目錄表
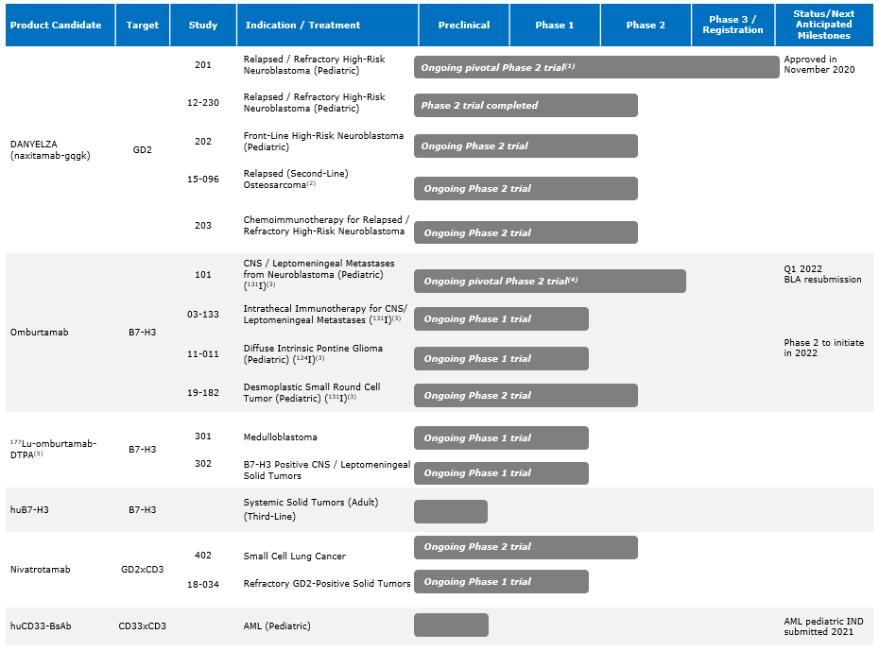
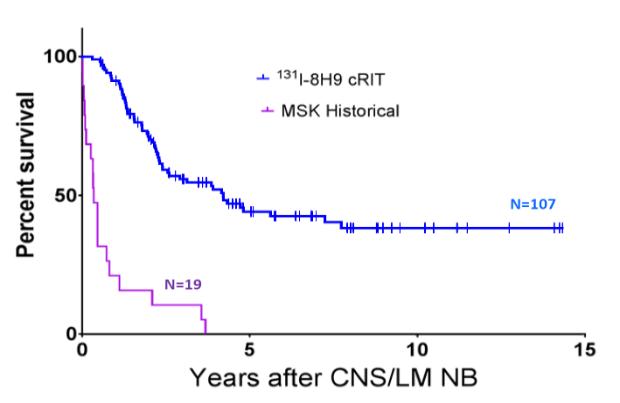
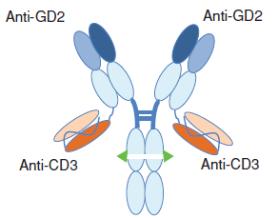
我們已經成功地為我們的2期試驗開設了IND,使用我們的候選GD2 BsAb產品nivatrotamab治療小細胞肺癌(SCLC)。此外,尼伐他單抗治療難治性GD2陽性成人和兒童實體瘤的1/2期試驗正在進行中。因此,我們的nivatrotamab計劃針對的是龐大的患者羣體。在臨牀前研究中,與現有的雙特異性構建物相比,nivatrotamab顯示了改善腫瘤結合、延長血清半衰期和顯著提高T細胞介導的殺傷力的潛力。此外,我們正在開發一種用於治療血液病的CD33 BsAb,CD33是一種表達在髓系細胞上的跨膜受體,我們預計將於2022年進入臨牀測試。我們還在通過後期臨牀前開發推進其他新型BsAbs的流水線。我們相信,我們的BsAbs有可能改善腫瘤結合,延長血清半衰期,並顯著提高T細胞介導的腫瘤細胞殺傷率,而不需要持續輸液。
SADA技術
2020年4月15日,我們與MSK和麻省理工學院(MIT)簽訂了一項許可協議或SADA許可協議,授予我們由MSK和MIT開發的某些專利和知識產權的全球獨家、可再許可的許可,以開發、製造和商業化許可產品,併為癌症診斷和癌症治療領域的所有治療和診斷用途提供服務SADA Technology。SADA許可協議涵蓋的專利和專利申請部分針對SADA技術,以及由MSK開發的一些SADA結構。
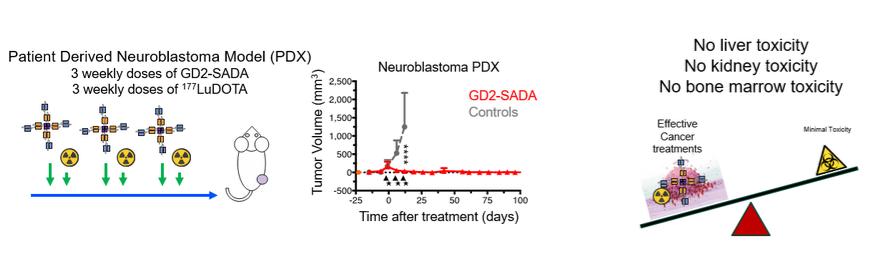
我們正在使用SADA技術來推進一系列基於SADA技術的抗體構建,在兩步法注射放射性有效載荷之前,雙特異性抗體片段與腫瘤結合。我們已經指定GD2-SADA用於GD2陽性實體瘤作為我們的第一個SADA構建,並於2021年12月提交了GD2-SADA的IND。我們相信,SADA技術可以潛在地提高放射性標記藥物在腫瘤中的療效,這些腫瘤在歷史上沒有表現出對放射性標記藥物的有意義的反應。

8