
| 收購正在進行的研究和開發 | ||||||||
| 折舊 | ||||||||
| 經營性租賃使用權資產攤銷 | ||||||||
| 基於股份的薪酬 | ||||||||
| 經營性資產和負債變動情況: | ||||||||
| 預付費用和其他流動資產 | ( | ) | ( | ) | ||||
| 應收所得税 | ( | ) | ( | ) | ||||
| 其他資產 | ( | ) | ||||||
| 應付帳款 | ( | ) | ||||||
| 應計費用和其他流動負債 | ||||||||
| 經營租賃負債 | ( | ) | ( | ) | ||||
| 遞延收入 | ( | ) | ( | ) | ||||
| 用於經營活動的現金淨額 | ( | ) | ( | ) | ||||
| 投資活動產生的現金流: | ||||||||
| 購買正在進行的研究和開發 | ( | ) | ( | ) | ||||
| 用於投資活動的現金淨額 | ( | ) | ( | ) | ||||
| 融資活動的現金流: | ||||||||
| 出售優先股和普通股認股權證所得款項,扣除發行成本 | ||||||||
| 出售普通股所得收益,扣除發行成本 | ||||||||
| 行使認股權證所得收益,扣除發行成本 | ||||||||
| 償還關聯方票據 | ( | ) | ||||||
| 融資活動提供的現金淨額 | ||||||||
| 現金淨減少 | ( | ) | ( | ) | ||||
| 現金,年初 | ||||||||
| 年終現金 | $ | $ | ||||||
| 補充披露非現金經營、投資和融資活動: | ||||||||
| 因修改租賃而引起的經營租賃使用權資產和負債的變化 | $ | $ | ||||||
| 從出售普通股所得款項中扣除的遞延發行成本 | $ | $ | ||||||
| 應付賬款和應計費用中的遞延發售成本 | $ | $ | ||||||
| 認股權證的淨結算額 | $ | $ | ||||||
| 應計費用中的正在進行的研究和開發 | $ | $ | ||||||
| 應付賬款和應計費用中的普通股發行成本 | $ | $ | ||||||
見 合併財務報表附註
| 95 |
十四行詩 生物治療控股公司
合併財務報表附註
1. 業務的組織和描述
業務説明
十四行詩 生物治療公司(“之前的十四行詩”)於2015年4月6日註冊為新澤西州的一家公司。Pre Sonnet於2020年4月1日完成了與公開持股的強啼克利爾控股公司(“強啼克利爾”)的合併。合併後,強啼克利爾更名為Sonnet BioTreateutics Holdings,Inc.(“Sonnet”或“公司”)。十四行詩是一家臨牀階段、專注於腫瘤學的生物技術公司,擁有創新單一或雙功能生物藥物的專有平臺。稱為FHAB™ (完全人白蛋白結合),該技術利用完全人類單鏈抗體片段(ScFv)與人血清白蛋白(“hSA”)結合並在其上“搭便車” ,從而將其輸送到靶組織。
十四行詩的主要專有資產SON-1010是白介素12(IL-12)的完全人類版本,共價鏈接到FHAB 結構,Sonnet正在尋求實體腫瘤適應症的臨牀開發,包括卵巢癌、非小細胞肺癌和頭頸癌。2022年3月,FDA批准了Sonnet用於SON-1010的研究新藥(IND)申請 。這使得該公司能夠在2022年第二個日曆季度啟動一項針對實體腫瘤腫瘤學患者的美國臨牀試驗(SB101)。2021年9月,公司成立了一家全資擁有的澳大利亞子公司SonnetBio Pty Ltd(“子公司”), 用於進行某些臨牀試驗。十四行詩獲得批准,並在2022年第三季度啟動了一項針對健康志願者的澳大利亞臨牀研究(SB102)。SB101和SB102研究的臨時安全性和耐受性數據於2023年4月報告。
2023年1月,Sonnet宣佈與羅氏就SON-1010與泰唑珠單抗(Tecentriq®)的臨牀評估達成合作協議。 兩家公司已簽訂主臨牀試驗和供應協議,以及輔助質量和安全 協議,以研究SON-1010和阿替唑單抗在鉑耐藥卵巢癌(“PROC”)患者環境中的安全性和有效性。此外,兩家公司將分別提供SON-1010和阿特唑珠單抗,用於1b期/2a期安全性、劑量遞增和療效研究(SB221)。這項由兩部分組成的研究的第一部分於2023年6月由澳大利亞當地人類研究倫理委員會根據CT-2023-CTN-01399-1批准,並已通知治療用品管理局。2023年8月,FDA接受了SB221的IND。該試驗包括第1部分中修改的3+3劑量遞增設計,以確定具有固定劑量的阿替唑單抗的SON-1010的最大耐受劑量(MTD)。PROC的臨牀益處將在一個擴展組中得到確認,以確定推薦的第二階段劑量(RP2D)。研究的第二部分隨後將通過隨機比較研究SON-1010單一療法、其與阿替唑單抗的聯合使用,或PROC的護理標準(SOC),以顯示概念驗證(POC)。
作為正在進行的成本削減評估的一部分,所有SON-1010的抗病毒開發都已暫停。
該公司於2020年4月通過收購Relipment Treeutics SA的已發行股份,獲得了其最先進的化合物SON-080的全球開發權,SON-080是白介素6(IL-6)的完全人類版本。十四行詩正在推進SON-080治療化療引起的周圍神經病變(CIPN)和糖尿病周圍神經病變(DPN)的目標適應症。十四行詩獲準在2022年第三季度與CIPN的SON-080一起啟動前美國1b/2a階段研究。監督這項研究的數據安全監測委員會預計將在2024年第一個日曆季度召開會議。在完成DSMB審查後,Sonnet預計將公佈CIPN研究的初步安全數據 。根據公司於2021年5月與新加坡新生命治療私人有限公司(“新生命”) 簽訂的許可協議,十四行詩和新生命將共同負責在DPN中開發SON-080。目標將是分析數據,並考慮在評估CIPN安全數據後啟動第二階段研究。
SON-1210(IL12-F)HAB-IL15),十四行詩的主要雙特定構建體,結合了FHAB含有全人IL-12和全人白介素15(“IL-15”)。這種化合物正在開發用於實體腫瘤的適應症,包括結直腸癌。2023年2月,該公司宣佈成功完成了兩項使用SON-1210對非人類靈長類動物進行的IND毒理學研究。十四行詩 準備啟動SON-1210的監管授權流程,等待任何合作活動的結果。
| 96 |
十四行詩 生物治療控股公司
合併財務報表附註
SON-1410(IL18-F)HAB-IL12)是白細胞介素18(“IL-18”)和IL-12治療實體瘤的雙特異性組合。細胞系開發和工藝開發正在進行中,早期實驗性藥物供應適合配方和分析方法開發活動。經過2023年的一些延遲之後,活動將持續到2024年,有可能產生一種適合臨牀前研究和後續人體研究的藥物。
公司已完成SON-3015(抗IL6-F)序列確認HAB-抗轉化生長因子β)。已生成早期雙特效藥物 ,並正在儲存以備將來使用體內老鼠研究。十四行詩已選擇擱置SON-3015開發 計劃以降低費用。
流動性
自成立以來,該公司因運營而出現經常性虧損和負現金流,預計在可預見的未來,主要由於其潛在產品候選產品的研發成本,運營將產生虧損。公司
相信其現金為$
2023年10月26日,公司通過Chardan Capital Markets,LLC和Ladenburg Thalmann & Co. Inc完成了普通股和某些認股權證的公開發行。作為承銷商,淨收益為
公司計劃在未來通過股權或債務融資、合作伙伴關係、合作或其他來源 獲得額外資本,以開展公司計劃的開發活動。如果在需要時無法獲得額外資本,則公司可能 需要延遲或縮減其運營,直到收到此類資金。各種內部和外部因素將影響公司的候選產品是否以及 何時被批准上市和成功商業化。公司候選產品的監管批准和 市場接受度、開發和商業化這些候選產品的時間長度和成本 和/或在批准過程的任何階段的失敗將對公司的財務狀況 和未來運營產生重大影響。
自成立以來, 的運營主要包括組織公司、獲得融資、通過研究和 開發開發技術以及進行臨牀前研究。本公司面臨與其產品正在開發中的公司相關的風險。 這些風險包括需要額外的資金來完成其研究和開發、實現其研究和開發 目標、維護其知識產權、招聘和留住熟練人員以及依賴 管理層的關鍵成員。
| 97 |
十四行詩 生物治療控股公司
合併財務報表附註
2. 重要會計政策摘要
a. 陳述的基礎
隨附的合併財務報表已按照美國公認會計原則(“美國會計準則”)編制。 GAAP”)。這些註釋中對適用指南的任何引用均指財務會計準則委員會(“FASB”)頒佈的會計準則 編碼(“ASC”)和會計準則更新(“ASU”)中的美國GAAP。
b. 整固
合併財務報表包括本公司及其全資子公司的賬目。所有公司間賬户和 交易已在合併中取消。
c. 預算的使用
根據美國公認會計原則編制綜合財務報表需要管理層作出影響綜合財務報表及附註所報金額的估計及假設 。反映在這些合併財務報表中的重大估計和假設 包括研發費用的應計費用。根據情況、事實和經驗的變化,定期審查估計和假設 。估計的變化記錄在它們被知道的時間段 。實際結果可能與管理層的估計不同。
d. 重新分類
上期的某些 金額已重新分類,以符合本期列報。
e. 細分市場信息
運營部門被定義為企業的組成部分,其獨立的離散信息可供首席運營決策者或決策小組在決定如何分配資源和評估業績時進行評估。公司在一個細分市場中查看其 運營並管理其業務。
f. 金融工具的公允價值
管理層 認為,由於該等金融工具的短期性質,本公司金融工具(包括應付賬款)的賬面價值大致為公允價值。
g. 財產和設備
財產和設備按成本入賬,並在資產的估計使用年限內採用直線法折舊。未延長預計使用壽命或改善資產的維修和維護支出 在發生時計入費用。在報廢或出售時,處置資產的成本及相關累計折舊和攤銷將從賬目中扣除,由此產生的任何損益將計入綜合經營報表。
| 98 |
十四行詩 生物治療控股公司
合併財務報表附註
h. 長期資產減值準備
當事件或環境變化表明資產的賬面價值可能無法收回時,公司將審查長期資產,如財產和設備的減值。將持有和使用的資產的可回收性是通過將資產的賬面價值與該資產預期產生的未貼現未來現金流量進行比較來衡量的。如果一項資產的賬面金額
