
附錄 99.1
1 公司概述總裁兼首席執行官 Dinesh V. Patel 博士 2023 年 7 月 4 日

前瞻性陳述 2 本陳述和隨附的口頭陳述包含根據1995年《私人證券訴訟改革法》的安全港提案發表的前瞻性陳述。除本簡報中包含的歷史事實陳述外,所有陳述,包括關於我們未來的經營業績和財務狀況、業務戰略、候選產品、資本資源、候選產品的潛在市場、我們與未來潛在合作安排相關的計劃、美國食品藥品監督管理局(“FDA”)的行動或決定對我們的業務或候選人的影響、臨牀試驗的註冊情況、對我們的任何潛在影響的陳述與冠狀病毒相關的業務-19,根據我們與Janssen Biotech, Inc. 的合作協議,我們可能收到里程碑式的付款和特許權使用費,這是前瞻性聲明。在某些情況下,你可以通過 “預期”、“相信”、“繼續”、“可以”、“估計”、“預期”、“打算”、“可能”、“計劃”、“可能”、“可能”、“預測”、“應該”、“將” 或這些術語或其他類似的經驗表達的負面等術語來識別前瞻性陳述。本演示文稿中發表的前瞻性陳述涉及已知和未知的風險、不確定性和其他重要事實,這些事實可能導致我們的實際業績、業績或成就與前瞻性陳述所表達或暗示的任何未來業績、業績或成就存在重大差異。這些前瞻性陳述存在風險和不確定性,包括主角向美國證券交易委員會提交的文件中討論的陳述,包括最近提交的10-K表和10-Q表格定期報告的 “風險因素” 和 “管理層對財務狀況和經營業績的討論和分析” 部分以及其中以引用方式納入的文件中隨後提交的文件。由於前瞻性陳述本質上會受到風險和不確定性的影響,其中一些風險和不確定性無法預測或量化,有些是我們無法控制的,因此你不應該依賴這些前瞻性陳述作為對未來事件的預測。我們的前瞻性陳述中反映的事件和情況可能無法實現或發生,實際結果可能與前瞻性陳述中的預測存在重大差異。除非適用法律要求,否則我們不打算公開更新或修改此處包含的任何前瞻性陳述,無論是由於任何新信息、未來事件、情況變化還是其他原因造成的。本演示文稿涉及正在接受臨牀研究且尚未獲得美國食品藥品監督管理局批准上市的產品。根據聯邦法律,它們目前僅限於調查用途,對於調查目的,沒有就其安全性或有效性作出任何陳述。此處包含的商標是其所有者的財產,僅供參考。此類使用不應被解釋為對此類產品的認可。本演示文稿中包含的任何內容均不是,也不應解釋為主持人、主角或主角或主角的任何董事、員工、經紀人或副手的推薦、承諾或陳述。本演示文稿並不聲稱包羅萬象,也不包含您可能想要的所有信息。
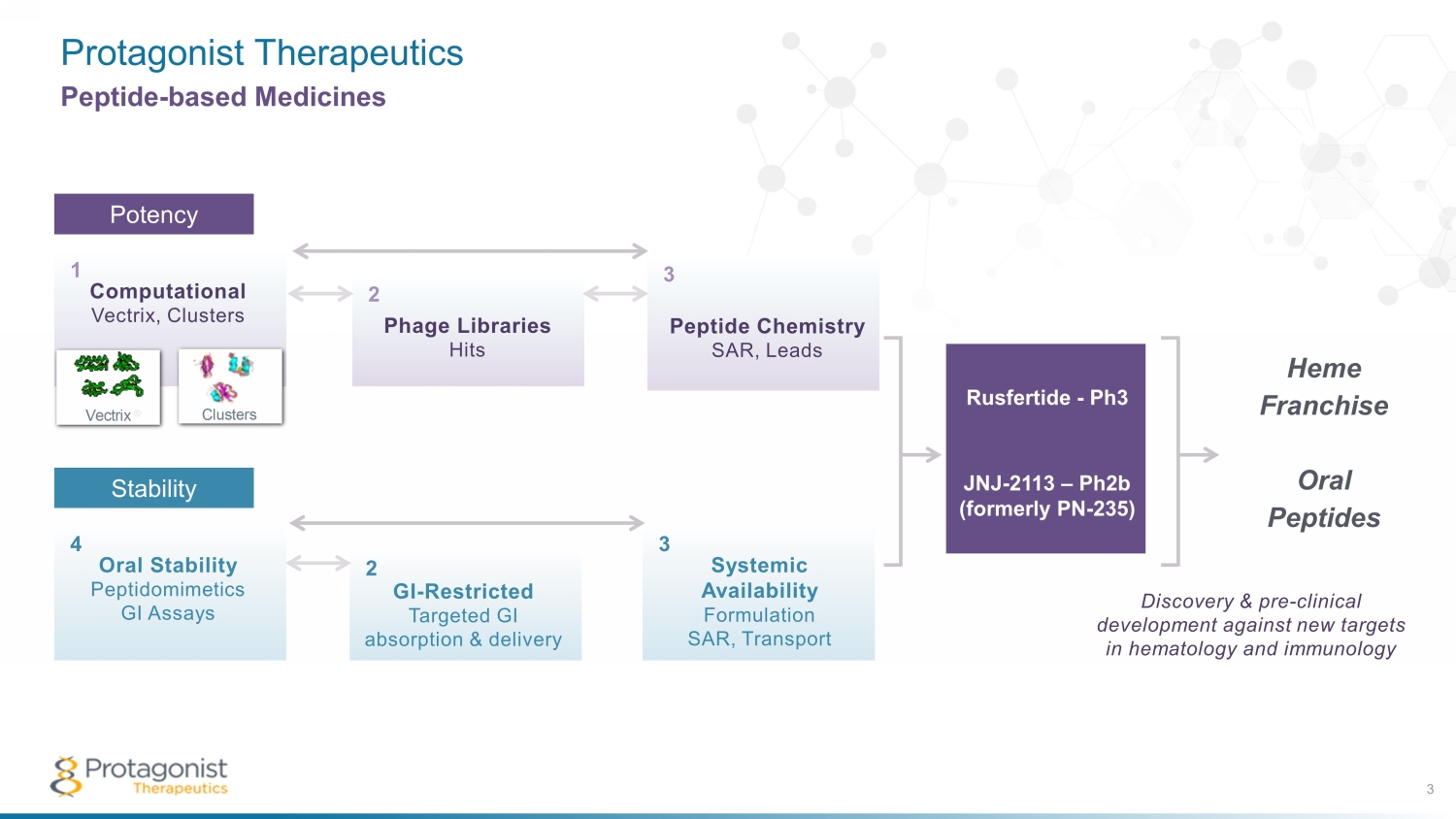
Proganist Therapeutics 3 基於肽的藥物效力肽化學 SAR,Leads 噬菌體庫命中計算 Vectrix,Clusters Vectrix® Clusters 1 2 3 效力口服穩定性 peptidometics GI 檢測 GI 限制靶向 GI 吸收和遞送系統可用性配方 SAR、Transport 4 5 6 穩定性計算向量、聚類 1 肽化學 SAR,引領噬菌體庫命中計算向量 Vectrix® 集羣 1 2 3 POTENCY 口服穩定性 Peptidomimetics GI 檢測 GI 受限的靶向 GI 吸收和輸送系統可用性配方 SAR、Transport 4 5 6 穩定性肽化學 SAR、Leads 3 噬菌體庫命中 2 個穩定性口服穩定性 peptidometics GI 檢測 4 全身可用性配方 SAR、Transportide 3 GI-限制靶向 GI 吸收和遞送 2 Rusfertide-Ph3 JNJ-2113 — P h2b(前身為 PN-235)血紅素特許經營口服肽發現和針對血液學和免疫學新靶標的臨牀前開發

正在進行的 PV 中的 Rusfertide 的 ph3 研究 | 與 Janssen 合作開發口服 IL-23R 拮抗劑 4 • JNJ-2113*(前身為 PN-235)— 與 Janssen 合作 — 口服 IL-23R 拮抗劑 — 多項臨牀研究 ▪ FRONTIER 1:ph2B 銀屑病研究** ▪ FRONTIER 2:延伸 FRONTIER 1 ▪ SUMMIT:ph2A 牛皮癬研究** ▪ 多項 1 期研究 ** — 計劃中的新研究 ▪ 牛皮癬的 3 期研究 ▪ 潰瘍性結腸炎的 2b 期研究 • Rusfertide − 可注射的 hepcidin 激素模仿物 − 驗證:真性紅細胞增多症 (PV) 中的 pH3 研究 − REVIVE:PH2 PV 研究隨機提款** − 太平洋:Ph2 高 HCT PV 研究** − 孤兒和快速通道狀態 − 血液學疾病的總銷售潛力達數十億美元 − 全資資產現金狀況強勁,現金流將持續到 2025 年底 *也稱為 JNJ-77242113 **已完成的主角療法

發現、臨牀前和臨牀開發 5 • 主要重點是完成 Rusfertide 在真性紅細胞增多症中的第 3 期驗證研究 • 今年將向已完成 REVIVE 研究的患者開放單獨的 2 年延期(PTG-300-21)研究 — 總治療持續時間長達 5 年 • 正在進行的發現項目 — 口服 hepcidin — 血液學的新靶點 — 專注於 “口服方法” 的免疫學新靶標 '作為強有力的差異化因素正在進行的項目和未來計劃
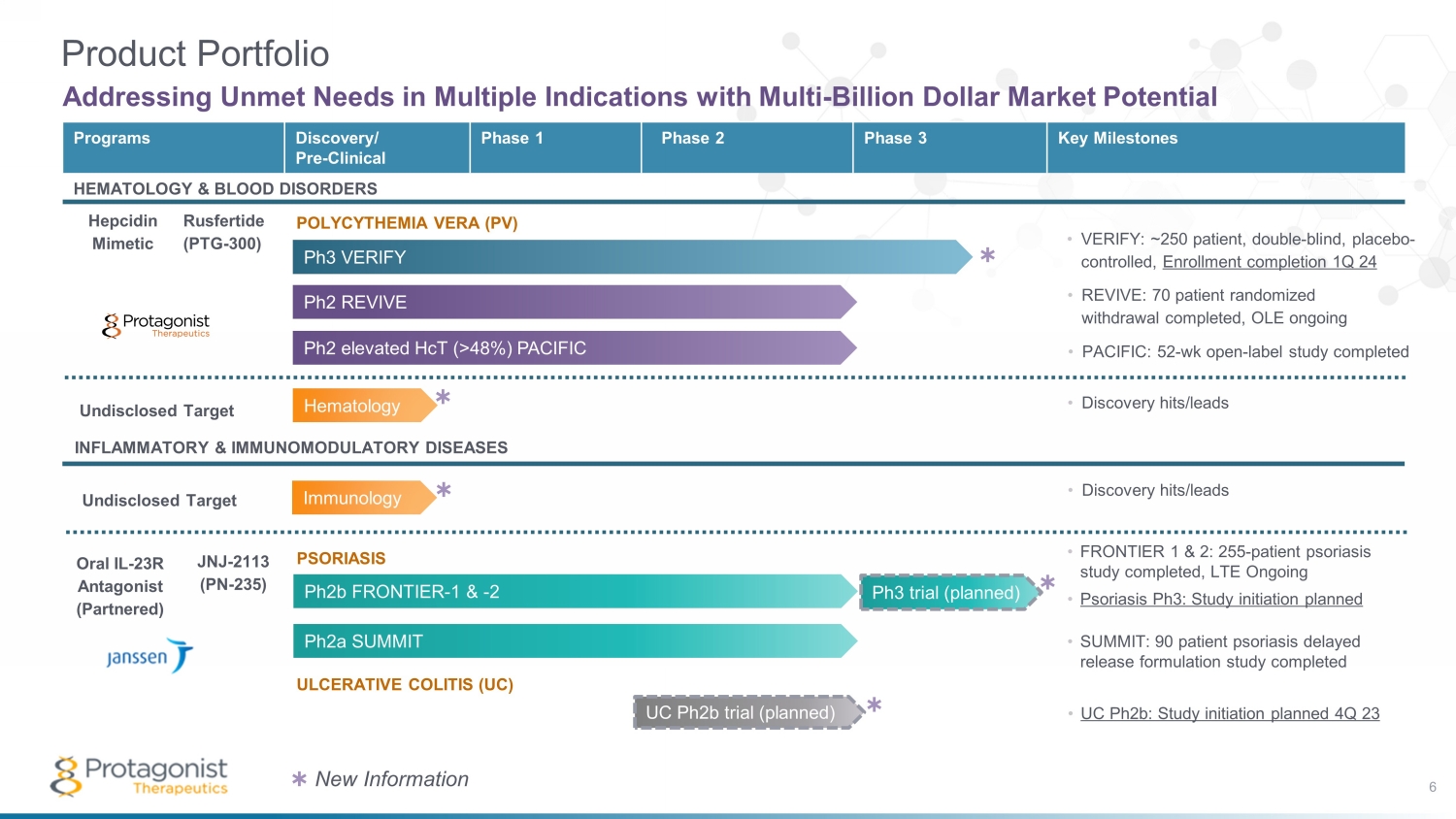
UC ph2b 試驗(計劃中)炎症和免疫調節疾病口服 IL-23R 拮抗劑(合作)JNJ-2113(PN-235)血液學 ph3 試驗(計劃中)項目發現/臨牀前 1 期 2 期關鍵里程碑血液學和血液疾病 Hepcidin 模仿魯斯費爾蒂德 (PTG-300) ph2b FRONTIER-1 和 -2 產品組合滿足多種未滿足的需求具有數十億美元的市場潛力 ph2A SUMMIT Ph2 升高 hcT (> 48%) PACIFIC Ph2 REVIVE PH3 驗證真性紅細胞增多症 (PV) 潰瘍性結腸炎 (UC) 牛皮癬 • FRONTIER 1 &2:255 — 患者牛皮癬研究已完成,LTE 正在進行中 • 牛皮癬 Ph3:計劃啟動研究 • 驗證:約 250 名患者,雙盲,安慰劑對照,入組完成 1Q 24 • REVIVE:70 名患者隨機戒斷已完成,OLE 正在進行 • 峯會:90 名患者牛皮癬延遲釋放配方研究已完成 • UC ph2b:計劃啟動 4Q 23 6 • D iscovery 命中/領導 • Discovery 命中/領先 Immunology ✱ ✱ ✱ ✱ ✱ 新信息 ✱ 未公開的目標未公開的目標

Rusfertide (PTG-300):Hepcidin 模仿物解決了真性紅細胞增多症中未滿足的需求
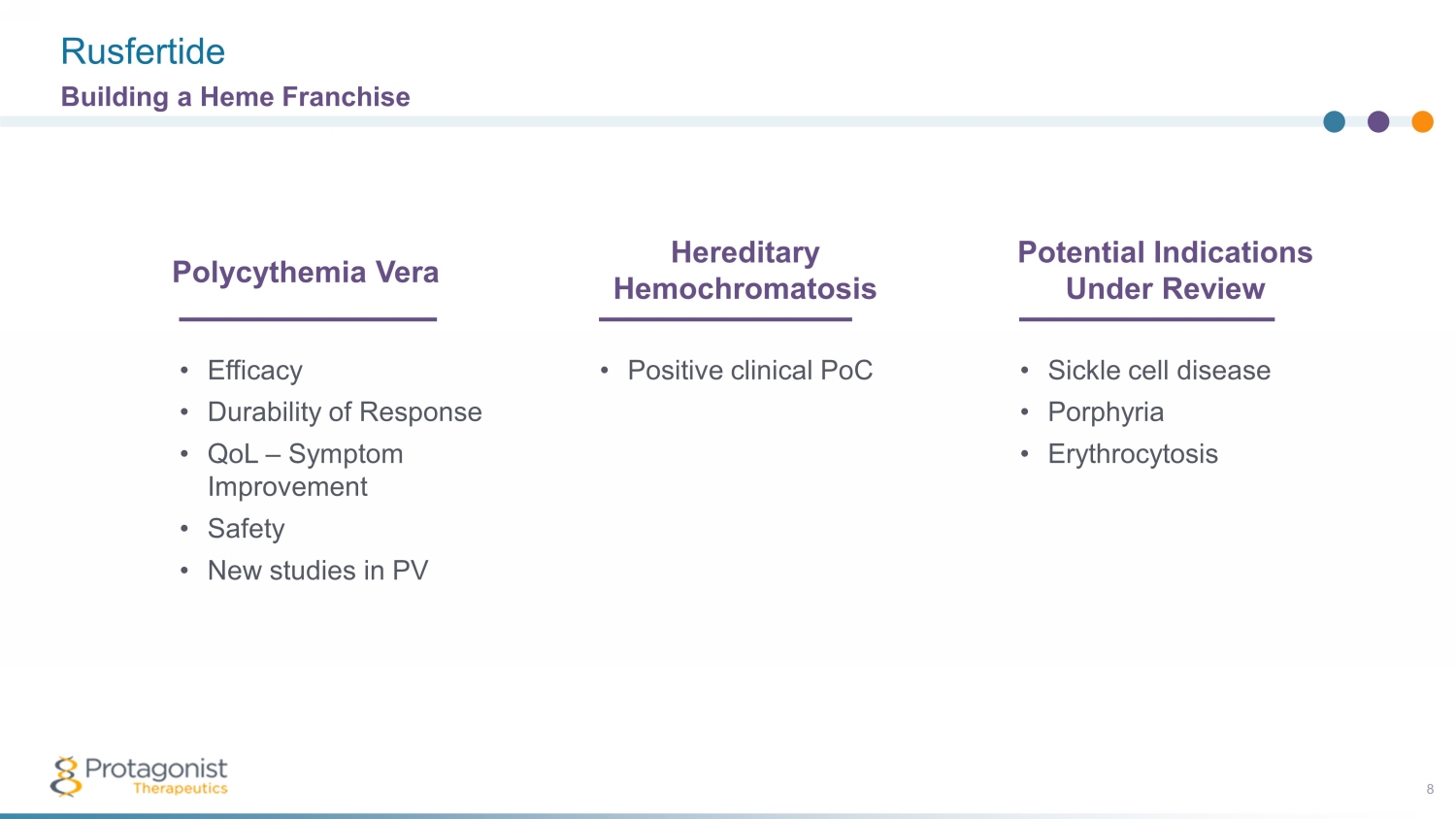
Rusfertide 8 建立血紅素特許經營權 • 功效 • 反應耐久性 • QoL — 症狀改善 • 安全性 • PV 真性紅細胞增多症的新研究 • 臨牀陽性 PoC 遺傳性血色素沉着症 • 鐮狀細胞病 • 卟啉病 • 紅細胞增多症潛在適應症正在審查中
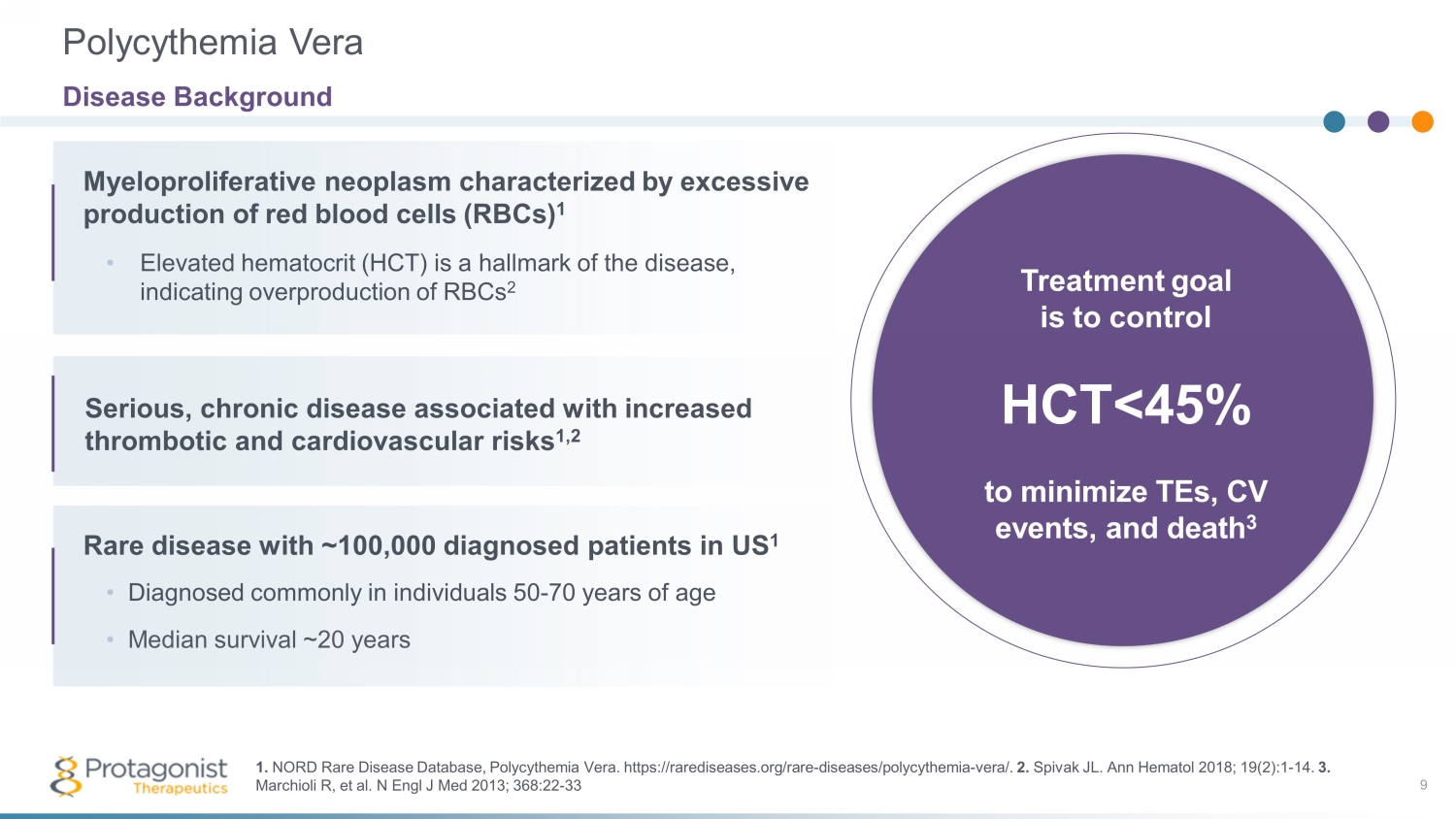
以紅細胞 (rbcs) 過量產生為特徵的骨髓增生性腫瘤 1 治療目標是控制 HCT

