アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
1934年証券取引法第13条又は15(D)条に基づいて提出された |
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 識別番号) |
(主にオフィスアドレスを実行) |
(郵便番号) |
登録者の電話番号、市外局番を含む:(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
同法第12条(G)に基づいて登録された証券:ありません
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してください。はい、そうです☐
登録者がこの法第13節または第15節(D)節に基づいて報告を提出する必要がないかどうかを再選択マークで示す。はい、そうです☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13または15(D)節に提出されたすべての報告を提出したかどうか、および(2)このような提出要求を過去90日以内に遵守してきたかどうかを、再選択マークで示す
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
大型加速ファイルサーバ |
|
☐ |
|
|
☒ |
|
|
|
|
|
|||
非加速ファイルサーバ |
|
☐ |
|
規模の小さい報告会社 |
|
|
|
|
|
|
|
|
|
新興成長型会社 |
|
|
|
|
|
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する☐
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する☐
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す☐
登録者が空殻会社であるか否かをチェックマークで示す(同法第12 b-2条で定義される)。はい、そうです
2022年6月30日現在、登録者の非関連会社が保有する投票権と無投票権普通株の総時価は、登録者普通株のその日のナスダック全世界精選市場における最終報告販売価格に基づいて、1株当たり額面0.001ドル、約0.001ドルである$
2023年2月25日現在、登録者が発行した普通株の数は
引用で編入された書類
登録者は、2022年12月31日までの財政年度終了後120日以内に証券取引委員会に提出する2023年株主総会の最終委託書の内容の一部を、引用により本年度報告の第3部Form 10−Kに組み込む予定である。
カタログ表
|
|
ページ |
前向き陳述に関する特別説明 |
II |
|
リスク要因をまとめる |
四 |
|
|
|
|
第1部 |
|
|
第1項。 |
業務.業務 |
1 |
第1 A項。 |
リスク要因 |
52 |
項目1 B。 |
未解決従業員意見 |
122 |
第二項です。 |
属性 |
122 |
第三項です。 |
法律訴訟 |
122 |
第四項です。 |
炭鉱安全情報開示 |
122 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
123 |
第六項です。 |
保留されている |
124 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
125 |
第七A項。 |
市場リスクの定量的·定性的開示について |
142 |
第八項です。 |
財務諸表と補足データ |
142 |
第九項です。 |
会計と財務情報開示の変更と相違 |
142 |
第9条。 |
制御とプログラム |
142 |
プロジェクト9 B。 |
その他の情報 |
144 |
プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示 |
144 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
145 |
第十一項。 |
役員報酬 |
145 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
145 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
145 |
14項です。 |
チーフ会計士費用とサービス |
145 |
|
|
|
第4部 |
|
|
第十五項。 |
展示·財務諸表明細書 |
146 |
プロジェクト16 |
表格10-Kの概要 |
148 |
i
前向きな特別説明について呉昌俊は声明した
このForm 10-K年間報告書は前向きな陳述を含んでいる。我々は1995年の個人証券訴訟改革法と他の連邦証券法における安全港条項に基づいてこのような前向きな声明を行った。前向きな陳述は歴史的事実でもなく、未来の業績の保証でもない。逆に、それらは私たちの現在の業務の未来、未来の計画と戦略、私たちの臨床開発スケジュールと結果、その他の未来条件に対する信念、期待と仮定に基づいている。“目標”、“予想”、“信じる”、“考慮”、“継続”、“可能”、“見積もり”、“期待”、“目標”、“予定”、“可能”、“進行中”、“計画”、“可能”、“潜在”、“予測”、“プロジェクト”、“求める”、“すべき”、“目標”、“目標”、“意志”、“将”またはこれらの用語の否定または他の同様の表現は、すべての前向き陳述がこれらの識別語を含むわけではないが、前向き陳述を識別することを意図している。
これらの前向きな陳述には、他に加えて、以下の態様に関する陳述が含まれる
II
このような展望的な陳述は経営陣の現在の予想に基づいている。これらの陳述は約束でも保証でもなく、既知と未知のリスク、不確定要素、および他の重要な要素に関連しており、これらのリスク、不確定要素および他の重要な要素は、私たちの実際の結果、業績または成果が展望性陳述と明示的または暗示する任意の未来の結果、業績または業績と大きく異なることを招く可能性がある。実際の結果が現在の予想と大きく異なる要素を招く可能性がある要素は、臨床試験の開始、実行と完成、臨床試験データの獲得可能性時間をめぐる不確実性、規制機関が行っている議論と行動、私たちの開発活動、および第1部1 A項で議論している他の要素を含む。“リスク要因”あなたは、本報告書のこれらのリスク要因および他の警告声明を、それらが本報告書に出現するにもかかわらず、すべての関連する展望的声明に適用されるものとみなさなければならない。リスク要因は網羅的ではなく、本報告書の他の部分には、私たちの業務および財務業績に悪影響を及ぼす可能性のある他の要素が含まれている可能性がある。このような不確実性を考慮して、あなたは未来の事件の予測としてこのような前向きな陳述に依存してはいけない。法的要求がない限り、私たちは未来に新しい情報があっても、これらの前向きな陳述を任意の理由で更新または修正する義務がない。
本10−K表年次報告で使用されるものは、他の説明や文意が別に言及されていない限り、用語“私たち”、“私たち”、“私たち”および“会社”は、ATEA製薬会社およびその子会社を意味する。本年度報告でForm 10−K形式で出現したすべてのブランド名または商標は,それぞれの所有者の財産である。
三、三、
まとめリスクF俳優
私たちの業務は、第1部1 A項で述べたリスクと不確実性を含む多くのリスクと不確実性に直面している。本年度報告表格10−Kにおける“リスク要因”。私たちの業務に影響を与える主なリスクと不確定要素は以下の通りです
四
v
第1部
第1項業務.業務.
