目錄表
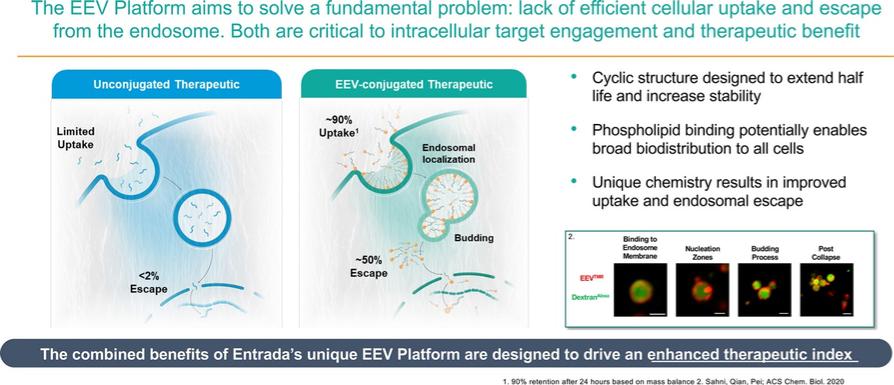
| ● | 主動攝取和藥物釋放:EEV通常避免被困在細胞膜中,而是被早期的內體吸收到細胞中。EEVS然後使早期內體中的囊泡萌發,我們認為這大大增加了到達細胞內預定靶點的治療水平。 |
我們相信,與現有的治療方法相比,我們的EEV平臺可以提供有意義的優勢,包括:
| ● | 廣闊的潛在治療指標基於臨牀前研究中的觀察。我們相信EEV治療候選方案可以吸引不同器官和組織的靶點,與類似劑量的非結合治療方案相比,細胞內靶點暴露高達50倍。 |
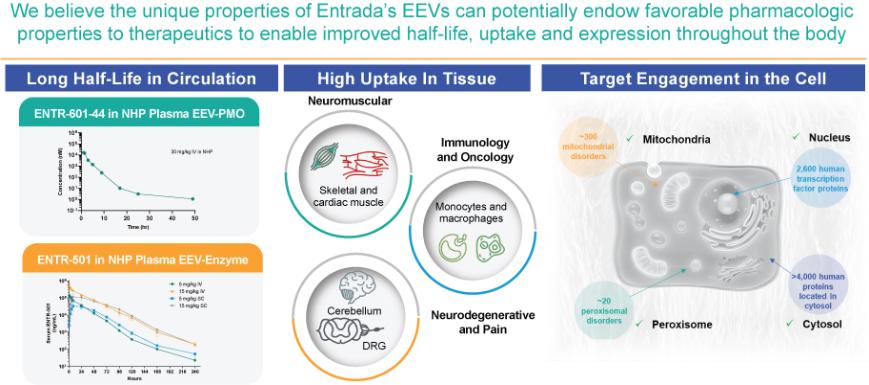
| ● | 跨多個醫療設備的潛在效用由於EEVS能夠促進大小從1 kDa到600 kDa的專有候選治療藥物在細胞內的攝取,包括寡核苷酸、肽、抗體和更大的多聚體蛋白。 |
| ● | 對多種疾病的潛在適用性我們認為EEVS可以通過與磷脂雙層結合進入細胞,磷脂雙層是人體所有細胞、組織和器官共同的。這可能意味着有能力實現對多種疾病的潛在治療候選藥物的系統和具體交付。 |
| ● | 多條送貨路線可能包括靜脈(IV)、肌肉(IM)、皮下(SQ)和鞘內(IT)注射,以提供我們的EEV候選治療方案併產生功能結果。 |
| ● | 模塊化方法支持高效地將開發擴展到多個治療領域,包括罕見疾病和免疫學的寡核苷酸療法,腫瘤學的基於抗體的蛋白質降解物和罕見疾病的酶替代療法。 |
| ● | 一種簡單且可擴展的結構,旨在將臨牀前開發轉化為臨牀開發由於EEVS已經有效地製造到了臨牀規模,而EEVS的小尺寸可能會限制免疫原性的風險。此外,ENTR-501計劃中的急性和慢性毒理學研究表明,在耐受性良好的非人靈長類動物(NHP)中提供臨牀相關劑量的潛力。 |
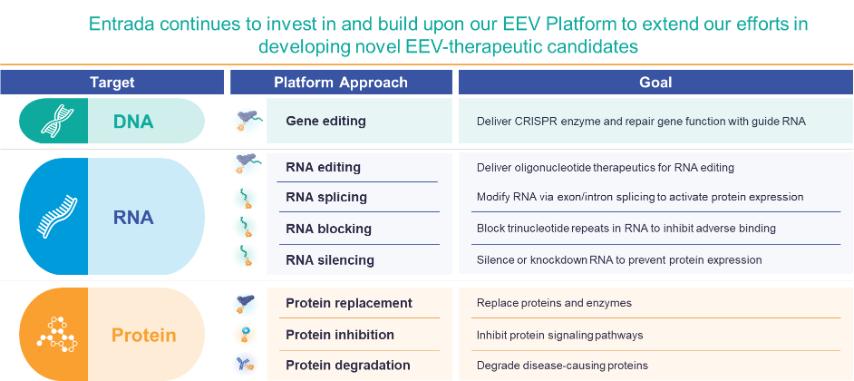
通過我們的EEV平臺,我們的目標是創建一個多樣化和不斷擴大的寡核苷酸、抗體和基於酶的程序的開發組合,如下圖所示。

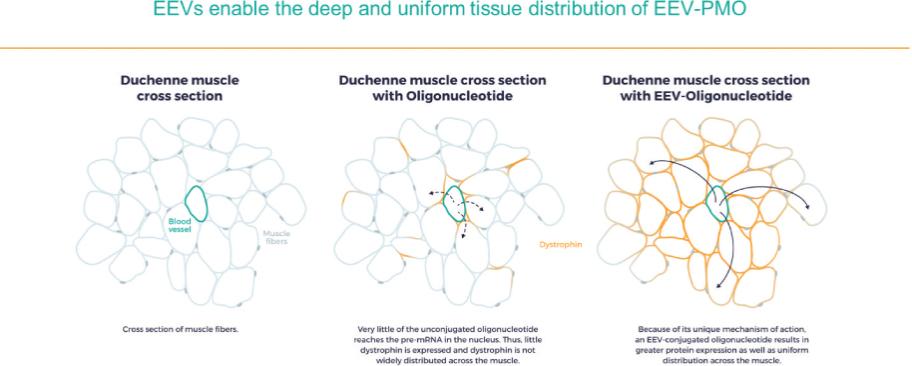
神經肌肉疾病
在神經肌肉疾病方面,我們最初專注於開發DMD的疾病修正治療方法。DMD是一種單基因X連鎖疾病,由DMD基因突變引起,DMD基因編碼Dstrophin蛋白。我們估計,大約每3,500至5,000名活男嬰中就有一名發生DMD,
9