真性紅細胞增多症 • 包括疲勞和注意力集中問題在內的繁瑣症狀 2 — 84% 的患者報告疲勞,23% 的患者報告説由於症狀而卧牀整天 3 • 影響患者報告的日常生活活動並降低工作效率 2 當前 SOC 4 10 維持 HCT 45% 4 • 目前的護理標準 (SOC) 方法不足以控制 HCT 和症狀管理 5 加拿大皇家銀行沒有可用的藥物選擇-靶向 HCT 的特定機制 • Rusfertide 是一種模仿物Hepcidin,一種調節鐵穩態和紅細胞增多的天然激素 1.Marchioli R 等人N Engl J Med 2013;368:22-33。2.Mesa R 等人BMC Cancer。2016;16,167。3.MPN Landmark Survey,PV 報告(2017 年)。4.Verstovsek S,等人美國真性紅細胞增多症患者的真實世界治療和血栓形成事件。Ann Hematol。2023 年 3 月;102 (3): 571-581。doi: 10.1007/s00277-023-05089-6。Epub 2023 年 1 月 13 日PMID:36637474;PMCID:PMC9977710。2 1 3 4

PV 的當前治療選擇和潛在侷限性 11 細胞還原療法可能導致不耐受和/或血細胞比容控制不足 Hydrokyurea Hydrea® 通常用作高危患者的第一線治療患者仍會經歷 HCT > 45%,1 可能導致劑量增加和需要放血術經常因血細胞計數、AE、疾病進展或依從性差而停止 2 患皮膚癌的風險 3 25% 的患者產生耐藥性或者不耐受的 2,4 幹擾素 Pegasys®,Besremi® 幹擾素已經存在很長時間了已用完——治療中的標籤;Besremi 是第一款獲準用於 PV 5 的幹擾素產品起作用緩慢,中位反應時間為 1.2 至 1.4 年 6 在 PROUD — PV 研究 7 黑匣子警告嚴重神經精神、自身免疫、缺血性和傳染性疾病 6 Ruxolitinib Jakafi® 僅獲準用於羥基脲或耐藥性不耐受患者 7 改善脾腫大,脾腫大是疾病進展的潛在標誌 8 潛在的嚴重副作用包括血小板減少、中性粒細胞減少和貧血 7 患皮膚癌的風險 7 23% 的患者在治療開始後的平均兩年內被發現停用魯索替尼 9 1.Chornenki NLJ 等人Leuk Res. 2018 年 7 月;70:62-66。2.Parasuraman S 等Exp Hematol Oncol。2016;5:3 3。Hydrea FDA 標籤。4.Altomare I 等人Clin 淋巴瘤 Myeloma Leuk。2021;21 (11): e915-e921。4.Gilreath JA 等人Blood。2018;132(補編 1)。5.Besremi FDA 標籤。6.Gisslinger H 等人Lancet Haematol。2020;7 (3): e196-e208。7.Jakafi FDA 標籤。8.Coltoff R 等人Clin 淋巴瘤 Myeloma Leuk。2020;20 (10):697-703。9.Tremblay,D. Leuk Res.2021;109。
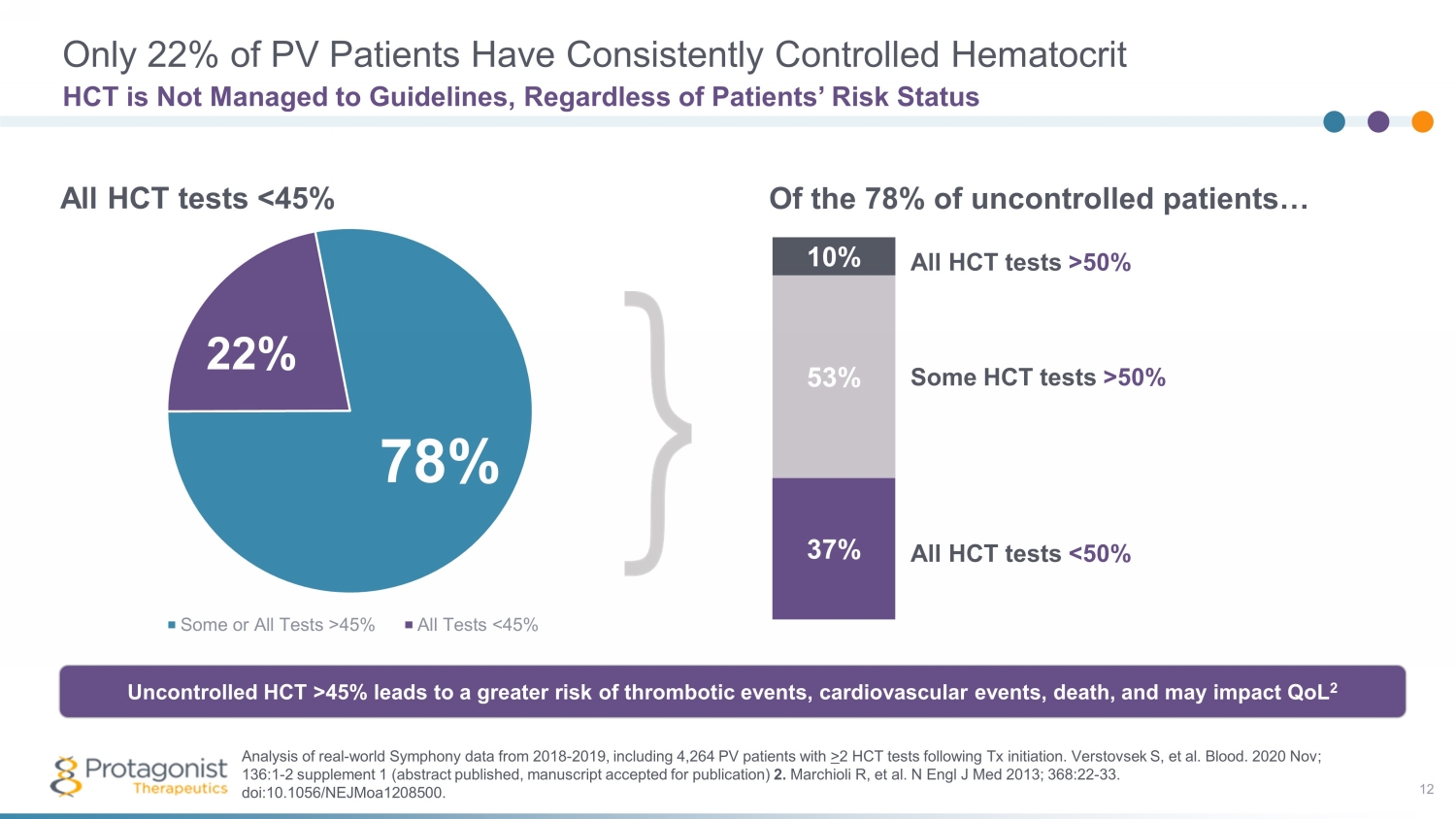
只有 22% 的 PV 患者持續控制血細胞比容 12 HCT 未達到指導方針,無論患者的風險狀況如何,部分或全部測試 > 45% 所有測試 50% 一些 HCT 檢測 > 50% 所有 HCT 檢測 45% 會增加血栓形成事件、心血管事件、死亡的風險,並可能影響 QoL 2 對 2018-2019 年真實世界 Symphony 數據的分析,包括 4,264 名 PV 患者 Tx 啟動後接受超過 2 次 HCT 檢測。Verstovsek S 等人Blood。2020 年 11 月;136:1-2 補編 1(摘要已出版,m anuscript 已獲準出版)2.Marchioli R 等人N Engl J Med 2013;368:22-33。doi: 10.1056/nejmoa1208500。
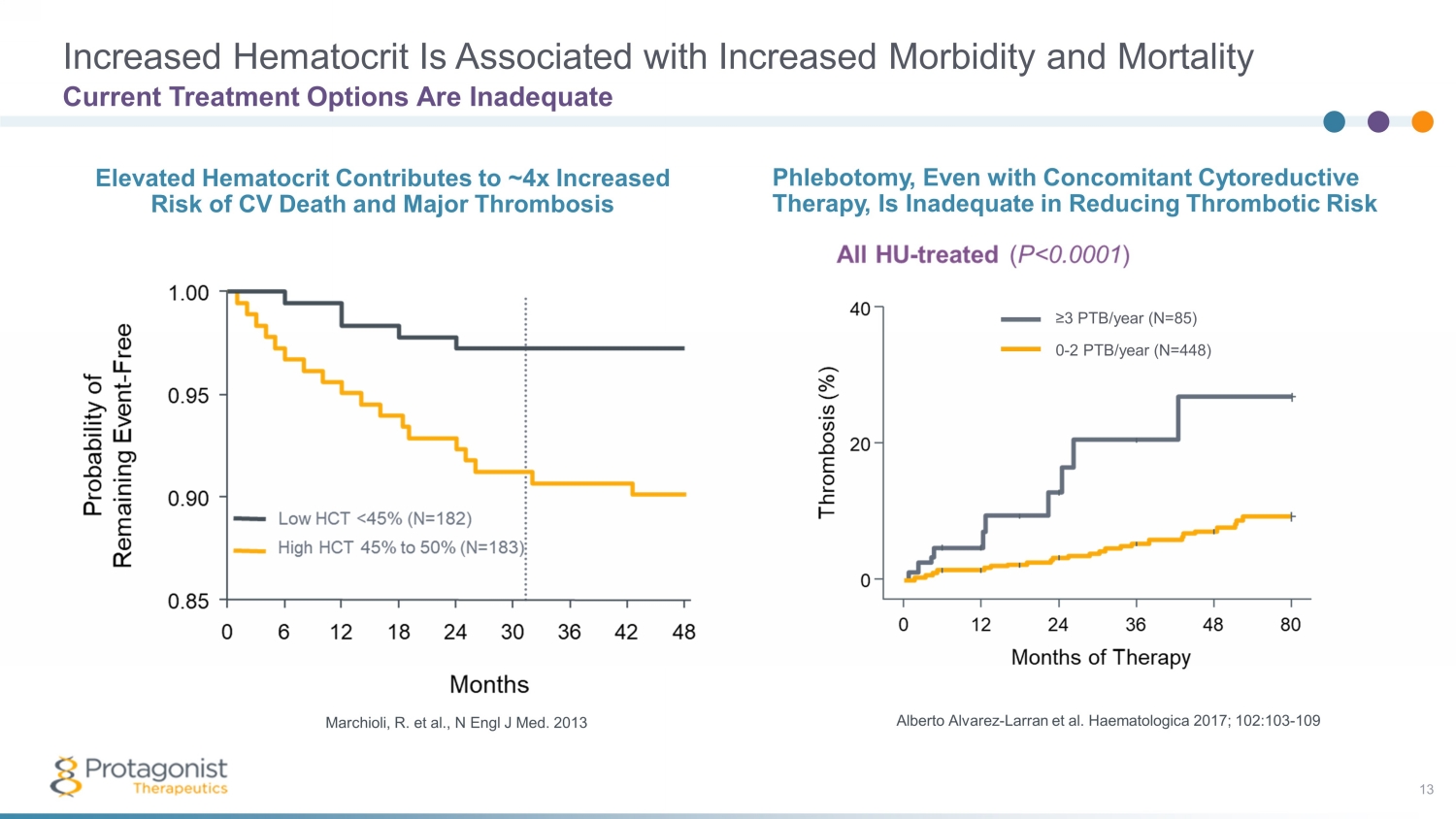
血細胞比容增加與發病率和死亡率增加有關 13 Alberto Alvarez-Larran 等人Haematologica 2017;102:103-109 Marchioli,R. 等人,N Engl J Med. 2013 ≥3 ptb/年 (N=85) 0-2 ptb/年 (N=448) 放血,即使同時進行細胞還原療法,也不足以降低血栓形成風險血細胞比容升高導致心血管死亡和嚴重血栓形成風險增加約 4 倍選項不足
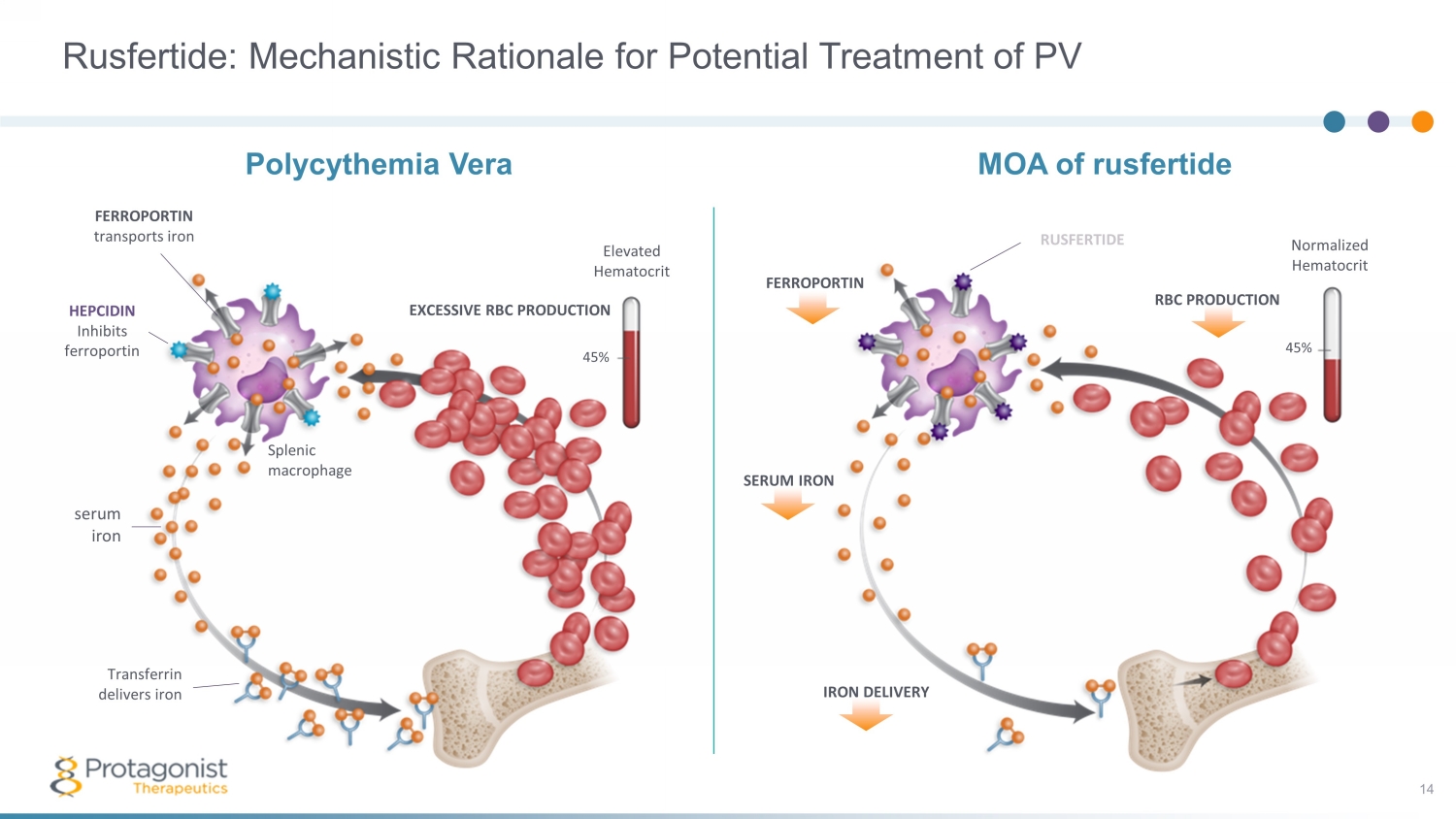
FERROPORTIN 標準化血細胞比容 45% 血清鐵輸送 RUSFERTIDE RBC 生產的 MOA RUSFERTIDE:可能治療 PV 14 的機制理由 FERROPORTIN 轉運鐵 HEPCIDIN 抑制 ferroportin 血清鐵轉鐵蛋白輸送過量紅細胞增生 45% 血細胞比容升高 veric Splenic 巨噬細胞