概要
著者らは臨床段階の生物製薬会社であり、深刻なウイルス感染患者の生活を改善するために、抗ウイルス治療薬の発見、開発と商業化に専念している。我々は新冠肺炎の治療に用いられる主要候補製品であるフェニフブビルを開発しており,重症急性呼吸症候群コロナウイルス2(“SARS−CoV−2”)とその変種に感染することによる疾患である。フェニルニホブビルとルザスビルの併用によるC型肝炎(C型肝炎)の治療も開発されている。
新冠肺炎は全世界の健康危機を引き起こし、数百万人の人が死亡し、多くの生存者の医療問題が頭から離れない。新冠肺炎の予防と治療において多くの迅速な進展を得たが、現在ワクチンと治療選択の制限のため、アメリカと全世界の大量のハイリスク群に対する医療需要はまだ満足されていない。われわれの新冠肺炎戦略の核心は,ベネホブビルを単一療法の開発とし,それを新冠肺炎療法の一部とすることであり,ベネホブビルと別の抗ウイルス薬を組み合わせ,これらの現在のワクチンや治療が依然として適用されていないハイリスク患者に専念することである。我々の目標は,依然としてSARS−CoV−2感染により入院·死亡しやすい個人に安全で有効かつ便利な治療選択を提供することである。
ワクチンや治療法があっても,新冠肺炎は米国で3番目の死亡原因であり,心臓病や癌に次ぐ。アメリカ疾病コントロール·予防センターの報告によると、2023年2月15日まで、アメリカでは毎日400人を超える人が新冠肺炎或いは関連合併症で死亡した。この人たちの75%以上の人の年齢が65歳以上だ。また,最近,2023年2月15日現在,60歳以上が米国の現在の新冠肺炎に関連する入院患者の70%を占めることが報告されている。
米国政府は最近、新冠肺炎に関連する公衆衛生緊急事態の発表を終了する計画を発表したが、新冠肺炎は今後しばらく深刻な地方的脅威になると予想される。新冠肺炎がまだ地方的脅威になる可能性がある原因は:(1)症状出現前のウイルス伝播;(2)全世界のワクチンの発売にばらつきがある;(3)ワクチンの持続的な躊躇;(4)自然感染とワクチン接種による免疫持続時間は限られている;(5)あるSARS-CoV-2変種に対するワクチンの効力は限られている;(6)薬物と薬物の相互作用、安全性と耐性などの既存の抗ウイルス薬物の局限性を内服する;(7)ワクチンの伝播に対する不確定な影響;(8)ウイルスは進化し続け、内因性免疫とワクチン誘導の免疫を逃避する;(9)マスク着用や社交距離のようなウイルス伝播の緩和行動を減少させる.
SARS-CoV-2変種の絶えずの出現はより大きな伝播性を持ち、更に深刻な疾病を招く可能性があり、更に普通の人群中の他のウイルス緩和行為の減少、及び公衆衛生緊急休暇の終了に関連する結果が予想され、現在の治療方法はこれらの患者にとって限られており、特にウイルスと関連疾病の影響を受けやすい。これらの要因を考慮して,我々はこれらの脆弱な患者のニーズを満たすために潜在的な治療法としてbemnifosbuvirを開発している。
新冠肺炎が引き続き深刻な全世界風土病になることに伴い、私たちはアメリカが引き続き最も重要な商業市場を占めることに伴い、新冠肺炎治療市場は今後数年間も数十億ドルの機会になると信じている。アメリカでは、新冠肺炎商業市場はすぐに単一の政府支払者からより伝統的な支払者ルート、例えば連邦医療保険、連邦医療補助、個人商業保険に移行することが予想される。これらの第三者支払人の精算を決定する主な考慮要因はコスト/価値分析であり,これはある程度入院の経済的負担によって推進され,特にハイリスク群であると予想される。
1
ベニホブビル
著者らのチームは数十年の革新的な抗ウィルス治療で得られた専門知識と経験を利用して、Bemnifosbuvirを設計し、1種の研究中の、独自の、有効かつ選択的なヌクレオチドポリメラーゼ阻害剤は、単一療法のすべての薬物として開発することができ、他の抗ウィルス薬物と共同開発することもできる。Bemnifosbuvir(AT-527)は著者らの内部発見計画に由来し、この計画は独特なヌクレオチドステントを新型二プロドラッグと結合し、目的はウイルス複製の中心酵素を抑制することである。このような二プロドラッグ部分を利用する方法は、一本鎖RNA(“ssRNA”)ウイルスの複製を防止し、同時に宿主細胞への毒性を回避することを目的とした経口抗ウイルス製品を創出するために、フェニニホブビルの活性代謝物を最大限に形成できると信じている。非臨床研究において、著者らはすでにフェニニホブビルが独特な作用機序を有し、RNA依存性RNAポリメラーゼ(RdRp)鎖の終結とSARS-CoV-2ウイルス及びその変異体のNidVirus RdRp関連ヌクレオチドトランスフェラーゼ(Niran)を含むことを証明した。このようなユニークな二重作用機序によりこれらの高度に保守的な部位を標的にすることにより,bemnifosbuvirは高い耐性障害を生じる可能性がある。さらにここでは体外培養我々が行った研究では,フェニニホブビルはすべてのテストされたオミク戎亜変種を含む新冠肺炎変種における抗ウイルス活性を維持した。
新冠肺炎の臨床研究
2022年11月、著者らは世界的、多中心、無作為、二重盲検、プラセボ対照の3期臨床試験であるSunISE-3を開始した。日の出3日には,少なくとも1,500名の軽中度新冠肺炎を有するハイリスク非入院患者に対して,フェニホブビル(1日2回,計5日間)の評価を行っている。この試験は米国,ヨーロッパ,日本,世界の他地域の臨床試験地点で行われる。患者群は疾患進展リスクが最も高い群から構成され、80歳の患者80歳、1つ以上の主要なリスク因子を有する65歳患者および18歳の免疫機能低下患者を含み、これらはすべて新冠肺炎接種状態と関係がない。