超過其估計的未貼現未來現金流量,則就該資產的賬面價值超過該資產的估計公允價值的金額確認減值費用。有幾個
i. 遞延發售成本
與股權發行有關的法律成本和其他成本在收到時計入遞延發行成本,並從股權發行的收益
中扣除。如果放棄融資,遞延發行成本將被計入費用。截至2023年9月30日,該公司擁有
j. 激勵性應收税款
子公司
有資格參加澳大利亞研發税收激勵計劃。作為該計劃的一部分,子公司
有資格獲得澳大利亞税務局的現金退款,以支付子公司在澳大利亞的研發費用的一定比例。
k. 協作收入
協作 安排可能包含多個組成部分,其中可能包括(I)許可證;(Ii)研發活動;以及(Iii)製造和供應某些材料。根據這些安排支付的款項可能包括不可退還的付款、預付款、在完成重大監管和開發活動時的里程碑付款、銷售里程碑和產品銷售的版税。 可變對價金額受到限制,直到收入很可能在未來一段時間內不存在重大逆轉風險。
在確定公司履行合作安排義務時應確認的適當收入金額時,公司執行以下步驟:(I)確定合同中承諾的貨物或服務;(Ii)確定承諾的貨物或服務是否是履約義務,包括它們是否能夠區分;(Iii)交易價格的計量,包括可變對價的限制;(Iv)將交易價格分配給履約義務 ;以及(V)確認收入為公司履行每項履約義務。
| 99 |
十四行詩 生物治療控股公司
合併財務報表附註
在評估合同義務是否代表不同的履約義務、將交易價格分配給合同內的履約義務、確定何時履行了履約義務以及評估 可變對價的確認時,公司應用重大判斷。如果在公司根據合同條款履行其履約義務之前收到對價,則合同負債被記為遞延收入。預計在資產負債表日後12個月內確認為收入的遞延收入 歸類為流動負債。於2021年5月,本公司與新生活訂立許可協議(“新生活協議”)。有關新生命協議的進一步討論,見附註6。
l. 研發費用
研究和開發費用包括與公司生物製藥 產品開發相關的所有直接和間接成本。這些費用包括人員成本、諮詢費以及向第三方支付的研發和製造服務費用。 這些成本在發生時計入費用。
在報告期結束時,公司根據合同中定義的進度衡量標準,將向第三方服務提供商支付的款項與相關項目完成的估計進度進行比較。公司在準備時考慮的因素 估計包括服務提供商產生的成本、實現的里程碑以及與其服務提供商的努力相關的其他標準。隨着獲得更多信息,這些估計可能會發生變化。根據向服務提供商付款的時間和公司估計所提供的服務取得的進展,公司將記錄與這些成本相關的預付費用或應計負債。向代表公司執行研究和開發服務的第三方支付的里程碑預付款在提供服務時計入費用。或有發展或監管里程碑 付款在相關的或有事項解決後予以確認。
m. 外幣
交易 以美元以外貨幣計價的交易因匯率變化而產生的損益計入交易發生期間的經營活動,並在合併經營報表的匯兑損失項目中報告。
本公司根據授予日的估計公允價值計量授予員工和非員工的股權分類股份獎勵,並確認該等獎勵在必要服務期(即相應獎勵的歸屬期間)內的補償費用。本公司對發生的沒收行為進行核算。對於基於服務的歸屬條件的股票獎勵, 公司在服務期內以直線方式確認薪酬支出。本公司在其綜合經營報表中對以股份為基礎的薪酬 費用進行分類,其方式與對獲獎者的工資成本進行分類或對獲獎者的服務付款進行分類的方式相同。
| 100 |
十四行詩 生物治療控股公司
合併財務報表附註
o. 所得税
公司採用資產負債法核算所得税。遞延税項資產及負債按可歸因於現有資產及負債的賬面金額與其各自計税基礎之間的差額,以及營業虧損及信貸結轉之間的差額而產生的估計未來税項後果確認。遞延税項資產及負債以制定税率計量,預期適用於預計收回或結算該等暫時性差額的年度的應税收入。當遞延税項資產的部分或全部很可能無法變現時,可提供估值準備。本公司確認其所得税申報單上已採取或預期採取的不確定税收狀況的好處 如果這種狀況更有可能持續下去的話。
p. 反向股票拆分
2023年8月31日,該公司向特拉華州國務卿提交了經修訂的公司註冊證書修正案證書,該證書實現了
基本 每股淨虧損的計算方法是淨虧損除以每個 期間已發行普通股的加權平均股數(以及可以很少或不需要對價行使的潛在普通股)。
稀釋後每股虧損包括可能行使或轉換普通股認股權證及股票期權等證券所產生的影響(如有),而該等證券將會導致發行普通股的增量股份。對於稀釋每股淨虧損,普通股的加權平均股數與每股基本淨虧損相同,這是因為當淨虧損存在時,稀釋證券 不包括在計算中,因為影響是反稀釋的。
| 101 |
十四行詩 生物治療控股公司
合併財務報表附註
| 9月30日 | ||||||||
| 2023 | 2022 | |||||||
| 普通權證 2021年8月 | ||||||||
| 承銷商 擔保2021年8月 | ||||||||
| 私人 認股權證 | ||||||||
| 強安提克利爾 認股權證 | ||||||||
| C系列認股權證 | ||||||||
| 系列 3認股權證 | ||||||||
| 未授予的 限制性股票單位和獎勵 | ||||||||
| 普通權證 2023年2月 | ||||||||
| 承銷商 擔保2023年2月 | ||||||||
| 普通股私募認股權證2023年6月 | ||||||||
| 配售 代理人認股權證2023年6月 | ||||||||
r. 最近的會計聲明
最近採用了
2021年5月,FASB發佈了ASU 2021-04,每股收益(主題260),債務修改和清償(分主題470-50),補償-股票補償(主題718),以及衍生工具和對衝-實體自有股權的合同(分主題815-40):發行人對獨立股權的某些修改或交換的會計 -分類書面看漲期權。ASU 2021-04中的修訂提供了 指導,以澄清和減少發行人對修改或交換獨立股權分類 書面看漲期權(例如,權證)的會計處理的多樣性,這些期權在修改或交換後仍保持股權分類。2022年10月1日採用ASU 2021-04對合並財務報表沒有任何影響。
2021年11月,財務會計準則委員會或FASB發佈了會計準則更新第2021-10號,政府援助 (主題832):企業實體關於政府援助的披露,或ASU 2021-10,提高了大多數企業實體接受政府援助的透明度 要求披露:(1)接受的政府援助的類型;(2)對這種援助的核算;以及(3)援助對企業實體財務報表的影響。本指導意見 適用於2021年12月15日之後發佈的年度財務報表。該指導意見於2022年10月1日採用,並未對我們的合併財務報表產生實質性影響。
| 102 |
十四行詩 生物治療控股公司
合併財務報表附註
3. 應計費用和其他流動負債
應計費用和其他流動負債包括:
| 9月30日 | ||||||||
| 2023 | 2022 | |||||||
| 薪酬 和福利 | $ | $ | ||||||
| 研發 | ||||||||
| 專業費用 | ||||||||
| 其他 | ||||||||
| $ | $ | |||||||
4. 租契
本公司於2019年12月簽訂為期36個月的新澤西州普林斯頓寫字樓租約,租期自2020年2月1日起生效。
於2022年5月,本公司修訂現有租賃協議,將租期延長約三年,
已計入租約修訂。經營租賃使用權資產和負債在修改之日重新計量,導致兩項餘額均增加#美元。
截至2023年9月30日和2022年9月30日止年度的租賃費用 構成如下:
| 租賃費用 | 2023 | 2022 | ||||||
| 運營 租賃費 | $ | $ | ||||||
| 可變 租賃費 | ||||||||
| 租賃總成本 | $ | $ | ||||||
於2023年9月30日,加權平均剩餘租期為
截至2023年9月30日和2022年9月30日的年度與經營租賃有關的現金流量信息如下:
| 為計量租賃負債所包括的金額支付的現金: | 2023 | 2022 | ||||||
| 運營 來自運營租賃的現金流 | $ | $ | ||||||
截至2023年9月30日,不可取消租賃的未來 最低租賃付款如下:
| 財年 年 | ||||
| 2024 | $ | |||
| 2025 | ||||
| 2026 | ||||
| 未貼現的租賃付款合計 | ||||
| 減去: 計入利息 | ( | ) | ||
| 租賃負債合計 | $ |
| 103 |
十四行詩 生物治療控股公司
合併財務報表附註
5. 承付款和或有事項
法律程序
本公司不時地參與其正常業務過程中出現的各種訴訟、索賠和其他法律程序。雖然這些問題的結果不確定,但管理層預計解決這些問題的最終成本不會對公司的綜合財務狀況、經營業績或現金流產生重大不利影響。
許可證 協議
於2012年7月,本公司與XOMA(US)LLC
(“XOMA”)訂立發現合作協議(“合作協議”),據此,XOMA向本公司授予非排他性、不可轉讓許可及/或權利,以使用與發現、優化及開發抗體及相關蛋白質有關的某些
材料、技術及相關資料,並據此開發及商業化產品。公司有義務向XOMA支付或有里程碑付款,總額為
$
於2015年8月,本公司與默克KGaA(“ARES”)的全資附屬公司Ares Trading訂立許可協議(“ARES許可協議”)。根據ARES許可協議的條款,ARES已向該公司授予可再許可的全球獨家專利使用費許可,用於研究、開發、使用和商業化使用阿特克沙金(“阿特沙金”)的產品,阿特克沙金是一種治療周圍神經疾病和血管併發症的低劑量人類IL-6製劑。根據ARES許可協議,公司將按其銷售產品的淨銷售額向ARES支付個位數的高額特許權使用費。版税 按產品和國家/地區支付,直至(I)在該國家/地區首次商業銷售後的指定時間段內和(Ii)該產品在該國家/地區被有效索賠的最後日期(以較晚者為準)為止。
於2019年1月,本公司與Sartorius Stedim
Cellca GmbH(“Cellca”)訂立框架服務及許可協議(“Cellca協議”),根據該協議,Cellca已授予本公司一項全球性、非排他性、永久性及不可轉讓的
許可證,以開發、製造或已製造、使用、銷售、進口、出口及/或以其他方式商業化基於Cellca生產指定轉基因細胞系及為該等細胞系開發上游生產工序的產品。Cellca協議
有效,除非任何一方發出六個月的通知終止,或如果出於正當理由終止,則給予14天通知。
| 104 |
十四行詩 生物治療控股公司
合併財務報表附註
於2021年10月,本公司與Brink Biologics Inc.