Rusfertide for Polycthemia veria 15 三項臨牀研究正在進行中 • Ph3 VERIFY 研究(n=250):− 研究仍在繼續,預計將於 2024 年第一季度完成註冊——與 FDA 和 CHMP(歐盟)商定了 3 期驗證協議 • 2 期太平洋研究:− 已完成為期16周的研究;• Ph2 REVIVE 研究(n=70):− 臨牀更新在 ASH 2021、ASCO 2022、EHA 2022、ASH 2022 上發佈” 在 ASCO 2022 上公佈的最新第 2 階段數據顯示了中斷和恢復給藥的影響 − 隨機戒斷研究已完成 • Ph2 PACIFIC 研究 (n=20):− 高血細胞比容 (HCT > 48%) 16 周研究已完成;52 周開放——標籤研究於 2023 年第二季度完成 • Rusfertide 具有孤兒藥名稱和快速通道狀態
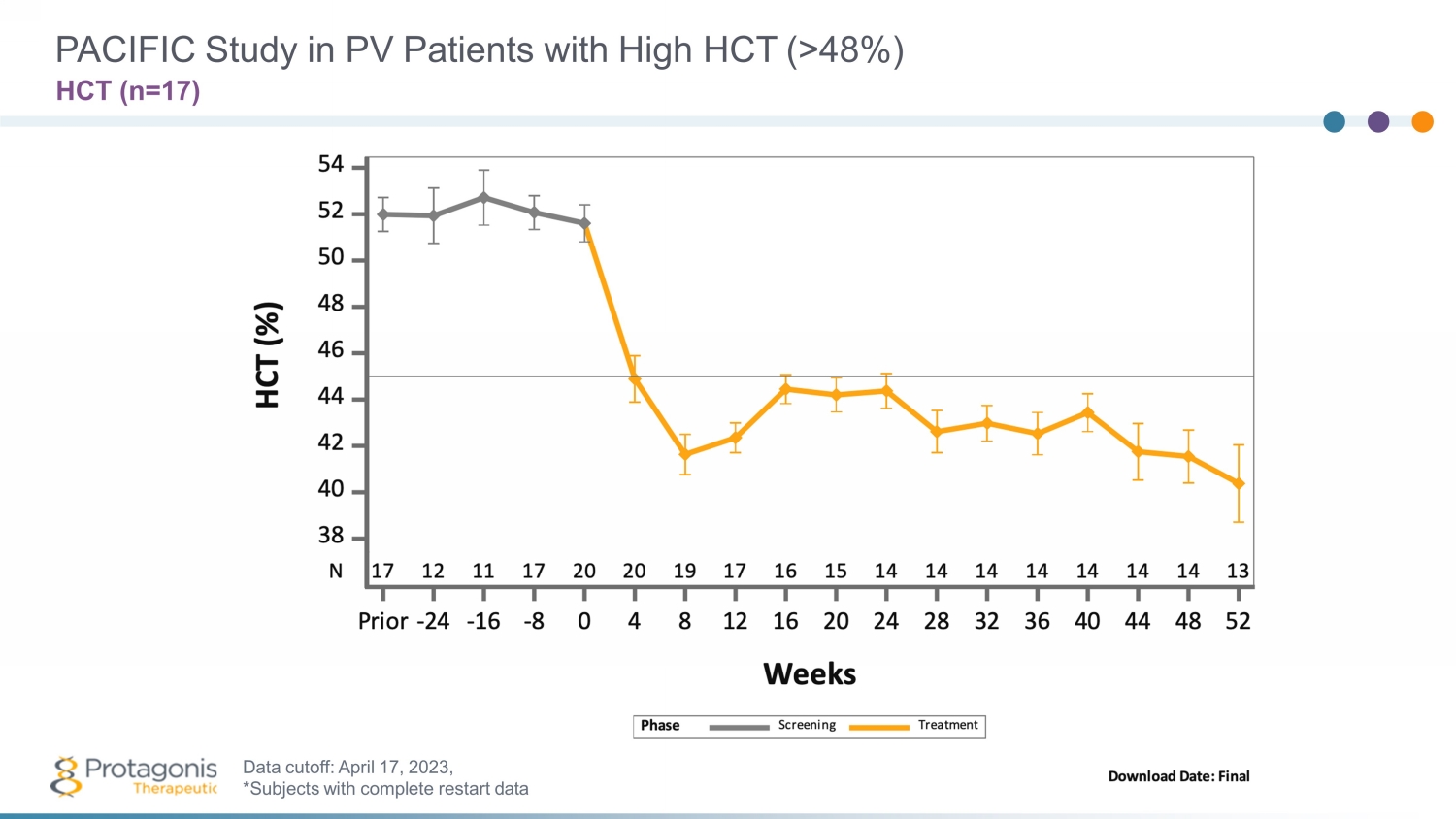
太平洋對高 HCT (> 48%) HCT (n=1 7) PV 患者的研究:202年 4 月 1 日 7 日 3,*具有完整重啟數據的受試者
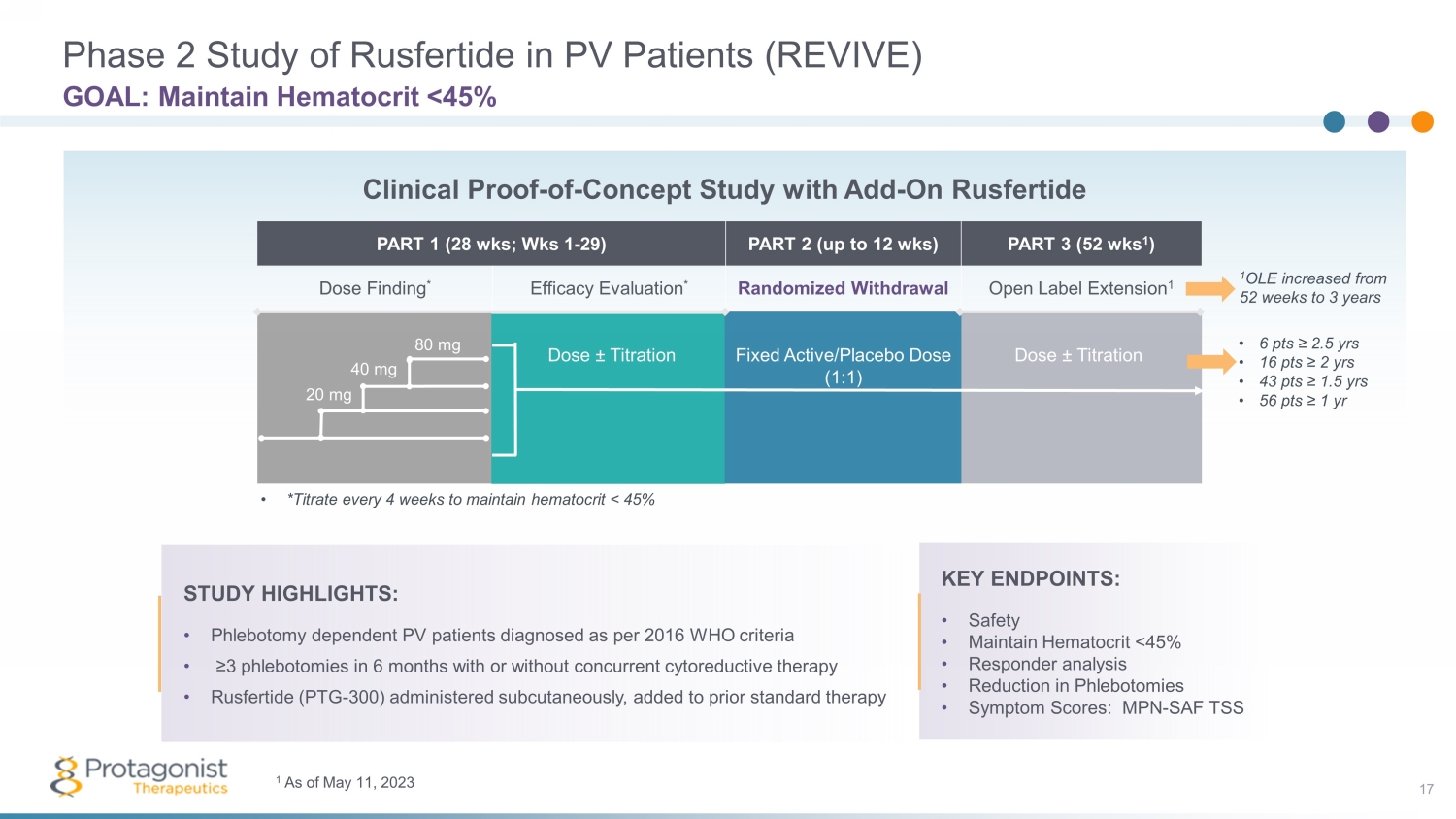
第 1 部分(28 周;Wks 1-29)第 2 部分(最多 12 周)第 3 部分(52 周 1)劑量發現 * 療效評估 * 隨機戒斷開放標籤延長 1 PV 患者 Rusfertide 第 2 期研究(REVIVE)17 目標:維持血細胞比容
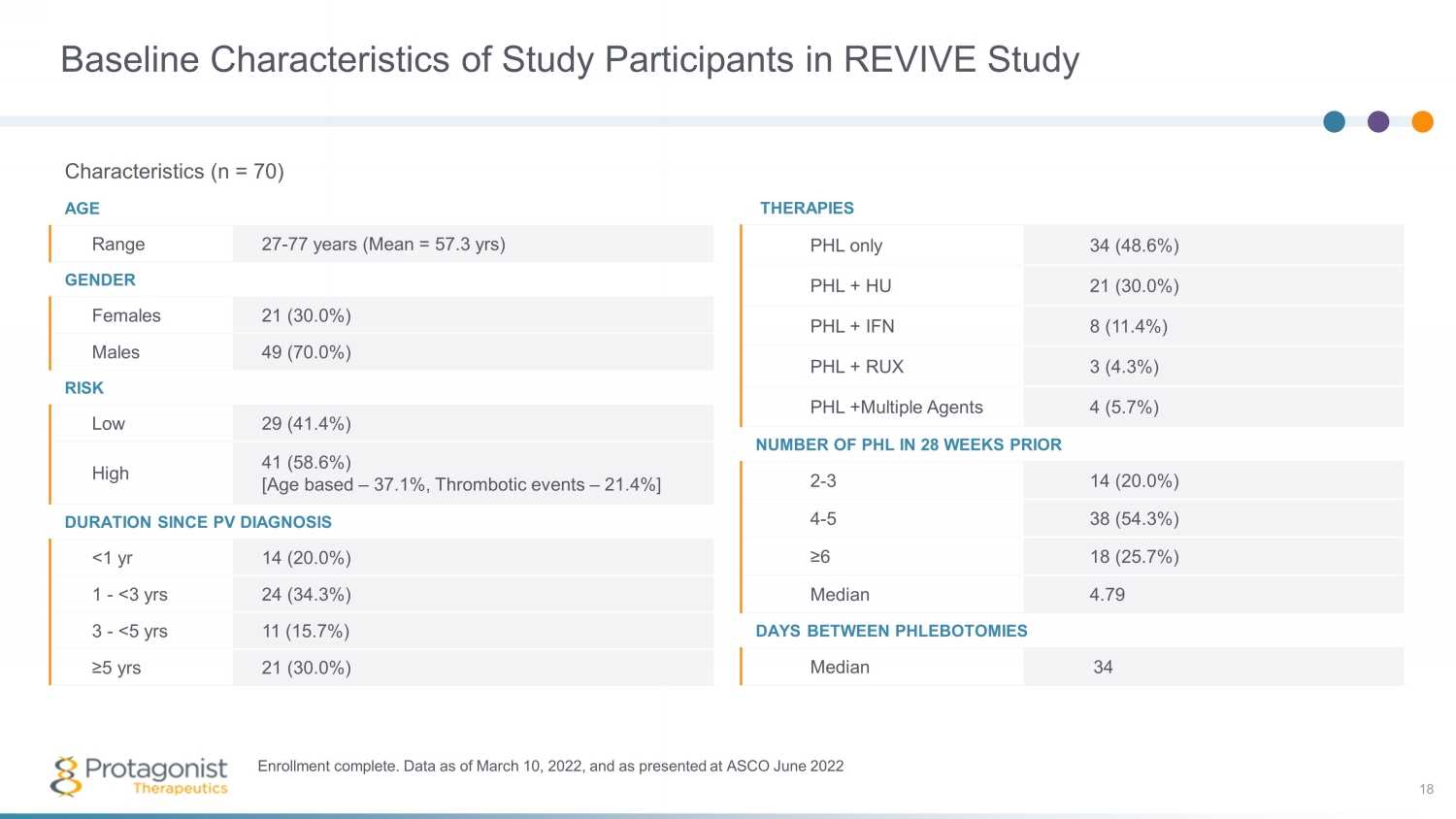
REVIVE 研究參與者的基線特徵研究特徵 (n = 70) 年齡範圍 27-77 歲(平均值 = 57.3 歲)性別女性 21 (30.0%) 男性 49 (70.0%) 風險低 29 (41.4%) 高 41 (58.6%) [基於年齡 — 37.1%,血栓形成事件 — 21.4%]PV 診斷後的持續時間
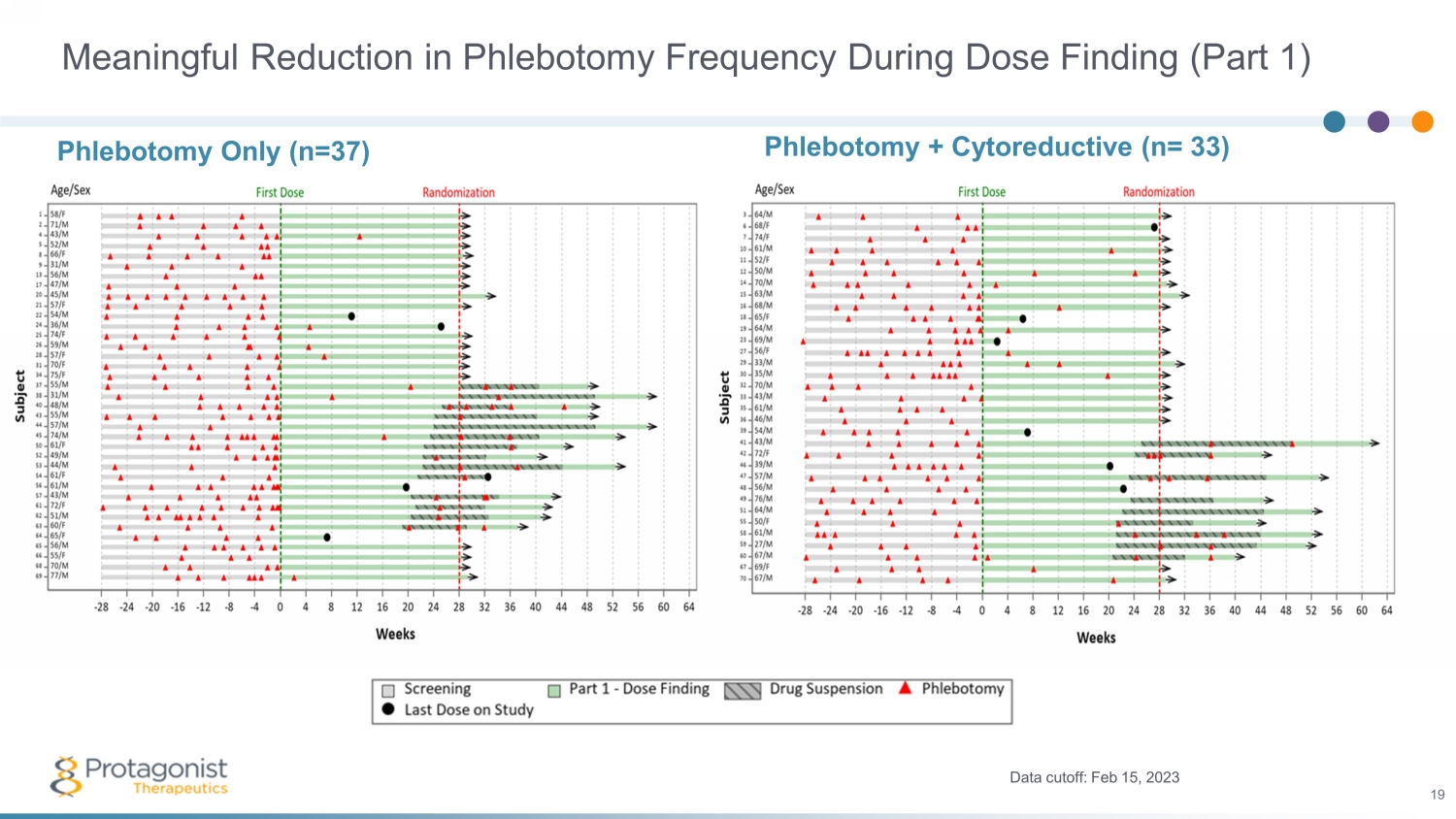
劑量發現期間放血頻率有意義地降低(第 1 部分)放血 + 細胞還原(n= 33)數據截止日期:2023 年 2 月 15 日 19 僅限放血(n=37)

第 1 部分(28 周;Wks 1-29)第 2 部分(最多 12 周)第 3 部分(52 周 1)劑量發現 * 療效評估 * 隨機戒斷開放標籤擴展 1 隨機戒斷的解盲,REVIVE 研究的第 2 部分 20 具有高度統計學意義的結果劑量 ± 滴定固定活性/安慰劑劑量 (1:1) 劑量 ± 滴定 • *每 4 周滴定以維持血細胞比容 t
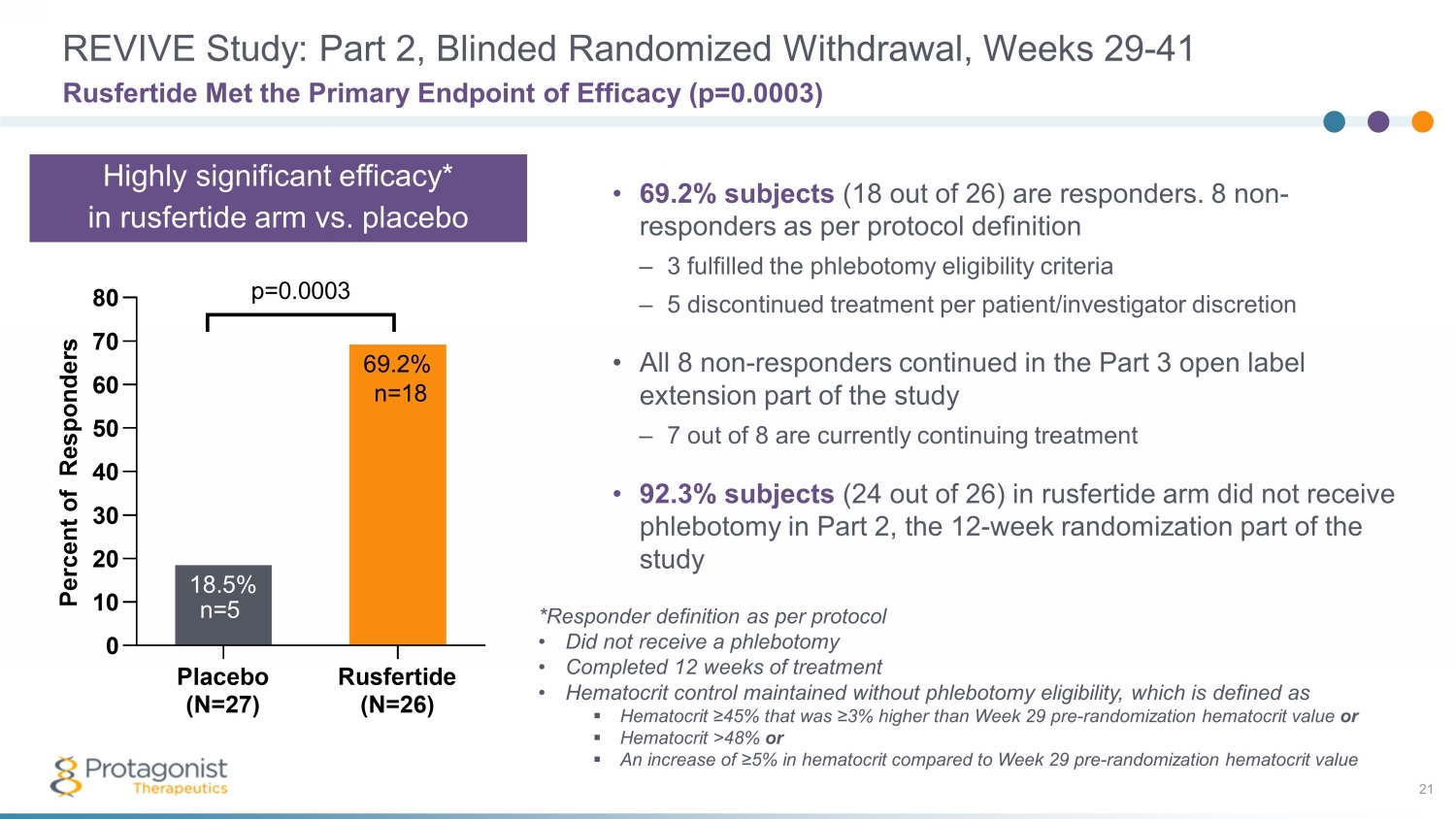
REVIVE 研究:第 2 部分,盲目隨機戒斷,第 29-41 周 21 • 69.2% 的受試者(26 人中的 18 人)是應答者。根據方案定義,8 名非反應者符合放血資格標準 — 每位患者/研究者自行決定 5 人已停止治療 • 所有 8 名非應答者繼續參與研究第 3 部分開放標籤延期部分 — 8 名受試者中有 7 名目前正在繼續治療 • 92.3%(在研究的 12 周隨機分組部分 Rusfertide 手臂中,有 26 人中有 24 人沒有接受放血術 Rusfertide 遇見了主要療效終點 (p=0.0003) 安慰劑 (N=27) Rusfertide (N=26) 0 10 20 30 40 50 60 70 80 69.2% P e e r c e n t o f R e s p o n d r s p=0.0003 n=5 n=18 *Responder 根據方案定義 • 未接受放血術 • 已完成 12 周的治療 • 血液在沒有放血資格的情況下維持控制血細胞比容,其定義為 ▪ 血細胞比容 ≥ 45%,比第 29 周隨機化前血細胞比容值高 ≥ 3% 或 ▪ 血細胞比容 > 48% 或 ▪ 與第 29 周隨機化前血細胞比容值相比,血細胞比容增加 ≥ 5% 高與安慰劑相比,rusfertide arm 的療效顯著*
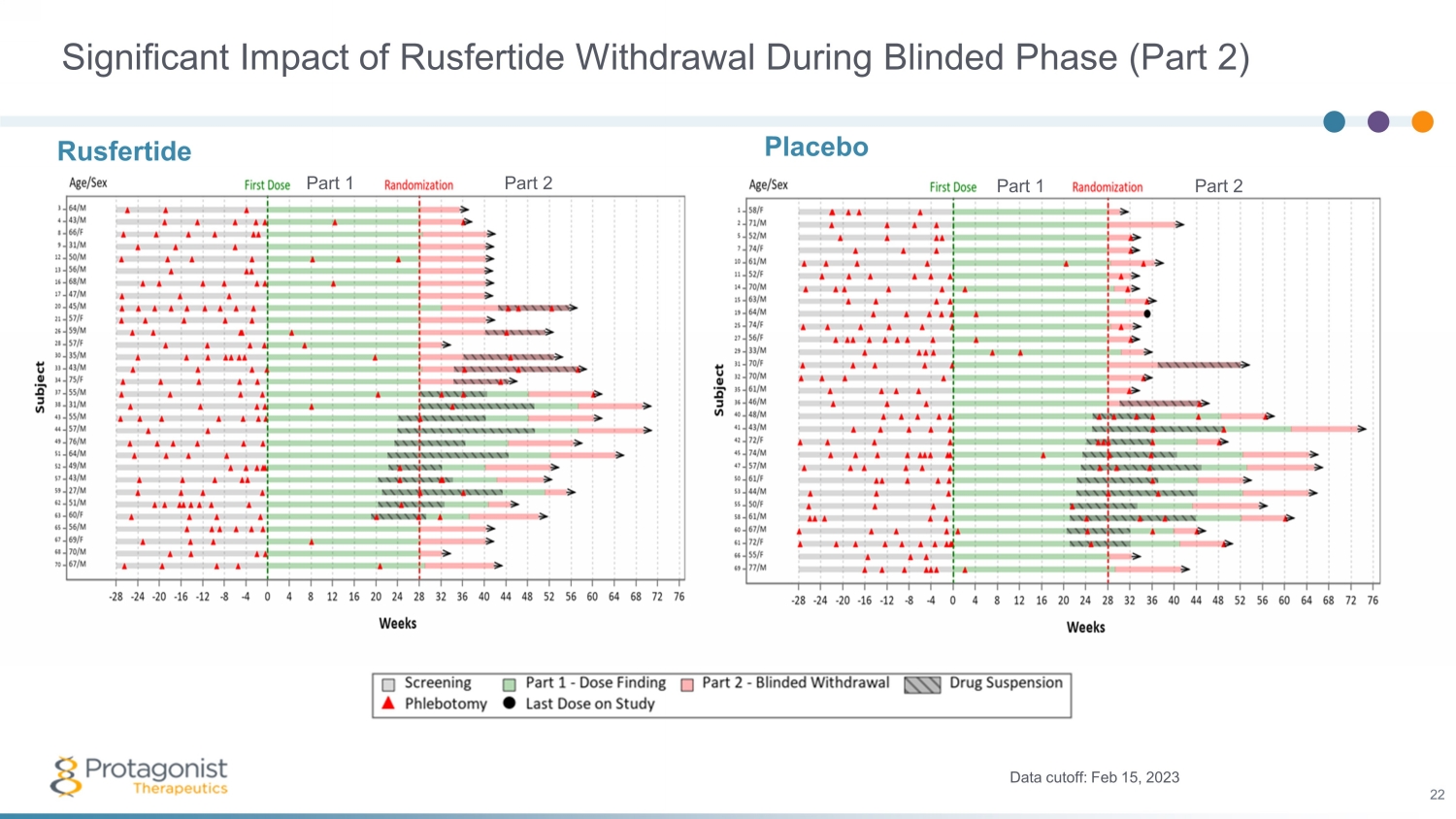
盲期 Rusfertide 戒斷的重大影響(第 2 部分)安慰劑數據截止:2023 年 2 月 15 日 22 Rusfertide 第 1 部分第 2 部分第 1 部分 2 部分
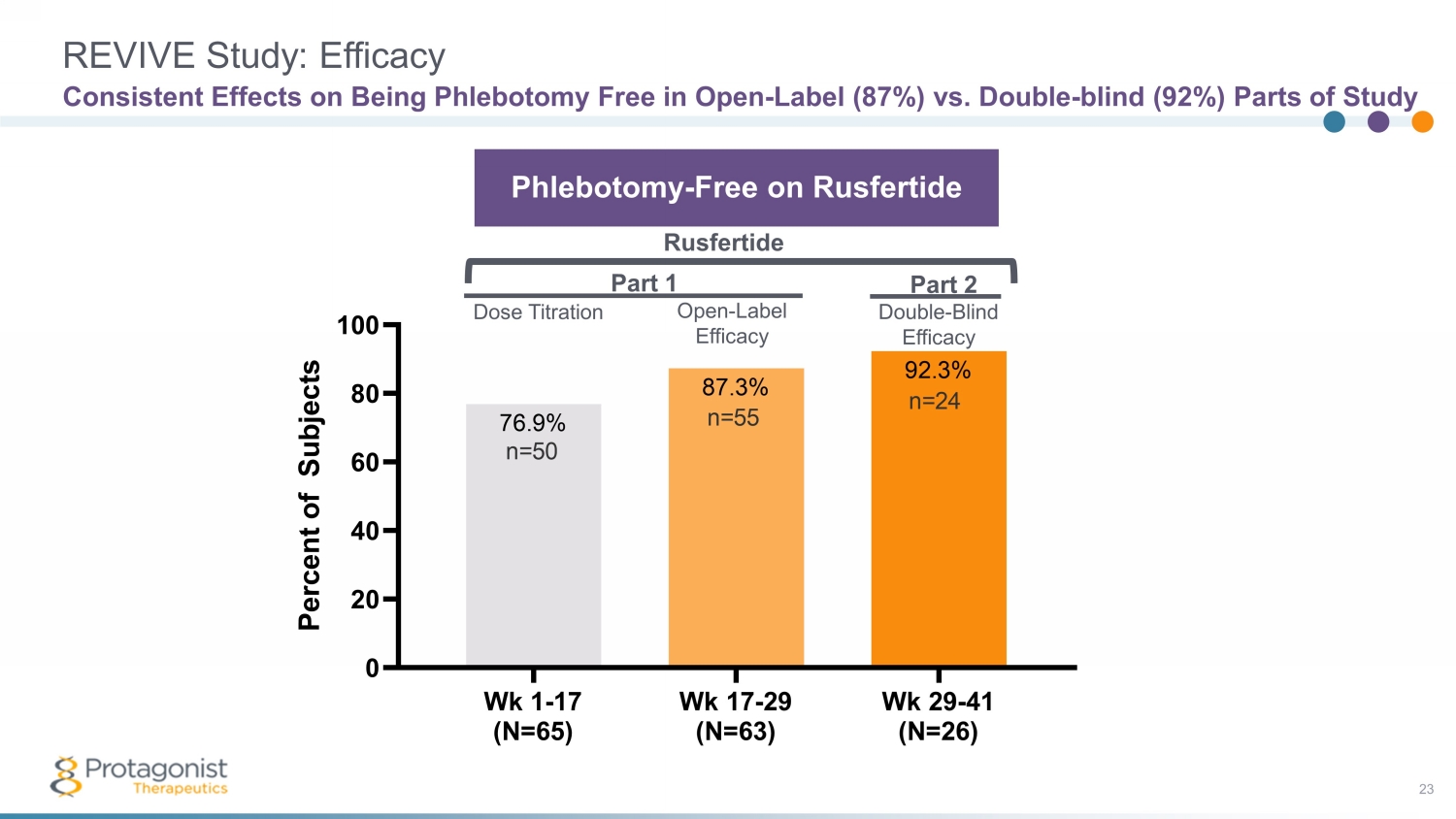
REVIVE 研究:功效 23 對開放式放血術的影響一致 — 標籤 (87%) vs 雙盲 (92%) 研究部分 Wk 1-17 (N=65) Wk 17-29 (N=63) Wk 29-41 (N=26) 0 20 40 60 80 100 92.3% 87.3% 76.9% P e r c e n t o f s u b e c t s n=9% P e r c e n t s 24 n=50 n=55 第 1 部分第 2 部分放血術——在 Rusfertide Rusfertide 劑量滴定上免費開放——標籤功效雙重——盲目功效

