目錄表
西利奧腫瘤活性細胞因子的主要特徵
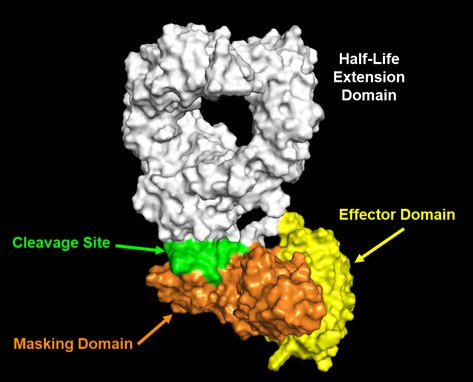
我們相信,我們的GPS平臺的上述特點實現了以下主要優勢:
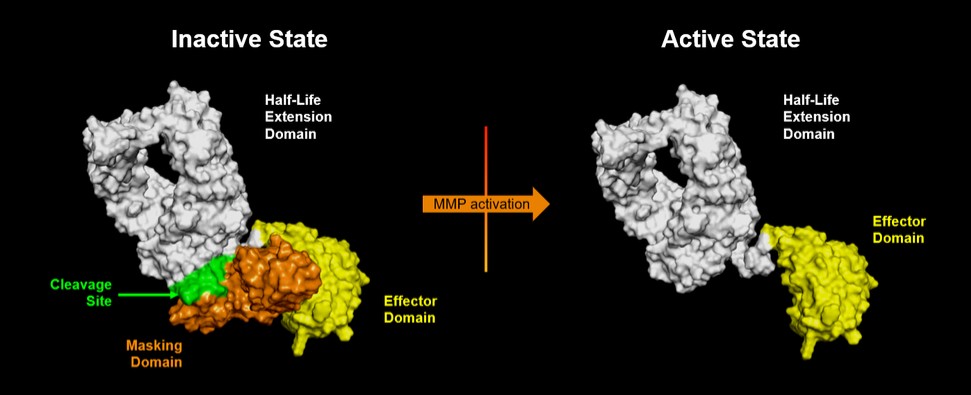
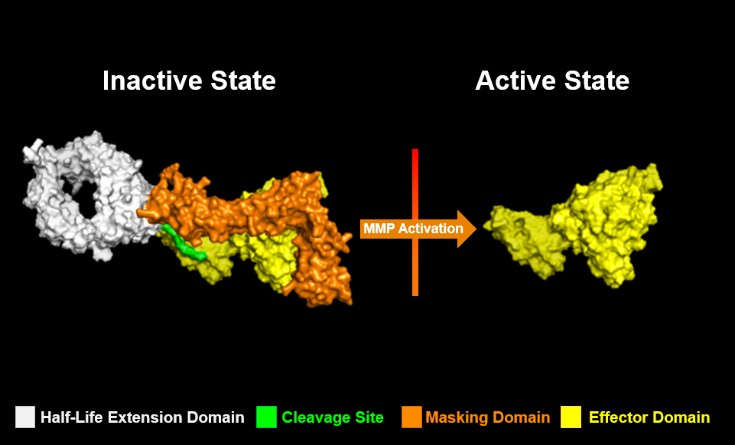
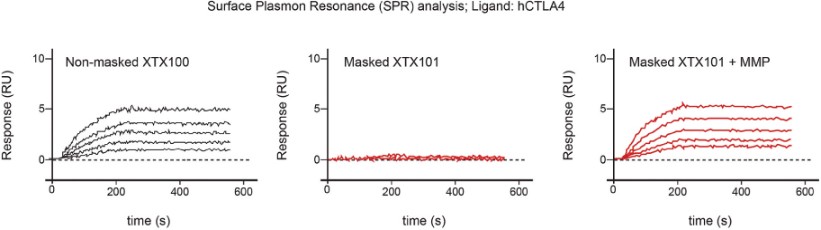
· | 掩蔽利用多個分子內相互作用,將腫瘤微環境外的活動風險降至最低,從而將毒性風險降至最低; |
· | 設計活性分子,以便在腫瘤微環境中揭開面紗,促進有效的、局部的抗腫瘤免疫反應; |