(“Brink”)訂立非獨家許可協議(“Brink協議”),據此Brink授予本公司非獨家、不可轉讓許可及有限權利
再許可若干材料及相關資料以發展以細胞為基礎的分析,以進行產品生產及商業化所需的批次、質量控制、穩定性、效力、效力或任何其他類型的分析。在產品開發階段,公司
有義務每年支付約$的產品開發許可費
於2022年2月,本公司與InvivoGen
SAS(“InvivoGen”)訂立生物材料許可協議(“InvivoGen協議”),據此InvivoGen已向本公司授予全球非獨家許可,允許本公司使用某些
報告電池進行研究、開發及/或質量控制。InvivoGen協議的初始期限為三年
,在公司書面通知並支付約歐元的情況下,可再延長兩次三年期限
於2022年3月,本公司與ProteoNic
B.V.(“ProteoNic”)訂立材料轉讓及許可協議(“ProteoNic協議”),據此ProteoNic已向本公司授予非排他性、不可轉讓、不可再許可的
許可證(ProteoNic協議另有規定者除外),包括用於產生本公司細胞系所用載體的質粒及DNA序列,以供本公司用於產品的研究、開發及商業化。
許可證將持續至任何一方終止。公司發生了一筆$
| 105 |
十四行詩 生物治療控股公司
合併財務報表附註
研究和開發協議
於2021年12月,本公司與Navigo Proteins GmbH(“Navigo”)訂立研發協議(“Navigo協議”),根據該協議,Navigo將執行特定的評估及開發程序,以評估某些 材料以確定其商業潛力。根據Navigo協議的條款,公司已授予Navigo使用某些技術進行評估和開發活動的免版税、 非獨家、全球、不可再許可、不可轉讓的權利和許可,而Navigo已授予公司(I)研究、開發、使用、銷售、分銷、進口或以其他方式商業利用某些材料的獨家、全球、永久、不可撤銷、可再許可、可轉讓、 免版税的權利和許可,以及(Ii)非獨家、全球、永久、可再許可、製造或已經制造此類材料的權利和許可證不可轉讓 。公司發生了一筆$簽署Navigo協議時,技術訪問費為100萬歐元,當時將其記為收購的正在進行的研發,並在截至2022年9月30日的年度綜合運營報表中計入研發費用 。公司有義務向Navigo支付2023年3月修訂的或有里程碑付款,總額最高可達$在實現《導航協定》中概述的某些評價和發展里程碑的基礎上達到100萬美元。 2023年實現了某些評價里程碑,總額為#美元。5,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,
僱傭協議
公司已與其高級管理人員和某些員工簽訂僱傭合同,規定在公司無故終止僱傭或員工有充分理由終止僱傭的情況下,提供遣散費和延續福利,兩者均符合合同中的定義 。此外,如果在控制權變更後終止僱傭關係,無論是由公司 無故終止,還是由員工出於正當理由終止,員工初始股票期權授予的任何未授予部分將立即歸屬 。
6. 協作收入
根據新生命協議,本公司於馬來西亞、新加坡、印度尼西亞、泰國、菲律賓、越南、文萊、緬甸、馬來西亞、新加坡、印度尼西亞、泰國、菲律賓、越南、文萊、緬甸開發及銷售含有特定重組人IL-6、SON-080(“化合物”)的藥物製劑(該等製劑為“產品”)以預防、治療或緩解人類糖尿病周圍神經病變(“DPN領域”)。老撾人民民主共和國和柬埔寨(“專屬領土”)。 新生活有權擴大(1)專屬許可證的範圍,包括預防、治療或緩解化療引起的人類周圍神經病(“CIPN領域”),該選項是非專屬的,於2021年12月31日到期;和/或 (2)許可證的地域範圍,包括人民Republic of China、香港和/或印度,該選項是獨家的 ,於2021年12月31日到期。
該公司將保留在世界任何地方生產化合物和產品的所有權利。本公司與新生活將訂立 後續供應協議,根據該協議,本公司將按雙方協商的條款向新生活供應產品,以供其在專屬地區的DPN油田進行開發及商業化。該公司還將協助轉讓對新生命能夠從許可證中受益的某些臨牀前和臨牀開發技術訣竅。
| 106 |
十四行詩 生物治療控股公司
合併財務報表附註
新人壽將承擔並負責在專屬區域內的DPN領域進行臨牀研究和其他非臨牀研究以及其他開發和監管活動以及產品商業化的費用和責任。
New Life向該公司支付了$
新生命協議將按產品逐個國家/地區繼續有效,並將於最後到期國家/地區的最後到期產品的特許權使用費期限屆滿時終止,但須受以下條件限制:(I)各方的提前解約權 ,包括因另一方重大違約或破產或破產而提前終止的權利,以及(Ii)本公司的回購權和新生命的 回退權(定義如下)。
此外,New Life授予本公司回購本公司授予New Life的權利的獨家選擇權,而公司 授予New Life按待商定的條款在專屬地區的一個或多個國家回購DPN現場產品的權利,該等選擇權將於適用產品的第三階段試驗開始時終止。
收入 確認
該公司首先根據ASC 808評估了新生命協議,協作安排(“ASC 808”),以確定新生活協議或新生活協議內的賬户單位是否代表基於雙方的風險、回報和活動的合作安排。本公司適用ASC 606的相關指導意見,與客户簽訂合同的收入 (“ASC 606”),以評估與新生活的合作安排的適當會計處理。根據本指南,公司根據該安排確定了以下義務:(I)許可在專屬區域內現場開發、營銷、進口、使用產品並將其商業化(“許可”);以及(Ii)轉讓專有技術和臨牀 開發和監管活動(“研發活動”)。擴大CIPN領域和地區以及未來供應協議的選項 為可選採購,作為單獨的合同入賬。該公司評估了這些 個單獨的合同,沒有確定有任何實質性權利在場。公司確定許可證和研發服務 彼此沒有區別,因此將這些重大承諾合併為一項履約義務。
公司確定單一履約義務的初始交易價格為$
| 107 |
十四行詩 生物治療控股公司
合併財務報表附註
協作
從單一績效義務中獲得的收入將在研發服務的估計績效中確認。公司
確認了$
7. 股東虧損額
2023年賽事
公司於2022年8月15日與BTIG,LLC訂立市場銷售協議(“BTIG”)(“2022年銷售協議”)。
根據2022年銷售協議,本公司可不時透過BTIG作為銷售代理及/或委託人發售其普通股
股份,總髮行價最高可達$
2023年2月10日,該公司通過Chardan Capital Markets LLC和Benchmark Investments LLC的部門EF Hutton作為承銷商完成了普通股和某些認股權證的公開發行,總收益為$
普通股認股權證可立即以$的價格行使。
在
另外,購買認股權證
2023年6月30日,公司通過Chardan Capital Markets,LLC作為配售代理,完成了登記直接發行普通股(及其普通股等價物)和同時私募若干普通股認股權證,總收益為$
| 108 |
十四行詩 生物治療控股公司
合併財務報表附註
普通股認股權證從2023年12月30日起可行使,價格為$
在
另外,購買認股權證
2022年活動
於2022年8月,本公司與數名認可投資者訂立證券購買協議(“優先SPA”),以發行及出售(I)合共其第三系列可轉換優先股的股票,陳述價值$每股,
(Ii)其第4系列可轉換優先股的股票,陳述價值$每股,以及(Iii)系列3認股權證,最多購買
系列3和系列4可轉換優先股的
股票沒有投票權,但作為單一類別的普通股股東有權就修訂後的公司公司註冊證書進行投票,以實現普通股流通股的反向拆分
。系列3可轉換優先股的每股股票有權在折算後的基礎上投票,以及
私募於2022年8月15日結束,股東投票通過了
在截至2022年9月30日的年度內,公司共銷售了根據與BTIG的2022年銷售協議
發行普通股,總收益為$
此外,在截至2022年9月30日的年度內,本公司發出限制性股票單位歸屬時的普通股。
| 109 |
十四行詩 生物治療控股公司
合併財務報表附註
普通認股權證
截至2023年9月30日,以下股權分類認股權證及相關條款尚未到期:
| 未償還認股權證 | 演練 價格 | 過期日期 | ||||||||
| 普通權證 2021年8月 | $ | |||||||||
| 承銷商 擔保2021年8月 | $ | |||||||||
| 強安提克利爾 認股權證 | $ | |||||||||
| C系列認股權證 | $ | |||||||||
| 系列 3認股權證 | $ | |||||||||
| 普通權證 2023年2月 | $ | |||||||||
| 承銷商 擔保2023年2月 | $ | |||||||||
| 普通股私募認股權證2023年6月 | $ | |||||||||
| 配售 代理人認股權證2023年6月 | $ | |||||||||
| 總計 | ||||||||||
在截至2023年9月30日的年度內,認股權證淨額結算後,發行了普通股。
在截至2023年9月30日的年度內,認股權證是以現金方式行使的。本公司收到了最低限度的收益,以換取發行普通股。
在截至2023年9月30日的年度內,
2020年4月,本公司通過了《2020年綜合股權激勵計劃》(以下簡稱《計劃》)。2023年1月1日,根據該計劃授權的股份總數 增至。有幾個自2023年9月30日起,可根據本計劃發行的股票。 該計劃將根據該計劃可發行的股票數量增加普通股流通股的4%,每年1月1日。該計劃允許根據本計劃的條款授予基於股票的獎勵,包括股票期權、限制性股票單位和獎勵、股票增值權和其他被認為適當的獎勵類型。獎勵條款由公司董事會決定。
| 110 |
十四行詩 生物治療控股公司
合併財務報表附註
受限 股票單位和獎勵
. . . .
在2023年1月,在2022年12月授予的RSU中,有1個被註銷,隨後作為本公司普通股的限制性股票(“限制性股票獎勵”或“RSA”)重新發行。RSA的歸屬條件與2022年12月簽發的原始RSU相同。該公司將此作為股票薪酬調整進行會計處理,結果為$將在剩餘的授權期內確認的增量 費用。
任何未授權的RSU或RSA將在服務終止時被沒收。RSU或RSA的公允價值等於授予日公司普通股的公允市場價值。RSU和RSA費用在授權期內按直線攤銷。
| 截至9月30日的年度, | ||||||||
| 2023 | 2022 | |||||||
| 研發 | $ | $ | ||||||
| 常規 和管理 | ||||||||
| $ | $ | |||||||
下表總結了該計劃下的RSU活動:
| 加權 | ||||||||
| 平均助學金 | ||||||||
| RSU | 日期 公允價值 | |||||||
| 2021年10月1日的未歸屬餘額 | $ | |||||||
| 授與 | $ | |||||||
| 既得 | ( | ) | $ | |||||
| 被沒收 | ( | ) | $ | |||||
| 2022年10月1日的未歸屬餘額 | $ | |||||||
| 授與 | $ | |||||||
| 既得 | ( | ) | $ | |||||
| 被沒收 | ( | ) | $ | |||||
| 截至2023年9月30日的未歸屬餘額 | $ | |||||||
截至2023年9月30日,與未歸屬RSU相關的未確認補償支出總額為$,預計將在加權平均期間內 確認.
下表總結了該計劃下的RSA活動:
| 加權 | ||||||||
| 平均助學金 | ||||||||
| RSA | 日期 公允價值 | |||||||
| 2022年10月1日的未歸屬餘額 | ||||||||
| 授與 | $ | |||||||
| 截至2023年9月30日的未歸屬餘額 | $ | |||||||
截至2023年9月30日,與未授權RSA相關的未確認補償支出總額為$,預計將在加權平均期間內 確認.
| 111 |
十四行詩 生物治療控股公司
合併財務報表附註
9. 所得税
截至2023年9月30日,該公司擁有$
由於國税法所有權條款的變更,本公司淨營業虧損結轉的可獲得性 可能會受到未來期間應納税收入的年度限制,這可能會大大限制此類結轉的最終使用 。本公司並無分析其股權融資對實益擁有權的歷史或潛在影響 ,因此尚未確定結轉的淨營業虧損是否受《國內收入法典》第382節的限制。在有限制的範圍內,遞延税項資產將會減少,但估值免税額會有抵消性的減少。
當存在不確定的税收頭寸時,本公司確認税收頭寸的税收優惠,以使其更有可能實現 。有關税務優惠是否更有可能實現的決定是基於税務狀況的技術優點以及可獲得的事實和情況的考慮。本公司在其綜合經營報表的所得税撥備內確認因任何未確認的税收優惠而應計的利息和罰金。未記錄未確認的 税收優惠。
造成遞延税金的暫時性差異的税收影響如下:
| 9月30日 | ||||||||
| 2023 | 2022 | |||||||
| 遞延 納税資產: | ||||||||
| 淨營業虧損結轉 | $ | $ | ||||||
| 研究和開發信用結轉 | ||||||||
| 攤銷 | ||||||||
| 基於股份的薪酬 | ||||||||
| 運營 租賃負債 | ||||||||
| 應計費用和其他 | ||||||||
| 第 163(J)節不允許的利息支出 | ||||||||
| 財產 和設備 | ||||||||
| 遞延税項資產總額 | ||||||||
| 減去: 估值免税額 | ( | ) | ( | ) | ||||
| 遞延 納税義務: | ||||||||
| 財產 和設備 | ( | ) | ||||||
| 運營 租賃使用權資產 | ( | ) | ( | ) | ||||
| 淨額 遞延税項資產 | $ | $ | ||||||
| 112 |
十四行詩 生物治療控股公司
合併財務報表附註
在截至2023年9月30日和2022年9月30日的年度內,公司未記錄任何所得税支出或福利。合併財務報表中反映的按法定聯邦所得税率計算的所得税(費用) 福利與所得税的對賬如下:
| 截至9月30日的年度, | ||||||||
| 2023 | 2022 | |||||||
| 美國聯邦法定利率 | ( | )% | ( | )% | ||||
| 州税,扣除聯邦福利的淨額 | ( | ) | ( | ) | ||||
| 更改估值免税額 | ||||||||
| 研究和開發信用 | ( | ) | ( | ) | ||||
| 永久性差異 | ( | ) | ||||||
| 國外 税率差異 | ||||||||
| 國家NOL | ||||||||
| 其他 | ||||||||
| 實際所得税率 | % | % | ||||||
2022年8月,美國頒佈了《2022年通脹削減法案》(IRA)。愛爾蘭共和軍包含一些與税務有關的規定
,這些規定將在2022年12月31日之後的納税年度生效,包括公司替代最低税額為
10. 後續事件
公司評估了從資產負債表日期到2023年12月14日的後續事件,2023年12月14日是可以發佈合併財務報表的日期 。有關10月產品的信息,請參閲注1。
2023年12月,該公司的銷售申請獲得初步批准,最高可達$
| 113 |
第 項9.會計和財務披露方面的變更和分歧。
沒有。
第 9A項。控制和程序。
關於我們的信息披露控制評估的結論
我們 維持披露控制和程序,旨在提供合理的保證,確保在我們根據1934年證券交易法(經修訂的《證券交易法》)提交的定期報告中披露的重大信息在美國證券交易委員會規則和表格中指定的時間段內被記錄、處理、彙總和報告,並提供合理的保證 此類信息被積累並傳達給我們的管理層、我們的首席執行官和我們的首席財務官, 以便及時就所需披露做出決定。我們在管理層(包括我們的主要高管和首席財務官)的監督和參與下,對我們的披露控制和程序的設計和操作 的有效性進行了評估,這符合《交易法》規則13(A)-15(E)的定義。根據這項評估,我們的首席執行官和首席財務官得出結論,截至2023年9月30日,我們的披露控制程序和程序是有效的。
管理層財務報告內部控制年度報告
我們的管理層負責按照《交易法》規則13a-15(F) 和15d-15(F)的規定,建立和維護對財務報告的充分內部控制。財務報告內部控制是根據美國公認會計原則,為財務報告的可靠性和為外部報告目的編制財務報表提供合理保證的過程。
對財務報告的內部控制包括以下政策和程序:(1)與維護記錄相關的政策和程序,這些記錄合理、詳細、準確和公平地反映交易和資產處置;(2)提供合理的保證,確保交易按必要記錄,以便根據美國公認會計原則編制財務報表,並且僅根據管理層和董事的授權進行收支;以及(3) 就防止或及時發現可能對TIS財務報表產生重大影響的未經授權獲取、使用或處置我們的資產提供合理保證。
在我們管理層(包括首席執行官和首席財務官)的監督和參與下,我們根據 框架中確定的標準對財務報告內部控制的有效性進行了 評估內部控制--綜合框架(2013)由特雷德韋委員會贊助組織委員會發布。 根據這項評估,我們的管理層得出結論,我們對財務報告的內部控制自2023年9月30日起有效。
由於其固有的侷限性,財務報告的內部控制可能無法防止或發現錯誤陳述。此外,對未來期間進行任何有效性評估的預測可能會受到以下風險的影響:由於 條件的變化,控制可能會變得不充分,或者政策或程序的遵守程度可能會惡化。
本 年度報告不包括我們獨立註冊會計師事務所的認證報告,因為我們是“非加速申請者”,可能會利用適用於加速申請者的上市公司的各種報告要求的某些豁免,包括但不限於不需要遵守薩班斯-奧克斯利法案 第404(B)節的審計師認證要求。
財務報告內部控制變更
在截至2023年9月30日的第四季度內,我們對財務報告的內部控制(根據《交易法》第13a-15(F)和15d-15(F)條的定義) 沒有發生任何變化,這對我們對財務報告的內部控制產生了重大影響,或很可能會對其產生重大影響。