REVIVE 研究:持續的療效 24 Rusfertide 與安慰劑相比,在多種結果下顯著延遲了事件發生時間 28 30 32 34 36 38 40 42 0 20 40 60 80 100 Responder Weeks p e r c e t s p
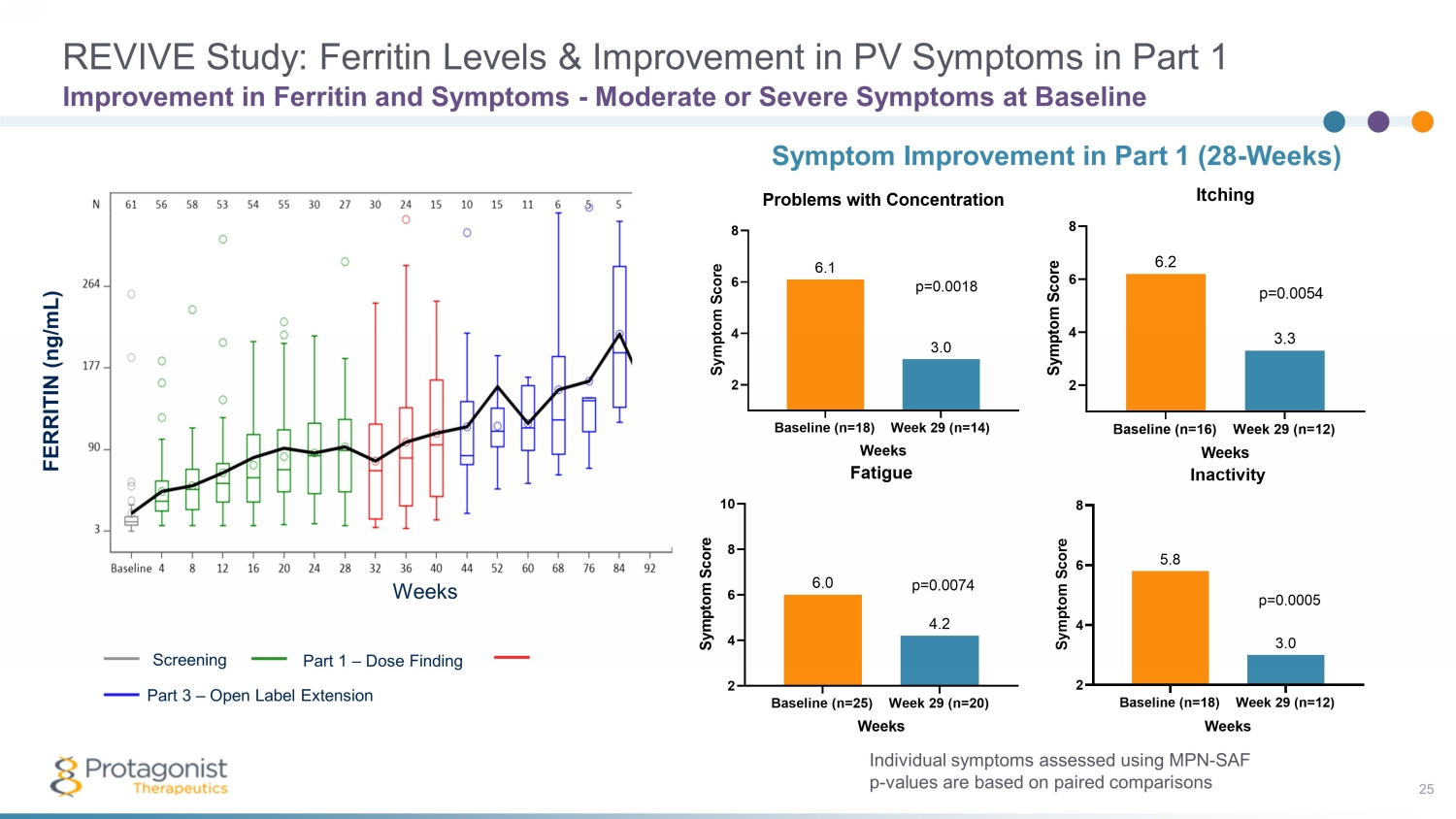
REVIVE 研究:第 1 部分中的鐵蛋白水平和 PV 症狀的改善 25 鐵蛋白和症狀的改善——基線時的中度或重度症狀使用 MPN 評估的個體症狀——SAF p——值基於配對比較第 1 部分(28-周)基線 (n=25) 第 29 周 (n=20) 2 4 6 8 10 4.2 6.0 疲勞周 S y m p t o r e p=0.0 0074 基線 (n=18) 第 29 周 (n=14) 2 4 6 8 3.0 6.1 注意力周的問題 S y m p t o m s c o r e p=0.0018 篩查第 3 部分 — 開放標籤擴展第 1 部分 — 劑量發現周 FERRITIN(納克/毫升)
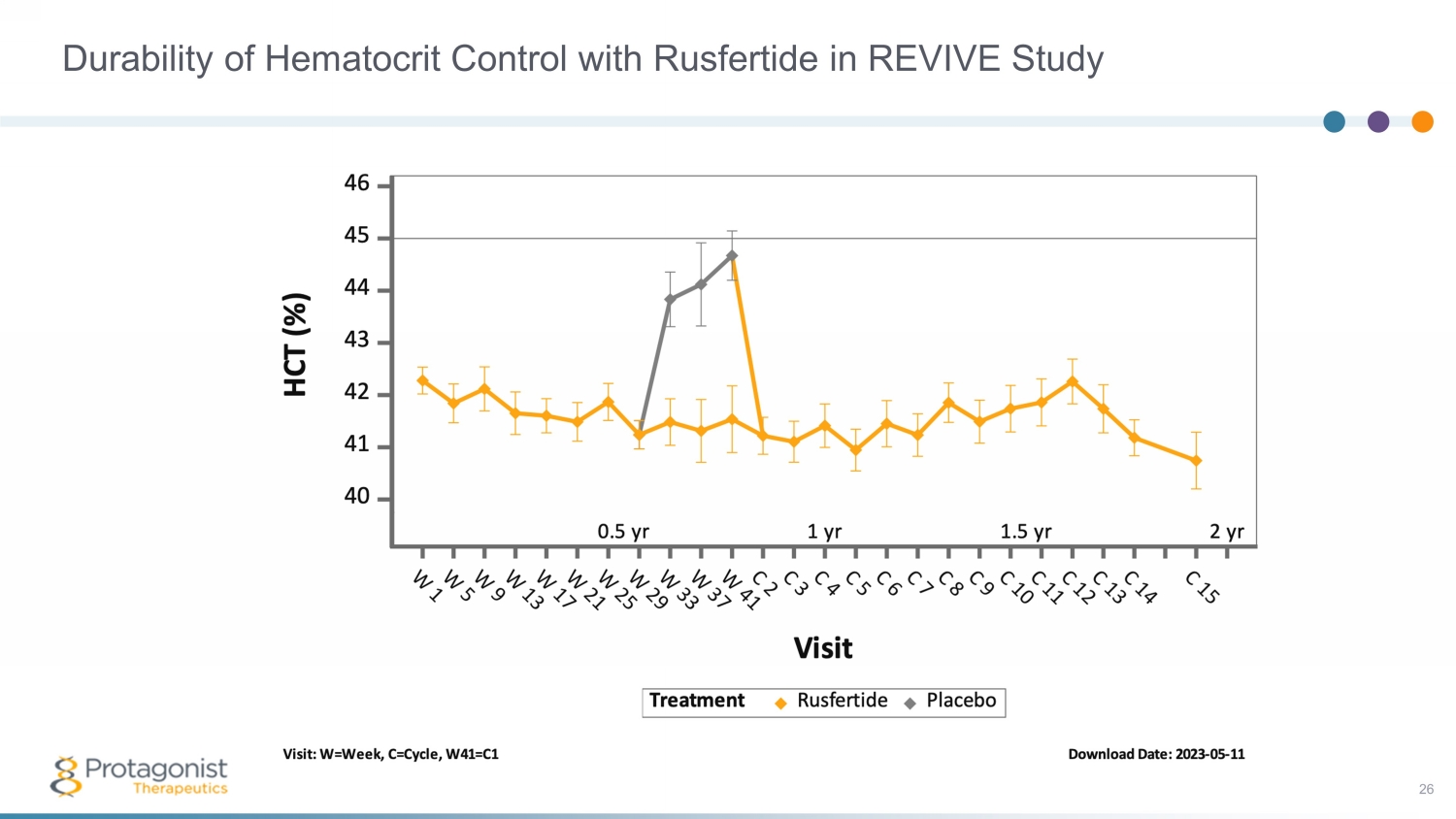
REVIVE 研究 26 中使用 Rusfertide 控制血細胞比容的耐久性
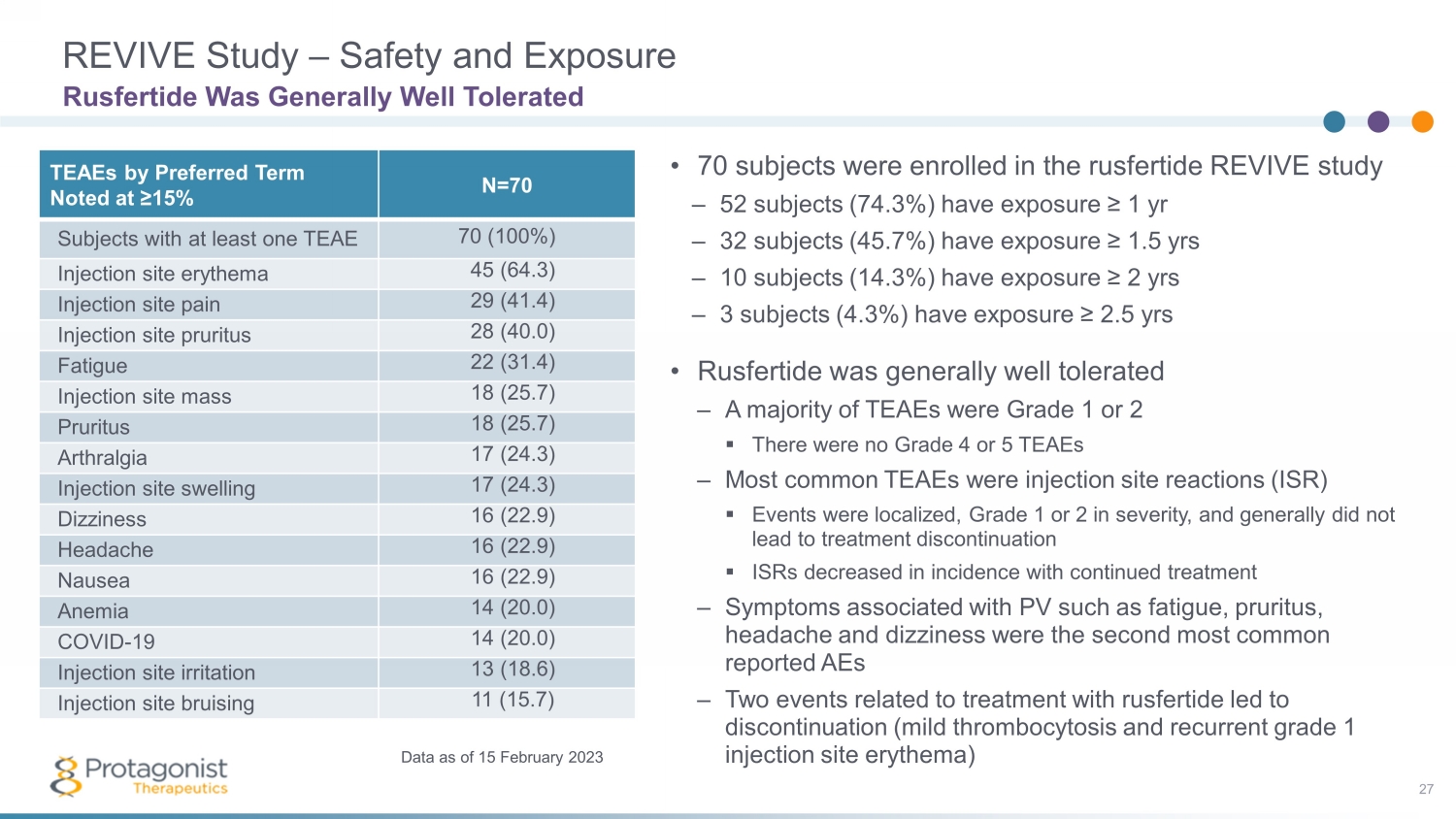
REVIVE 研究 — 安全與暴露 27 • 70 名受試者參加 rusfertide REVIVE 研究 — 52 名受試者(74.3%)的暴露 ≥ 1 年 — 32 名受試者(45.7%)的暴露 ≥ 1.5 年 — 10 名受試者(14.3%)暴露 ≥ 2.5 年 • Rusfertide 總體耐受性良好 — 大多數 TEAE 都是 1 級或 2 級 ▪ 沒有 4 或 5 級 TEAE — 最常見的 TEAE 是注射部位反應 (ISR) ▪ 事件是局部性的,嚴重程度為 1 或 2 級,通常不會導致停藥 ▪ ISR 降低在持續治療的發生率中 — 與 PV 相關的症狀,例如疲勞、瘙癢、頭痛和頭暈,是報告的第二常見的 AE — 兩起與魯斯費肽治療有關的事件導致停藥(輕度血小板增多和復發 1 級注射部位紅斑)Rusfertide 的耐受性總體良好 TEAE 按首選術語表示為 ≥ 15% N=70 受試者至少有一個 TEAE 70 (100%) 注射部位紅斑 45 (66) 4.3) 注射部位疼痛 29 (41.4) 注射部位瘙癢 28 (40.0) 疲勞 22 (31.4) 注射部位質量 18 (25.7)瘙癢 18 (25.7) 關節痛 17 (24.3) 注射部位腫脹 17 (24.3) 頭暈 16 (22.9) 頭痛 16 (22.9) 噁心 16 (22.9) 貧血 14 (20.0) COVID-19 14 (20.0) 注射部位刺激 13 (18.6) 注射部位瘀傷 11 (15.7) 截至 2023 年 2 月 15 日的數據
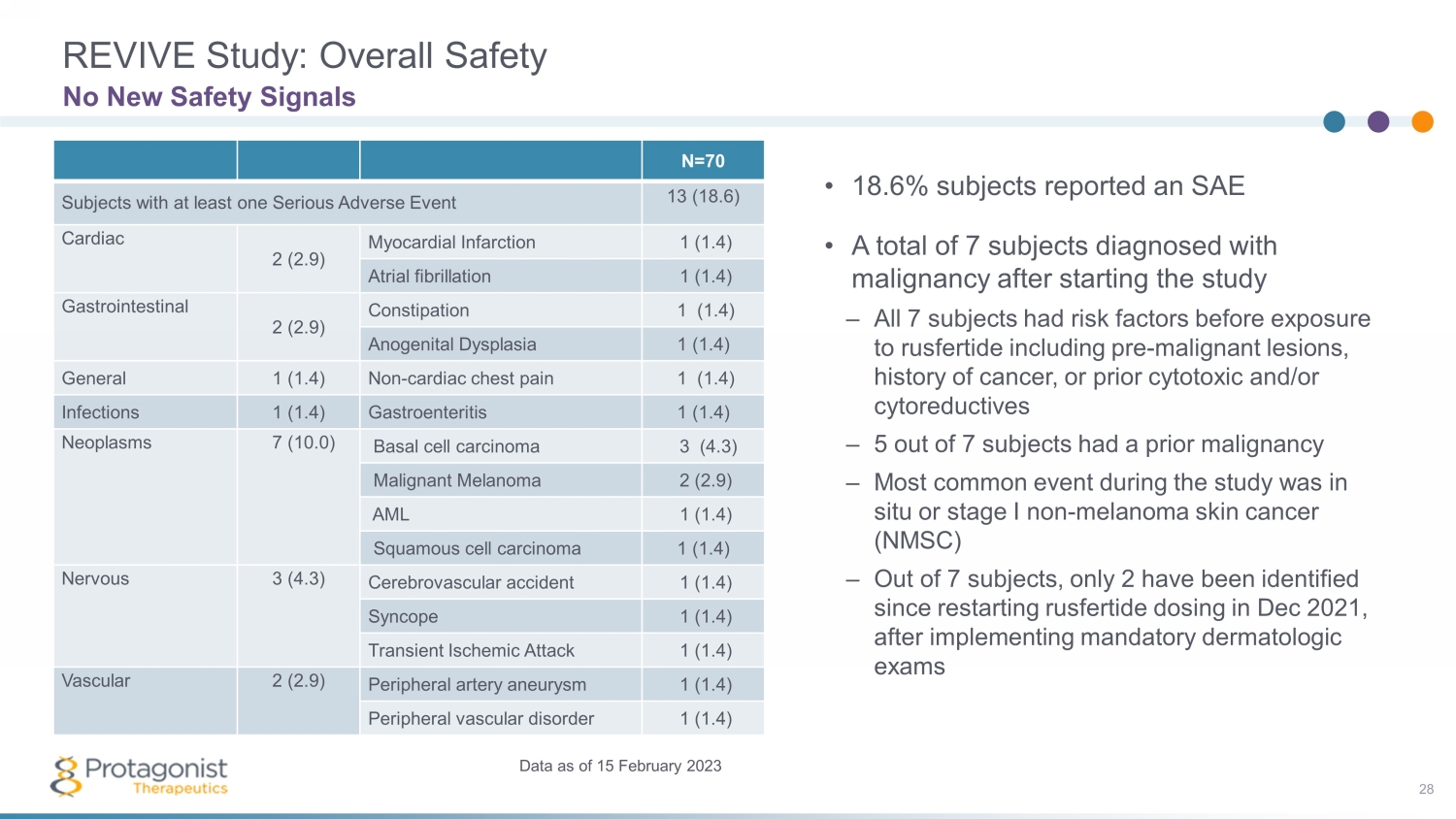
REVIVE 研究:總體安全性 28 截至 2023 年 2 月 15 日沒有新的安全信號數據 • 18.6% 的受試者報告了 SAE • 共有7名受試者在開始研究後被診斷出患有惡性腫瘤——所有 7 名受試者在接觸魯斯費肽之前都有危險因素,包括惡性病變前病變、癌症史或先前的細胞毒性和/或細胞還原藥——研究期間最常見的事件是原位或非黑色素 — 腫瘤皮膚癌 (NMSC) — 在7名受試者中,自重啟rusfertide以來,只有2人被確認在實施強制性皮膚科檢查後,2021 年 12 月給藥 N=70 至少有一次嚴重不良事件的受試者 13 (18.6) 心臟 2 (2.9) 心肌梗死 1 (1.4) 心房顫動 1 (1.4) 胃腸道 2 (2.9) 便祕 1 (1.4) 肛門生殖器發育不良 1 (1.4) 非心臟性胸痛 1 (1.4) 感染 1 (1.4) 胃腸炎 1 (1.4) 1.4) 腫瘤 7 (10.0) 基底細胞癌 3 (4.3) 惡性黑色素瘤 2 (2.9) AML 1 (1.4) 鱗狀細胞癌 1 (1.4) 神經 3 (4.3) 腦血管意外 1 (1.4) 暈厥 1 (1.4) 短暫性缺血發作 1 (1.4) 血管 2 (2.9) 外周動脈瘤 1 (1.4) 外周血管疾病 1 (1.4)

