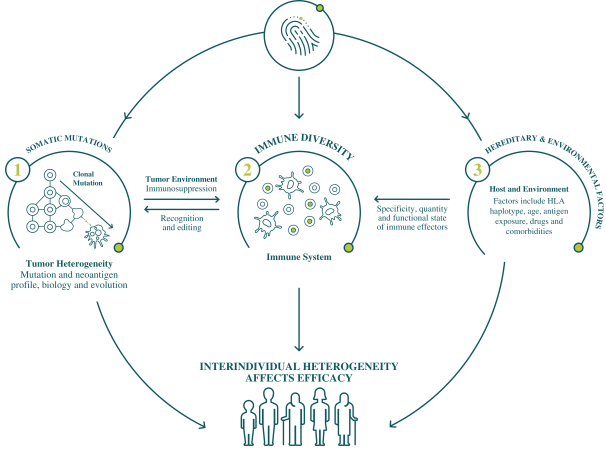
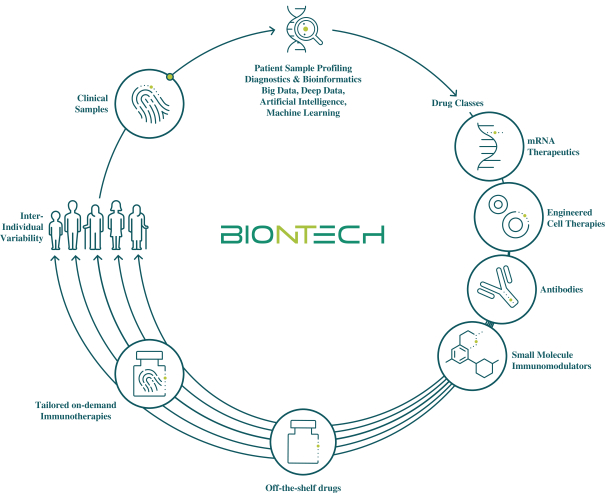
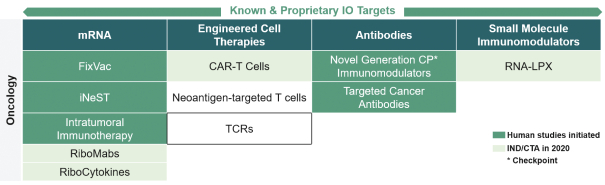
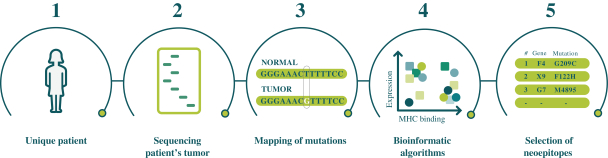
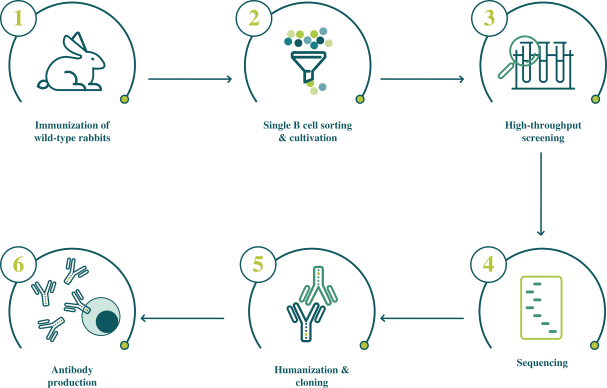
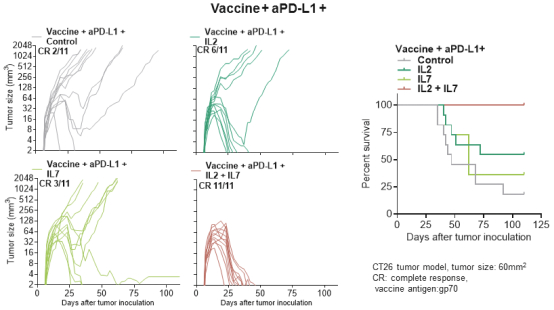
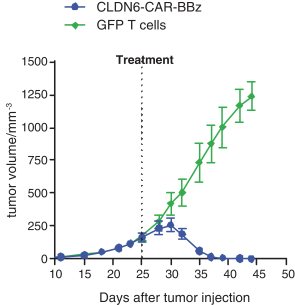
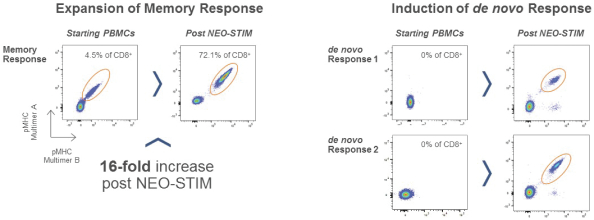
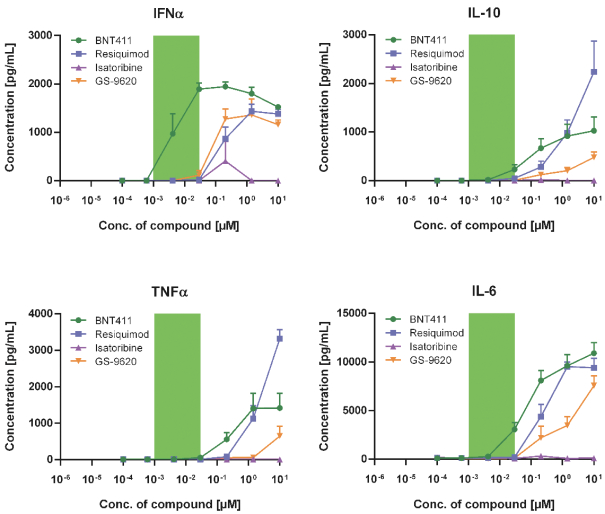
配股最多7,505,596股普通股,包括以美國存托股份為代表的普通股

我們向普通股東提供認購新普通股的權利,並通過紐約梅隆銀行、我們的託管人和ADS 權利代理向美國存托股份(ADS)持有人提供根據供股或供股認購新ADS的不可轉讓權利。若干普通 股東已不可撤銷地同意在本次發售中不轉讓或行使其購買總計5,616,407股普通股的權利,代表我們普通股的5,500,000股新美國存託憑證以承銷 公開發售或包銷發售的方式發售,價格與本次供股中發售的新普通股和新美國存託憑證相同。因此,我們預計在供股中發行最多1,889,189股新普通股,包括以美國存託憑證為代表的 股普通股。每一股新的ADS代表一股新的普通股,每股沒有面值。這些美國存託憑證在納斯達克全球精選市場上市,代碼是BNTX。2020年7月22日,納斯達克全球精選市場上美國存託憑證的最新報告售價為104.12美元/ADS。我們的普通股沒有在任何國家的證券市場或交易所上市。
向美國存託憑證持有人提供