第 9B項。其他信息。
2023年12月11日,Sonnet BioTreateutics Holdings,Inc.(“本公司”)董事會薪酬委員會(“委員會”)根據本公司的“2020綜合股權激勵計劃”(“本計劃”)批准了對公司某些高管的限制性股票單位(“RSU”)獎勵(使用公司標準格式的RSU獎勵協議,其副本作為本公司的附件10.10存檔並納入本文作為參考)。每個RSU代表在歸屬時獲得一股公司普通股的權利,受下述條款和條件的限制。授權書於2024年1月1日生效,但須視乎根據該計劃可供授出的股份是否可供授出,並視乎有關承授人於該日期繼續受僱於本公司而定 承授人可於2023年12月31日或之前通知本公司行政總裁,條件是該承授人選擇接受限制性股票獎勵而非RSU,在此情況下,承授人將獲得採用本公司先前批准表格的限制性股票獎勵 。
首席執行官Pankaj Mohan博士獲得了RSU獎,相當於公司普通股的60,433股。首席財務官Jay Cross獲得了RSU獎,相當於公司普通股的11,622股。首席科學官John Cini獲得了RSU獎,相當於公司普通股的15,108股。首席技術官Susan Dexter獲得了相當於11,622股公司普通股的RSU獎勵。首席醫療官Rick Kenney獲得了相當於公司普通股10,169股的RSU獎勵。每個RSU獎項將於2025年1月1日100%授予。如果受讓人停止連續服務(如本計劃所定義),截至停止服務之日仍未歸屬的任何RSU將被沒收。
第 9C項。披露妨礙檢查的外國司法管轄區。
不適用 。
| 114 |
第 第三部分
項目 10.董事、行政人員和公司治理
董事
下表列出了有關本公司現任董事的某些信息。董事任期至下一屆年度股東大會及繼任者選出並獲得資格為止。
| 董事 | 年齡 | 第一年 變成了 董事 | ||
| Pankaj 莫漢博士 | 59 | 2020 | ||
| Nailesh 巴特 | 51 | 2020 | ||
| 艾伯特·戴爾內斯 | 61 | 2020 | ||
| 唐納德·格里菲斯 | 75 | 2020 | ||
| 拉古 拉奧 | 61 | 2020 | ||
| 洛莉 麥克尼爾 | 51 | 2022 |
以下是目前擔任本公司董事的個人的簡要簡歷,其依據是該等人士向本公司提供的資料。
Pankaj Mohan博士
Pankaj Mohan博士於2015年創立十四行詩,此後一直擔任其董事會成員,並在合併結束時被任命為我們的董事會(“董事會”)的主席。莫漢博士於2018年6月出任十四行詩主席,並於2019年1月出任十四行詩首席執行官,並於合併完成時被任命為總裁兼本公司首席執行官。2011年1月至2018年6月,他擔任他於2011年創立的腫瘤生物製劑公司(現為Outlook治療公司;納斯達克:OTLK)首席執行官兼董事長總裁。在此之前,莫漢博士曾在百時美施貴寶公司擔任生物工藝和產品開發的業務運營和投資組合管理主管,並在基因泰克公司擔任生物工藝工程部門的董事 負責人。在此之前,莫漢博士曾在禮來公司擔任高級經理。1993年5月至1996年4月,莫漢博士在英國倫敦大學學院生物化學工程高級中心擔任助理教授(講師/研究員)。Mohan博士擁有英國伯明翰伯明翰大學化學工程學院的生化工程博士學位、英國倫敦米德爾塞克斯大學商學院的財務管理碩士學位、杜克大學福庫商學院的高管管理課程(AMP)以及印度羅基印度理工學院的化學工程學士學位。他也是一本關於生物過程操作的行業參考書(McGraw Hill)的作者。公司相信莫漢博士能夠為董事會做出寶貴的貢獻,因為他對生物製藥行業有廣泛的瞭解,而且他以前擔任高管的經驗。
Nailesh 巴特
Nailesh Bhatt自2018年7月以來一直在Sonnet董事會任職,並在合併結束時被任命為我們的董事會成員。自2018年1月以來,巴特先生一直擔任VGYAAN PharmPharmticals LLC的首席執行官兼董事會成員,VGYAAN PharmPharmticals LLC是一家專注於開發臨牀關鍵藥物並將其商業化的公司。在此之前,巴特先生於2001年11月創立了Proximare公司,並擔任董事的總經理。 Proximare是一家專注於製藥行業的戰略諮詢公司。自2018年4月以來,巴特先生還擔任Azity製藥公司的董事會成員。2015年6月,巴特先生創立了Proximare生命科學基金。巴特先生在波士頓大學攻讀文學學士學位,主修生物學。公司相信巴特先生能夠為董事會做出寶貴的貢獻,因為他在製藥行業與財富500強公司的初創公司合作多年的經驗。
| 115 |
艾伯特·戴爾內斯
Albert Dyrness has served on Sonnet’s board of directors since October 2019, and was appointed to our Board at the closing of the Merger. Mr. Dyrness is a recognized biopharmaceutical industry expert in bio-process engineering with expertise in upstream, downstream, and fill/finish processes. Since July 2019, Mr. Dyrness has been the Managing Director of ADVENT Engineering Services, Inc., a Trinity Consultants Company, which serves as its life-sciences division. In 1988, Mr. Dyrness Co-Founded ADVENT Engineering Services, Inc., an engineering consulting firm serving the energy and life sciences industries. Starting with only 4 employees in the San Francisco Bay Area, ADVENT has grown to a staff of over 130 engineers with offices in Toronto, Canada, Singapore, Raleigh, North Carolina, Portland Oregon, Boston, Massachusetts, Irvine and San Ramon, California. In 2016, Mr. Dyrness became President and Chief Technical Officer of ADVENT and, in 2017, guided the company to a merger with Trinity Consultants, a 700-person engineering consulting firm. He also served as a member of the board of directors of Oncobiologics, Inc. (now Outlook Therapeutics, Inc.; Nasdaq: OTLK) from December 2015 to September 2017. In 1986, Mr. Dyrness graduated from the Massachusetts Institute of Technology where he studied mechanical engineering and entrepreneurism. The Company believes Mr. Dyrness is capable of making valuable contributions to the Board due to his years of experience in a Nasdaq-listed public company, along with years of entrepreneurial experience, including in the biopharmaceutical industry.
Donald Griffith,CPA
Donald Griffith,CPA自Sonnet於2015年4月成立以來一直在Sonnet董事會任職,2015年4月至2018年6月擔任Sonnet董事會主席,並在合併結束時被任命為董事會成員。Griffith先生自2019年1月1日起擔任Sonnet的財務 總監,自合併以來擔任我們的總監。在擔任財務總監之前,他於2015年4月至2016年12月擔任Sonnet的 首席執行官兼首席財務官。在此之前,Griffith先生是Oncobiologics Inc.的首席財務官、董事兼祕書。(now Outlook Therapeutics; Nasdaq:OTLK),2011年至2018年。Griffith先生在財務和會計方面擁有超過 40年的經驗,是Stolz & Griffith,LLC(新澤西州會計師事務所)的創始人和合夥人。本公司相信,Griffith先生憑藉其多年的財務 和製藥行業經驗,能夠為董事會做出寶貴貢獻。
Raghu Rao
Raghu Rao has served on Sonnet’s board of directors since November 2019, and was appointed to our Board at the closing of the Merger. Mr. Rao is a serial entrepreneur, strategic business advisor and angel investor. Mr. Rao has founded, scaled and had successful exits with several high-technology companies. In his 33-year career, Mr. Rao has advised clients on the strategy and roll-out of high-profile projects, such as USA.gov, TSA Screening Gateway, Cancer.gov and other eGovernment initiatives. As the Vistage Princeton Chair, from July 2012 to March 2017, Mr. Rao ran three high-performing peer advisory boards for middle-market CEOs and business leaders of companies with total revenues exceeding $2 Billion. As the Chairman & President of InfoZen from August 1995 to July 2008, Mr. Rao has managed over $1 Billion in U.S. Federal Government contracts. Mr. Rao is a 20-year Charter Member of The Indus Entrepreneurs (TiE.org) and a 5-year patron of the Indiaspora. He has held board positions at several companies including Cellix BioSciences (Jan 2016 - Jan 2017), Paper Battery Company (Jan 2009 - Dec 2018), Kovid Group (Feb 2016 - Oct 2017) , WizNucleus (Jun 2010 - present) and InfoZen (Aug 1995 - Jul 2008). Mr. Rao is active in social entrepreneurship and community service. He co-founded the Hindu Jewish Coalition in December 2012 and Forum for Religious Freedom in March 2007, to preserve religious diversity worldwide. He has held non-profit board positions at the Infinity Foundation (New Jersey), Arsha Vidya Gurukulam (Pennsylvania) and the Family Services Agency (Maryland). Mr. Rao has an MBA in Finance from George Washington University (Dec 1991), an M.S. in Computer Science from Virginia Tech (Dec 1986), and a B.Tech. in Electrical Engineering from Indian Institute of Technology Madras (June 1984). The Company believes Mr. Rao is capable of making valuable contributions to the Board due to his 15 years of experience as an executive, along with 25 years of entrepreneurial experience, including in the biotech industry.
| 116 |
Lori McNeill
Lori McNeill has served on our Board since September 2022 and as Chairperson of our Business Advisory Committee since September 2019. Ms. McNeill is the founder and Chief Executive Officer of McNeill Consulting, LLC since 2016, a management consulting company focused on developing leaders to be more effective and ensuring that change management initiatives are seamless. Ms. McNeill has over twenty years’ experience in the healthcare industry, thirteen of which were at Pfizer Inc., which included working as the Chief of Staff of Global Operations in the Integrated Health Business unit. From 2020 to 2021, Ms. McNeill was the Chief Operating Officer and Chairperson of the board of directors of Global PPE, Inc., a worldwide supplier of personal protective equipment and safety supplies focused on healthcare and government entities to fight the COVID-19 pandemic. She has been recognized by several institutions: Top 100 Global Women in Leadership - Global Council for the Promotion of International Trade, 2021; Changemakers Summit Award Winner, 2021; The State of Women in Leadership - Cover article for HR.com, 2020; and Pfizer International Innovation Excellence Award, 2011 and is currently Global Chairperson of Womenomics. The Company believes Ms. McNeill is capable of making valuable contributions to the Board due to her over 20 years of experience in the healthcare industry, including in senior leadership positions.