• HCT 快速控制 (48%) 太平洋研究 • 使用或不使用細胞還原劑的 Rusfertide 治療似乎耐受性良好 — 2022 年 12 月在 ASH 上發佈了安全更新;未觀察到新的安全信號 1 • 大約 250 名患者,隨機、安慰劑對照 Ph3 VERIFIDI 研究,以確認療效和安全性 — 正在執行,2024 年第一季度完成註冊 Rusfertide 摘要 29 一項用於治療多細胞的研究性可注射 Hepcidin 模仿物 Themia Vera • 需要經常放血 + 細胞還原劑的 PV 患者已經接受了 rusfertide 治療超過 2 年在REVIVE研究中,受試者基本上沒有放血——快速、持續和持久的血細胞比容控制——在所有類別的患者中都具有強勁的療效——Rusfertide給藥被中斷並導致效果喪失;重新開始恢復治療益處——症狀評分的積極改善——53名患者,1:1 隨機化研究的第二部分完成 1.Pemmaraju、Naveen 等“Revive 中給藥 Rusfertide 後不良事件的亞組分析:對患有真性紅細胞增多症的 Pati ent 的第二階段研究。”Blood 140。補充 1 (2022):6835-6837。
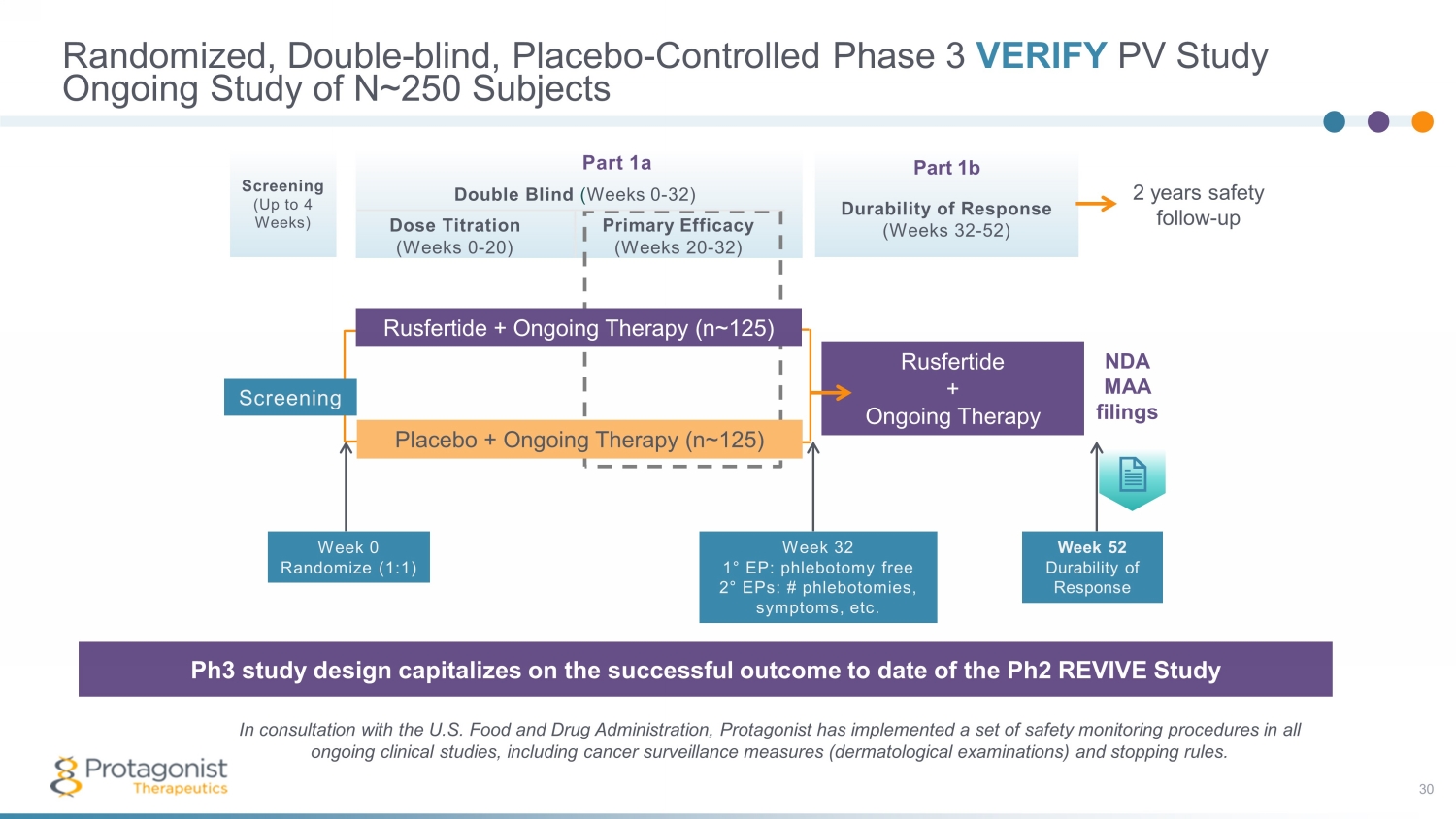
第 1a 部分雙盲(第 0-32 周)劑量滴定主要療效(第 0-20 周)(第 20-32 周)第 1b 部分反應的耐久性(第 32-52 周)篩查(最多 4 周)隨機、雙盲、安慰劑——對照 3 期 VERIVE 研究 30 正在進行的對 N~250 名受試者的研究 3 期研究設計利用了 60 多名患者開放標籤 REVIVE 研究的成功結果 Protaginest 與美國食品藥品監督管理局協商,在所有正在進行的臨牀研究中實施了一套安全監測程序,包括癌症監測措施 (皮膚病學檢查) 和停止規則.Ph3 研究設計利用 Ph2 REVIVE 研究第 32 周 1° EP 迄今為止的成功結果:無放血 2° eP:# 放血、症狀等。安慰劑 + 持續治療 (n~ 125) Rusfertide + 持續治療 Rusfertide + 持續治療 (n~ 125) 第 52 周反應耐久性 NDA 備案第 0 周隨機化 (1:1) 篩查 2 年安全關注-向上
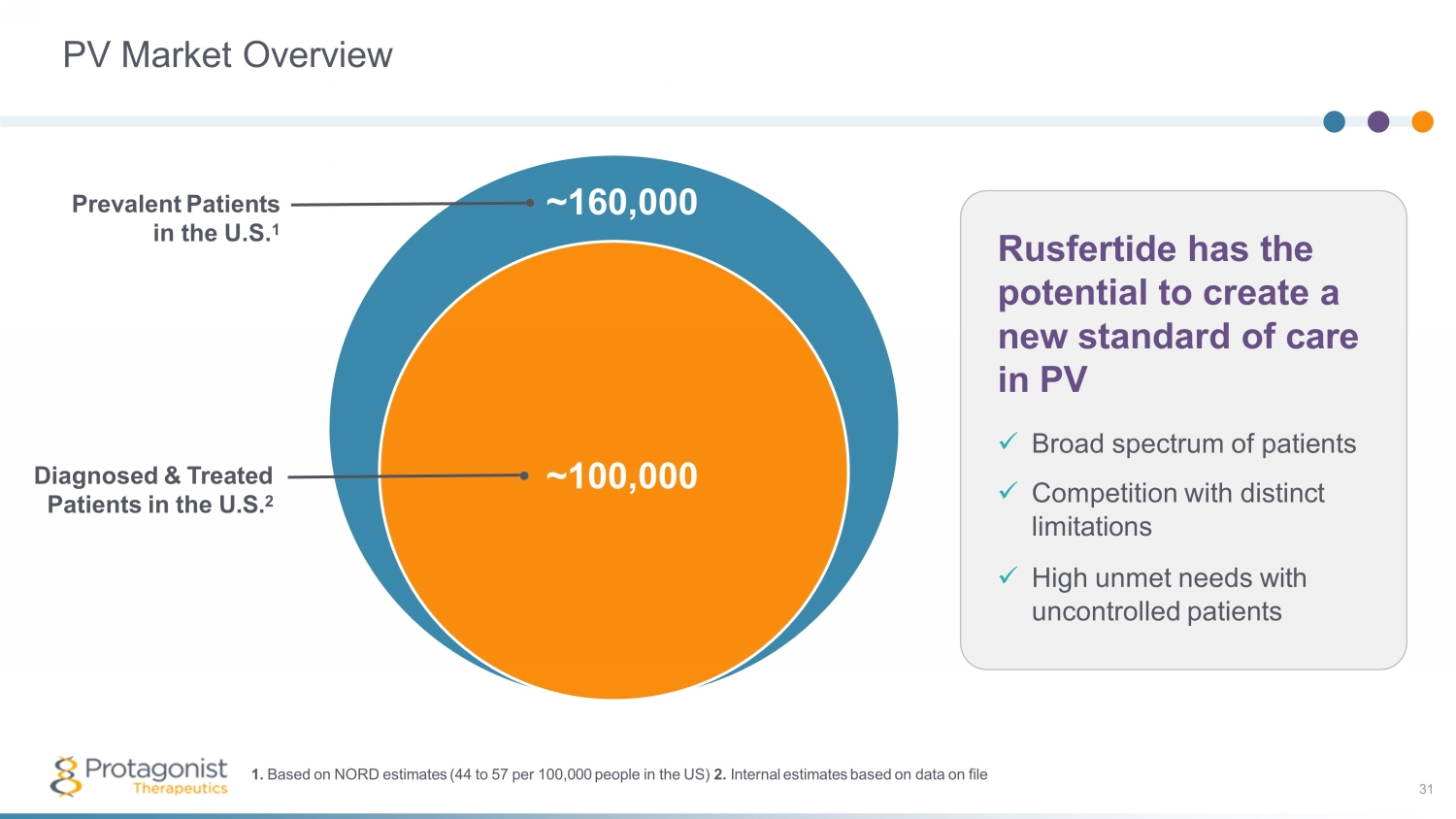
光伏市場概述美國約有100,000名被診斷和治療的患者 2 Rusfertide 有可能在 PV 中創造新的護理標準 x 廣泛的患者 x 具有明顯侷限性的競爭 x 不受控制的患者的高未滿足需求 ~ 160,000 美國流行患者 1 1.根據NORD的估計(美國每10萬人中有44至57人)2.基於文件 31 數據的內部估算
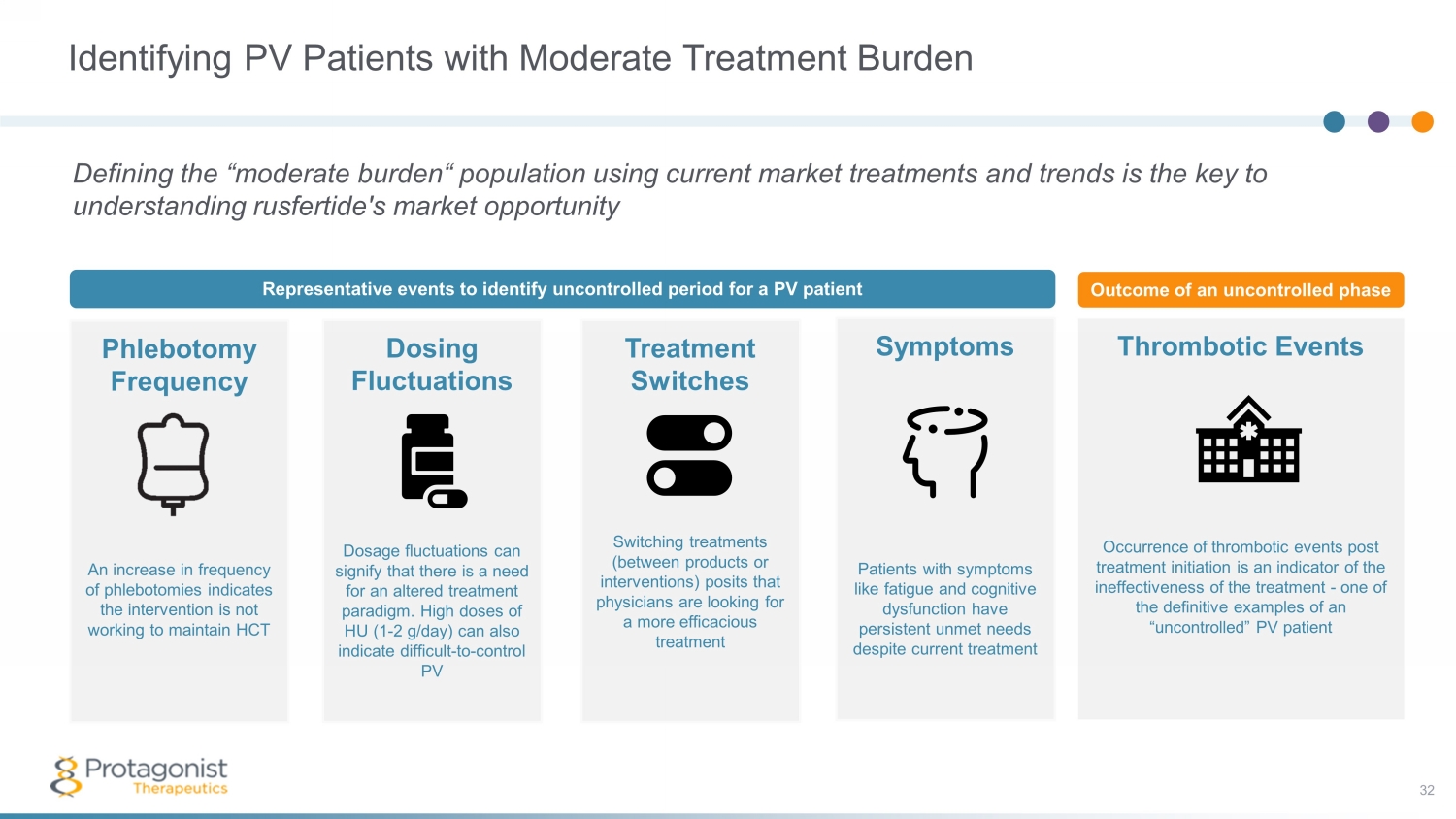
識別治療負擔中等的PV患者使用當前的市場治療方法和趨勢定義 “中等負擔” 人羣是瞭解rusfertide的市場機會放血頻率的關鍵放血頻率的增加表明幹預措施無法維持HCT劑量波動劑量波動可能表明需要改變治療模式。高劑量 HU(1-2 g/day)也可能表明難以控制 PV 治療開關切換治療(在產品或幹預措施之間)假設醫生正在尋找更有效的治療方法血栓形成事件治療開始後出現血栓形成事件表明治療無效,這是 “不受控制” 的光伏患者症狀的權威例子之一儘管當前有治療代表性事件,但患有疲勞和認知功能障礙等症狀的患者的需求一直未得到滿足到識別 PV 患者的不受控制的時期 32 期不受控制的結果

Rusfertide 療法的潛在商業定位是為中等治療負擔的患者提供的 Rusfertide 療法的潛在商業定位低負擔不經常放血 (

JNJ-2113(前身為 PN-235):IL-23 受體拮抗劑牛皮癬和其他伊利諾伊州口服靶向研究療法——23 介導疾病 34

根據強生公司2022年年度報告,主角——伊利諾伊州詹森·奧拉爾——23R Antagonist Collaboration 35 1 Stelara® 在2022年創造了97億美元的銷售額,Tremfya® 在2022年創造了27億美元的銷售額。Ste lara® 和 Tremfaya® 不是 Protaginest — Janssen 合作的一部分。合作概述 — 於 2017 年與 I&I 市場領導者 Janssen Biotech 1 — JNJ-2113(前身為 PN-235)使用主角的專有肽發現平臺共同發現 — 主角完成了臨牀前和第 1 期研究 — Janssen 負責進一步開發和商業化合作經濟學 — 主角有資格獲得高達 8.55 億美元的開發和銷售里程碑及以上——JNJ-2113 和其他合作化合物的分層特許權使用費最近臨牀數據;開發現狀 — 2B 期 FRONTIER 1 數據在 WCD 2023 上發表(2023 年 7 月)— 計劃進行 3 期牛皮癬研究 — 計劃進行 2b 期 UC 研究(2023 年第四季度)JNJ-2113 潛在的最佳口服藥物 — 有可能將伊利諾伊州23市場擴展到口服療法

伊利諾伊州-23R 拮抗劑市場概述 36 億美元的口服藥物市場機會伊利諾伊州-23R 拮抗劑的巨大市場潛力——相關適應症包括牛皮癬、銀屑病關節炎、IBD(潰瘍性結腸炎、克羅恩病)— 牛皮癬患病率 = 1.25億美國患者 1(800 萬美國 1)— 30% 患銀屑病關節炎 — 牛皮癬主要市場銷售額預計增長至 132 億美元(2020 年)2 — 預計牛皮癬主要市場銷售額將增長至 25.32 億美元 2030年B 2 — IBD(2020)的主要市場銷售額為142億美元 2 — 預計到2029年將增長至249億美元 2 口服藥劑預計將為市場增長做出貢獻 —在接受現行護理標準治療的患者中,有很大一部分患者——25%的牛皮癬患者接受了生物製劑治療 1 — 儘管療效很強,但生物製劑與安全問題、反應喪失、給藥不便有關,這凸顯了安全有效的口服選擇的必要性 3、4 1。評估 2022 年製藥公司;2.2020 年 GlobalData;3.Levin、E. 等人J Dermatol Treat;25 (1);78-82;4.Piragine E. 等人J Clin Med 2022;11 (6);1506

牛皮癬治療概述 JNJ-2113 是潛在的第一、最佳、唯一的同類候選藥物 • 生物靶標:IL-23 受體 • 藥物遞送:口服 • 化學模式:肽可注射 IL-23 拮抗劑在牛皮癬中表現出強大的療效和安全性 JNJ-2113(前身為 PN-235)口服 IL-23 受體拮抗劑 • 口服 IL-23 途徑抑制劑有可能將市場大幅擴大到可注射生物製劑之外 • Pan-JAK 和選擇性 JAK1/3 抑制劑有 “黑匣子警告” 已在 IBD 中獲批,但由於風險原因未獲準用於牛皮癬-好處 • TYK2 抑制劑 Sotyktu 最近被批准用於牛皮癬 • 最有效的口服藥物,但不如 IL-23 和 IL-17 拮抗劑那麼有效 • 沒有像其他 JAK 抑制劑那樣出現黑匣子警告,但長期安全經驗有限 • TYK2 抑制劑是小分子方法,進入門檻低,有多個競爭對手 • BMS/ Sotyktu,takeda/tak279 37