美國存託憑證持有者將 在下午5:00收到每一個已登記ADS的ADS版權。(紐約市時間)2020年7月24日。31 ADS權利持有人將有權以每ADS 93美元的價格認購和購買一個新的ADS。 要認購新的美國存託憑證,ADS權利持有人必須向紐約梅隆銀行支付該認購價。部分美國存託憑證將不會發行。ADS轉播權將於上午12:01到期。(紐約時間)2020年8月14日。請參閲向美國存託憑證持有人提供的配股説明。
向普通股持有人要約
普通股持有者將在晚上11點59分之後的一分鐘內,以每股登記在冊的普通股換取一股普通股權利。(德國美因茨 時間)2020年7月29日。31股普通股將使此類權利的持有人有權認購和購買一股新普通股,認購價為每股新普通股80.32歐元,相當於每股新ADS的 美元價格,根據2020年7月22日的有效匯率換算。或者,普通股持有者可以改為支付每股新普通股93美元,這是每股新ADS的美元價格。不會發行 股零碎普通股。認購新普通股的權利將在晚上11點59分後一分鐘到期。(德國美因茨時間)2020年8月14日。請參閲向 普通股持有人提供配股的説明。
全球服務
如本招股説明書所述,供股和包銷發售是最多7,505,596股普通股(包括由美國存託憑證代表的普通股 )的單一全球發售的一部分。
投資我們的普通股和代表我們普通股的美國存託憑證涉及高度風險。請參閲本招股説明書第22頁開始的風險因素。
我們是一家新興的成長型公司,也是一家根據美國聯邦證券法定義的外國私人發行人,因此,我們有資格 降低上市公司的披露要求。有關更多信息,請參閲招股説明書摘要和作為新興成長型公司和外國私人發行商的影響。
美國證券交易委員會(Securities And Exchange Commission)和任何州證券委員會都沒有批准或不批准這些證券,也沒有確定本 招股説明書是否真實或完整。任何相反的陳述都是刑事犯罪。
配股中的普通股和美國存託憑證預計將於2020年8月27日左右交付。
經銷商經理
| 摩根大通 |
美國銀行證券 |
貝倫伯格 |
日期為2020年7月23日的招股説明書