· | 及早考慮並將製造和開發方面納入分子設計,以促進臨牀用高質量藥物產品的生產; |
· | 適當延長非活性(掩蔽)細胞因子的半衰期,以支持患者按照與其他生物製劑一致的時間表給藥;以及 |
· | 局部激活在腫瘤微環境中具有較短半衰期的細胞因子分子,將釋放的細胞因子在腫瘤微環境外表現出活動的風險降至最低,因此,進一步降低了毒性風險。 |
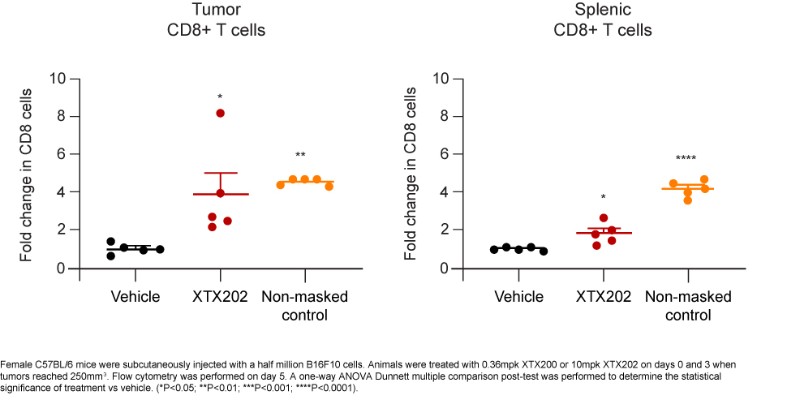
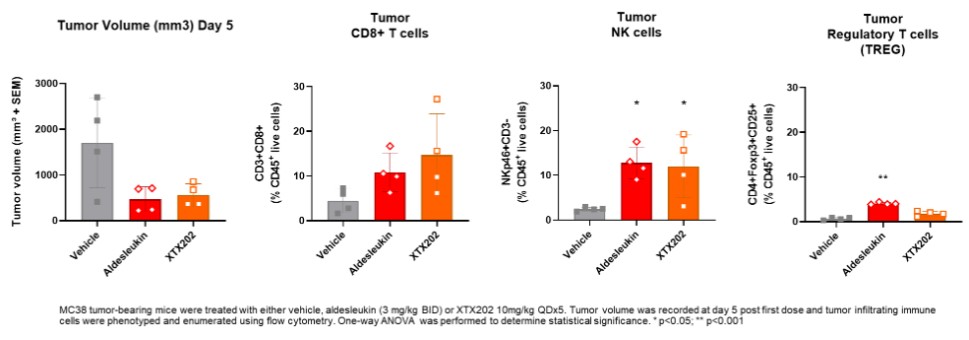
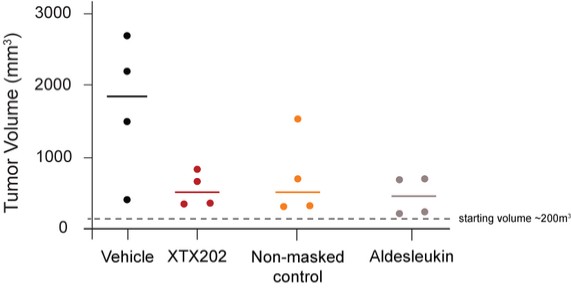
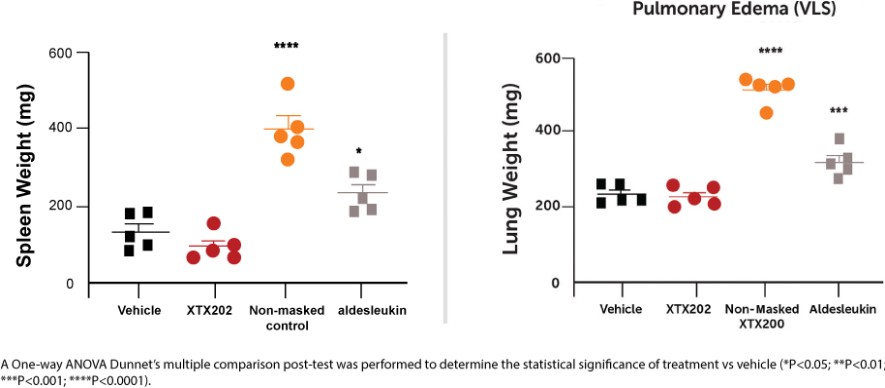
