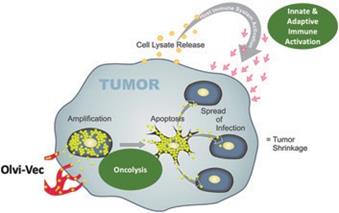
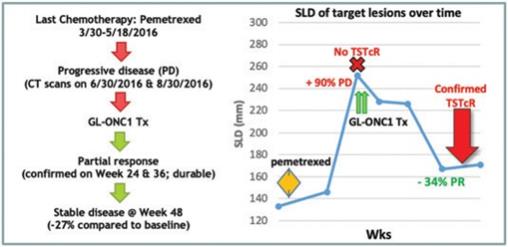
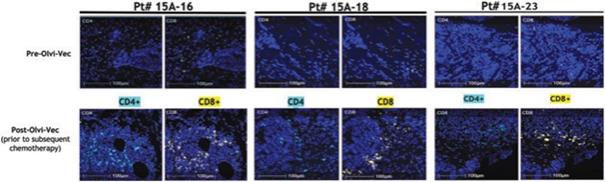
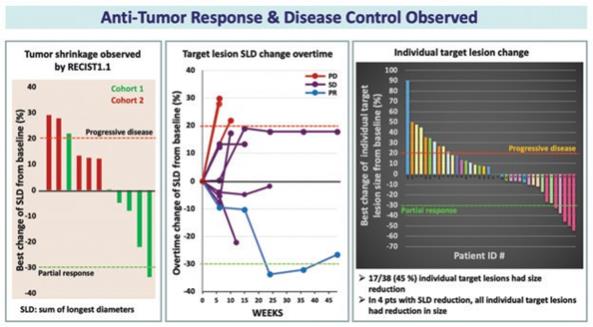
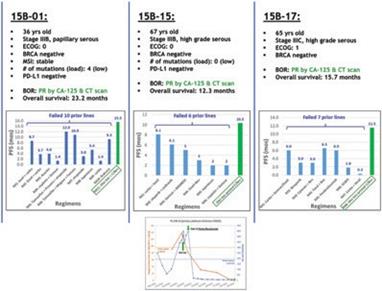
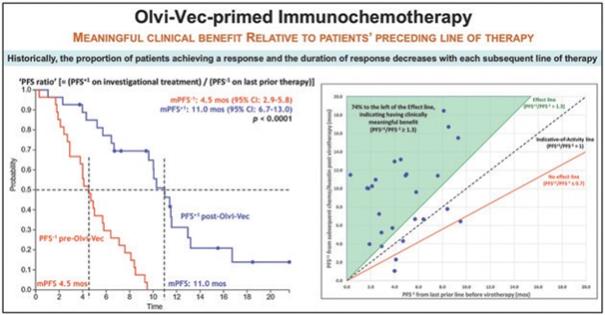
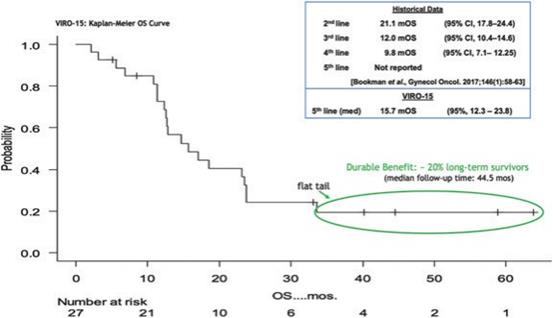
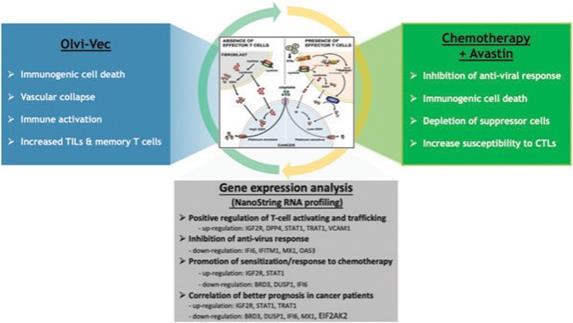
下表總結了我們的臨牀開發流程:
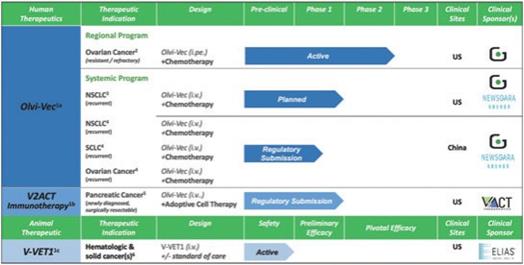
| 1 | 商業權利 |
1a Genelux: 全球(不包括大中國);Newsoara(大中國)
1bV2ACT免疫療法:全球(不包括大中國)
1cELIAS:全球
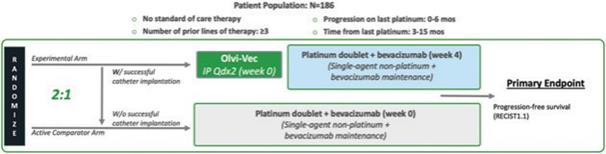
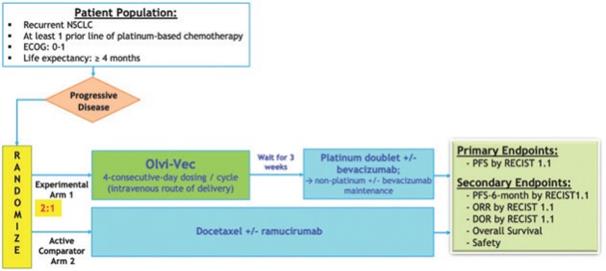
| 2 | 我們在我們的3期臨牀試驗中招募了第一名患者。 |
| 3 | 根據我們之前完成的對實體腫瘤患者靜脈注射Olvi-Vec的第一階段臨牀試驗的結果,我們計劃啟動Olvi-Vec治療復發NSCLC的第二階段臨牀試驗。 |
