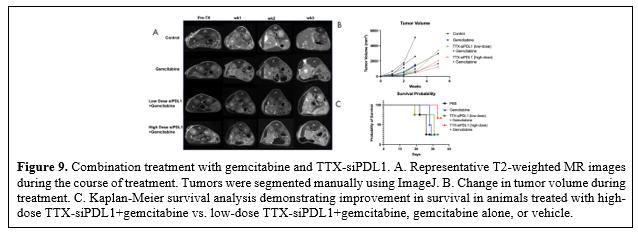
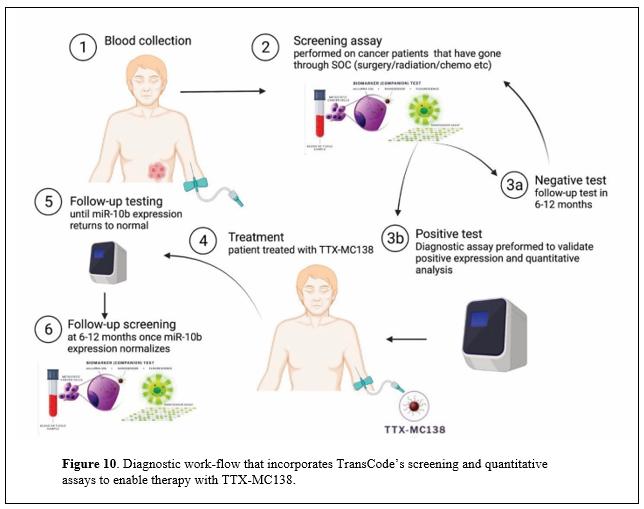
目錄
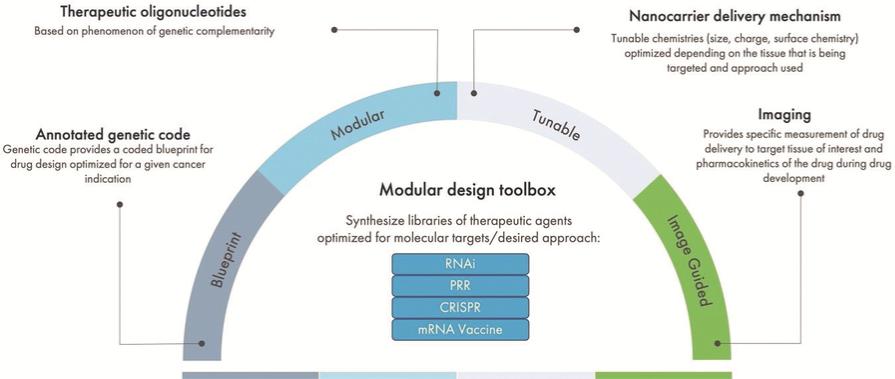
大小,表面塗層和電荷,親水性和疏水性,以及通過摻入靶向多肽進行抗原靶向。
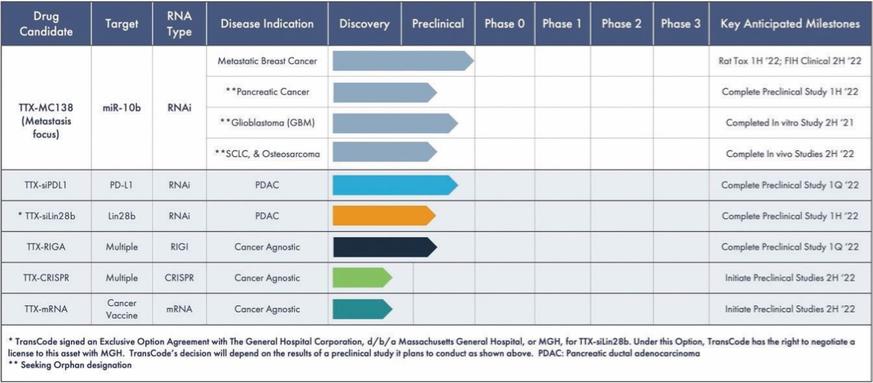
●遺傳密碼 - 我們的藥物開發方法利用了我們迅速增長的關於人類基因組的知識和對基因組的註釋 - ,即關於不同基因負責什麼的知識,特別是在癌症中。有了這些知識,我們就可以利用基因組的編碼性質來設計與感興趣的遺傳目標相對應的特定寡核苷酸。一旦我們確定了癌症靶點的密碼,我們就可以使用與該靶點相協調的特定寡核苷酸來開發治療候選藥物,並有可能重寫關於癌症的故事。這就是代碼轉換的意思是 - 來更改代碼。在確定遺傳目標之後,我們可能會嘗試設計最優方法從siRNA、反義寡核苷酸和非編碼RNA模擬或基於mRNA的癌症疫苗,到基於CRISPR的基因修復和替換平臺或模式識別受體,如RIG-I。
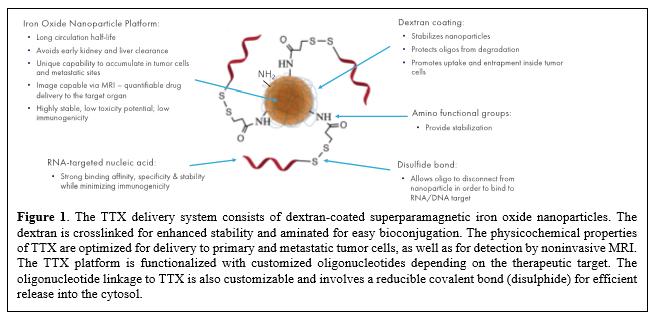
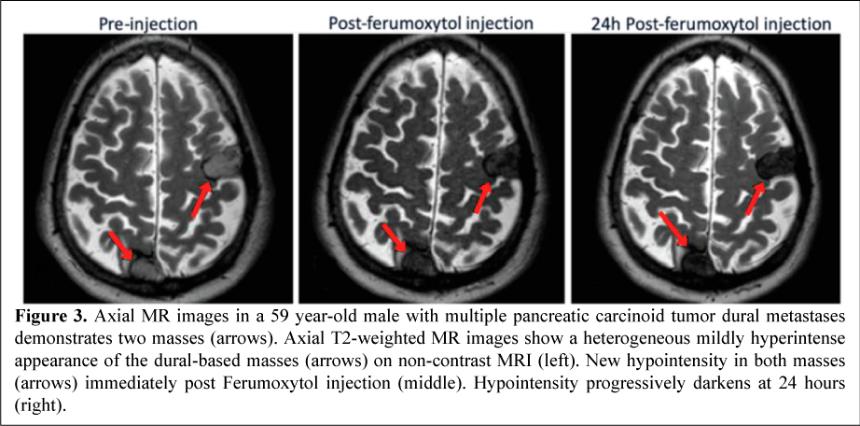
●圖像制導 - 因為我們的候選產品天生就可以使用非侵入性成像進行檢測,所以我們可以監控它們向感興趣組織的傳輸,並測量它們的生物利用度。使用磁共振成像(MRI)監測遞送的能力可以有助於評估和控制到達目標組織的寡核苷酸的數量。