“日の出3号”の設計目的は,ベネホブビルを単一療法として評価(予備分析)することであるが,抗ウイルス薬をベネホブビルとともに治療を受ける比較的小さい患者のサブセットにおける併用療法の効果も探索する(二次分析)。この試験には、受けた看護基準(“SOC”)から得られた2つの集団が含まれる:1)“看護支援集団”(承認された抗ウイルス治療を受ける資格のない患者、または現地で抗ウイルス薬を有さない患者)は、単一療法としてベンフォビルに投与された患者(予備分析)および2)“併用抗ウイルス集団”を評価し、SOCが他と互換性のある抗ウイルス薬による新冠肺炎に対する治療を含む場合、併用治療の状況を評価する(二次分析)。患者は、Bemnifosbuvir 550 mg Bidプラスローカル利用可能SOCまたはプラセボBIDプラスローカル利用可能SOCを1:1のランダム割合で5日間受け入れる。
日の出-3研究の主な終点は、支持性看護集団中の少なくとも1300人の患者のすべての原因が入院または死亡して29日目までであり、この集団においてプラセボと比較して臨床的意義のある入院/死亡の減少を検出する能力があることである。試験参加患者の中で最も疾患進展リスクの高い患者を増加させることにより,入院率/死亡率は~4−6%を目標とした。1つの中期分析は1つの独立したデータ安全監視委員会(“DSMB”)によって行われ、60%の患者が研究のARMに登録した後、支持性看護群に組み入れられる。各支持性看護患者群と連合抗ウイルス群中の副次的な終点は新冠肺炎合併症、受診、症状リバウンド/再発とウイルス負荷リバウンドを含む。
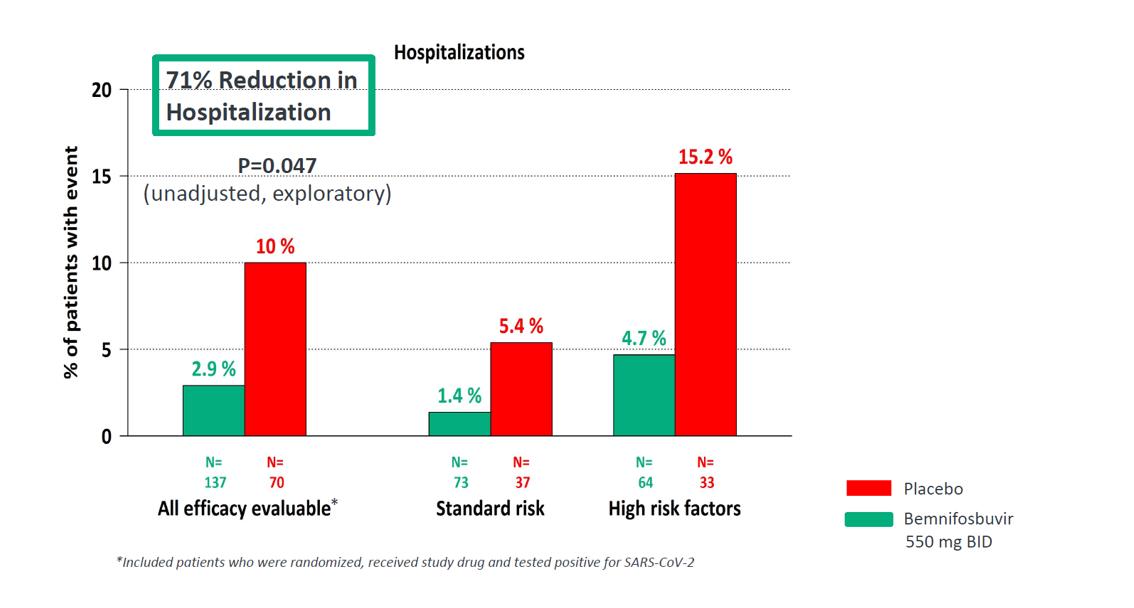
我々が日の出−3号の設計を支持するフェニホブビルの以前に研究したデータは、MONINGSKYと呼ばれる第3段階臨床試験の結果を含み、この試験は第1段階薬物相互作用(DDI)研究の結果と共に閉鎖された。MORNINGSKY研究の主な終点である症状緩和時間は達していないが,MORNINGSKYの結果,プラセボ(70)と比較してフェニホブビルを服用した患者の入院期間は71%(2.9%対10%)減少した(p=0.047,無調整,探索性;副次的終点)。MORNINGSKY試験に参加した患者は広範な外来群を含み、その中の47%はハイリスク患者であり、28%はワクチン接種患者であり、56%の患者はベースライン血清陽性患者であった。中のサブグループ分析では
2
40歳以上の患者では,MONINGSKY試験でベネホブビルを服用した患者の入院率は82%と減少した。
これまでに5つの臨床DDI研究が完了し,ベネホブビルに関連するDDIの潜在力は全体的に低く,ベネフォブビルとCYP 3 Aを基質とする薬物や外排や肝臓摂取トランスポーター感受性基質としての薬物の併用投与は投与量の調整を必要としないことが証明された。チトクロームP 3 Aは多種の薬物と補充剤を代謝する酵素であり、その敏感な外排基質と肝臓摂取トランスポーターは多くの薬物の細胞輸送を調節しており、これらの薬物は通常新冠肺炎のハイリスク患者に処方される。
これらのDDI研究では,BemnifosbuvirはCYP 3 A 4(ミダゾラム),P−糖タンパク質(ジゴキシン,シクロスポリン,カルバマゼピン),乳癌耐性蛋白と有機陰イオントランスポーターポリペプチド1 b 1(ロシュバスタチン)の指標薬物とともに使用されている。薬物相互作用の低可能性から,ベネフォブビルは通常他の疾患に用いられる常用処方薬とともに使用される可能性があり,特に疾患が重篤な新冠肺炎に発展する高リスク脆弱患者群である可能性が信じられている。
日の出3号の臨床試験を行うとともに,候補蛋白加水分解酵素阻害剤製品の発見に努めており,フェニニホブビルと併用し,免疫反応が生じず併用治療が必要な特定の新冠肺炎患者集団の治療に用いることができる。私たちはすでに行いました体外培養ベニホブビルとネマレビルを含むプロテアーゼ阻害剤類の抗ウイルス薬を併用した場合,付加的な抗ウイルス作用があることが明らかになった。私たちは、共同治療を受けた患者のサブセットから行われる日の出−3号臨床試験から得られるデータは、Bemnifosbuvirといくつかの他の現在許可されている抗ウイルス治療との組み合わせを評価する第1の臨床データであると信じている。
総合療法
多種の異なる作用機序を有する直接作用抗ウイルス薬物を利用して連合治療を行うことは既定の策略であり、多くの生命を脅かすウイルス疾患の治療において歴史上の成功を得ており、ヒト免疫不全ウイルス(HIV)、B型肝炎ウイルス(B型肝炎ウイルス)とC型肝炎ウイルスを含む。核種(T)類似体は多くの成功した併用療法の柱である。有利なことに,薬物の組み合わせはウイルス複製周期における複数の点に対して同時に抗ウイルス活性を増加させる効果があり,単一の薬物を用いることで時間の経過とともに形成される可能性のある薬剤耐性にも対抗することができる。
C型肝炎ウイルスの臨床研究
慢性C型肝炎ウイルス感染の治療にはbemnifosbuvirとRuzasvirの併用が進められており,Ruzasvirは研究中のNS 5 A阻害剤である。全世界で約5800万人が慢性C型肝炎ウイルスを患っており,その中で米国は約240万人である。世界保健機関(WHO)は、全世界で毎年150万例の発病があり、死亡は399,000人であると報告した。C型肝炎の発病率の上昇に伴い、治療を受けた新しい患者の数量を相殺し、今後数年のアメリカのC型肝炎の流行率は変わらないと予想される。
著者らはbemnifosbuvirとruzasvirの結合は現在の看護標準を改善する可能性があると信じ、肝硬変を合併或いは合併しないC型肝炎ウイルス感染患者に差別化された短い治療コース、汎遺伝子型プロテアーゼ保留方案を提供する。