執行官員
下表載列有關本公司現任行政人員的若干資料:
| 執行官員 | 年齡 | 位置 和辦公 | ||
| Pankaj Mohan博士 | 59 | 總裁 和首席執行官 | ||
| Jay 克羅斯 | 53 | 首席財務官 | ||
| John K. Cini博士 | 71 | 首席科學官 | ||
| Susan 德克斯特 | 68 | 主任 技術官 | ||
| 理查德·肯尼,醫學博士。 | 65 | 首席醫療官 |
以下是目前擔任本公司高管的個人的簡要簡歷,其依據是此等人士向本公司提供的信息。
Pankaj Mohan博士
請參閲導向器下的 説明。
Jay 克羅斯
Jay 克羅斯於2019年5月加入十四行詩,此後一直擔任該公司的首席財務官和首席業務官,並在合併完成時被任命為公司首席財務官。在加入十四行詩之前,克羅斯先生於2015年11月至2019年3月在查爾丹資本的醫療保健投資銀行團隊擔任董事董事總經理,期間專注於生物製藥。在此之前,從2014年5月至2015年6月,克羅斯先生在Alere Financial Partners擔任董事,並於2011年5月至2013年10月在Balyasny Asset Management擔任高級分析師 。他於1999年在Hambrecht&Quist的醫療保健股票研究團隊擔任負責生物技術的助理分析師,開始了他的金融職業生涯。克羅斯先生擁有耶魯大學醫學院的碩士學位和華盛頓與李大學的心理學學士學位。
約翰·K·西尼,博士。
John K.Cini博士於2015年共同創立了十四行詩,此後一直擔任該公司的首席科學官,並在合併結束時被任命為該公司的首席科學官,負責監督和指導該公司的發現和開發計劃。他的職責包括監督癌症和免疫腫瘤學靶標的選擇過程以及概念驗證測試。在加入十四行詩之前,他於2011年1月至2015年4月在腫瘤學公司擔任發現與發展科學部總裁副主任。Cini博士已成功地將20多種新型單抗產品從發現階段推向了IND階段。他擁有多項新產品和配方專利及應用。他還通過早期發現、研發和生產、臨牀試驗和商業化,直接參與了幾種成功的新型生物製品。之前的職位包括在美德士(2010年被百時美施貴寶收購)擔任董事高管,在強生(生物科技整形外科和藥物研究)擔任發現科學領導職務,以及拜耳。西尼博士在系統生物學方面的專業治療領域包括腫瘤學、免疫腫瘤學、炎症、骨質疏鬆症、傷口癒合、外科粘連和細胞老化。西尼博士擁有北得克薩斯大學生物化學博士學位。
| 117 |
蘇珊·德克斯特
蘇珊·德克斯特自2019年5月以來一直以首席技術官的身份擔任十四行詩的合同顧問,並於2020年4月1日合併完成時被任命為公司的全職首席技術官。她在Sonnet的角色是管理藥物開發的運營 ,從細胞系開發到藥物物質和臨牀藥物產品的cGMP製造, 啟動人體臨牀試驗的監管提交,以及標籤/包裝和分發到臨牀倉庫的供應鏈。所有活動均受FDA的生物藥物化學制造和控制(“CMC”)管轄。她還負責藥品供應和非臨牀動物研究的管理,以支持與首例人類研究相關的監管文件。她來到十四行詩 ,在生物技術科學、製造和商業發展方面擁有30多年的經驗, 直接參與了三家初創公司和多項併購活動。她在CMC生物製藥工藝開發方面的專業知識範圍從細胞生產線開發到通過商業製造的工藝開發。從2008年9月到合併完成,德克斯特女士在萊瑟姆生物製藥集團(“萊瑟姆生物製藥”) 擔任董事經理,負責產品開發服務,管理與臨牀前毒理學研究相關的活動和學科,以及與CMC相關的活動,包括IND備案、cGMP活動的質量監督和其他CMC供應鏈活動。她從Xcellerex,Inc.來到LBG,Xcellerex,Inc.是一家CDMO和生物處理一次性使用技術的開發商。2004年4月至2008年9月,她擔任Xcellerex的首席商務官。在加入Xcellerex之前,從1998年7月到2004年4月,她在陶氏化學公司的CDMO擔任業務發展副總裁,在Dexter女士的推動下於2000年收購了Collaborative BioAlliance,以及Assoc。Celltech Biologics的業務發展董事,被龍沙生物收購, 生物CDMO。1986年至1998年7月,她在Celltech/Lonza工作。德克斯特女士擁有位於華盛頓特區的美國大學免疫學和市場營銷學榮譽專業的雙學位,以及哈佛大學頒發的“律師談判”和“非金融經理的金融”證書。她也是倫敦大學學院生物工程系榮譽退休教授,在1999至2006年間,為畢業生、博士和研究生專業人員講授“生物製藥設施的項目管理”的榮譽課程講座和研討會。自2015年以來,她一直擔任Sartorius Stedim Biotech的非執行董事會成員,自2019年以來擔任薪酬委員會成員,自2022年以來擔任審計委員會成員。2023年2月,蘇珊被任命為英國倫敦一家公司Virocell的董事會成員,該公司是一家技術開發商和CDMO公司,致力於製造用於細胞和基因治療的病毒載體。
理查德·肯尼,醫學博士。
Richard Kenney,醫學博士自2021年4月起擔任公司首席醫療官。Kenney博士在生物製劑的預防階段開發以及臨牀前、臨牀階段和商業化疫苗和免疫療法的商業化戰略和企業管理方面擁有20多年的經驗。作為ClinReg Biologics的總裁,他以多種身份為生物製劑的臨牀和法規事務以及醫學監測和藥物警戒提供戰略諮詢。因此,Kenney博士還擔任 公共衞生疫苗馬爾堡疫苗項目的CMO。他曾在X-VAX Technology 擔任首席開發官,在此之前,他曾在Immune Design和Crucell Holland擔任首席醫療官,領導臨牀開發和法規 事務小組。Kenney博士是FDA的研究員/評審員超過六年,並在杜克大學和NIH進行了研究生培訓。博士 肯尼獲得了學士學位。喬治·華盛頓大學化學博士來自哈佛醫學院
第16(A)節實益所有權報告合規性
1934年《證券交易法》第 16(a)條(經修訂)要求公司董事和高管、高級職員以及持有公司註冊類別股本證券10%以上的受益所有人 向SEC提交所有權和所有權變更報告。證券交易委員會的規定要求這些人員向公司提供他們提交的所有第16(a)節表格的副本。
僅根據公司對提供給公司的表格3、4和5副本的審查,公司認為其所有董事、 執行官和任何其他適用的股東在截至2023年9月30日的 財政年度內及時提交了《交易法》第16(a)節要求的所有報告,但以下情況除外:應於2022年12月16日提交的表格4於2022年12月22日提交給Nailesh Bhatt,Albert Dyrness,Raghu Rao,Lori McNeil,Pankaj Mohan,John Cini,Jay Cross,Susan Dexter和Rick Kenney; Pankaj Mohan、John Cini、Susan Dexter和Rick Kenney於 2023年2月17日提交了應於2023年1月10日提交的表格4; Donald Griffith於 2023年2月17日提交了應於2022年12月13日提交的表格4。
| 118 |
商業行為和道德準則
公司已採用適用於其董事、高級職員和員工的《商業行為和道德準則》。《商業行為和道德準則》 的目的是阻止不法行為,併為公司的董事、管理人員和員工 提供指導,幫助他們認識和處理道德問題,提供報告不道德或非法行為的機制,併為公司的誠實和責任文化做出積極貢獻 。公司https://www.sonnetbio.com/如果公司對《商業 行為和道德準則》進行任何實質性修訂或授予任何豁免,包括對 其董事或執行官的《商業行為和道德準則》條款的任何默示豁免,公司將在其網站上或在表格8-K的當前 報告中披露此類修訂或豁免的性質。
審計委員會
董事會已成立審計委員會,目前由Bhatt先生(主席)、Dyrness先生和Rao先生組成。審計委員會的 主要職能是監督和審查:公司的合併財務報表和公司提供的其他財務 信息的完整性,公司遵守法律和監管要求的情況,公司的內部會計和財務控制系統 ,獨立審計師的聘用、資格、業績、薪酬 和獨立性,關聯方交易,並遵守公司的商業行為和道德準則。
審計委員會的每名成員都是“獨立的”,因為該術語是根據美國證券交易委員會的適用規則和納斯達克股票市場的適用規則來定義的。董事會已確定,每名審計委員會成員在財務和審計事務方面擁有足夠的知識,可在審計委員會任職。董事會認定Rao先生為“美國證券交易委員會適用規則及納斯達克證券市場適用規則所界定的審計委員會財務專家”。公司董事會已通過了審計委員會章程,可在https://www.sonnetbio.com/.上查看
項目 11.高管薪酬
彙總表 薪酬表
下表顯示了在2023財年擔任本公司首席執行官的每位高管、截至2023年9月30日擔任本公司高管的兩名薪酬最高的高管以及至多另外兩名本應獲得披露信息的個人在2023財年獲得的薪酬 。下表中所列人員在本文中稱為“指定的執行官員”。
彙總表 薪酬表
| 姓名 和主要職位 | 年 | 薪金 ($) | 獎金 ($) | 庫存 獎項 ($)(1) | 選擇權 獎項 ($)(1) | 所有 其他 補償 ($) | 總計 ($) | ||||||||||||||||||||
| Pankaj Mohan博士 | 2023 | 538,998 | 309,924 | 95,724 | - | 78,074 | 1,022,720 | ||||||||||||||||||||
| 總裁 和首席執行官 | 2022 | 559,729 | 218,680 | 144,061 | - | 82,923 | 1,005,393 | ||||||||||||||||||||
| 約翰·西尼,博士。 | 2023 | 417,750 | 146,490 | 23,931 | - | 38,240 | 626,411 | ||||||||||||||||||||
| 首席科學官 | 2022 | 413,048 | 113,899 | 36,015 | - | 52,014 | 614,976 | ||||||||||||||||||||
| Jay 克羅斯 | 2023 | 388,725 | 178,813 | 15,829 | - | 35,882 | 619,250 | ||||||||||||||||||||
| 首席財務官 | 2022 | 403,676 | 127,649 | 30,128 | - | 43,358 | 604,811 | ||||||||||||||||||||
| (1) | 代表根據財務會計準則委員會(“FASB”)會計準則編纂(“ASC”)主題718計算的2023年和2022年贈款的公允價值總額。此計算不會對與基於服務的歸屬相關的沒收進行任何 估計,但假設執行人員將為全額歸屬的 獎勵執行必要的服務。 |
| 119 |
Narrative 薪酬彙總表披露
僱傭協議
每名被提名的執行幹事的僱用協議或安排的具體條款如下所述。
十四行詩 於2018年12月31日與Mohan博士訂立經修訂的僱傭協議(“Mohan協議”),列明其擔任首席執行官的僱用條款,該協議由本公司於合併完成時承擔。根據僱傭協議,Mohan博士有權(其中包括)(I)每年490,000美元的總基本工資,(Ii)有資格獲得相當於本公司從戰略交易中獲得的毛收入5.4%的獎金,以及(Iii)對於上一條款中的獎金 低於基本工資的50%的任何年度,額外發放基於業績的現金獎金,使該年度的現金獎金總額達到董事會確定的基本工資的50%。僱傭協議應根據其條款終止。根據莫漢博士的僱傭協議,如果他在“控制權變更”之前或之後的2個月內或之後的12個月內被無故終止或因“好的 原因”而被終止,他有權獲得(I)18個月的基本工資,(Ii)相當於他在終止發生當年的績效獎金的獎金,除以12,然後 乘以18,以及(Iii)如果他及時根據COBRA繼續投保,則支付繼續投保所需的COBRA保費,直至(A)終止日期後的最早18個月,(B)他有資格獲得與新就業或自僱相關的基本等值醫療保險的日期 ,或(C)他不再有資格享受COBRA繼續保險的日期。 如果莫漢博士被無故終止或因與“控制權變更”不一致的“充分理由”而被解僱, 他有權獲得(I)18個月的基本工資,(Ii)他被終止時所在業績年度的任何績效獎金, 和(Iii)如果他及時根據COBRA繼續承保,支付繼續承保所需的眼鏡蛇保費,直至(A)終止日期後18個月,(B)他有資格獲得與新就業或自僱相關的實質等值醫療保險之日,或(C)他不符合眼鏡蛇繼續承保資格之日。
