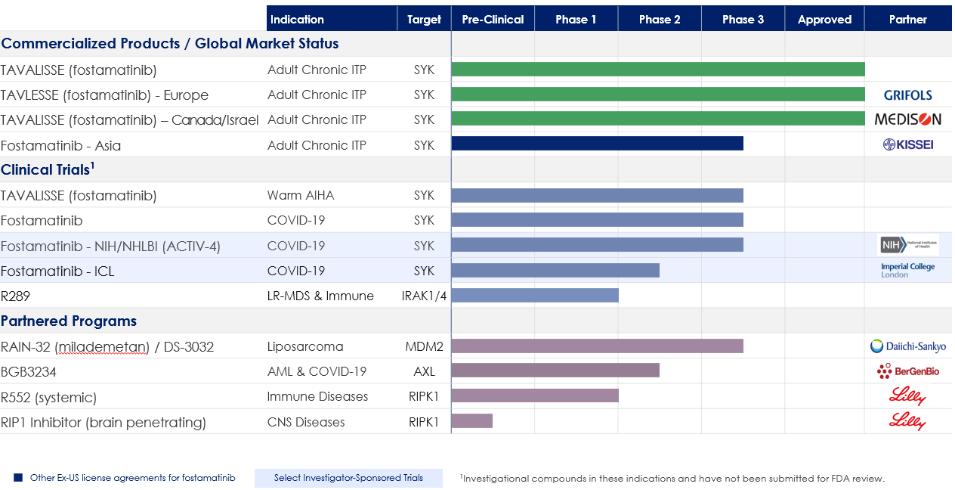
2020年1月,歐盟委員會批准了在英國脱離歐盟後在整個歐盟(EU)和英國有效的福斯塔替尼的集中營銷授權(MA),用於治療其他治療無效的成人患者的慢性免疫性血小板減少症。有了這一批准,我們在2020年2月收到了一筆20.0百萬不可退還的付款,其中包括$17.5在EMA批准福斯塔替尼的第一個適應症的銷售授權申請(MAA)時應支付百萬美元,以及$2.5根據我們與Grifols的合作協議條款,支付100萬可抵免的預付版税。上述里程碑式的付款被分配給與Grifols合作協議中的不同業績義務。
我們根據ASC 606對這項協議進行了説明,並在協議開始時確定了以下明確的履行義務:(A)許可的授予,(B)與我們正在進行的針對ITP患者的長期開放標籤擴展研究相關的研究和監管服務的表現,以及(C)與我們在AIHA進行的第三階段研究相關的研究服務的表現。2020年10月,我們簽訂了許可地區的商業供應協議。我們得出的結論是,這些履行義務中的每一項都是不同的。我們的評估基於以下幾點:(I)我們的評估認為Grifols可以通過使用自己的資源開發和商業化底層產品而從許可證本身受益,以及(Ii)製造服務本質上不是高度專業化的,可以由其他供應商執行。在執行我們與Grifols的協議後,我們確定預付費用$5.0百萬美元,這是美元中不可退還的部分30.0百萬預付手續費,代表交易價格。在2020年第一季度,我們修改了交易價格,將25.0根據我們的協議不再可以退還的預付款中的100萬美元和20.0收到的付款不再受限制。我們根據對相對獨立銷售價格的最佳估計,將更新後的交易價格分配給協作協議中的不同履行義務,具體如下:(A)對於許可,我們使用調整後的市場評估方法估計獨立銷售價格,以估計其在許可區域的獨立銷售價格;(B)對於研究和監管服務,我們使用成本加預期利潤率方法估計獨立銷售價格。由於調整後的交易價格,調整在累積追趕的基礎上記錄,並作為2020年第一季度合作的合同收入的一部分記錄。
剩餘的未來可變對價為#美元277.5與未來監管和商業里程碑有關的100,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000美元與未來的監管和商業里程碑相關的收入受到了充分的限制,因為考慮到這些未來的里程碑的成功的內在不確定性,累積收入很可能會發生重大逆轉。在使用輸入法的各個臨牀項目期間,我們確認與研究和監管服務相關的收入。對於基於銷售的里程碑和特許權使用費,我們確定許可證是與特許權使用費或基於銷售的里程碑相關的主要項目。因此,吾等將於(I)相關銷售發生時或(Ii)已獲分配部分或全部特許權使用費的履約義務已獲履行(或部分獲履行)時確認收入。我們將在每個報告期內以及在不確定事件得到解決或情況發生其他變化時重新評估交易價格。
截至2021年12月31日和2020年12月31日,剩餘遞延收入為美元0.7百萬美元和美元1.6百萬美元,分別與研究服務的業績有關。在截至2021年12月31日的年度內,我們確認了0.9與研發服務相關的收入為100萬美元。我們還確認了$2.0將藥品供應交付給Grifols進行商業化的收入為100萬美元。在截至2020年12月31日的年度內,我們認識到$39.9與知識產權許可權利有關的收入為百萬美元,$3.8與所執行的研究服務相關的收入為100萬美元。此外,我們認識到$0.7將藥品供應交付給Grifols進行商業化以及$0.5如上文所述,與Grifols行使其將更多領土包括在內的選擇權有關的100萬美元。於截至2019年12月31日止年度內,我們確認$4.7與許可權以及執行的研究和監管服務相關的收入為100萬美元。