| 4 | Newsoara已向中國醫療產品協會提交了IND和方案。 |
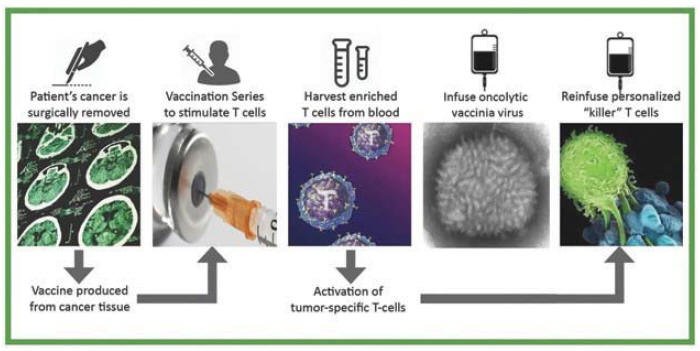
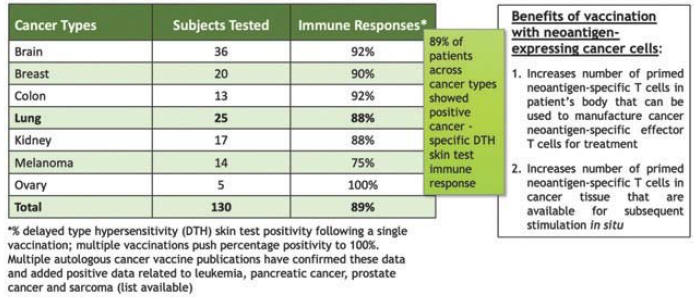
| 5 | V2ACT具有此候選產品的活動IND。1b/2a期臨牀試驗尚未計劃啟動。 |
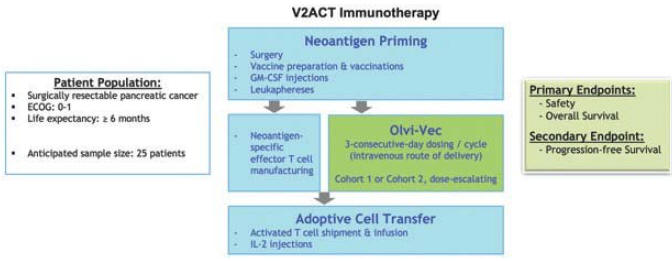
| 6 | 伊萊亞斯正在開發一種療效試驗。 |
我們是由洛馬林達大學的一個學術團隊於2001年創立的,該團隊由Aladar A.Szalay博士領導,Aladar A.Szalay博士是監測基因調控以及使用發光蛋白質或蛋白質融合進行全細胞和活體成像方面的國際公認的領導者。我們組建了一支經驗豐富的業務領導團隊,在腫瘤治療方面擁有豐富的經驗,包括推動候選產品從臨牀前研究到臨牀開發和商業化。Thomas D.Zindrick,J.D.,首席執行官兼董事長總裁此前曾在阿米泰克治療解決方案公司擔任總裁兼首席執行官和董事 職位,並在安進公司擔任過各種高管管理職位,包括協理副總裁總裁、總法律顧問和首席合規官,並在陶氏化學公司擔任責任日益增加的法律職位。董事首席獨立董事詹姆斯·L·泰裏曾在雅培擔任過多個高管職務,其中包括環球醫藥執行副總裁總裁,曾在Sugen,Inc.擔任總裁一職,還曾在百時美施貴寶公司和輝瑞公司擔任過管理職務。
3