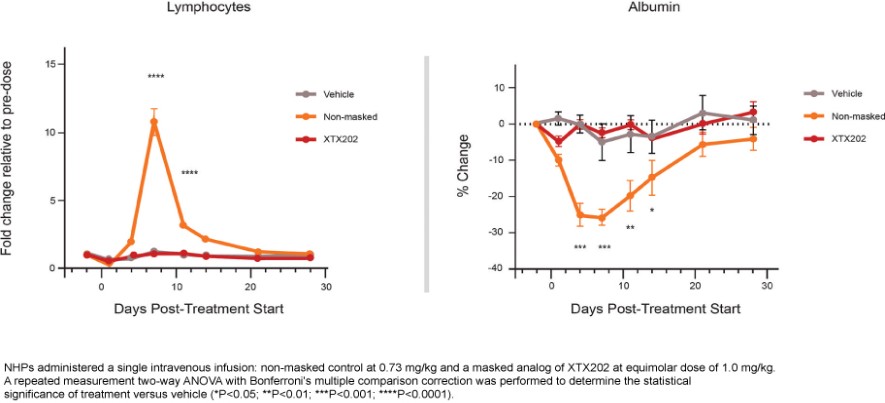
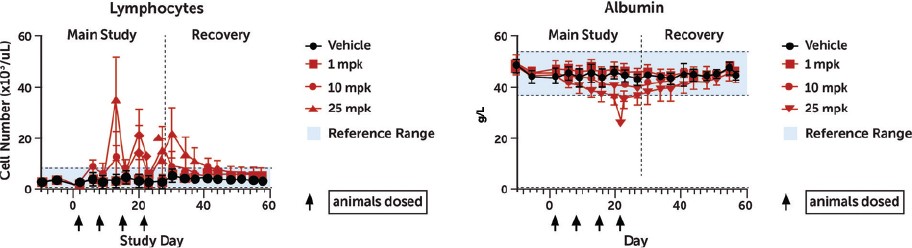
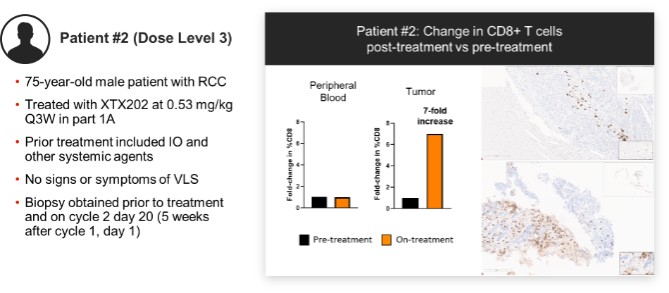
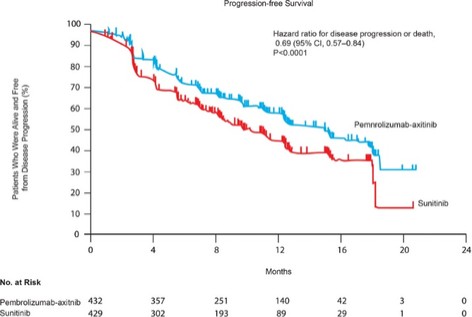
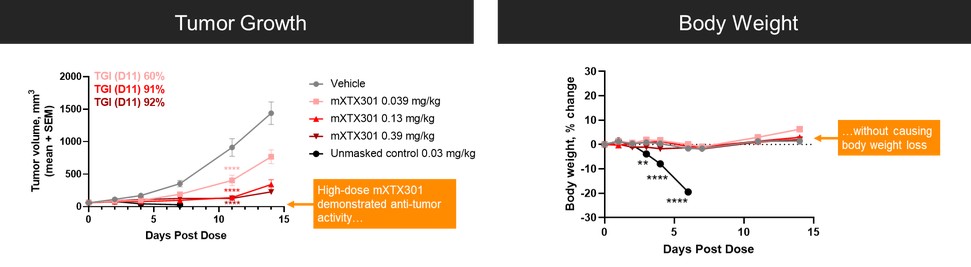
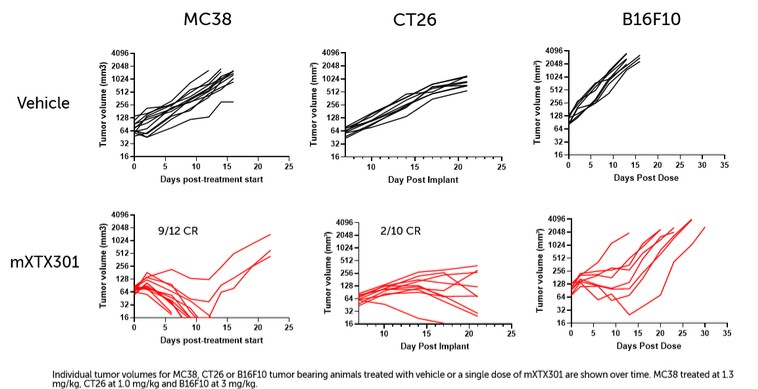
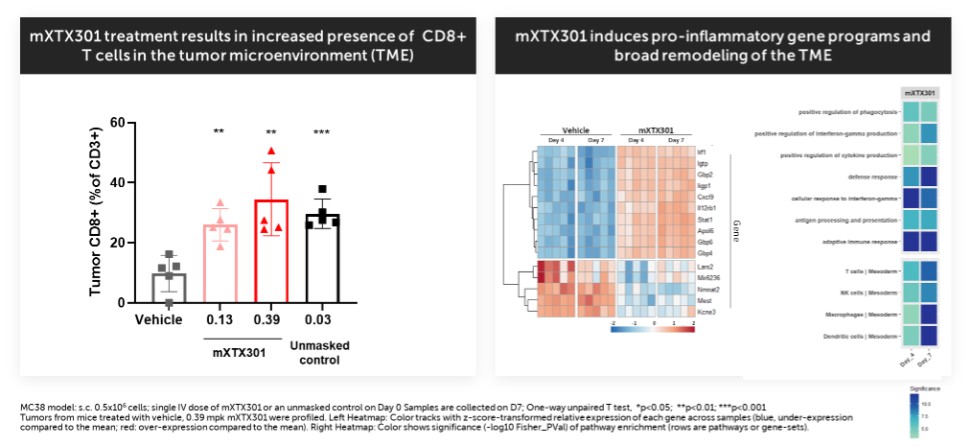