十四行詩 於2020年1月10日與Cini博士訂立經修訂的僱傭協議(“Cini協議”),列明其擔任首席科學官的僱用條款,該協議由本公司於合併完成時承擔。根據僱傭協議,Cini博士有權(其中包括)(I)370,000美元的年度總基本工資,(Ii)有資格獲得相當於本公司從戰略交易中獲得的毛收入1.1%的獎金,以及(Iii)對於上一條款中的獎金 低於基本工資的35%的任何年度,額外發放一筆基於業績的現金獎金,使該年度的現金獎金總額最高可達基本工資的35%,由董事會決定。僱傭協議應根據其條款終止。根據Cini博士的僱傭協議,如果他在“控制權變更”之前或之後的2個月內或之後的12個月內因“原因”或“正當理由”而被解僱,他有權獲得(I)12個月的基本工資和(Ii)如果他及時根據COBRA繼續承保,支付繼續承保所需的COBRA保費 直到(A)終止日期後18個月,(B)他有資格獲得與新就業或自僱相關的基本同等的健康保險之日,或(C)他不符合COBRA續保資格的日期 。如果Cini博士在“原因”或不符合“控制權變更”的“充分理由”下被解僱,他有權獲得(I)9個月的基本工資和(Ii)如果他及時繼續在COBRA下承保,支付繼續承保所需的 COBRA保費,直到(A)終止日期後12個月,(B)他有資格獲得與新就業或自僱相關的實質上同等的醫療保險之日,或(C)他沒有資格獲得COBRA繼續承保之日。
十四行詩 於二零二零年一月十日與克羅斯先生訂立僱傭協議(“交叉協議”),列明其擔任首席財務官的聘用條款 ,該協議由本公司於合併完成時承擔。根據僱傭協議,江樂士先生有權(其中包括)(I)年度基本薪金總額365,000美元及(Ii)有資格獲得董事會釐定的最高達基本薪金40%的以工作表現為基礎的現金花紅。僱傭協議將根據其條款終止 。根據克羅斯先生的僱傭協議,如果他在“控制權變更”之前或之後的2個月內或12個月內因“正當理由”而被解僱,他有權 獲得(I)12個月的基本工資,(Ii)發生解僱的業績年度的任何績效獎金,以及(Iii) 如果他及時根據COBRA繼續投保,支付繼續投保所需的COBRA保費,直至(A)終止日期後18個月 ,(B)他有資格獲得與新就業或自僱相關的基本等值健康保險的日期,或(C)他沒有資格享受眼鏡蛇延續保險的日期。如果克羅斯先生因“原因”或不符合“控制權變更”的“充分理由”而被解僱,他有權獲得 (I)9個月的基本工資,(Ii)發生解僱的業績年度的任何績效獎金,以及(Iii)如果他 及時繼續在COBRA下投保,支付繼續投保所需的COBRA保費,直到(A)終止日期後12個月 ,(B)他有資格獲得與新就業或自僱相關的基本同等的醫療保險之日,或(C)他不符合《眼鏡蛇》續保資格的日期。
| 120 |
其他 協議
本公司於2020年4月1日與Dexter女士訂立僱傭協議(“Dexter協議”),列明其擔任首席技術官的僱傭條款。根據僱傭協議,德克斯特女士有權(其中包括)(I)每年310,000美元的基薪總額及(Ii)有資格獲得董事會釐定的高達基薪35%的績效現金花紅。僱傭協議應根據其條款終止。根據德克斯特女士的僱傭協議,如果她在“控制權變更”之前或之後的2個月內因“原因”或“正當理由”而被解僱,她有權獲得(I)12個月的基本工資,(Ii)她被解僱所在業績年度的任何績效獎金,以及(Iii)如果她及時根據COBRA繼續承保,則支付繼續承保所需的COBRA保費 ,直至(A)終止日期後18個月,(B)她有資格獲得與新就業或自僱相關的基本同等醫療保險的日期,或(C)她不符合COBRA延續保險資格的日期。如果德克斯特女士在沒有“原因”或不符合“控制權變更”的情況下被解僱,她有權獲得(I)他9個月的基本工資,(Ii)她被解僱所在業績年度的任何績效獎金,以及(Iii)如果她及時根據COBRA繼續承保,支付COBRA保費,以繼續承保,直到(A)終止日期後12個月,(B)她有資格獲得與新就業或自僱相關的實質上同等的醫療保險之日,或(C)她不符合COBRA繼續承保資格的日期。
財政年度末未償還的 股權獎勵
下表逐個列出了有關購買普通股的未行使期權、尚未授予每位指定高管且截至2023年9月30日已發行的普通股和普通股的限制性股票的某些信息。
2023財年末未償還的 股權獎勵
| 股票 獎勵 | ||||||||
| 名字 | 股權 激勵計劃獎勵:尚未授予的未賺取的股份、單位或其他權利的數量(#) | 股權 激勵計劃獎勵:尚未授予的未賺取的股份、單位或其他權利的市場或派息價值($) | ||||||
| Pankaj 莫漢博士 | 3,343 | (1) | 9,680 | |||||
| 約翰·西尼,博士。 | 843 | (1) | 2,420 | |||||
| Jay 克羅斯 | 705 | (2) | 2,025 | |||||
| (1) | 每個 限制性股票獎勵在2024年1月1日100%授予。 |
| (2) | 每個 限制性股票單位(“RSU”)獎在2024年1月1日100%授予 |
| 121 |
董事 薪酬
非員工 董事薪酬政策
與合併相關的是,董事會批准了董事非僱員董事的新薪酬政策。除報銷 出席董事會和委員會會議的合理費用外,本政策還規定以下現金 補償:
● 每個非員工董事有權從我們那裏獲得35,000美元的年費;
● 我們的審計委員會主席將從我們那裏獲得15,000美元的年費;
● 我們的薪酬委員會主席將從我們那裏獲得10,000美元的年費;
● 我們的提名和公司治理委員會主席將從我們那裏獲得8,000美元的年費;以及
● 審計委員會、薪酬委員會和提名和公司治理委員會的每位非主席成員將分別從我們那裏獲得7,500美元、5,000美元和4,000美元的年費。
每位加入董事會的非僱員董事將獲得一項初始期權授予,可在合併完成時購買公司已完全稀釋的已發行普通股的0.080%,這將在三年內每年授予33%,第一個歸屬日期發生在授予日期的一年 週年紀念日。每位非員工董事還將獲得年度期權授予,在合併完成時購買公司0.040%的完全稀釋後已發行普通股,並將在授予日期一週年或下一次年度股東大會中較早的日期授予100%。一旦控制權發生變更,如本公司的股權激勵計劃所界定,這些期權相關股份的100%將在控制權變更之前歸屬並可行使。
除下表所列的 外,在2023財年,非僱員董事未收到任何現金或股權薪酬:
董事 薪酬
| 名字 | 賺取的費用 或 已支付 在 現金中(美元) | 庫存 獎項 ($)(1) | 選擇權 獎項 ($)(1) | 所有 其他 補償 ($) | 總計 ($) | |||||||||||||||
| Nailesh 賓館(2) | 54,000 | 3,845 | - | - | 57,845 | |||||||||||||||
| Albert Dyrness(3) | 55,500 | 3,845 | - | - | 59,345 | |||||||||||||||
| Donald 格里菲斯(4) | - | 3,278 | - | 134,048 | 137,326 | |||||||||||||||
| Raghu 拉奧(5) | 116,500 | 3,845 | - | - | 120,345 | |||||||||||||||
| Lori 麥克尼爾(6) | 70,000 | 3,845 | - | - | 73,845 | |||||||||||||||
| (1) | 代表 根據FASB ASC Topic 718計算的2023年授予的總授予日公允價值。此計算 不對與基於服務的歸屬相關的任何沒收估計生效,但假設執行官將執行 所需的服務,以授予充分。 |
| (2) | 先生 截至2023年9月30日,Bhatt共持有171個限制性股票單位。 |
| (3) | 先生 截至2023年9月30日,Dyrness共持有171個限制性股票單位。 |
| (4) | 先生 Griffith自2019年1月1日起擔任Sonnet的財務總監,自合併以來擔任我們的財務總監。 上表“所有其他薪酬”項下的金額代表Griffith先生 2023財年。見下面與格里菲斯先生的就業協議的描述。格里菲斯先生總共持有146 限制性股票單位,截至2023年9月30日。 |
| (5) | 先生 截至2023年9月30日,Rao共持有171股限制性股票單位。 |
| (6) | 女士 截至2023年9月30日,McNeill共持有171股限制性股票。 |
與董事達成的其他 協議
Sonnet 於2019年1月1日與Griffith先生簽訂了一份僱傭協議,規定了其擔任財務總監的僱傭條款。 根據僱傭協議,Griffith先生有權(除其他事項外)(i)按比例計算的年基本工資總額為150,000美元,以及(ii)有資格獲得相當於工資總額25%的目標獎金。就業協議沒有具體期限,構成 隨意就業。
| 122 |
第 項12.某些實益所有人和管理層的擔保所有權及相關股東事項。
安全 某些受益所有者和管理層的所有權
下表列載截至2023年12月11日本公司普通股實益擁有權的若干資料:(I)本公司每名現任董事;(Ii)每名被點名的行政人員;(Iii)所有現任行政人員及董事作為一個整體;及(Iv)本公司所知實益擁有本公司普通股已發行股份超過5%(5%)的每名人士。
就下表而言,實益所有權是根據適用的美國證券交易委員會規則確定的,信息為 不一定表示實益所有權用於任何其他目的。除表內附註另有註明外,本公司相信表中所列的每名個人或實體對該個人或實體實益擁有的所有本公司普通股股份擁有獨家投票權及投資權(或與其配偶分享該等權力)。根據美國證券交易委員會的規則,根據可於2023年12月11日或之後60天內行使的期權(“目前可行使的期權”)可發行的本公司普通股被視為已發行股票,因此包括在表中點名的個人或實體報告為實益擁有的股份數量中,並用於計算該個人或實體實益擁有的普通股的百分比。然而,在計算任何其他個人或實體實益擁有的普通股的百分比時,這些股票不被視為流通股。
下表所列每個個人或實體實益擁有的普通股的百分比是基於截至2023年12月11日已發行和已發行的普通股3,069,516股,加上該個人或實體持有的任何因行使目前可行使的期權而可發行的股份。
第 個標題 班級 | 受益人姓名或名稱及地址* | 受益所有權的金額和性質 | 班級百分比 | |||||||
| 任命了 名高管和董事: | ||||||||||
| Pankaj Mohan博士 | 131,322 | (1) | 4.2 | % | ||||||
| Nailesh 巴特 | 1,396 | (2) | ** | |||||||
| 艾伯特·戴爾內斯 | 1,316 | (3) | ** | |||||||
| 唐納德·格里菲斯 | 468 | (4) | ** | |||||||
| 拉古 拉奧 | 47,117 | (5) | 1.5 | % | ||||||
| 洛莉 麥克尼爾 | 171 | (6) | ** | |||||||
| 約翰·K·西尼博士。 | 1,830 | ** | ||||||||
| Jay 克羅斯 | 1,087 | (7) | ** | |||||||
| 全體 現任執行幹事和董事(10人) | 187,064 | 5.9 | % | |||||||
| 5% 持有者 | ||||||||||
| 約翰·馬基 | 231,115 | 7.5 | % | |||||||
| (*) | 除非 除非另有説明,地址為Sonnet BioTherapeutics,Inc.,100俯瞰中心,套房102,普林斯頓,新澤西州,08540。 |
| (**) | 不到1%。 |
| (1) | 包括 (i)Mohan家族辦公室持有的3,021股普通股,Mohan博士與 Swati Mohan,他的配偶,以及(ii)Mohan博士的孩子Pankhuri Mohan個人持有的25股普通股, 莫漢博士與潘庫裏·莫漢分享投票和處置權。包括68,750股普通股,目前可發行於 認股權證的行使。 |
| 123 |
| (2) | 包括 171個限制性股票單位,將以普通股股份結算,並於2023年12月11日起60天內歸屬。 |
