目錄
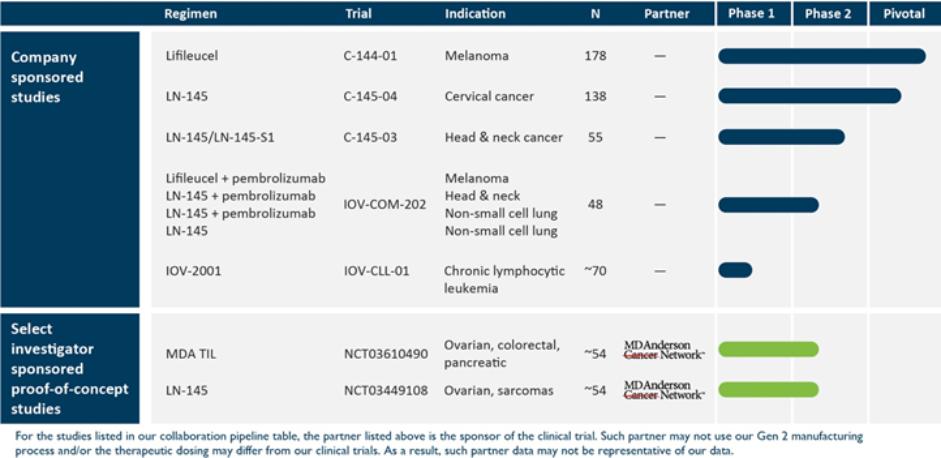
在2019年4月,我們結束了IOV-Lun-201臨牀試驗,研究TIL治療非小細胞肺癌,並結合抗PD-L1治療,啟動上述IOV-COM-202試驗的隊列3A和3B,以適應我們的臨牀發展計劃,以反映非小細胞肺癌治療前景的進展。
作為我們與MD安德森癌症中心(MDACC)合作項目的一部分,2018年開始了兩項第二階段的試驗。這兩項試驗都是由MDACC贊助的。第一次試驗,2017-0672或NCT 03449108,旨在允許調查我們生產的LN-145,使用我們的製造工藝治療軟組織肉瘤、骨肉瘤和耐鉑卵巢癌患者。與MDACC合作進行的第二次審判NCT 03610490也在進行中。這項試驗正在治療鉑耐藥卵巢癌、胰腺癌和結直腸癌患者。本試驗使用MDACC製造的TIL,使用Urelumab,一種4-1BB激動劑抗體,作為製造過程的一部分。使用這個製造過程獲得的數據可能不能代表我們使用第二代製造過程的數據。
我們目前的產品候選管道和選定的調查員贊助的概念證明研究總結如下:
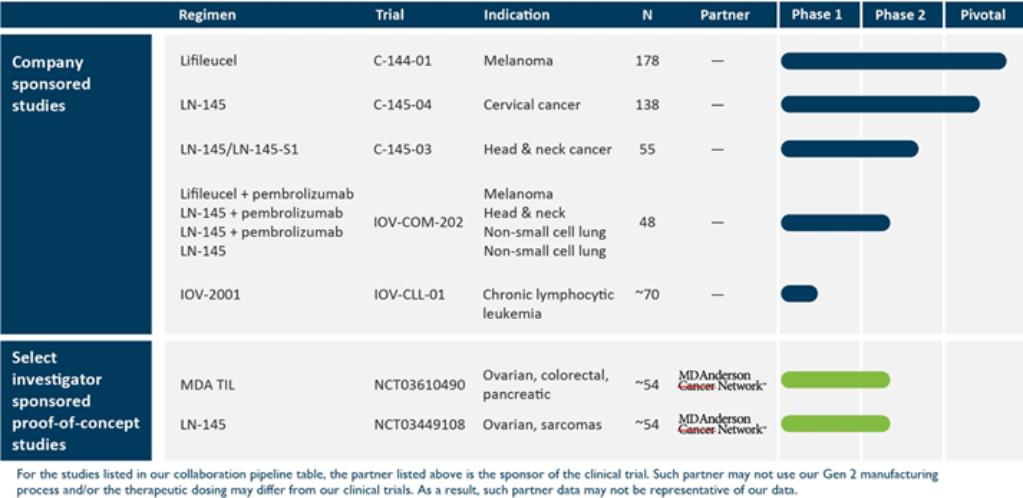
我們還與加拿大的一個機構--蒙特利爾大學中心醫院--開展了合作,根據該機構,Chum同意進行一項臨牀研究,該研究使用一位夥伴合作者使用Chum開發的工藝製造的Pd-1選定的TIL產品進行臨牀研究。我們也正在開發一種Pd-1精選的TIL產品,由Iovance公司生產.該產品,稱為LN-145-S1,將在C-145-03試驗中對頭頸部癌症患者進行初步測試。
我們開發了第三代製造工藝,稱為第三代。第三代比第二代更短。我們在頭頸部癌症的C-145-03試驗中使用第三代。
我們目前擁有與我們的第二代製造工藝有關的多種癌症的成分和治療方法的美國專利,包括美國專利10,130,659,10,166,257,10,272,113,10,363,273,10,398,734,10,420,799,10,463,697和10,537,595。我們擁有和授權的知識產權組合還包括與TIL、骨髓浸潤和外周血淋巴細胞療法、製造方法、在TIL治療和製造中使用共刺激分子、穩定和短暫的轉基因TIL療法以及治療患者亞羣的方法有關的專利申請。
我們以前與Roswell Park癌症研究所(RPCI)合作,使用我們的LN-145產品與抗Pd-1抗體pbrolizumab聯合治療膀胱癌患者。由於入學人數不足,這一審判於2019年結束。
6