JNJ-2113 (PN-235) 38 多項臨牀研究正在進行和計劃中 • 計劃中 — 中度至重度斑塊狀牛皮癬的 3 期研究 — 潰瘍性結腸炎的 2b 期研究 • FRONTIER 1 — 255 名患者 2b 期安慰劑——中度至重度斑塊狀牛皮癬的對照研究 — 口服片劑量,qd and bid — Topline 數據摘要;2023 年 3 月 7 日 • FRONTIER 2 — 長期延期研究;正在進行中 • 峯會 — 90 名患者 2 期安慰劑——中度至重度斑塊狀牛皮癬的對照研究 — 每日口服一次延緩釋放片劑 — 4 月完成2023 年 10 月 10 日 • 其他 — 針對健康志願者的第一階段研究;已完成(由 Protagonist 撰寫)— NCT05062200:針對健康日本和中國參與者的第一期研究;已完成 — NCT05703841:針對健康中國成年參與者的第一期研究;已完成

JNJ-2113 特徵 • 高效(個位數 picomolar)口服 IL-23R 拮抗劑:— >1000 — 與第一代候選藥物(PBMC、phospho-STAT3 測定)相比更強或更高的靶標親和力——23 mAB • 口服穩定性高:體外穩定性:>24 小時糞便(人類、cyno 和大鼠)體內穩定性:> cynos 24 小時後糞便恢復 25% • 臨牀前驗證-概念:— 在伊利諾伊州口服給藥實現臨牀前 PoC-23-誘導的大鼠耳部皮膚炎症模型 — 抑制與全身 IL 類似-23 mAB • NHV 的 1 期研究:— 抑制 IL — 23 種與批准的 IL 相當的途徑相關生物標誌物——23 個 maB 臨牀前、1 期和 2b 期數據支持強有力的臨牀開發計劃 • 牛皮癬的 2b 期 FRONTIER1 研究:— 牛皮癬的最佳口服藥物的潛力 39 ISID — Fourie A 等全系統靶向 IL-23 途徑的同類口服肽。摘要在國際皮膚病學調查學會上發表;2023 年 5 月 WCD — Bissonnette R 等人口服 JNJ — 77242113 治療中度至重度斑塊狀牛皮癬的第二階段、隨機、安慰劑對照、劑量範圍研究:FRONTIER 1.最新摘要在世界皮膚病學大會上發表;2023 年 7 月
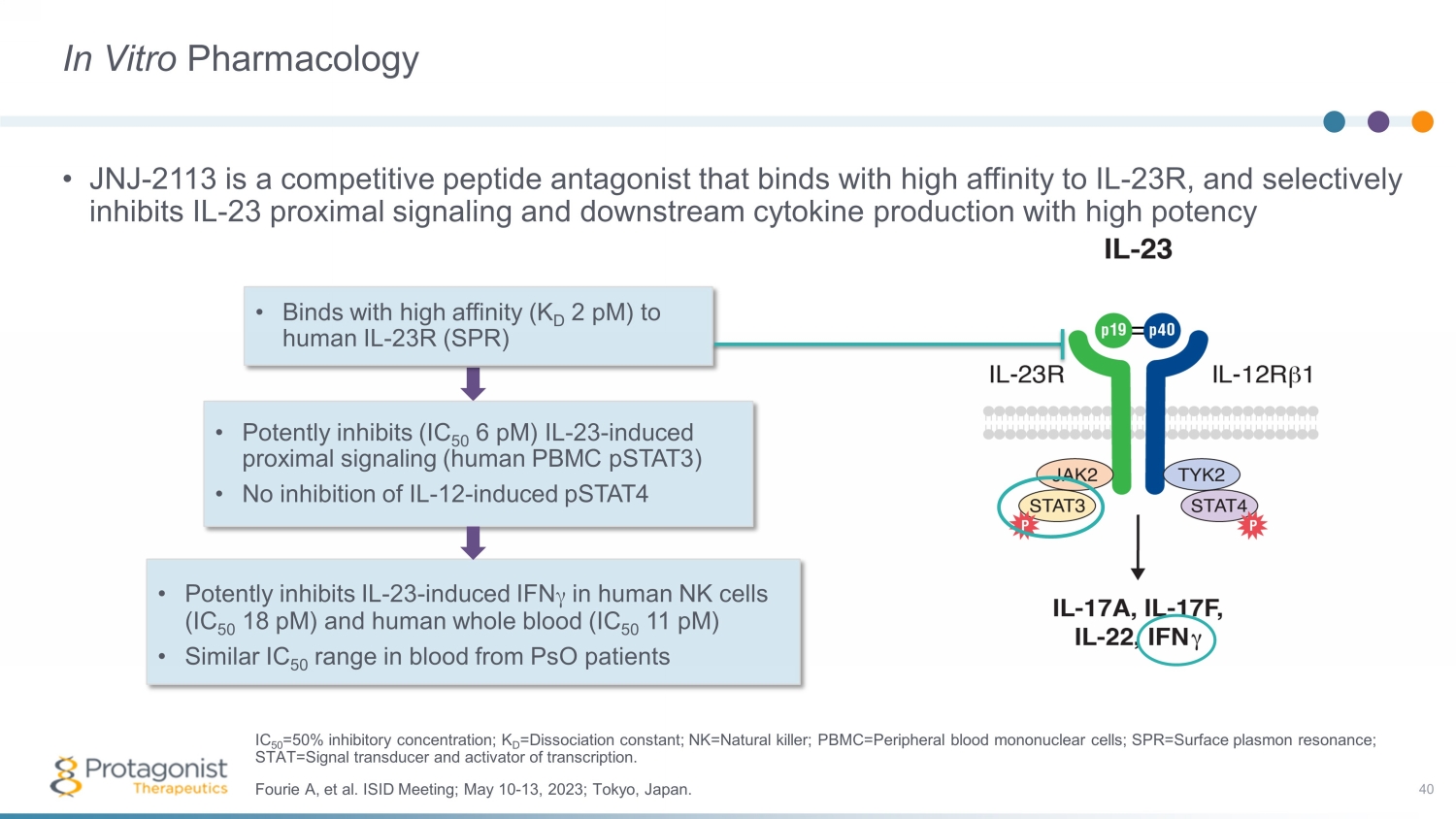
體外藥理學 • JNJ-2113 是一種競爭性肽拮抗劑,它與 IL-23R 具有高親和力,選擇性地抑制 IL-23 近端信號傳導和下游細胞因子的產生 40 • 與人類 IL-23R (SPR) 的高親和力 (K D 2 pM) 結合 • 有效抑制 (IC 50 6 pM) IL-23-誘導的近端信號傳導(人類 PBR)mc pstat3) • 不抑制 IL-12-誘導的 pstat4 • 有效抑制人類 NK 細胞 (IC 50 18 pM) 和人類全血 (IC 50 11 pM) 中的 IL-23-誘導的 IFN γ• 血液中的類似 IC 50 範圍psO 患者 Fourie A 等人ISID 會議;2023 年 5 月 10 日至 13 日;日本東京。IC 50 = 50% 抑制濃度;K D =解離常數;nk=自然殺手;pbmc=外周血單核細胞;spr=表面等離子體共振;stat=信號換能器和轉錄激活劑。