2023年第2四半期に,bemnifosbuvirとruzasvirを併用して単純なC型肝炎ウイルス感染患者を治療する第2段階臨床試験を開始する予定であり,肝硬変なしでも代償性肝硬変でも。この研究は汎遺伝子組み合わせの安全性と有効性を評価することを目的としており、この組み合わせは1日550 mg(“bemnifosbuvir”)と治療8週間後に毎日180 mgのルザスビル(Ruzasvir)を服用することを含む。約60名の患者を含む全遺伝子型の約280名のC型肝炎ウイルスに感染した未治療患者が,この第2段階の臨床試験に参加する予定である。研究の主な終点は安全性と治療後12週間の持続ウイルス学的応答(“SVR”)である。他のウイルス学的終点はウイルス学的失敗、治療後24週間のSVRと薬剤耐性を含む。
3
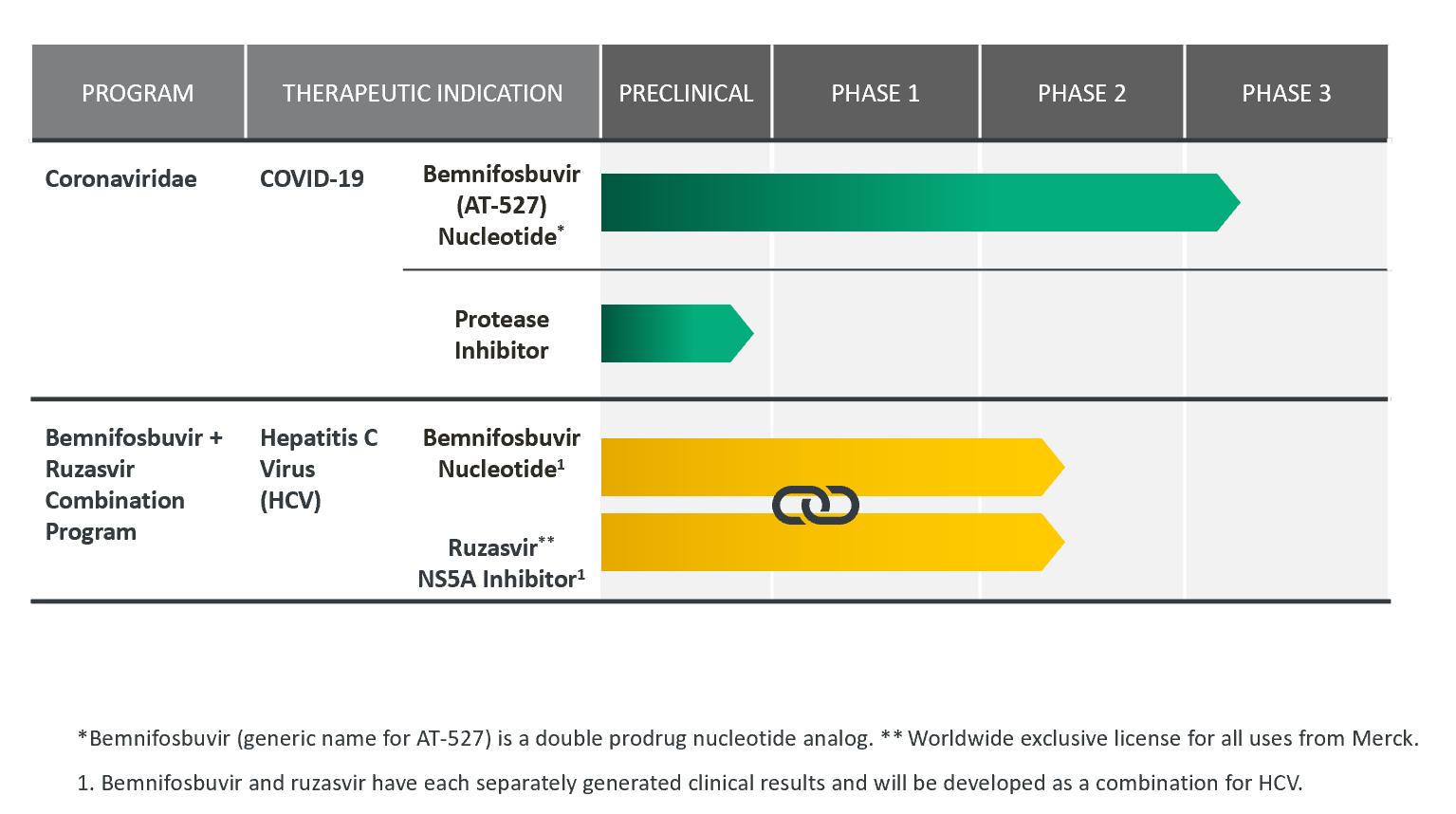
私たちの発展のパイプは
次の表に我々の経口抗ウイルス候補製品パイプラインをまとめた。私たちは私たちのすべての候補製品をすべての適応で商業化する完全な世界的権利を持っている。
デング熱と呼吸器合胞体ウイルス
2023年2月,AT−752を第2段階臨床試験に進めた後,デング熱の治療および予防にAT−752をさらに開発しないことにした。この行動をとったのは,患者登録予定期間が長く,感染直後に抗ウイルス薬の使用に成功した挑戦を含む期待される臨床操作課題であり,現在の診断テストでは不可能であり,大量のコストを含む個々のデング熱治療や予防のための抗ウイルス薬のさらなる開発に関する推定資源負担があるからである。
我々は最近,呼吸器合胞体ウイルス(“RSV”)を治療する候補製品を決定するために,我々の発見作業を継続しないことにした。この行動は私たちの管理チームのより集中を促進し、私たちの他の資源を私たちが計画しているより先進的な治療適応に配置するためです。
私たちは私たちが現在の計画を推進するための十分な資本を持っていると信じている。2022年12月31日現在、私たちは6.467億ドルの現金、現金等価物、有価証券を持っている。私たちの現在の計画によると、これらの財務資源は、現在と計画中の臨床プロジェクトを重要な転換点に推進し、重要な転換点を通過し、2026年まで私たちの活動に資金を提供することができると予想される。
私たちの戦略
重篤あるいは生命に危険なウイルス感染を発見,開発,商業化する新しい抗ウイルス療法の世界トップとなることを目標としている。私たちは以下の戦略を実施することでこの目標を達成するつもりだ
著者らの専門知識と経験、特に著者らの核種(T)類似物の深い知識を用いて、新しい或いは差別化された直接作用抗ウィルス薬物を発見と開発し、これらの薬物は満たされていない医療需要を満たす或いは現在の看護標準を改善する可能性がある私たちはデモンストレーションコースを持つチームを作り専門知識と経験を利用しています
4
高効率、成功的に発見、開発、全世界の監督管理の承認を得て、革新の直接作用経口抗ウィルス薬物を商業化した記録. 著者らのチームは満足されていない患者の需要、ウイルス学、薬物化学、特に核化学と最適化、薬物発見、臨床前と臨床開発、監督管理事務と商業化の面で非常に特殊な専門知識を持っている。私たちはこの分野の専門知識に頼っています
さらに私たちはこれらの専門知識を利用しています
ビンニホブビル(AT-527)を新冠肺炎の単一療法として開発し,現在の治療法の主要な限界を解決し,特定の患者群に対する併用療法を探索した。Bemnifosbuvirは研究、経口、非突然変異、非奇形、直接作用の抗ウィルス薬物であり、著者らの3期日の出-3臨床試験で単一療法として評価し、そして現地で使用可能なSOCの一部として他の抗ウィルス薬物と併用する。MORNINGSKYからの支持的データでは,bemnifosbuvir ARMを服用した患者の入院リスクはプラセボを服用した患者より71%(p=0.047,未調整,探索性;副次的終点)であった。40歳以上の患者に対する亜群分析では,入院期間の減少幅が82%と大きかった。
我々は現在最も満足されていない医療ニーズを解決するために,ニュー冠肺炎のためにフェニフブビルを開発している。具体的には,最も脆弱な患者群を対象としており,重篤な新冠肺炎や死亡に発展する最大のリスクに直面しており,現在彼らにとって治療選択が最も少ない。現在利用可能な経口抗ウイルス薬物は深刻な制限が存在し、ある患者群で使用される適合性を最大限に減少或いは除去し、しかも単クローン抗体は新冠肺炎変種と亜変種に対して有効ではなくなった。これらの制限には常用処方薬のDDISが含まれている てんかん薬、抗精神病薬、抗凝固剤など。そのほか、現在利用可能なワクチンも局限性が存在し、免疫力の低下と特定の人群で免疫反応を開始できなかったことを含む。
我々は,潜在的低リスクDDISを有するフェニルニホブビルの潜在製品プロファイルに対して,新冠肺炎の経口抗ウイルス薬に対する満足されていない需要を満たすと信じている。臨床研究で実現すれば,ベネフォブビルが承認されれば,この潜在的な状況は,ベネホブビルを新冠肺炎治療の単一療法と併用経口療法の礎にする可能性があると信じている。
本ニホブビルを単一療法として開発したほか,特定免疫不全群に対する新冠肺炎併用療法の開発を進めている離体するBemnifosbuvirと許可された直接作用抗ウイルス薬との組み合わせは、酵素阻害剤を含み、追加の抗ウイルス活性を示しており、我々は、将来のBemnifosbuvirとの併用治療のために、我々の内部酵素阻害剤計画を引き続き推進している。