在候選產品的設計階段使用MRI可以指導藥物設計、給藥時間表、路線和劑量,並可以在候選治療方案在給定患者失敗的情況下提出替代方案。這在藥物開發過程中至關重要,因為它應該允許我們優化藥物設計,以最大限度地發揮治療效果。
下圖總結了我們的模塊化設計方法:
我們的團隊
在代碼轉換方面,我們被驅使改變癌症作為一種治療方式和在改善患者結果方面的治療方式。我們相信RNA療法的潛在能力是為患者提供疾病完全消退而不會復發,而不是目前的規範,即給患者額外的幾個月的生存時間。我們由一支經驗豐富的敬業科學家和專家團隊領導,他們在RNA和藥物開發的基礎領域擁有數十年的經驗,包括使用反義寡核苷酸或ASO的RNA藥物開發和沉默RNA方法。我們的聯合創始人兼首席執行官邁克爾·達德利在醫療器械、診斷和治療領域擁有40多年的行政領導經驗。Zdravka Medarova博士,我們的聯合創始人兼首席技術官,是一名訓練有素的遺傳學家和癌症生物學家。她是國際公認的癌症治療非編碼RNA領域的領導者,也是Transcode技術的發明者之一。她開發了核心TTX遞送平臺,並驗證了許多治療靶點。我們的聯合創始人安娜·摩爾博士以其在靶向成像和圖像引導治療方面的開創性研究而聞名國際。我們的首席財務官Tom Fitzgerald在擔任首席財務官和投資銀行家方面取得了30多年的成就,為生命科學、技術、金融和工業領域的公司從新興增長到扭虧為盈的財富500強公司提供服務。我們的研發副總裁兼首席科學家Peter Liu博士在生物製藥行業擁有超過20年的研發、經驗和領導能力,並在化學、寡核苷酸生物化學和
13