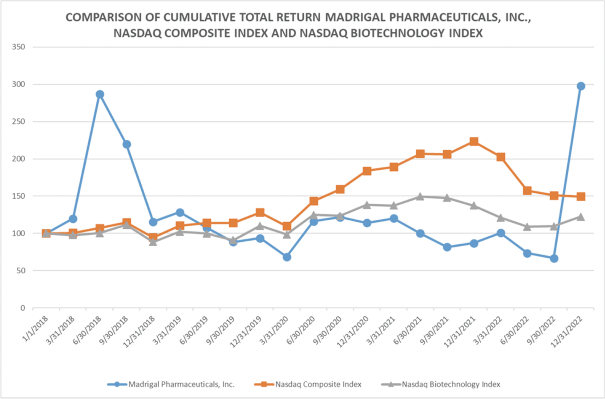
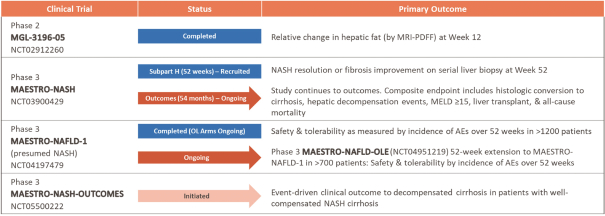
瑞美替隆在肝臟活檢患者中的3期對照研究-確認為NASH,於2019年3月啟動。這項研究的H部分招募了1000多名經活檢證實的NASH患者(至少一半患有F3(晚期)纖維化,其餘的F2或F1B(中度纖維化)與少數早期F1患者),隨機1:1:1接受每日一次的瑞美替隆80毫克、瑞美替隆100毫克或安慰劑治療。治療52周後,進行第二次肝臟活檢。活檢上的雙主要代理終結點是Nash解析≥2分NAS(NAFLD活動評分)降低,且沒有纖維化惡化或1分纖維化減輕,無NAS惡化。任何一個主要終點的實現都被認為是成功的試驗結果。一個關鍵的次要端點是降低低密度脂蛋白-C所有參加Maestro-Nash研究的患者(總共多達2000人)在最初的治療後繼續接受治療52周治療期間長達54個月,以累積和衡量肝臟臨牀結果事件,包括活檢進展為肝硬變(52周和54個月)和肝臟失代償事件,以及全因死亡率。2022年12月,我們報告了這項研究H部分的TOPLINE結果:與安慰劑相比,resmetirom獲得了兩個主要終點,每日口服劑量分別為80毫克和100毫克,如下文“關鍵進展”中所述。
2021年7月13日,我們宣佈了計劃中的第一名患者52周開放式標記物主動治療的推廣研究大師-NAFLD-1,命名為MAESTRO-NAFLD-開放標籤擴展(OLE)。OLE研究允許完成Maestro-NAFLD-1同意用瑞美替隆進行52周的積極治療,使被分配到安慰劑的兩名患者都能使用這種治療。Maestro-NAFLD-1以及服用瑞美替羅的患者Maestro-NAFLD-1
2022年8月,Madrigal在大約700名早期Nash肝硬變患者中發起了一項隨機、雙盲、安慰劑對照研究,以允許對肝臟失代償事件的進展進行非侵入性監測。一個積極的結果預計將支持RESMETROM用於非肝硬化性NASH的完全批准,有可能加速完全批准的時間表。此外,這項研究有可能支持在代償性納氏肝硬變患者中應用瑞美替隆的另一種適應症。
下表彙總了我們的resmetirom候選產品開發計劃的狀況:
8