目錄表
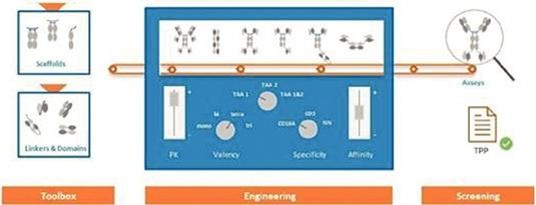
我們的研發渠道
我們正在開發一種治療癌症的天然細胞激活劑流水線,如下所示:

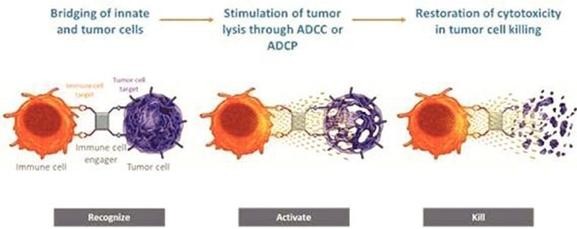
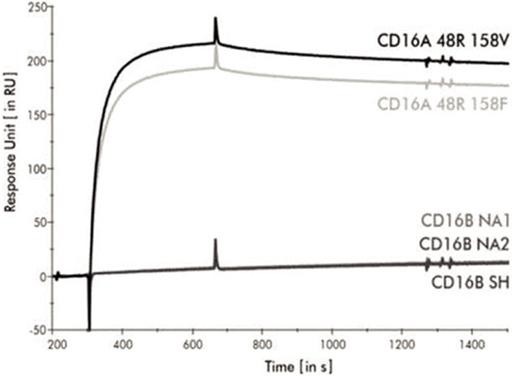
我們最先進的候選人AFM13是一流的ICE®專為治療某些CD30陽性(CD30+)惡性腫瘤而設計,包括霍奇金淋巴瘤和某些非霍奇金淋巴瘤。AFM13選擇性地結合CD30和CD16A,CD30是臨牀驗證的靶點,CD16A是NK細胞和巨噬細胞表面表達的整合膜糖蛋白受體,觸發信號下跌,導致CD30陽性腫瘤細胞的破壞。與傳統的全長抗體不同,AFM13不與CD16B結合,CD16B阻止與其他類型的細胞結合,E.g..、中性粒細胞,並在第158位與CD16A多態具有相同的親和力。此外,AFM13結合CD16A的親和力比單抗高約1000倍,從而顯著提高了臨牀前證明的效力和療效。AFM13在複發性/難治性外周T細胞淋巴瘤(PTCL)患者的2期研究(REDIRECT)中作為單一療法進行研究,目前正在與MD Anderson癌症中心合作進行的1/2a期臨牀研究中與過繼NK細胞聯合進行CD30+淋巴瘤患者的研究。重定向研究的背線數據已於2022年12月報告,該公司預計將在2023年的科學會議上提交該研究的全面數據。2022年11月,我們宣佈與Artiva BioTreateutics合作,目標是推動AFM13與Artiva的AB-101 NK細胞療法的結合開發成為一項潛在的註冊使能研究。我們預計在2023年上半年提交AFM13與AB-101聯合使用的IND,並在FDA批准IND的情況下,在2023年啟動臨牀研究。
我們的第二個候選基因AFM24是一種四價、雙特異性的表皮生長因子受體(EGFR)和CD16A結合的天然細胞結合蛋白。AFM24旨在解決與當前治療性抗EGFR單抗相關的侷限性,如毒性或治療耐藥性,同時還通過激活針對表達EGFR的實體瘤的固有免疫而不是抑制EGFR介導的信號轉導來提供更好的療效和安全性的潛力。AFM24目前正作為人類第一階段1/2a試驗的單一療法進行研究,並在兩項聯合臨牀試驗中研究AFM24與過繼NK細胞和PD-L1抑制劑的關係。
52