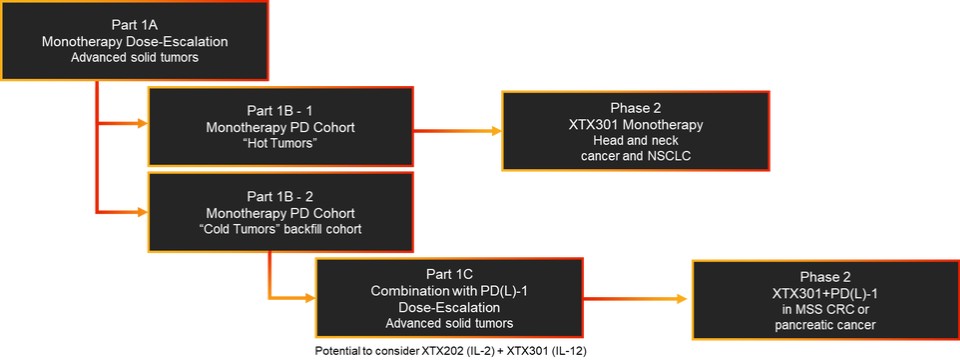
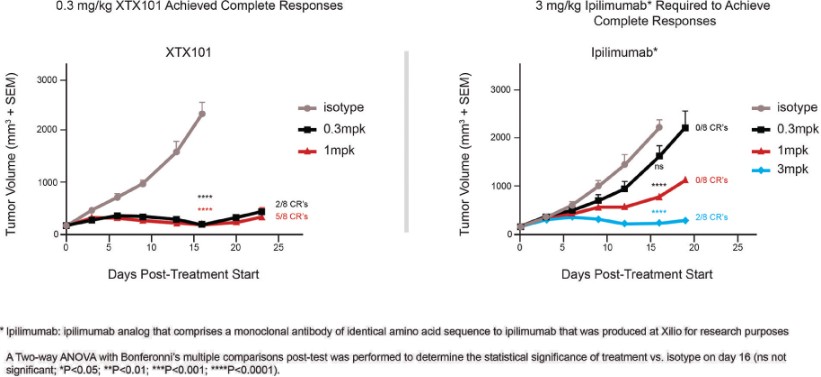
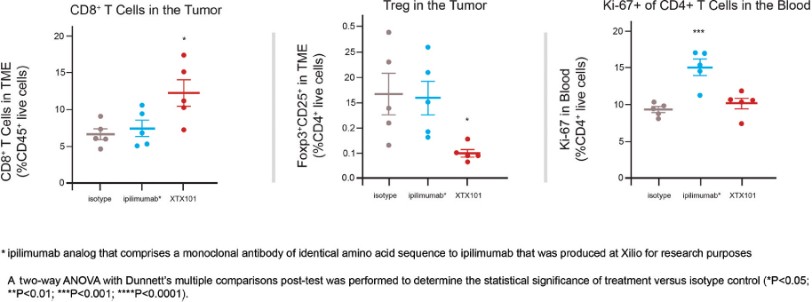
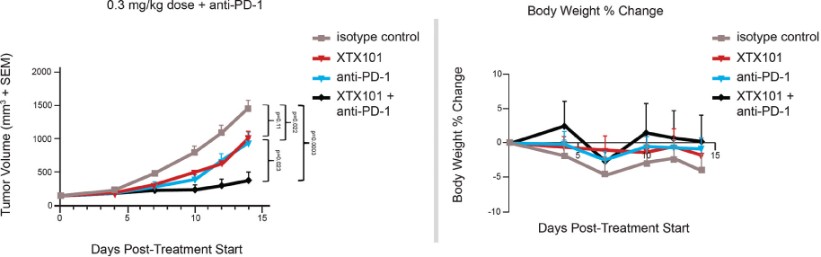
我們已經展示了我們的GPS平臺開發腫瘤激活的細胞因子和抗體的能力的臨牀前驗證和有希望的早期臨牀證據。在臨牀前研究中,我們的每一種最先進的候選產品都顯示出腫瘤激活的生物活性、腫瘤生長抑制以及腫瘤微環境外的最小或零毒性。這些臨牀前數據的重複性突出了我們的GPS平臺應用於多種結構不同的細胞因子、抗體和多功能分子的潛在廣度。在我們的XTX202(我們的腫瘤激活的IL-2)的第一階段試驗中,我們展示了我們的GPS平臺的初步臨牀驗證,兩名接受XTX202治療的患者的初步腫瘤內PD數據證明瞭這一點,這兩名患者都有可選的治療中腫瘤活檢,並且是截至2023年3月1日僅有的兩名獲得腫瘤活檢分析的患者。對於每個患者,腫瘤樣本的特徵是間質腫瘤浸潤性淋巴細胞(TIL)數量增加,這些TIL中CD8+效應T細胞的頻率增加。在腫瘤樣本時,這些變化發生在每個患者沒有CD8+效應T細胞外周變化的情況下,證明瞭腫瘤選擇性激活的初步證據。我們相信我們的GPS平臺可以應用於許多分子,這些分子具有治療癌症的潛力,但需要在腫瘤微環境內進行局部活動,以克服因腫瘤微環境外活動而產生的劑量限制性毒性。
10