口服劑量 JNJ-2113 可減輕大鼠 TNBS 誘發的結腸炎模型的體重減輕和結腸炎症 • 從 0.1 至 0.3 mg/kg/天的劑量開始出現統計學上的顯著影響 • 儘管血漿和皮膚中的暴露量遠低於胃腸道組織,但 JNJ-2113 的絕佳效力表明胃腸道之外還有全身活性 41 Fourie A 等ISID 會議;2023 年 5 月 10 日至 13 日;日本東京。gi=胃腸道;TNBS= 三硝基苯磺酸。ns=不重要,**p
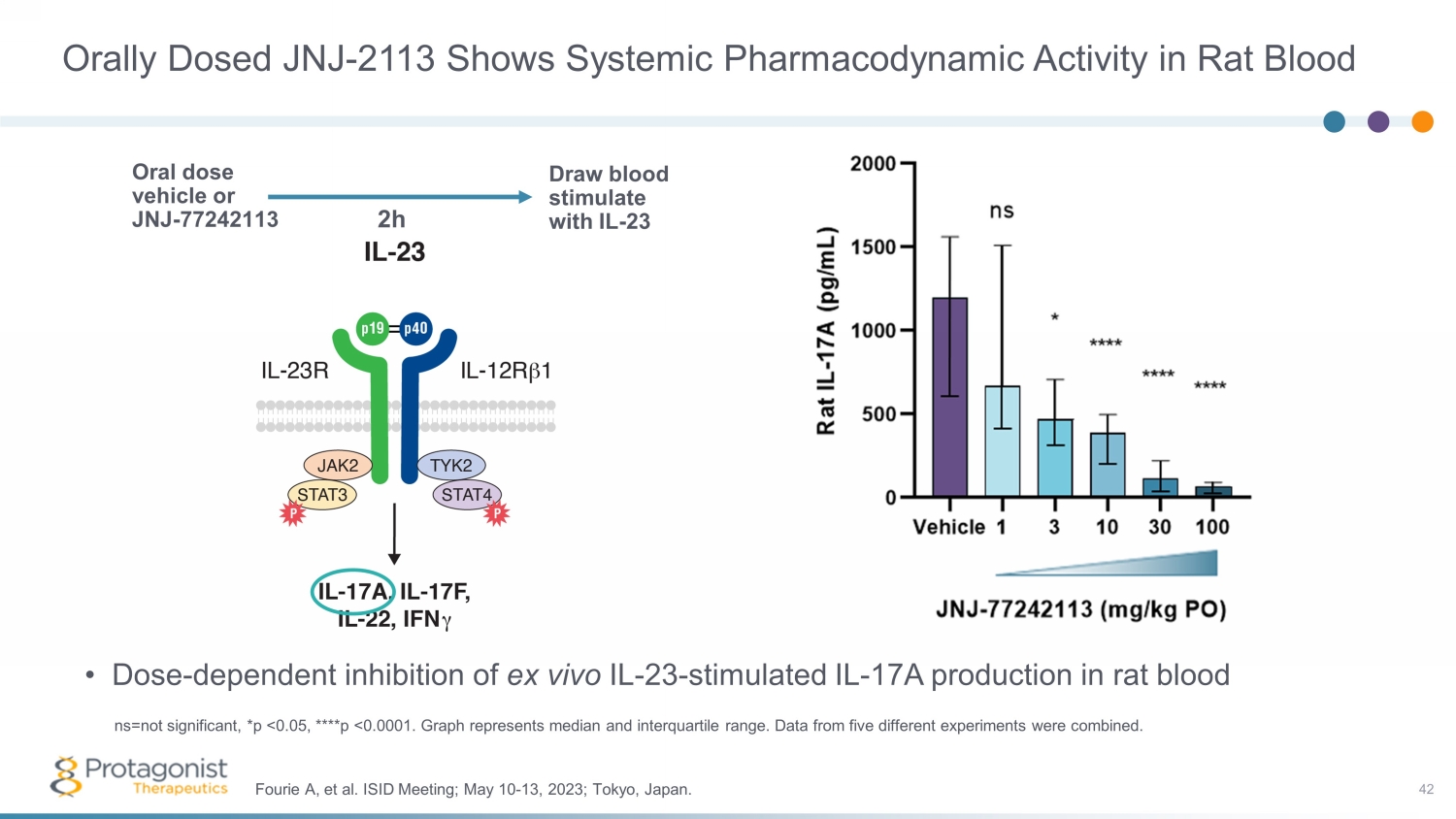
口服劑量 JNJ-2113 在大鼠血液中顯示出全身藥效學活性 • 劑量依賴性抑制體外 IL-23 — 刺激大鼠血中 IL-17A 的產生 42 Fourie A 等ISID 會議;2023 年 5 月 10 日至 13 日;日本東京。ns=不重要,*p
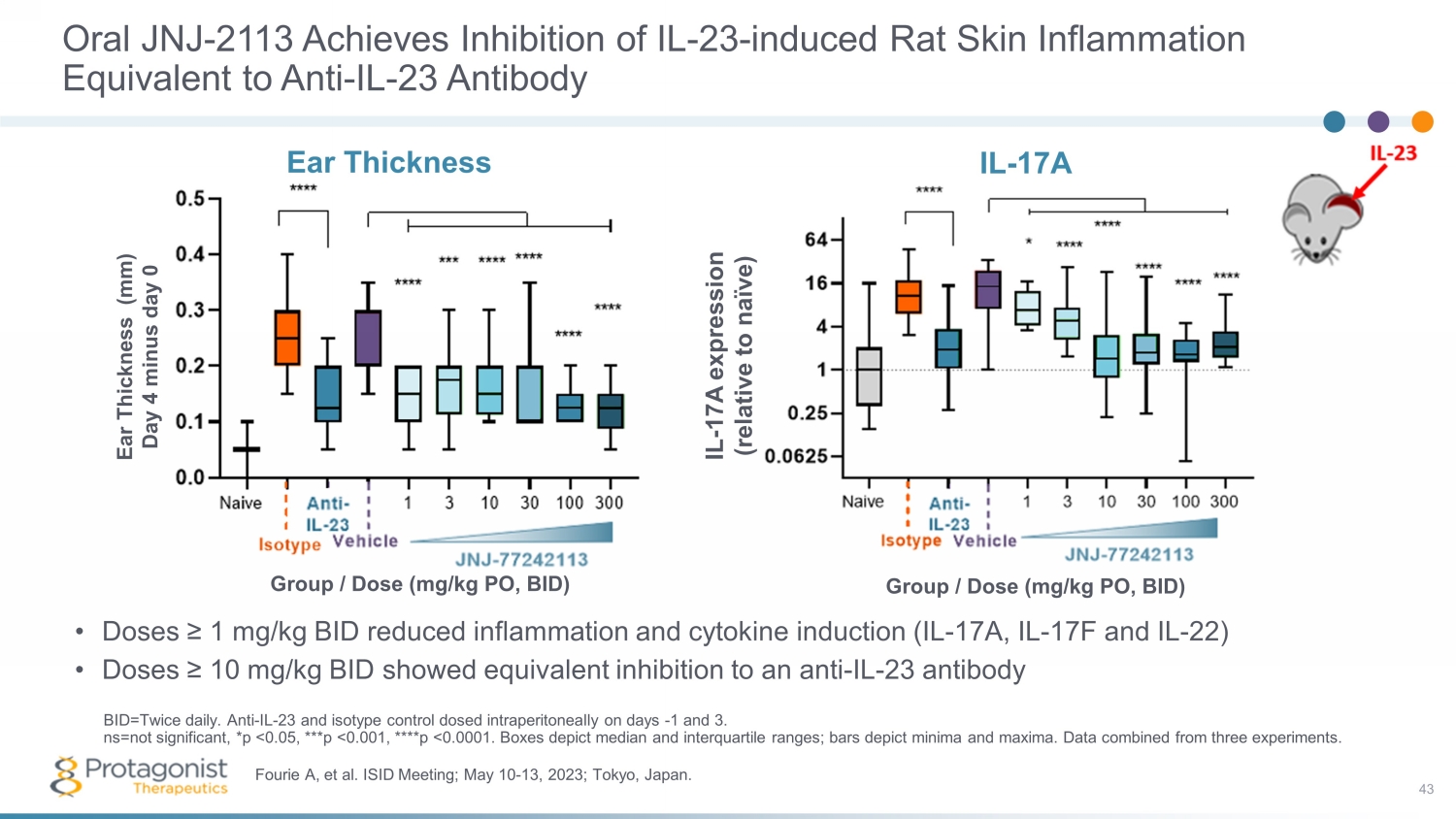
Oral JNJ-2113 抑制 IL-23 誘發的大鼠皮膚炎症相當於抗 IL-23 抗體 • 劑量 ≥ 1 mg/kg BID 可減少炎症和細胞因子誘導(IL-17A、IL-17F 和 IL-22)• 劑量 ≥ 10 mg/kg BID 顯示出與抗 IL-23 抗體 43 Fourie A 等同的抑制作用ISID 會議;2023 年 5 月 10 日至 13 日;日本東京。出價=每天兩次。Anti-IL-23 和同型對照在第 1 天和第 3 天腹腔內給藥。ns=不顯著,*p
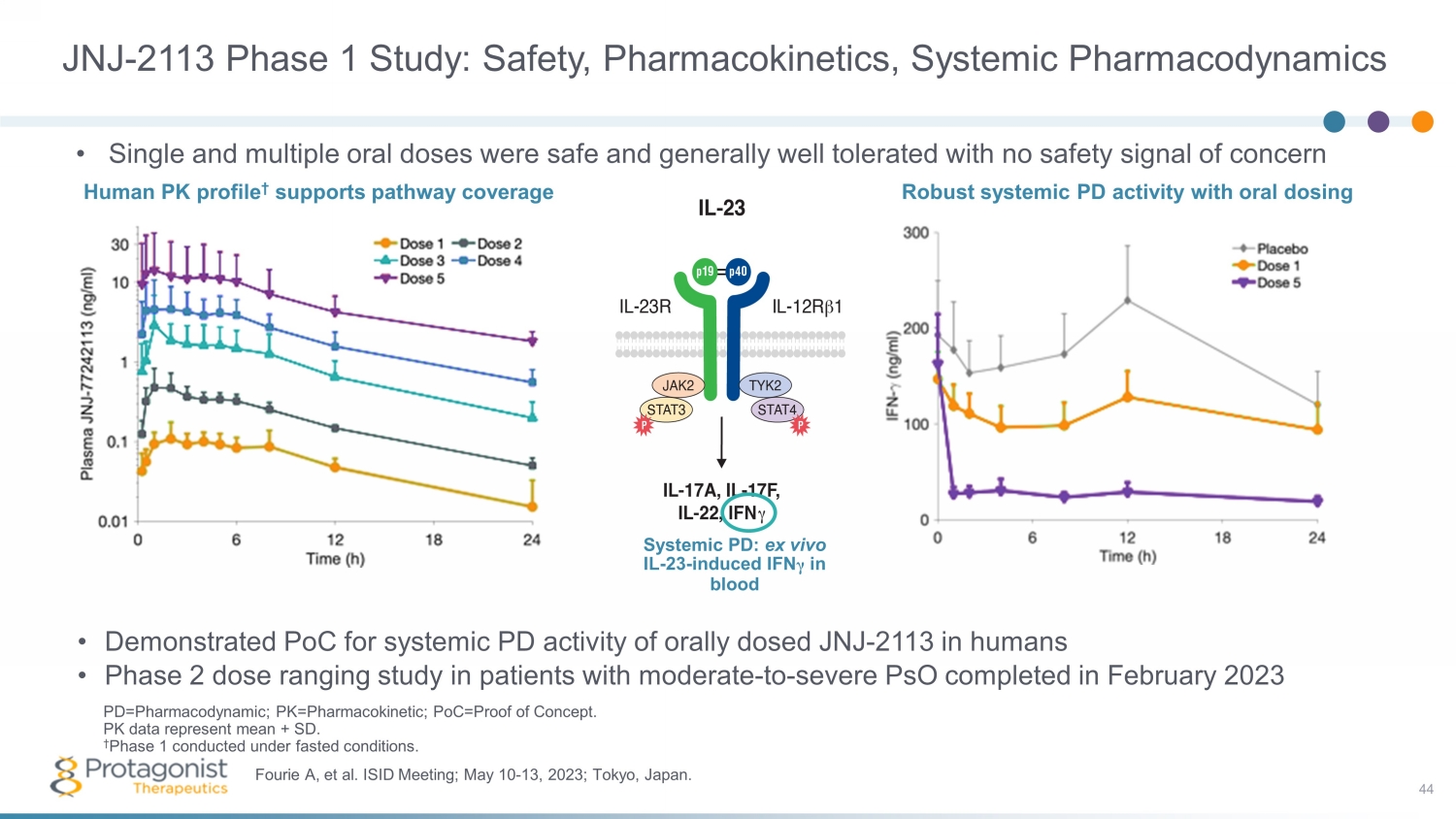
JNJ-2113 第 1 期研究:安全性、藥代動力學、全身藥效學 • 證實 PoC 可對人類口服 JNJ-2113 的全身 PoC 活性 • 針對中度至重度 psO 患者的第 2 期劑量範圍研究於 2023 年 2 月完成 44 Fourie A 等人ISID 會議;2023 年 5 月 10 日至 13 日;日本東京。pd=Pharmacydynamic;pk=Pharmacokinetic;poc=概念驗證。PK 數據表示平均值 + SD。† 第 1 階段在禁食條件下進行。• 單次和多次口服劑量是安全的,總體耐受性良好,沒有令人擔憂的安全信號 Human PK profile † 支持通路覆蓋全身 P D:體外 IL-23 — 在血液中誘導的 IFN γ通過口服給藥強勁的全身 PD 活性
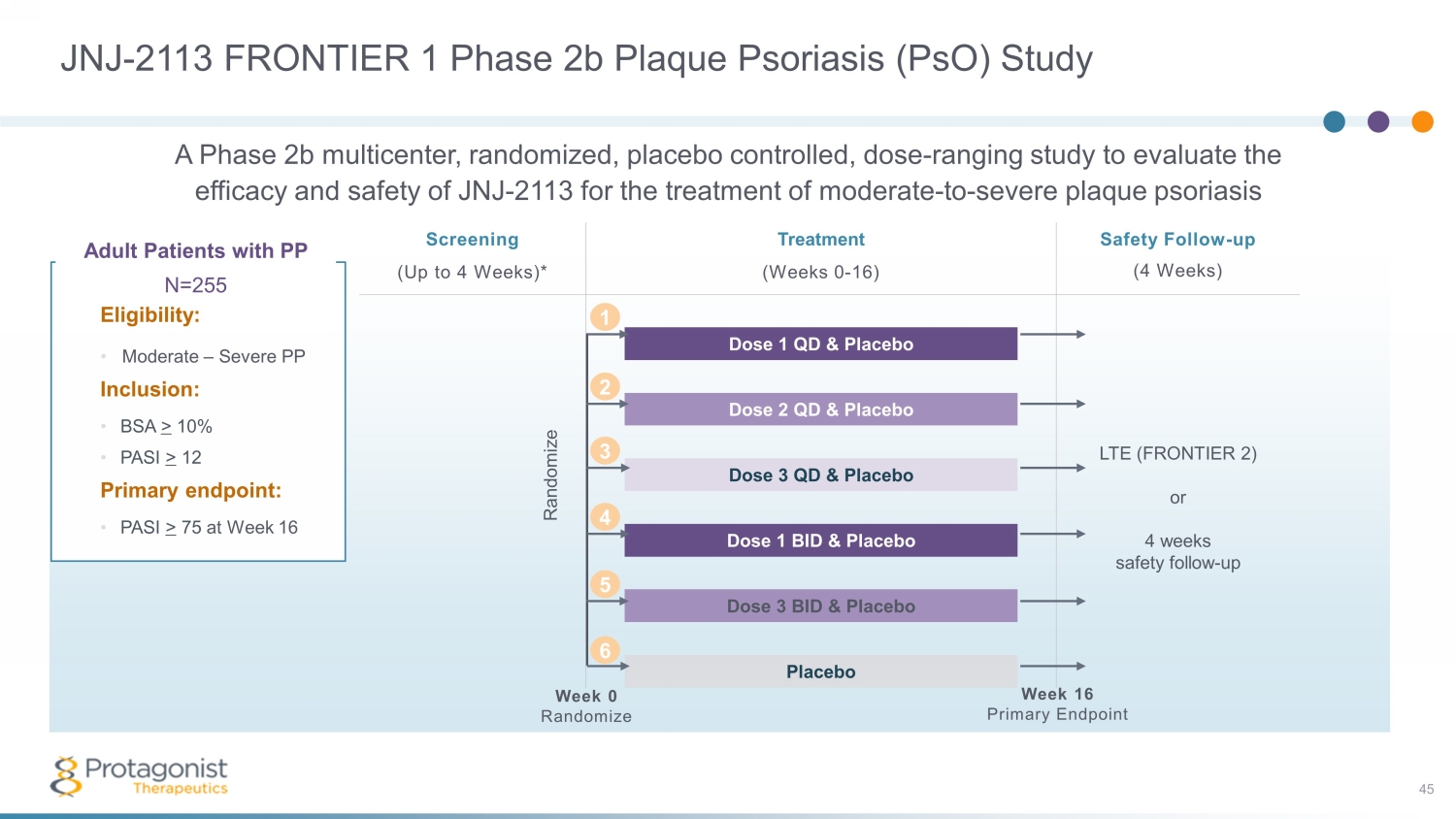
JNJ-2113 FRONTIER 1 期 2b 斑塊狀牛皮癬 (psO) 研究 45 名具有 PP N=255 資格的成年患者:• 中度 — 重度 PP 包含:• BSA > 10% • PASI > 12 主要終點:• 第 16 周篩查治療安全隨訪(4 周)*(第 0-16 周)劑量 1 QD 和安慰劑 LTE(FRONTIER 2)或 4 周安全隨訪-up dose 2 QD 和安慰劑劑量 3 QD 和安慰劑劑量 1 BID 和安慰劑劑量 3 BID 和安慰劑安慰劑第 0 周隨機化第 16 周主要終點隨機化 2 1 3 4 5 6 A phase 2b 多中心、隨機、安慰劑對照、劑量-評估JNJ-2113治療中度至重度斑塊狀牛皮癬的療效和安全性的範圍研究
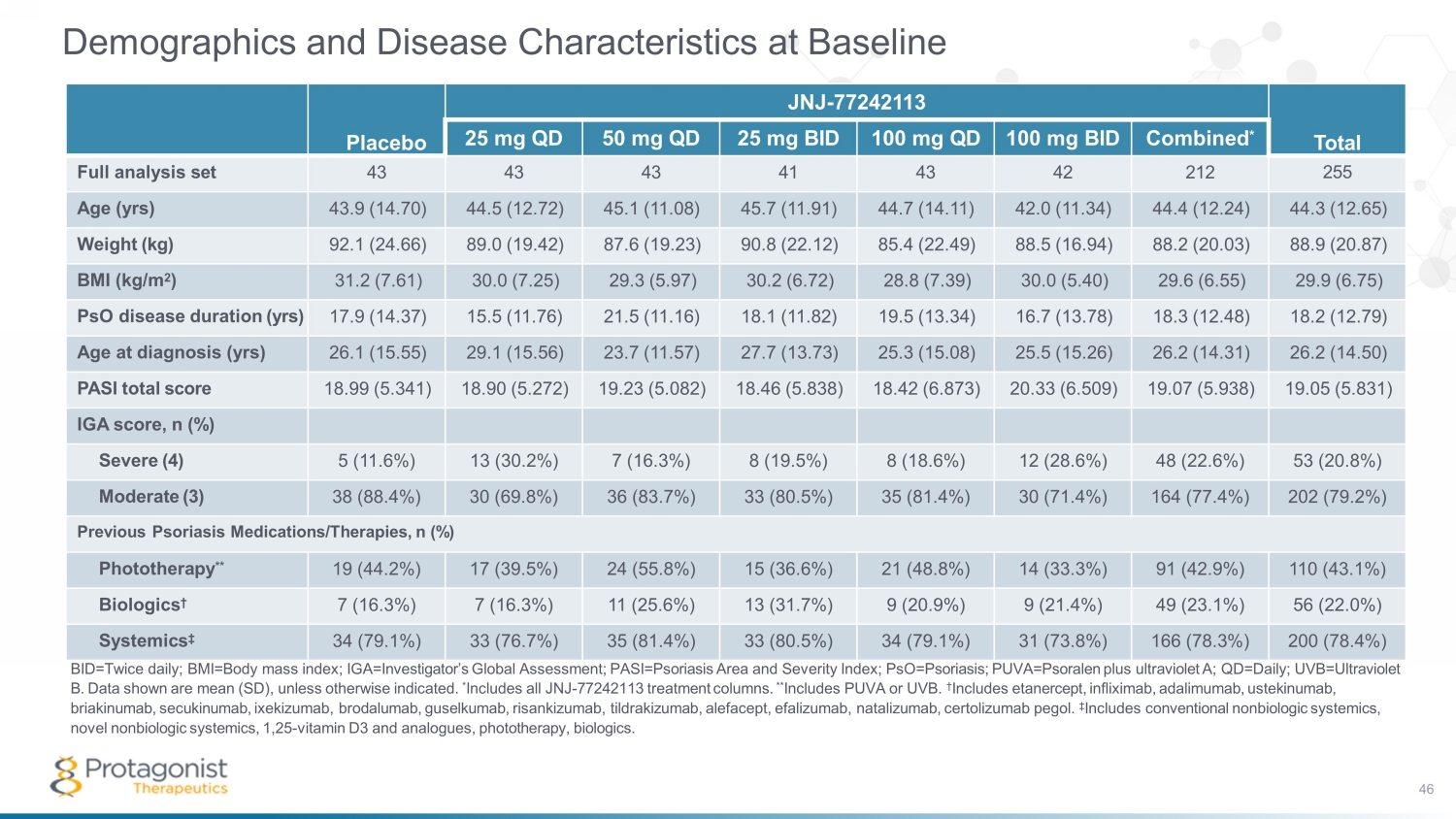
基線的人口統計學和疾病特徵 bid=每天兩次;bmi=體重指數;IGA=調查員的全球評估;pasi=牛皮癬面積和嚴重程度指數;pso=牛皮癬;puva=psoralen 加紫外線 A;qd=Daily;uvb=紫外線 B。* 包括所有 JNJ-77242113 治療專欄。** 包括 PUVA 或 UVB。† 包括依那西普、英夫利昔單抗、阿達木單抗、ustekinumab、briakinumab、secukinumab、ixekizumab、brodalumab、guselkumab、risankizumab、tildrakizumab、alefacept、efalizumab、natalizumab、certolizumab pegol。‡ 包括傳統的非生物系統藥、新型的非生物系統藥、1,25-維生素 D3 和類似物、光療、生物製劑。安慰劑 JNJ-77242113 總計 25 mg QD 50 mg QD 25 mg BID 100 mg QD 100 mg QD 100 mg BID 合併 * 完整分析套裝 43 43 43 42 212 255 年齡 43.9 (14.70) 44.5 (12.72) 45.1 (11.11) 42.0 (11.34) 44.3 (12.65) 重量 (kg) 92.1 (24.66) 89.0 (19.42) 87.6 (19.23) 90.8 (22.12) 85.4 (22.49) 88.5 (16.94) 88.2 (20.03) 88.9 (20.87) 體重指數 (kg/m 2) 31.2 (7.61) 30.0 (7.25) 29.3 (5.97) 30.2 (6.72) 28.8 (7.39) 30.0 (5.40)) 29.6 (6.55) 29.9 (6.75) psO 疾病持續時間 (年) 17.9 (14.37) 15.5 (11.76) 21.5 (11.16) 18.1 (11.82) 19.5 (13.34) 16.7(13.78) 18.3 (12.48) 18.2 (12.79) 診斷年齡 (年) 26.1 (15.55) 29.1 (15.56) 23.7 (11.57) 27.7 (13.73) 25.3 (15.08) 25.5 (15.26) 26.2 (14.50) PASI 總分 18.99 (5.341) 18.90 (5.272) 19.23 (5.082)) 18.46 (5.838) 18.42 (6.873) 20.33 (6.509) 19.07 (5.938) 19.05 (5.831) IGA 分數,n (%) 嚴重 (4) 5 (11.6%) 13 (30.2%) 7 (16.3%) 8 (18.6%) 12 (28.6%) 48 (22.6%) 53 (20.8%) 中等 (3) 38 (88%) 8.4%) 30 (69.8%) 36 (83.7%) 33 (80.5%) 35 (81.4%) 30 (71.4%) 164 (77.4%) 202 (79.2%) 以前的牛皮癬藥物/療法,n (%) 光療**19 (44.2%) 17 (39.5%) 24 (55.8%) 15 (36.6%) 21 (48.8%) 14 (33.3%) 91 (42.9%) 110 (43.1%) 生物製劑 † 7 (16.3%) 7 (16.3%) 11 (25.6%) 13 (31.7%) 9 (21.4%) 99 (23.1%) 56 (22.0%) Systemics ‡ 34 (79.1%) 33 (76.7%) 35 (81.4%) 33 (80.5%) 34 (79.1%) 31 (73.8%) 166 (78.3%) 200 (78.4%) 46
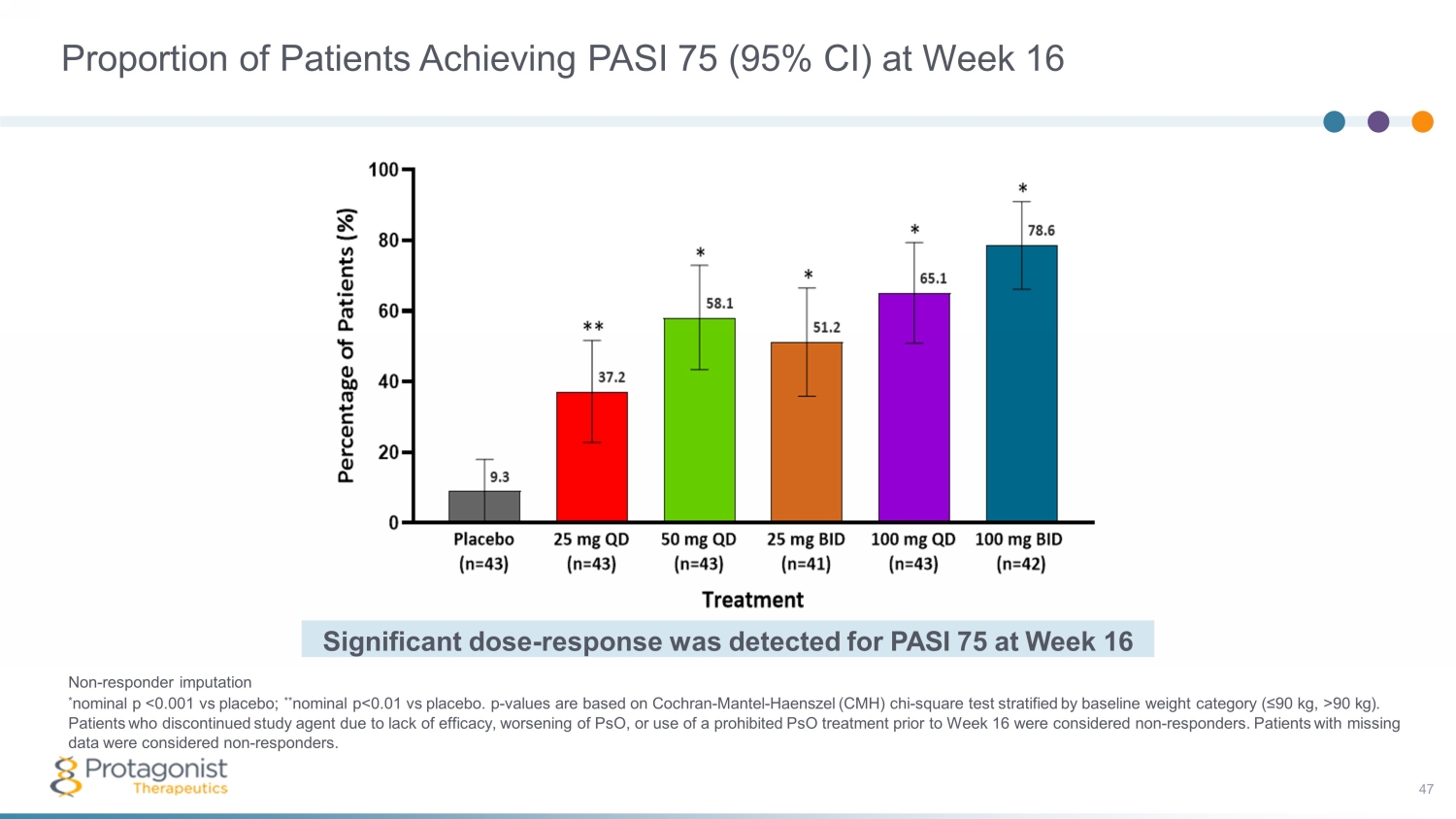
非響應者估算* 標稱值 p 90 kg)。在第16周之前因療效不足、psO惡化或使用違禁的psO治療而停用研究藥物的患者被視為非反應者。數據缺失的患者被視為非反應者。劑量顯著——在第 16 周檢測到 PASI 75 的反應在第 16 周達到 PASI 75(95% 置信區間)的患者比例 47
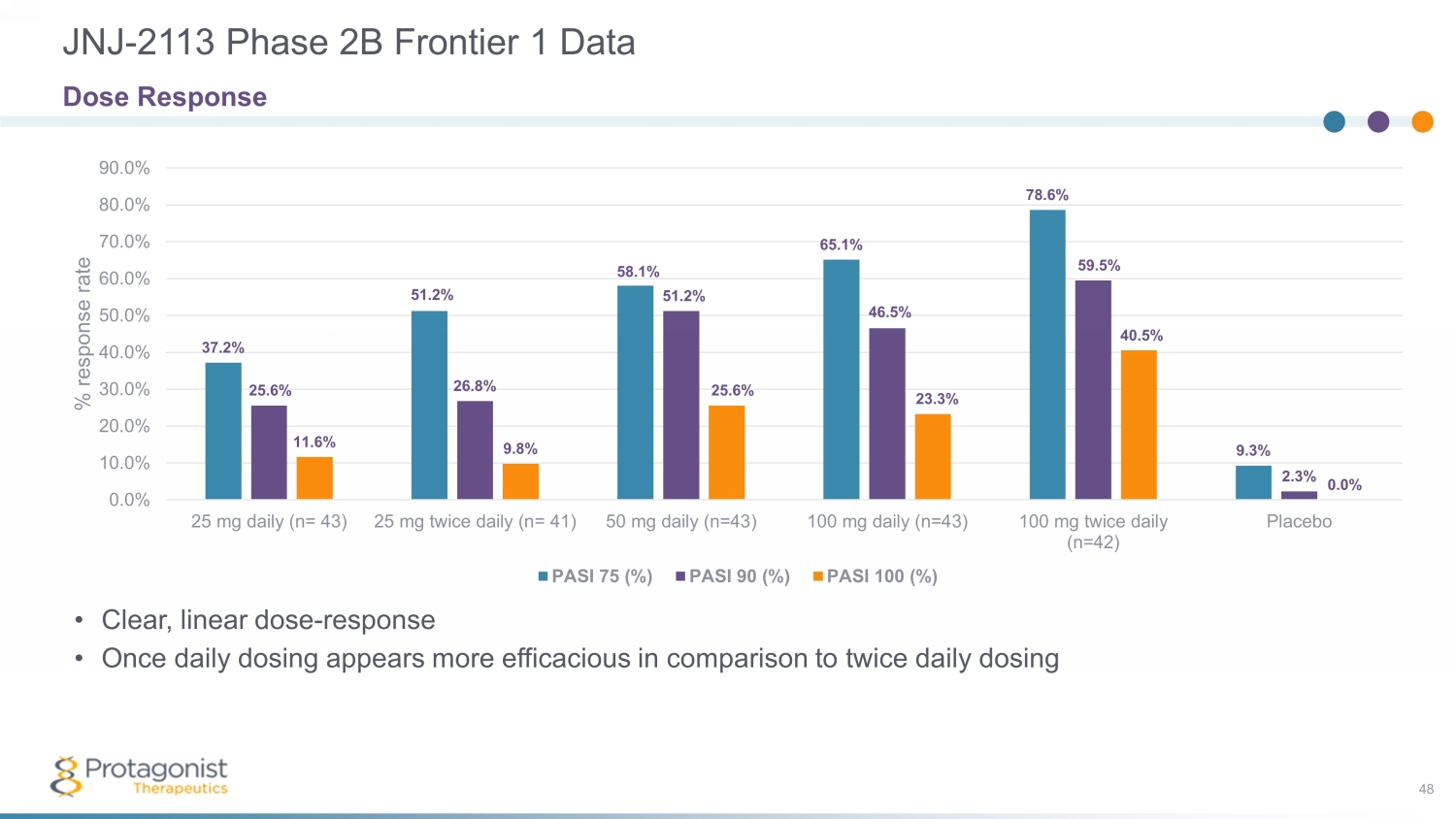
JNJ-2113 Phase 2B Frontier 1 數據 48 37.2% 51.2% 58.1% 65.1% 78.6% 9.3% 25.6% 25.8% 51.2% 56.5% 2.3% 9.8% 25.6% 20.0% 30.0% 50.0% 60.0% 70.0% 25毫克每天兩次 (n= 43) 25 毫克 (n= 43) 25毫克 (n= 43) 25毫克 = 41) 每日 50 mg (n=43) 每日 100 mg (n=43) 100 mg 每天兩次 (n=42) 安慰劑百分比反應率 PASI 75 (%) PASI 90 (%) PASI 100 (%) 劑量反應 • 清晰、線性劑量——反應 • 與每天兩次給藥相比,每日給藥顯得更有效
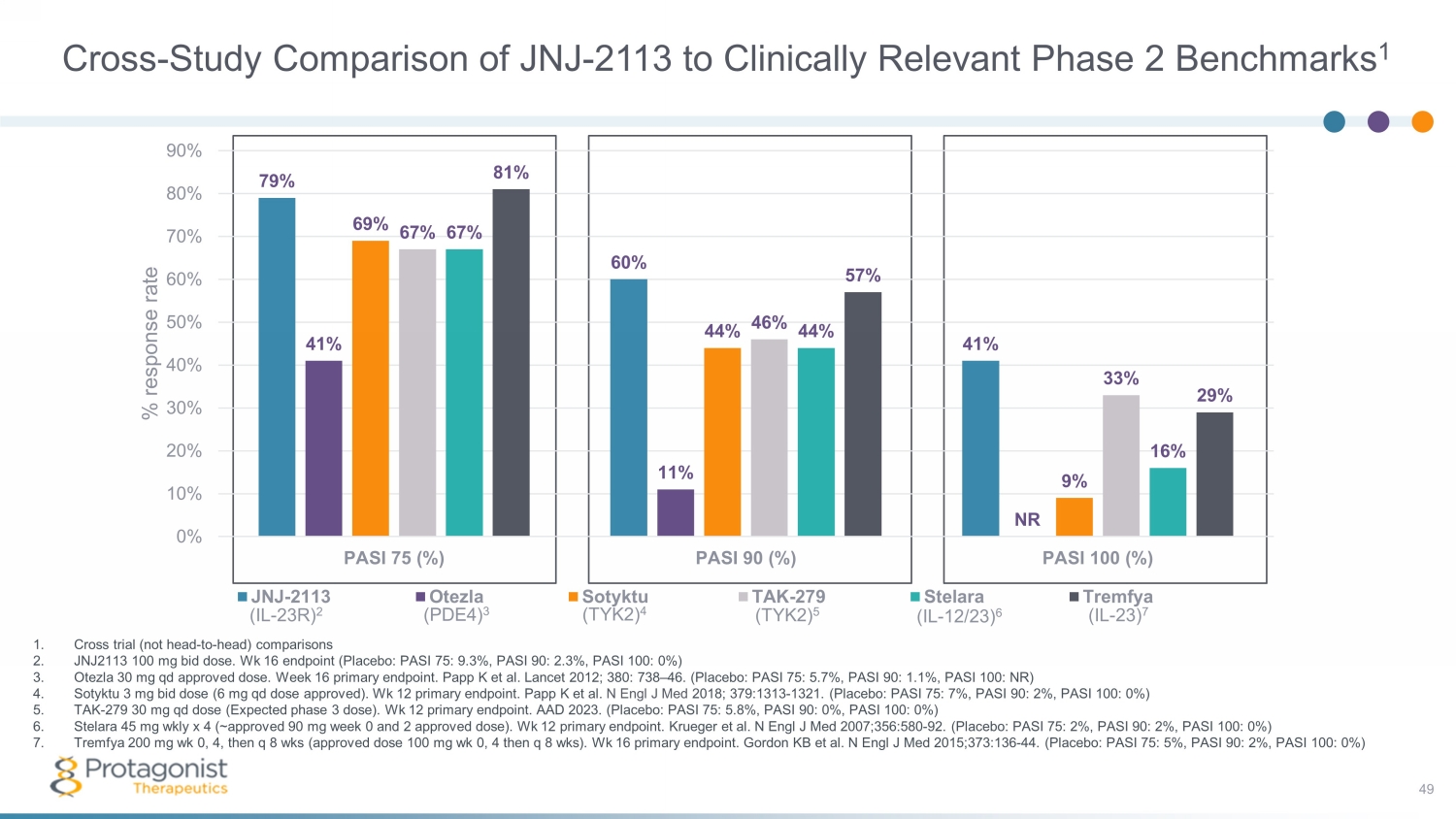
JNJ-2113 與臨牀相關的 2 期基準的交叉研究比較 1 49 1.交叉試驗(不是正面對頭)比較 2.J NJ2113 100 mg bid 劑量。Wk 16 終點(安慰劑:PASI 75:9.3%,PASI 90:2.3%,PASI 100:0%)3。Otezla 30 mg qd 批准劑量。第 16 周主終端節點。Papp K 等人。《柳葉刀》2012;380:738 — 46。(安慰劑:PASI 75:5.7%,PASI 90:1.1%,PASI 100:NR)4。sotyktu 3 mg bid 劑量(6 mg qd 劑量已獲批准)。第 12 周主終端節點。Papp K 等人N Engl J Med 2018;379:1313-1321。(安慰劑:PASI 75:7%,PASI 90:2%,PASI 100:0%)5。TAK-279 30 mg qd 劑量(預計第 3 階段劑量)。第 12 周主終端節點。AAD 2023。(安慰劑:PASI 75:5.8%,PASI 90:0%,PASI 100:0%)6。Stelara 45 mg wkly x 4(~批准的第 0 周 90 毫克和 2 次批准的劑量)。第 12 周主端點。Krueger 等人。N Engl J Med 2007;356:580-92。(安慰劑:PASI 75:2%,PASI 90:2%,PASI 100:0%)7。Tremfya 200 mg wk 0、4,然後 q 8 周(批准劑量 100 mg wk 0,4 然後 q 8 周)。第 16 周主端點。Gordon KB 等人N Engl J Med 2015;373:136-44。(安慰劑:PASI 75:5%,PASI 90:2%,PASI 100:0%)(IL-23R)2(PDE4)3(TYK2)4(TYK2)5(IL-23)6(IL-23)7 79% 60% 41% 41% 11% NR 69% 47% 44% 9% 67% 47% 0% 20% 30% 40% 50% 60% 70% 80% 90% PASI 75 (%) PASI 90 (%) PASI 100 (%)% 回覆率 JNJ-2113 Otezla sotyktu TAK-279 Stelara Tremfya
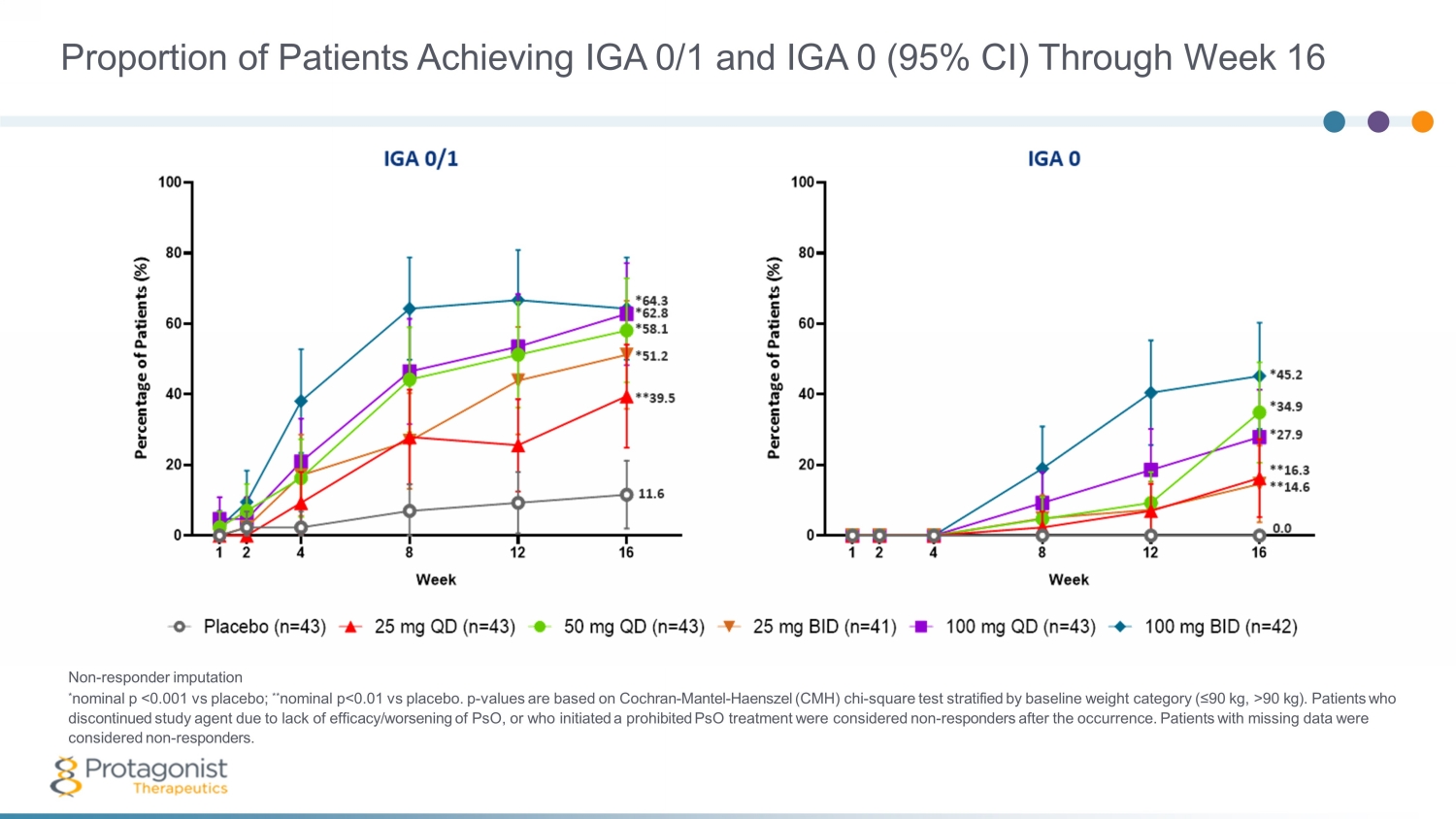
在第 16 周之前達到 IGA 0/1 和 IGA 0(95% 置信區間)的患者比例(非反應者歸因 * 標稱 p 90 kg)。由於psO療效不足/惡化而停用研究藥物的患者,或者開始了違禁的psO治療的患者,在發生後被視為無反應者。數據缺失的患者被視為非反應者。