C型肝炎ウイルスを治療するBemnifosbuvirとRuzasvirの汎遺伝子方案を提出し、看護標準を高める可能性があるそれは.直接作用のある抗ウイルス経口併用療法はC型肝炎ウイルスの治療に応用できるが、アメリカでは、まだ十分なサービスを受けていないC型肝炎患者の人口は引き続き増加している。発病率増加の大部分の原因はオピオイド危機、静脈投与とC型肝炎ウイルス再感染、特に若者の中である。メルク社とルザスビルによる臨床研究とわれわれが行ったベネホブビルの臨床研究はいずれも強力な抗ウイルス活性を示し,C型肝炎ウイルス感染患者に対して良好な耐性を示した。C型肝炎ウイルス複製抑制においても,フェニニホブビルとルザスビルの協同作用が認められた体外培養それは.私たちは学生募集を始める予定です
5
2023年第2四半期にフェニホブビルとルザスビルの組み合わせを評価する第2段階臨床試験 開発に成功し承認されれば,治療コースの短いbemnifosbuvirとruzasvirの組み合わせおよび酵素阻害剤のないレジメンが考えられる 米国と全世界で拡大しているC型肝炎ウイルス感染患者群に利益を与える可能性がある。
権利を保持し、有利な協力を選択的に求めることで、私たちの世界的な商業化カバー範囲を強化し、私たちの候補製品の価値を最大限に高める私たちは一般的に私たちの候補製品の世界的な開発と商業化の権利を維持するつもりで、私たちは製品の組み合わせの最大の潜在的価値を維持すると信じています。しかし、私たちが重要なビジネスインフラを構築する必要がなく、特定の市場専門知識や他の商業化資源を得る機会があると考えた場合、特に米国以外では商業化許可協定や協力を日和見的に締結することができるかもしれない。
許可内の機会に対する日和見主義的な態度を維持して、私たちのチャンネルを拡大する著者らは現在、第二世代プロテアーゼ阻害剤候補製品の潜在的な発見と臨床前に開発された内部研究活動に集中しているほか、第三者臨床段階の抗ウイルス薬候補薬を評価する際に日和見主義的な態度を維持する予定であり、これらの候補薬を許可して既存のルートを拡大する可能性がある。我々の科学的専門知識を利用して,許可内の機会を評価し続け,重大な満たされていない医療ニーズを満たすことができるようにしたり,既存の看護基準に基づいて実質的な改善を行うことができると予想される。
私たちのチームは
我々の管理チームは,生命を脅かすウイルス感染に対する抗ウイルス療法の発見,開発,商業化に豊富な経験を持っている。著者らの創業者、会長兼最高経営責任者Jean-Pierre Sommadossi博士は生物製薬業界で30年以上の科学、運営、戦略と管理経験を持っている。Sommadossi博士は180編以上の同業者評議の出版物を執筆し、抗ウイルスと癌治療に関連する135件以上の米国特許を持っている。Sommadossi博士は2014年にメルクに買収されたIdenix製薬会社(“Idenix”)の主要創業者であり、2012年にgilead Sciences,Inc.に買収されたPharmAsset,Inc.(“PharmAsset”)の共同創業者である。
著者らは経験豊富な管理と科学チームを結成し、抗ウィルス薬物開発領域で成功の記録があり、その中の多くの人は以前に協力したことがある。著者らのチームは核化学、生化学とウイルス学において豊富な専門知識を持っており、これらの専門知識を革新的な抗ウイルス療法の発見と開発に応用しており、Epivir、Sovaldi、Tyzeka、Valtrex、Wellferon、VIDAX、Reyataz、Sustiva、Mavyret、Xofluza、Relenza、Zerit、Zepatier、Epclusa、ハヴォニとVekluryを含む。私たちのチームのメンバーはアスリーカン、メルク、グラクソ·スミスクライン、CHIRON、ノファ国際株式会社、Biogen、F.Hoffmann La Roche、Abbvie、Bristol Myers Squibb、Shire、Bioaven Pharma、PharmAsset、Idienix、Valeant PharmPharmticals International、gilead Sciences、Inc.とAlnylam Pharmticalsで上級職を務めたことがあります。
抗ウイルス治療
ウイルスの背景について
ウイルスは細胞寄生虫であり、自身の生存と複製に必要な機序が乏しく、宿主細胞の複製過程でしか複製できない。ヒトなどのDNAを遺伝物質の基礎とする生物とは異なり,ウイルスはDNAやRNAを用いることができる。ウイルスの約70%がRNAウイルスですRNAウイルスは一本鎖(SsRNA)ウイルスまたは二本鎖(DsRNA)ウイルスであってもよく、具体的には遺伝物質として使用されるRNAタイプに依存する。
ウイルスには2つの主要成分がある:核酸(一本鎖或いは二本鎖RNA或いはDNA)と保護シェル(カプシド)。いくつかのウイルスはまた、カプシドの周囲に脂質二重層(エンベロープ)を有する可能性があり、カプシドは、ウイルスタンパク質を含む宿主細胞膜から誘導される追加の膜である。脂質二重層に包まれたウイルスをエンベロープウイルスと呼び,この二重層を持たないウイルスを非エンベロープウイルスと呼ぶ。エンベロープ単鎖RNAウイルスは深刻な人類ウイルス性疾患を引き起こすもっと普遍的な原因である。SARS-CoV-2はコロナウイルス科のコロナウイルスであり、C型肝炎ウイルスはフラビウイルスの一種であり、それらはすべて被膜の一本鎖RNAウイルスである。
6
ウイルスが付着蛋白を介して宿主細胞膜上の特定の受容体位置に付着すると,ウイルス感染やウイルス複製過程が開始される。ウイルス複製機序はウイルスがRNAウイルスかDNAウイルスかに依存する。ほとんどのDNAウイルスは、ウイルスゲノムを複製するため、またはメッセンジャーRNA(“メッセンジャーRNA”)を転写するために、宿主細胞タンパク質および酵素を使用して追加のDNAを製造する。RNAウイルスはそのRNAを合成ウイルスゲノムRNAとmRNAのテンプレートとして使用する。MRNAsはウイルスの複製と転写を担当する非構造蛋白をコードし、ウイルスの組み立てを担当する構造蛋白もコードする。最後に,新たに産生されたウイルス粒子(ウイルス粒子)が宿主細胞から放出され,感染と複製周期を繰り返す。RNAウイルスの治療は特に挑戦的である可能性があり、ウイルスRNAポリメラーゼが指導したRNA合成の誤り率は複製過程中の高い突然変異率を招き、抗ウィルス治療に変異と薬剤耐性挑戦をもたらす。