| (3) | 包括 171個限制性股票單位,將以普通股股份結算,並於2023年12月11日起60天內歸屬。 |
| (4) | 包括 146個限制性股票單位,將以普通股股份結算,並於2023年12月11日起60天內歸屬。 |
| (5) | 包括 31,250股普通股可於2023年12月11日起60天內行使認股權證時發行。包括 171個限制性股票單位,將以普通股股份結算,並於2023年12月11日起60天內歸屬。 |
| (6) | 包括 171個限制性股票單位,將以普通股股份結算,並於2023年12月11日起60天內歸屬。 |
| (7) | 不包括 705個限制性股票單位,將以普通股股份結算,並於2023年12月11日起60天內歸屬。 |
權益 薪酬計劃信息
下表提供了截至2023年9月30日的有關可能根據公司現有股權薪酬計劃(包括2020年綜合股權激勵計劃(“2020年計劃”))發行的公司普通股股份的信息。
| 權益 薪酬計劃信息 | ||||||||||||
| 計劃 類別 | (a) 證券數量 到 予發行 關於 未行使的期權、認股權證 和 權利 | (b) 加權 平均值 執行 價格 未償還 期權、認股權證 和 權利 | (c) 證券數量 剩餘的 可用於 未來 發行 在 權益下 薪酬 計劃 (不包括證券 第(A)欄反映了 ) | |||||||||
| 股權 證券持有人批准的薪酬計劃(1) | 2,326 | 不適用 | - | |||||||||
| 股權 未經證券持有人批准的薪酬計劃 | 不適用 | 不適用 | 不適用 | |||||||||
| 總計 | 2,326 | - | - | |||||||||
| (1) | 由於 RSU沒有行權價格,因此加權平均行權價格不反映與RSU結算相關的將發行的股份。截至2023年9月30日,除RSU外,我們的股權補償計劃中沒有未償還的期權、認股權證或權利。 |
| 124 |
第 項13.某些關係和關聯交易,以及董事獨立性。
除指定高管和董事的薪酬安排外,本公司還介紹了自2022財年開始以來本公司參與或將參與的每筆交易和一系列類似交易,其中:
| ● | 所涉金額超過或將超過12萬美元或較小報告公司過去兩個完整財政年度年底總資產平均值的百分之一,兩者以較小者為準;以及 |
| ● | 本公司任何董事、董事的被提名人、高管或持有超過5%的本公司普通股的 任何成員或上述人士的任何直系親屬曾經或將會擁有直接或間接的重大利益。 |
薪酬 本公司指定高管及董事的薪酬安排載於“高管薪酬”一節。
公開服務
我們的董事長兼首席執行官Pankaj Mohan購買了34,375股普通股和68,750股認股權證,以購買68,750股普通股 根據美國以每股1.60美元的價格進行的承銷公開發行,並伴隨着兩份認股權證。此次發售 於2023年10月27日結束。
董事的拉古·拉奧購買了15,625股普通股和31,250股認股權證,以購買31,250股普通股。此次發行於2023年10月27日結束。
賠償協議
公司已與每一位現任董事和高管簽訂了賠償協議。這些協議將 要求公司在特拉華州法律允許的最大範圍內賠償這些個人因向公司提供服務而可能產生的責任,並預支因起訴他們而產生的費用,因為他們 可能會得到賠償。該公司還打算與其未來的董事和高管簽訂賠償協議。
董事 獨立
公司目前由一個六人董事會管理。本公司已確定,巴特先生、戴爾尼斯先生、拉奧先生和麥克尼爾女士是“獨立的”,這一術語是根據納斯達克股票市場規則定義的。
| 125 |
第 項14.總會計師費用和服務
下表彙總了畢馬威會計師事務所、本公司的獨立註冊會計師事務所(以及合併前的十四行詩)在過去兩個會計年度各自每年提供的專業服務費用:
| 費用 類別 | 2023 | 2022 | ||||||
| 審計費用 | $ | 540,000 | $ | 435,000 | ||||
| 與審計相關的費用 | - | - | ||||||
| 税 手續費 | 61,565 | 33,500 | ||||||
| 所有 其他費用 | - | - | ||||||
| 總費用 | $ | 601,565 | $ | 468,500 | ||||
審計費用
代表與審計公司年度綜合財務報表和審查公司季度中期綜合財務報表相關的專業服務費用,以及與註冊報表、慰問函和同意書相關的費用。
與審計相關的費用
我們 在2023年或2022年沒有從我們的獨立審計師那裏產生任何與審計相關的費用。
税 手續費
税務 費用與税務合規、税務諮詢、税務規劃和税務準備服務相關。
所有 其他費用
與總會計師提供的產品和服務有關的費用,但不包括上述各節所報告的服務。
審計委員會負責任命、確定薪酬和監督獨立審計師的工作。審計委員會 需要審查和批准保留獨立審計師的提議,以執行其章程中概述的任何擬議的審計和非審計服務 。審計委員會沒有在其章程之外製定關於預先批准審計和非審計相關服務的政策和程序。根據《交易法》第10A條的要求,我們的審計委員會已授權畢馬威有限責任公司在2023年至2022年期間提供的所有審計和非審計服務以及為這些服務支付的費用。但是,如果符合《交易法》第10A(I)(1)(B)節的“最低限度”條款,則可免除為公司提供非審計服務的預先審批要求 。
審計委員會已考慮上述與審計相關的費用、税費和所有其他費用的提供是否符合保持畢馬威會計師事務所的獨立性,並已確定2023年和2022年的此類服務是兼容的。 所有此類服務均經審計委員會根據交易所法案S-X法規第2-01條批准,但 規則適用。
審計委員會負責與管理層審查和討論經審計的合併財務報表,與獨立註冊會計師討論上市公司會計監督委員會第1301號審計準則所要求的事項與審計委員會的溝通根據上市公司會計監督委員會的適用要求,收到獨立註冊會計師關於獨立註冊會計師與審計委員會就獨立性進行溝通的書面披露 ,並與獨立註冊會計師討論其獨立性,並建議董事會將經審計的合併財務報表納入本公司的10-K表格年度報告。
| 126 |
第四部分
第 項15.物證、財務報表附表。
(a)(1) 財務報表。作為本報告一部分提交的財務報表列於綜合財務報表索引 。
(a)(2) 財務報表明細表。表被省略是因為它們不適用,或者所需信息顯示在合併財務報表或附註中。
(a)(3) 展品。請參考《展品索引》。本年度報告以表格10-K的形式包含或通過引用併入本年度報告,並根據S-K法規第601項進行編號。
第 項16.表格10-K摘要。
沒有。
| 127 |
簽名
根據1934年《證券交易法》第13或15(D)節的要求,註冊人已正式促使本報告由經正式授權的簽署人代表其簽署。
十四行詩 生物治療公司 (註冊人) | |
| 日期: 2023年12月14日 | /s/ Pankaj Mohan,博士 |
| Pankaj 莫漢博士 | |
總裁 和首席執行官 (首席執行官和 及時 授權簽字人) | |
| 日期: 2023年12月14日 | /s/ 傑伊·克羅斯 |
| Jay 克羅斯 | |
首席財務官 (首席財務會計官 ) |
簽名 和委託書
通過這些陳述,茲確認以下簽名的每一位人士構成並任命Pankaj Mohan,Ph.D.和Jay Cross, 以及他們中的每一個人,他的真實和合法的代理人,代理人和代理人,具有完全的替代和重新替代的權力,以他的名義,地點和代替,以任何和所有身份,(i)採取行動,簽署並向證券交易委員會 提交本年度報告的任何和所有修訂,表格10-K,連同所有附表和附件,(ii)採取行動,簽署和存檔 與此相關的必要或適當的證書,文書,協議和其他文件,(iii) 採取任何和所有必要或適當的行動,儘可能完全出於所有意圖和目的,他可以或可以親自做,特此批准,批准和確認所有該代理人,代理人和實際代理人或其任何替代人可以合法地進行或導致進行。
根據 《1934年證券交易法》的要求,本報告已由以下人員代表註冊人 簽署,其身份和日期均已註明。
| 簽名 | 標題 | 日期 | ||
| /s/ Pankaj Mohan,博士 | 總裁,首席執行官兼董事長 | 2023年12月14日 | ||
| Pankaj 莫漢博士 | (首席執行官 ) | |||
| /s/ 傑伊·克羅斯 | 首席財務官 | 2023年12月14日 | ||
| Jay 克羅斯 | (首席財務會計官 ) | |||
| /S/ 阿爾伯特·戴爾尼斯 | 董事 | 2023年12月14日 | ||
| 艾伯特·戴爾內斯 | ||||
| /S/ 納伊萊什·巴特 | 董事 | 2023年12月14日 | ||
| Nailesh 巴特 | ||||
| /S/ 拉古拉奧 | 董事 | 2023年12月14日 | ||
| 拉古 拉奧 | ||||
| /S/ 唐納德·格里菲斯 | 董事 | 2023年12月14日 | ||
| 唐納德·格里菲斯 | ||||
| /S/ 洛裏·麥克尼爾 | 董事 | 2023年12月14日 | ||
| 洛莉 麥克尼爾 | ||||
| 128 |
展品索引
展品 不是的。 |
描述 | |
| 2.1 | 協議 和合並計劃,日期為2019年10月10日,由公司和公司之間,十四行詩子公司。和合並子公司(作為公司於2019年10月11日提交的表格8-K當前報告的附件2.1提交,並通過引用併入本文)。# | |
| 2.2 | 本公司、十四行子公司和合並子公司之間於2020年2月7日對合並協議和計劃的第1號修正案(作為2020年2月7日提交的公司當前8-K報表的附件2.1提交,並通過引用併入本文)。 | |
| 2.3 | Sonnet BioTreateutics,Inc.和Relipment Treateutics Holding SA之間的股票交換協議,日期為2019年8月9日(通過引用該公司於2019年11月27日提交給美國證券交易委員會的S-4表格註冊聲明的附件2.10納入)。# | |
| 3.1 | 經修訂的Sonnet BioTreateutics Holdings,Inc.公司註冊證書(通過引用公司於2020年12月17日提交給美國證券交易委員會的10-K表格年度報告的附件3.1合併而成)。 | |
| 3.2 | 3系列優先股的優先股、權利和限制指定證書(通過引用合併到我們於2022年8月15日提交給美國證券交易委員會的8-K表格當前報告的附件3.1)。 | |
| 3.3 | 4系列優先股的優先股、權利和限制指定證書(通過引用合併到我們於2022年8月15日提交給美國證券交易委員會的8-K表格當前報告的附件3.2)。 | |
| 3.4 | Sonnet BioTreateutics Holdings,Inc.於2022年9月16日修訂的註冊證書修正案證書(通過引用附件3.1併入我們於2022年9月19日提交給美國證券交易委員會的當前8-K表格報告中)。 | |
| 3.5 | 日期為2023年8月31日的Sonnet BioTreateutics Holdings,Inc.公司註冊證書修正案證書(通過引用附件3.1併入我們於2023年9月1日提交給美國證券交易委員會的當前8-K表格報告中)。 | |
| 3.6 | 修訂和重新修訂了Sonnet BioTreateutics Holdings,Inc.的章程(通過引用附件3.3併入我們於2022年8月15日提交給美國證券交易委員會的 Form 8-K的當前報告中)。 | |
| 4.1 | 普通股證書表格 (於2011年12月2日向美國證券交易委員會備案的我們的S-1表格註冊説明書附件4.1(註冊號: 333-178307))。 | |
| 4.2 | 2017年5月4日的認股權證表格 (通過引用附件4.2併入我們於2017年5月5日提交給美國證券交易委員會的當前8-K表格報告中)。 | |
| 4.3 | 剝離 實體認股權證,日期為2020年4月1日(通過引用附件4.1併入公司於2020年4月3日提交給美國證券交易委員會的當前8-K表格報告 )。 | |
| 4.4 | Sonnet BioTreateutics,Inc.轉換的Form (通過引用公司於2020年8月14日提交給美國證券交易委員會的Form 10-Q季度報告的附件4.2併入)。 | |
| 4.5 | A/B系列認股權證表格 (參考公司於2020年2月7日向美國證券交易委員會提交的S-4/A表格註冊説明書附件4.16)。 | |
