カタログ表
TIL治療過程、コア生検による腫瘍組織、その他の遺伝子組換えTIL療法を獲得し、多数の免疫チェックポイント遺伝子編集とサイトカイン結合系TIL療法、及びTIL治療過程と治療方案全体に関連する製造スケジュール、サンプル収集と支持治療を改善する潜在的な経路として、IOV-3001と呼ばれる新型インターロイキン2或いはIL-2類似体を含む。
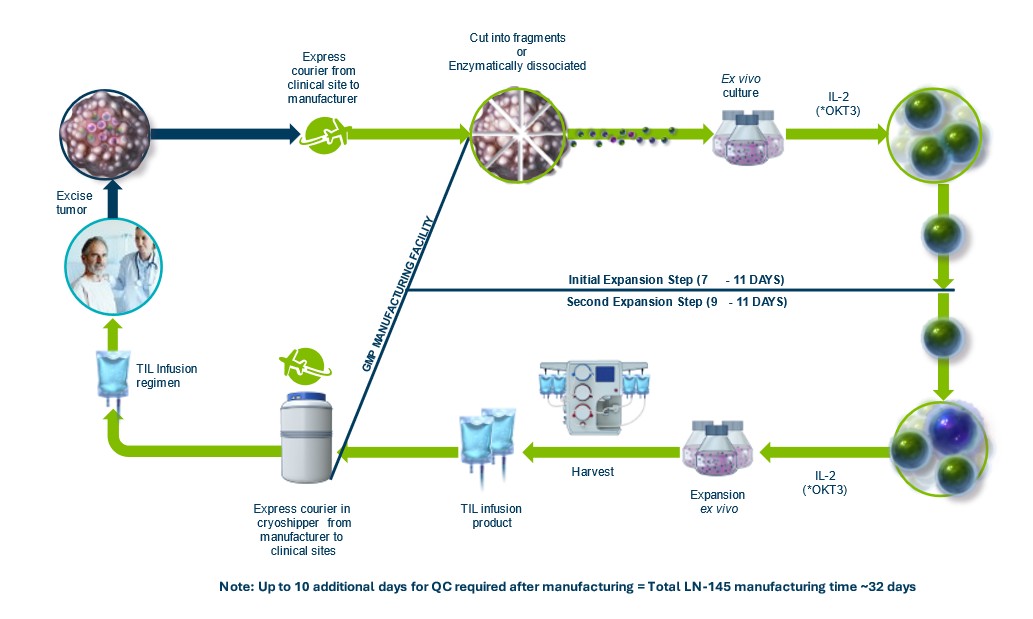
我々の現在の開発フローの要点を以下の図に示す

プラットフォーム技術と製造
著者らはT細胞に基づく免疫治療技術プラットフォームは多くの固形腫瘍タイプと血液癌に適用可能である。各プラットフォームは、各患者固有の異なる癌細胞を識別し、攻撃するために、患者固有の細胞を利用することに集中している。特定の腫瘍によく見られる単一あるいは少量の共通抗原標的に作用する細胞療法とは異なり,われわれのポリクローナルT細胞は患者や腫瘍特有の様々な新しい抗原に対する個人化療法である。多くの固体腫瘍免疫標的は患者特有であり、1%未満の患者だけが共有している。TIL治療は著者らが多種の末期固形腫瘍の中でT細胞に基づく免疫治療のリードプラットフォームである。血液癌に対して、著者らの末梢血リンパ球或いはPBL治療プラットフォームは患者の血液サンプルから収集したポリクローナルT細胞に基づいて、その後増幅と活性化を行う。
末期、転移性或いは切除不可能固形腫瘍におけるTILの臨床研究進展
これまでに国家癌研究所(NCI)を含む単一学術センターで行われたTIL治療臨床試験に基づいて、著者らはTILが末期黒色腫、子宮頸癌、NSCLCと頭頸部扁平上皮癌(HNSCC)を治療する全世界多中心2期臨床試験を研究した。われわれの臨床試験に関するより多くの情報を以下にまとめた。
抗PD-1末期黒色腫の後、著者らは著者らのC-144-01臨床試験においてLimiileucelを研究しており、この臨床試験は著者らが提出したBLA申請を支持し、Limiileucelの抗PD-1末期黒色腫後の治療のための使用を許可する可能性がある
PD-1治療に興味のない一線末期黒色腫患者の中で、著者らはIOV-COM-202臨床試験とTILVANCE-301第三段階臨床試験においてLiLieucelとPembrolizumabの連合応用を研究している。TILVANCE-301はランダムな3期臨床試験であり、末期一線黒色腫の登録を支持し、末期抗PD-1黒色腫を完全に承認した後の黒色腫の実証試験とすることを目的としている
5