截至研究結束時,按系統器官類別和首選術語 ae=不良事件;bid=每日兩次;qd=Daily;teae=Treation-Emergent 不良事件的患者人數。* 包括所有 JNJ-2113 治療專欄。無論患者實際經歷事件的次數是多少,任何給定事件都只計算一次。不良事件使用 MedDRA 版本 25.1 進行編碼。安慰劑 JNJ-77242113 25 mg QD 50 mg QD 25 mg BID 100 mg QD 100 mg QD 100 mg BID 組合 * 安全分析套裝,n 43 43 43 42 212 平均隨訪時間(周)15.03 15.70 15.75 16.20 16.07 15.90 AE ≥ 1 的患者,n (%) 22 (51.2%) 20 (46.5%) 26 (60.5%) 20 (40.5%) 20 (40.5%) 8.8%) 19 (44.2%) 26 (61.9%) 111 (52.4%) 系統器官類別/首選術語,n (%) 感染和侵擾 12 (27.9%) 15 (34.9%) 17 (39.5%) 14 (34.1%) 7 (16.3%) 11 (26.2%) 64 (30.2%) COVID-19 5 (11.6%) 5 (11.6%) 8 (19.0%) 5%) 3 (7.0%) 4 (9.5%) 23 (10.8%) 鼻咽炎 2 (4.7%) 1 (2.3%) 8(18.6%) 3 (7.3%) 1 (2.3%) 2 (4.8%) 15 (7.1%) 上呼吸道感染 1 (2.3%) 3 (7.0%) 0 0 2 (4.8%) 5 (2.4%) 胃腸道疾病 5 (11.6%) 3 (7.0%) 6 (14.0%) 4 (9.3%) 7 (16.7%) 24 (11.3%) 腹瀉 1 (2.3%)) 2 (4.7%) 4 (9.3%) 2 (4.9%) 1 (2.3%) 1 (2.4%) 10 (4.7%) 神經系統疾病 1 (2.3%) 0 3 (7.0%) 2 (4.9%) 3 (7.0%) 2 (4.8%) 10 (4.7%) 頭痛 1 (2.3%) 1 (2.4%) 3 (2.0%) 6 (2.4%) 1 (2.4%) 6 (2.4%) 8%) 呼吸道、胸腔和縱隔疾病 1 (2.3%) 1 (2.3%) 1 (2.3%) 0 3 (7.0%) 2 (4.8%) 7 (3.3%) 咳嗽 0 1 (2.3%) 1 (2.3%) 0 3(7.0%) 1 (2.4%) 6 (2.8%) 51
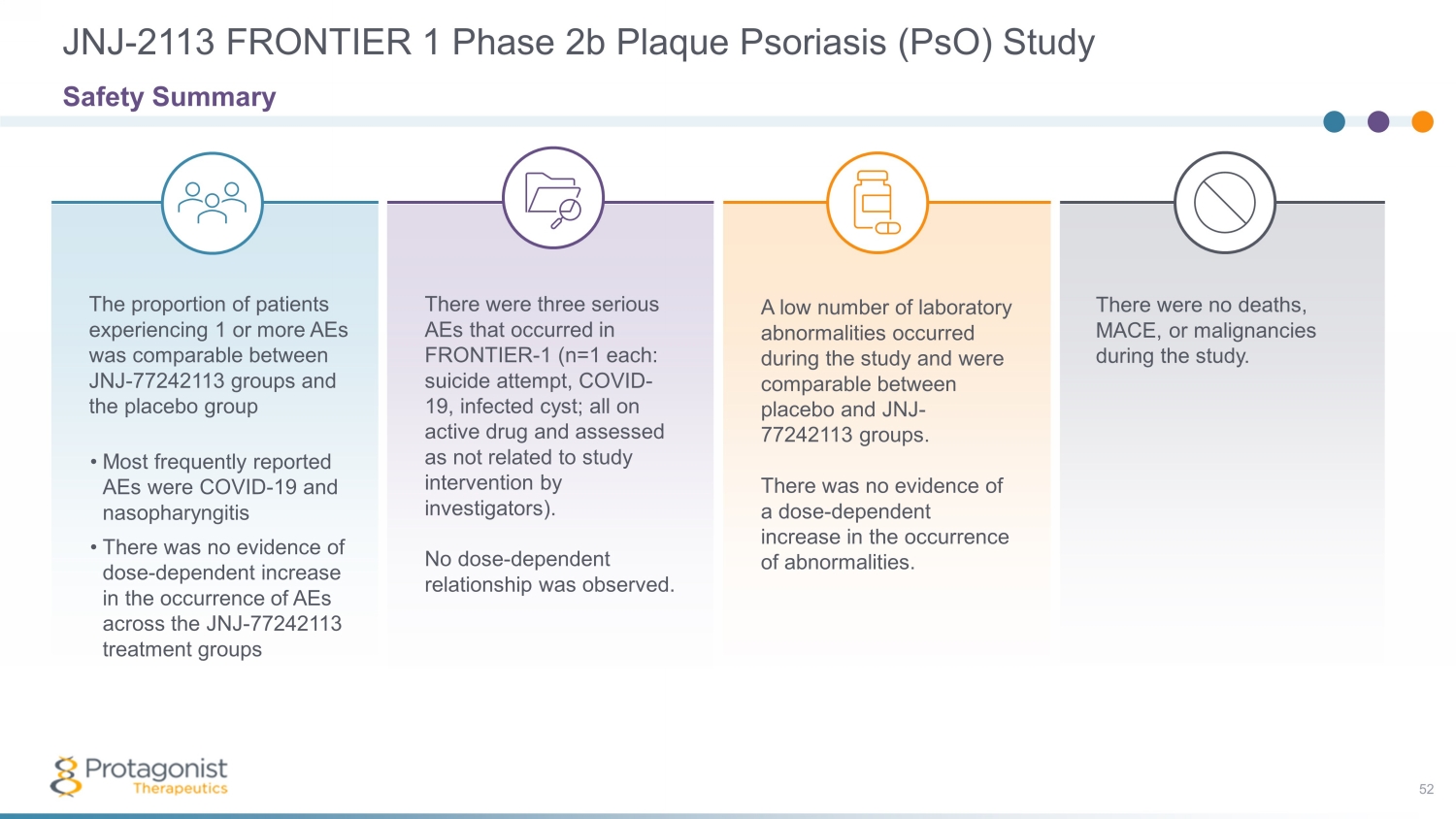
研究期間沒有出現死亡、MACE 或惡性腫瘤。研究期間出現的實驗室異常數量很少,安慰劑和JNJ-77242113組之間的實驗室異常數量相當。沒有證據表明異常發生率的劑量依賴性增加。FRONTIER中發生了三次嚴重的AE——1(各為n=1:自殺未遂、COVID-19、感染囊腫;全部服用活性藥物,研究人員評估為與研究幹預無關)。未觀察到劑量依賴關係。JNJ-77242113 組和安慰劑組之間出現 1 個或更多 AE 的患者比例相當 • 最常報告的 AE 是 COVID-19 和鼻咽炎 • 沒有證據表明 JNJ-77242113 治療組 AE 發生率的劑量依賴性增加 JNJ-2113 FRONTIER 1 phase 2b 牛皮癬斑塊 (psO) 研究 52 安全摘要
JNJ-2113 53 FRONTIER 1 的研究結果支持 JNJ-77242113 在牛皮癬以及其他 IL-23 介導的疾病適應症的 3 期研究中進一步發展 FRONTIER 1 期 2b psO 研究和後續步驟證實 PASI 分數(75、90、100)的劑量反應 JNJ-77242113 是同類首款口服 IL-23R 拮抗劑肽,其療效明顯更高在一項2b期研究中,所有劑量均為中度至重度斑塊psO的患者使用安慰劑 JNJ-77242113 在所有劑量下耐受性良好AE的數量與安慰劑組相當。未觀察到 AE 或實驗室異常中的劑量依賴關係 JNJ-77242113 有望作為一種有效的口服療法,例如在中度至重度斑塊狀牛皮癬中的療效和安全性 JNJ-2113(前身為 PN-235)是潛在的第一個、最好也是唯一的口服 IL-23 受體拮抗劑後續步驟 • 牛皮癬的 3 期研究:預計詹森將根據ph2b結果選擇最佳的QD劑量 • 2b期研究在潰瘍性結腸炎中
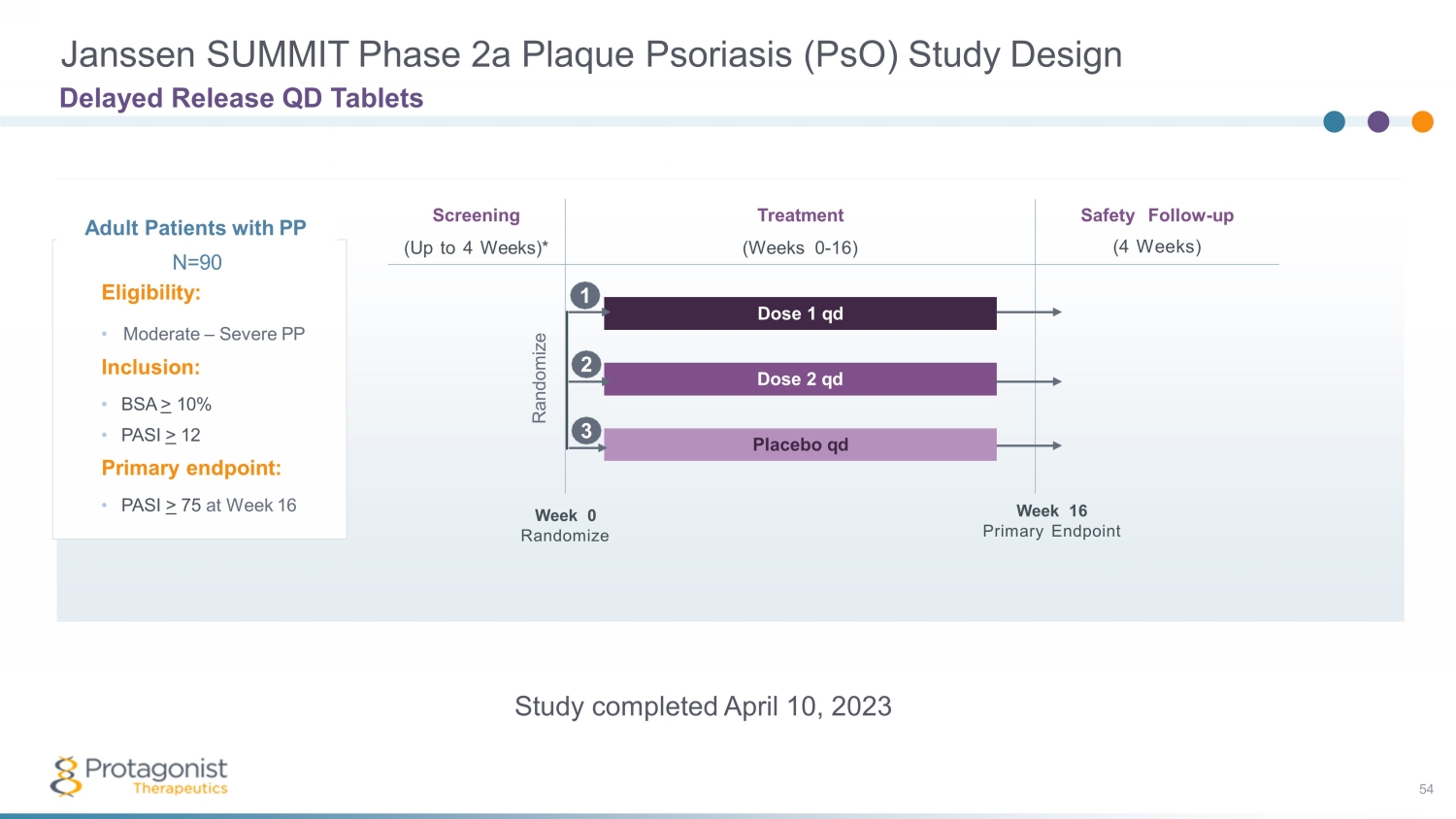
Janssen SUMMIT 2a 期斑塊狀牛皮癬 (psO) 研究設計延遲釋放 QD 片劑 PP N =9 0 資格:• 中度 — 重度 PP 包含:• BSA > 10% • PASI > 12 主要終點:• 第 16 周的 PASI > 75 安全隨訪(4 周)* 治療(第 0-16 周)劑量 1 qd 劑量 2 qd 安慰劑 qd 隨機周 0 第 16 周主要終點隨機化 2 1 3 研究於 2023 年 4 月 10 日完成 54

Royalty Janssen 里程碑現狀和展望 55 2023 年及以後 1.125 億美元主角仍有資格實現的未來開發和銷售里程碑總額預付和開發里程碑已達到 6% 至 10% • 向上分層 • 淨銷售額超過 40 億美元即將到來的潛在里程碑 Ph3 1° 終點實現了 1.15 億美元* 保密協議批准 5000萬美元* 第二個跡象 Ph2 啟動 1000 萬美元 Ph3 啟動費 1500 萬美元 * 不包含在當前的現金跑道預測中

56 財務狀況

財務亮點財務資源預測將持續到2025年全年現金和證券 *截至2023年3月31日**關注——在2023年4月發行時,現金和證券提供截至2023年3月31日的2025年全年財務資源預測 1 股截至2023年3月31日已發行約5720萬美元2.308億美元* + 1.08億美元** 1 基於我們目前的運營計劃和支出,包括2023年4月完成的發行的收益 57

58 謝謝