抗ウイルス標的であるウイルスポリメラーゼ
ウイルスポリメラーゼはすべてのRNAウイルスに存在する単一蛋白質であり、ウイルス複製の重要な酵素であり、抗ウィルス治療の魅力的な標的にさせる。そのほか、ウイルスポリメラーゼの核心構造特徴は異なるウイルス中で高度に保守され、このポリメラーゼに対する薬物はウィルス突然変異と薬剤耐性の影響を受けにくい。ウイルスとそのゲノム構成によると、ウイルスポリメラーゼには4つのタイプがある
ウイルスRNAポリメラーゼの合成はヒト宿主細胞では発生しないため、RNAウイルスに対する抗ウイルス薬物開発の重点は標的ウイルスRNAポリメラーゼの選択性薬物分子を識別することである。技術の進歩により、ウイルスRNAポリメラーゼの構造と機能を深く研究し、SARS-CoV-2症例でNidVirus RdRp関連ヌクレオチドトランスフェラーゼ(Niran)を確定し、より有効な新しい抗ウイルス療法の開発に道を開いた。
ウイルス耐性と変異体
直接作用する抗ウイルス薬の開発の主な課題の1つはウイルス耐性の出現である。薬剤耐性はウイルス遺伝子突然変異能力の機能であり、時間の経過とともに、ウイルスはある抗ウイルス療法にそれほど敏感ではない。校正能力に乏しいRNAウイルスの場合,突然変異率はDNAウイルスより大きく高く,宿主細胞の突然変異率よりも6桁高い可能性がある。
ウイルス突然変異のもう一つの期待と自然反復出現の結果は新しい変種の出現である。変種は原始ウイルスの新しい毒株であり、その遺伝子コードは原始ウイルスと唯一無二である。独特な遺伝子コードのため、変異ウイルスは多かれ少なかれ伝播性或いは毒力を有する可能性があり、そして原始ウイルスよりもっと深刻な疾病を招く可能性がある。また,変異の遺伝子コードの変化により,ワクチンや療法の有効性が時代遅れに低下する可能性がある。
2020年秋以降,SARS−CoV−2は迅速に変異することが証明され,600万個以上の変種が発見された。その中のいくつかの変異はWHOや疾患制御·予防センター(CDC)によって興味のある変異(VOI)として指定されており,伝播性が増加し,疾患がより深刻であり,ワクチンや抗体の有効性が低下したり,診断検出に失敗したりする証拠がある。しかしこれらの株は孤立した地域にしか出現していないかもしれません
7
他の国です。WHOや疾病管理センターではVOCもいくつか決定されており,それらの発現属性はVOISと類似しているが,より世界的に深刻な疾患を引き起こす可能性がある。従来識別されていたVOCにはアルファ,ベータ,ガンマ,デルタが含まれていたが,現在流通しているVOCはBA.1,BA.2,BA.3,BA.4,BA.5とその子孫スペクトル系である。
世界的には,2023年1月10日から2023年2月6日まで,99.6%のSARS−CoV−2配列がオミックのVOCであることが報告されている。オミックVOCでは,BA.5とその子孫が世界で主導的な地位を占めており,2023年1月16日から1月22日までに提出された全系列のうち,BA.5とその子孫は53.9%を占めている。オミック揮発性有機化合物の全世界での広範な伝播を考慮して、WHOはその変異体追跡システムにおいて新しいカテゴリー、即ち“監視している奥密克戎亜型”を追加し、これは優先的に注目と監視が必要かもしれない。現在モニタリング中の奥密克戎亜型には,BF.7,BQ.1,BA.2.75,CH.1.1,XBB,XB.1.5,XBFがある。
SARS-CoV-2の突然変異性質を考慮して、ウイルスの進化は継続し、より多くの変種が出現し、新しいと多様な健康挑戦をもたらすことが予想される。主要なSARS-CoV-2変異株の持続的な出現は新冠肺炎が大流行から地方性脅威に変化する重要な要素であり、このような脅威の下で、ウィルスは依然として伝播し、そして時々急増する。
核種(T)類似体とプロドラッグ
核酸は天然に存在するヌクレオシドとヌクレオチドと呼ばれる化合物からなり,細胞中の主な情報を持つ分子であり,タンパク質合成過程を指導することでヒトやウイルス遺伝物質の遺伝的特徴を決定する。核酸の2つの主なカテゴリーはDNAとRNAである。ヌクレオチド(T)類似体は、天然に存在するヌクレオシドおよびヌクレオチドの構造を模倣し、ウイルスポリメラーゼに対して直接、これらの類似体を新生核酸に誤って結合させ、ウイルス複製を抑制する合成化合物である。核(T)類似体は他のタイプの抗ウイルス療法と比較してウイルス耐性に高いバリアを有しており,これは生活ウイルス粒子を産生するために必要なポリメラーゼの構造が保存されているためである。
プロドラッグは生物活性が強くない化合物であり、ヌクレオシド類似物を通じて薬物放出を改善し、制限速度活性化ステップを迂回し、毒性を低下させ、経口バイオアベイラビリティと細胞膜透過性を高めることができる。ヌクレオシド(T)類似体のプロドラッグは、生命を脅かすウイルス感染(HIV、B型肝炎ウイルスおよびC型肝炎ウイルスを含む)を治療する単一薬物および併用薬物療法の柱となっている。
ベニホブビル
Bemnifosbuvirは研究中の、新規、独自の経口オルニチンヌクレオチド類似体の二プロドラッグである。より具体的には、多段活性化後に活性化される5‘-三リン酸代謝物AT-9010を代謝するホスファミドProTideの半硫酸塩AT-511であり、AT-9010はSARS-CoV-2およびC型肝炎ウイルス複製の阻害剤である。