| 4.6 | C系列認股權證的表格 (通過參考我們於2020年8月4日提交給美國證券交易委員會的當前8-K表格報告的附件4.1併入)。 | |
| 4.7 | 登記 本公司與其中所列若干投資者之間於2020年2月7日訂立的配股協議(以引用方式併入本公司於2020年2月7日提交予美國證券交易委員會的S-4/A表格註冊説明書附件4.17)。 |
| 129 |
| 4.8 | 證券説明 * | |
| 4.9 | 日期為2021年8月24日的預籌資權證表格 (通過參考2021年8月25日提交給美國證券交易委員會的公司當前報告的8-K表格的附件4.2併入)。 | |
| 4.10 | 日期為2021年8月24日的承銷商認股權證表格 (參照本公司於2021年8月16日向美國證券交易委員會提交的S-1/A表格註冊説明書的附件4.9併入)。 | |
| 4.11 | 日期為2021年8月24日的普通權證表格 (通過引用本公司2021年8月25日提交給美國證券交易委員會的8-K表格的附件4.4併入)。 | |
| 4.12 | 日期為2023年2月10日的預融資認股權證表格 (通過引用附件4.2併入我們於2023年2月13日向美國證券交易委員會提交的當前8-K表格報告中)。 | |
| 4.13 | 日期為2023年2月10日的承銷商認股權證表格 (通過引用我們於2023年2月13日向美國證券交易委員會提交的當前8-K表格報告的附件4.3併入)。 | |
| 4.14 | 日期為2023年2月10日的普通權證表格 (通過引用附件4.4併入我們於2023年2月13日向美國證券交易委員會提交的當前報告中的表格8-K)。 | |
| 4.15 | 日期為2023年6月30日的預融資認股權證表格 (通過參考2023年6月30日提交給美國證券交易委員會的當前8-K表格報告的附件4.1併入)。 | |
| 4.16 | 日期為2023年6月30日的授權書表格 (通過引用附件4.2併入2023年6月30日提交給美國證券交易委員會的當前8-K表格報告中)。 | |
| 4.17 | 2023年6月30日的配售代理認股權證表格(通過參考2023年6月30日提交給美國證券交易委員會的8-K表格當前報告的附件4.3併入)。 | |
| 10.1 | GEM Global Year Fund LLC SCS與Sonnet BioTreateutics,Inc.於2019年8月6日簽訂的普通股購買協議(通過引用2019年11月27日提交給美國證券交易委員會的公司S-4表格註冊説明書附件10.54納入該協議)。 | |
| 10.2 | GEM Global Year Fund LLC SCS與Sonnet BioTreateutics,Inc.於2019年9月25日簽訂的普通股購買協議修正案(通過引用2019年11月27日提交給美國證券交易委員會的公司S-4表格註冊説明書附件10.55而併入)。 | |
| 10.3 | GEM Global Year Fund LLC SCS,Sonnet BioTreateutics,Inc.和Chancleer Holdings,Inc.於2020年2月7日簽訂的普通股購買協議第 面函件和修正案2(通過引用2020年2月7日提交給美國證券交易委員會的公司註冊表格S-4中的附件10.60併入)。 | |
| 10.4 | Pankaj Mohan和Sonnet BioTreateutics,Inc.於2018年12月31日簽訂的僱傭協議(通過參考2020年2月7日提交給美國證券交易委員會的公司S-4表格註冊聲明的附件10.56而併入)。† | |
| 10.5 | John Cini和Sonnet BioTreateutics,Inc.於2020年1月10日簽訂的僱傭協議(通過引用附件10.58納入公司於2020年2月7日提交給美國證券交易委員會的S-4表格註冊聲明中的附件10.58)。† | |
| 10.6 | Jay Cross和Sonnet BioTreateutics,Inc.之間的僱傭協議,日期為2020年1月10日(通過引用附件10.57納入公司於2020年2月7日提交給美國證券交易委員會的S-4表格註冊聲明中的附件10.57)。† | |
| 10.7 | 蘇珊·德克斯特與公司簽訂的僱傭協議,日期為2020年4月1日(通過引用附件10.7併入公司於2020年4月3日提交給美國證券交易委員會的當前8-K表格報告中)。† |
| 130 |
| 10.8 | 唐納德·格里菲斯與Sonnet BioTreateutics,Inc.於2019年1月1日簽署的要約函(通過引用公司於2020年2月7日提交給美國證券交易委員會的S-4表格註冊聲明的附件10.59而併入)。† | |
| 10.9 | 十四行詩 生物治療控股公司2020年綜合性股權激勵計劃(通過引用公司於2020年5月20日提交給美國證券交易委員會的S-8表格註冊説明書的附件4.2納入)。† | |
| 10.10 | 限制性股票單位獎表格 (參考我們於2020年7月9日提交給美國證券交易委員會的8-K表格(文件號:001-35570)附件10.1併入)。† | |
| 10.11 | 許可證 ARES Trading SA和Relipment Treeutics SA之間的協議,日期為2015年8月28日(通過引用公司於2020年2月7日提交給美國證券交易委員會的S-4表格註冊聲明的附件10.51而併入)。* | |
| 10.12 | Discovery XOMA(US)LLC和Oncobiologics,Inc.於2012年7月23日簽署的合作協議(通過引用附件10.52併入該公司於2020年2月7日提交給美國證券交易委員會的S-4表格註冊聲明中)。* | |
| 10.13 | XOMA(US)LLC和Sonnet BioTreateutics,Inc.於2019年5月7日簽訂的Discovery合作協議修正案(合併於 參考該公司於2020年2月7日提交給美國證券交易委員會的S-4表格註冊聲明附件10.53)。* | |
| 10.14 | 證券購買協議,日期為2020年2月7日,由強第克利爾控股公司、十四網生物治療公司及其投資者 方簽署(通過引用2020年2月7日提交給美國證券交易委員會的公司S-4表格註冊説明書第10.64號附件納入)。 | |
| 10.15 | 由Sonnet BioTreateutics Holdings, Inc.和持有人(通過引用附件10.1併入我們於2020年8月4日提交給美國證券交易委員會的當前8-K表格(文件編號001-35570)中),於2020年8月3日由Sonnet BioTreateutics Holdings和持有人之間簽署的認股權證行使和綜合修訂協議表格 。 | |
| 10.16 | Sonnet BioTreateutics Holdings,Inc.的轉讓和假設僱傭協議,2020年4月1日生效(通過引用併入該公司於2020年12月17日提交給美國證券交易委員會的Form 10-K年度報告的附件10.16)。† | |
| 10.17 | Pankaj Mohan與本公司於2020年11月23日簽訂的高管聘用協議第1號修正案(通過引用併入本公司於2020年12月17日提交給美國證券交易委員會的Form 10-K年度報告附件10.17)。† | |
| 10.18 | 約翰·西尼與公司於2020年11月23日簽訂的高管聘用協議的第1號修正案(通過引用2020年12月17日提交給美國證券交易委員會的公司10-K表格年度報告的附件10.18納入)。† | |
| 10.19 | 賠償協議表格 (通過引用公司於2020年12月17日提交給美國證券交易委員會的10-K表格年度報告的附件10.19併入)。† | |
| 10.20 | 本公司與必和必拓於2020年2月5日簽訂的場外銷售協議(於2021年2月5日提交給美國證券交易委員會的8-K表格(文件編號001-35570)引用附件1.1併入其中)。 | |
| 10.21 | 許可證 Sonnet BioTherapeutics,Inc.與Sonnet BioTherapeutics,Inc.和New Life Therapeutics PTE,LTD(通過引用併入 我們於2021年5月17日向SEC提交的10-Q表格季度報告的附件10.1)。 | |
| 10.22 | 第一個 Sonnet BioTherapeutics,Inc.與Sonnet BioTherapeutics,Inc.於2021年6月11日簽署的許可協議修正案和New Life Therapeutics PTE,LTD(註冊成立 參考我們於2021年12月17日向SEC提交的10-K表格年度報告的附件10.22)。 | |
| 10.23 | 第二次 Sonnet Biotherapeutics CH SA、Sonnet BioTherapeutics,Inc.於2021年7月7日簽署的許可協議修正案新生活(New Life) Therapeutics PTE,Ltd.(通過引用併入我們於 年12月向SEC提交的10-K表格年度報告的附件10.23) 17,2021)。 | |
10.24
|
修正案 ARES TRADING SA和Sonnet BioTherapeutics CH SA(註冊成立 )於2021年11月1日簽訂的許可協議和和解協議 參考我們於2021年12月17日向SEC提交的10-K表格年度報告的附件10.24)。 |
| 131 |
| 10.25 | 在市場上 公司與BTIG簽訂的銷售協議,日期為2022年8月15日(通過引用納入我們當前報告的附件1.1 於2022年8月15日向SEC提交的8-K表格)。 | |
| 10.26 | 承保 公司與Chardan Capital Markets,LLC於2023年2月8日簽訂的協議(通過參考附件1.1合併 我們於2023年2月13日向SEC提交的8-K表格的當前報告)。 | |
| 10.27 | 表格 證券購買協議,日期為2023年6月28日,由本公司和 買方(通過引用納入我們提交的表格8-K的當前報告的附件10.1 於2023年6月30日與SEC簽署)。 | |
| 21.1 | 子公司 公司的 * | |
| 23.1 | 同意 畢馬威會計師事務所 * | |
| 24.1 | 功率 (見簽名頁)* | |
| 31.1 | 認證 根據1934年證券交易法頒佈的規則13 a-14(a)和15 d-14(a), 經修訂的 * | |
| 31.2 | 認證 根據1934年證券交易法頒佈的規則13 a-14(a)和15 d-14(a), 經修訂的 * | |
| 32.1 | 認證 根據18 U.S.C.第1350條,根據 年《薩班斯-奧克斯利法案》第906條通過 2002年 ** | |
| 32.2 | 認證 根據18 U.S.C.第1350條,根據 年《薩班斯-奧克斯利法案》第906條通過 2002年 ** | |
| 97.1 | 追回政策 * | |
| 101.INS | 內聯 XBRL實例文檔-實例文檔不顯示在交互式數據文件中,因為它的XBRL標記嵌入在 內聯XBRL文檔。* | |
| 101.SCH | 內聯 XBRL分類擴展架構文檔。* | |
| 101.CAL | 內聯 XBRL分類擴展計算鏈接庫文檔。* | |
| 101.DEF | 內聯 XBRL分類擴展定義鏈接庫文檔。* | |
| 101.LAB | 內聯 XBRL分類擴展標籤鏈接庫文檔。* | |
| 101.PRE | 內聯 XBRL分類擴展演示鏈接庫文檔。* | |
| 104 | 封面 頁面交互式數據文件(格式為內聯XBRL,包含在附件101中)。* |
| * | 隨函存檔。 |
| ** | 隨函提供。 |
| *** | 茲存檔;根據S-K法規第601(B)(10)項的規定,部分展品已被省略。任何遺漏部分的副本 將根據要求提供給證券交易委員會。 |
| # | 根據S-K法規第601(B)(2)項,本協議的附表和證物已被省略。如有任何遺漏的時間表和/或證物,應要求提供給美國證券交易委員會。 |
| † | 表示 管理合同或補償計劃、合同或安排。 |
| 132 |