我々の薬物化学者が設計したベネフォブビルは以下の重要な成分を含み、以下の目標の実現に努力している
8
これらの修正は、二重プロドラッグ法と共に、bemnifosbuvir以下の潜在的な有利な特徴および特徴を与える可能性があると考えられる
Bemnifosbuvirは二重機序を通じてウイルスRNAポリメラーゼに対して、これはウイルスの複製と転写に重要な高度に保存された酵素であるため、著者らはそれが受容体認識と宿主細胞膜融合過程を担当するスパイク蛋白突然変異を有する新たに出現した変異体に対抗する抗ウイルス活性を維持することが予想される。事実,過去および現在のVOCsに出現したポリメラーゼでは少数のアミノ酸置換(Y 273 H,P 323 LおよびG 671 S)がヌクレオシド三リン酸(NTP)結合部位やヌクレオチド(T)類似体の抵抗性部位から離れていた。それらは機能の異なるクラスターに属し、ヒト宿主の中で絶えず進化するウイルスに一般的な適応を提供し、薬剤耐性を産生する可能性はあまりない。また,フェニルフォブビル代謝経路中のすべての酵素は普遍的に存在する宿主細胞酵素であり,ウイルスがコードするタンパク質ではないことから,ウイルスの高い突然変異率はベンゼンブビルの活性化に影響しないと考えられる。
発展計画
SARS-CoV-2
背景
SARS-CoV-2は1種のコロナウイルスであり、コロナウイルス科に属し、嚢膜ウイルスであり、陽性の一本鎖RNAゲノムを有し、29種類のウイルス蛋白をコードする。それは現存する他の6種類のヒトコロナウイルスの一つであり、その中の4種類のウイルスは3分の1の普通風邪感染を招いた。
SARS-CoV-2は構造的に他の2種類の生命を脅かすコロナウイルスと類似している:SARS-CoVと中東呼吸症候群コロナウイルス(MERS-CoV-1)。
SARS-CoV-2は1種の球形ウイルスであり、4種類の異なる構造蛋白を持っている:刺突起蛋白、エンベロープ蛋白、膜糖蛋白と核カプシドタンパク質である。次の図に示すように,刺激性タンパク質が標的細胞表面のアンギオテンシン変換酵素2細胞受容体(“ACE 2”)に結合すると,感染周期が開始される。第二の細胞表面タンパク質、膜貫通セリンプロテアーゼ2(“TMPRSS 2”)は、ウイルス粒子を細胞に進入させ、そこでそのRNAを放出する。いくつかのRNAは宿主細胞の機序によって新しいタンパク質に翻訳される--これらのタンパク質は4種類の構造タンパク質と、いくつかの複製複合体を形成する非構造タンパク質(NSP)を含む。この複合体では,RdRpsは約30,000ヌクレオチドのRNAウイルスゲノムを触媒合成する。そしてタンパク質とRNAはゴルジ体中で新たなウイルス粒子に組み立てられ,細胞嘔吐作用により放出される。
9
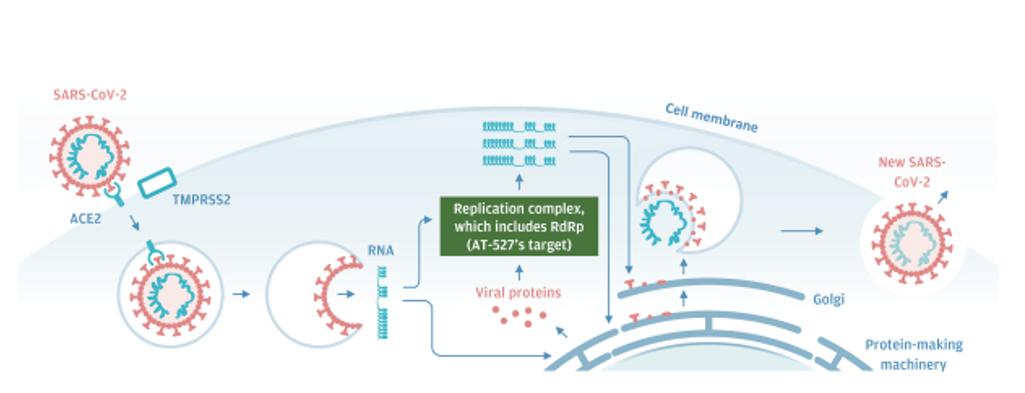
新冠肺炎−疾患概要
2019年のコロナウイルス病(“新冠肺炎”)はSARS-CoV-2及びその変種に感染することによる疾病であり、すでに全世界の大流行を引き起こし、2020年から迅速に世界を席巻し、そして免疫力の減弱とSARS-CoV-2変種の絶えずの出現により引き続き感染と疾病を招く。疾病管理センターのデータによると、2023年2月15日までに、米国だけで1億例以上の確定診断例が報告され、110万人以上が死亡した。世界保健機関は、世界で7.54億例以上の新冠肺炎確定例が報告され、700万人以上が死亡したと報告している。2022年、アメリカ疾病コントロール·予防センターは、新冠肺炎はアメリカの第三の死因であり、心臓病と癌に次ぐ、多くの死亡例は65歳以上の患者に発生すると報告した。高齢者やリスク要因のある人は新冠肺炎のより深刻な合併症を発症し,入院や死亡のリスクが高い。
SARS-CoV-2感染は無症状である可能性があり、一連の疾病を引き起こす可能性もあり、軽微な上気道感染から深刻な生命を脅かす敗血症と多臓器不全までを招く可能性がある。通常報告されている症状としては,発熱,咳,呼吸急,味覚喪失や嗅覚喪失,咽頭痛,疲労,頭痛,筋肉痛,胃腸(GI)障害がある。症状は通常2~3週間持続するが,多くの患者は数週間症状や新たな症状が出現し続け,現在では急性後新冠肺炎症候群,あるいは長期コロナウイルス感染と考えられている。新冠肺炎はすべての年齢層の人に影響を与える;しかし、免疫機能の低下、老年或いはいくつかの潜在的な疾患(例えば、慢性心肺と腎臓疾患;糖尿病、肥満と癌)を有する人の予後不良のリスクは増加する。
高齢者(合併症があるかないか)やどの年齢の免疫機能が障害されている人も,ウイルスに対して十分な免疫反応を生じる可能性が低いことを十分な証拠があり,ワクチンを接種しても十分な抗体反応に成功していないようである。また,これらの人の多くはリトナビルと薬物−薬物相互作用を有すると考えられる随伴薬を受け入れている可能性があり,ネマレビル/リトナビルの受け入れが禁止されていることを意味している。同じ集団の中で、Molnupiravirを使用したくないかなりの人がいる。なぜなら、その突然変異性と下流の結果につながる可能性があるからだ。ウイルスの進化や新しい変種の出現に伴い,モノクロナル抗体の用途も廃止された。この結果,これらの患者は現在有効な外来治療が得られず,治療としてレミッシビルの静脈内投与が必要である可能性が高く,より重篤な疾患で入院する可能性が高い。
米国政府は最近、新冠肺炎に関連する公衆衛生緊急事態の発表を終了する計画を発表したが、新冠肺炎は今後しばらく深刻な地方的脅威になると予想される。新冠肺炎がまだ地方的脅威になる可能性がある原因は、(1)症状出現前のウイルス伝播;(2)全世界のワクチン接種の不均衡;(3)持続的なワクチン躊躇;(4)自然感染とワクチン接種による免疫持続時間が限られている;(5)あるSARS-CoV-2変種に対するワクチン効力が限られている;(6)現在のワクチンの局限性がある
10
経口抗ウイルス薬、例えば薬物と薬物の相互作用、安全性の問題と耐性、(7)ワクチンの伝播に対する不確定な影響、(8)ウイルスは内因性免疫およびワクチンによって誘導される免疫の持続的な進化から逃避し、(9)マスク着用と社交距離のようなウイルス伝播緩和行動の減少。
新冠肺炎の予防と治療現状とその局限性
新冠肺炎の大流行が始まった時、この新しい疾病のワクチンと治療方案はすべてかつてない進展を得た。これらの進展を得たにもかかわらず、現在利用可能なワクチンと治療方法は依然として大きな局限性が存在し、自然獲得とワクチン産生に対する免疫力の減弱、一部の人群はワクチンに対して十分な免疫反応を産生できず、現在利用可能なSARS-CoV-2亜型に対するモノクロナル抗体は効力を欠く(これらの抗体は伝播性と中和抗体の能力を増加させた)。現在、経口抗ウィルス薬物の局限性はよく見られる処方薬物とのDDIS、例えばてんかん薬物、抗精神病薬物、抗凝固剤など、及び安全問題を含む。
そのため,新たな,安全,有効,便利,低薬物相互作用リスクの低い経口療法の開発が急がれており,これらの療法は単一療法としても併用療法の一部とすることができる。深刻な感染と伝播を予防する口腔治療が依然として切実に必要であり,特に現在の治療には限られた脆弱な患者を選択していると考えられる。これには、ワクチンを接種していない患者、既存のワクチンに無効な患者、ワクチン接種後に効力が低下した患者(この場合、免疫接種後3~6ヶ月以内に発生する可能性がある)、およびワクチン接種および既存の治療を禁止する患者が含まれる。新冠肺炎が地方的に流行するに伴い,持続的な変異による大流行が急増する可能性があり,この需要は長年続くと考えられる。
予防用ワクチン
いくつかのワクチンは承認されているか、または緊急使用許可(“EUA”)に従って許可されており、他のワクチンは新冠肺炎感染を防止するために開発されている。承認され許可されたワクチンには、ファイザー/バイオテクノロジー社のComirNatyやModerna社のmRNA-1273などのメッセンジャーリボ核酸ワクチンが含まれており、各一価ワクチンは、元の毒株によって引き起こされる症状のある新冠肺炎の予防に許可されている。2020年12月以来、これらのワクチンはアメリカと世界で発売されている。最近,2022年8月にファイザー/バイオテクノロジーとModernaがそれぞれ生産した二価遺伝子ワクチンが米国食品と薬物管理局の許可を得て,オミック亜型BA.4あるいはBA.5による症候性新冠肺炎の予防に用いられている。
ワクチンが疾病と伝播に対する持続的な免疫保護を産生する能力は現在、多種の要素によって制限されている
11
新冠肺炎治療におけるモノクロナル抗体の応用
2020年11月から2022年まで、FDAはいくつかのモノクロナル抗体を新冠肺炎の予防と/或いは治療に使用することを許可した。しかし、モノクロナル抗体による新冠肺炎の治療はずっと制限され、現在アメリカでは許可されておらず、原因は以下の通りである
新冠肺炎に対する抗ウイルス薬
ワクチンの補充として、抗ウイルス療法は新冠肺炎の治療のために承認または許可されている。アメリカではVeklury®(Redesivir)は、重度新冠肺炎の治療が進行する高リスク外来患者を含む新冠肺炎の治療のために承認されたRdRp阻害剤である。また、ラグフリオ(Molnupiravir)は、軽度から中度の新冠肺炎成人患者を外来治療する経口直接作用抗ウイルス薬、及びパシクロビル(リトナビル助剤ニマレビル)、軽度~中度の新冠肺炎成人患者を外来治療する経口蛋白加水分解酵素阻害剤であり、アメリカと世界の多くの他の国と地域で欧州連合A項で使用されることが許可されている。
現在許可または承認されている抗ウイルス療法の制限は:
12
我々の新冠肺炎戦略は
著者らはBemnifosbuvirを開発しており、研究中の経口新型抗ウイルス製品候補薬であり、新冠肺炎の治療に用いられている。
早い時期に終了した3期MORNINGSKY臨床試験のデータは症状緩和時間の主な終点に達していないが,bemnifosbuvir(n=137)を服用した患者はプラセボ(n=70)を服用した患者と比較して入院期間が71%(2.9%対10%)減少した(p=0.047,無調整,探索性;副次的終点)ことが示された。MORNINGSKY試験に参加した患者は広範な外来群を含み、その中の47%はハイリスク患者であり、28%はワクチン接種患者であり、56%の患者はベースライン血清陽性患者であった。40歳以上の患者に対する亜群分析では,MORNINGSKY試験でbemnifosbuvirを服用した患者の入院期間は82%減少した。入院患者の全世界第二段階研究においても臨床的メリット(全因死亡率)の傾向が観察された。疾患進展の低背景率により研究は最初の設計では完成できなかったにもかかわらず,研究中の3例の死亡はすべてプラセボを服用した患者で発生したが,フェニホブビルを服用した患者では死亡しなかった。また,Bemnifosbuvirは5項目1期臨床研究でDDISのリスクが低いことを示した すべての試験に対するVOCの抗ウイルス活性体外培養研究によると、変異原性あるいは催奇性はない体外培養研究では,その作用機序に鑑み,高い耐性障害が存在する。
単一療法として,フェニフブビルは現在の治療法の主要な限局性と持続的に満たされていない医療ニーズを解決する潜在力があり,特に治療選択が限られている高リスク患者に対して可能性があると信じている。我々はすでにSunISE-3を開始し、これは世界的な3期無作為、二重盲検、プラセボ対照臨床試験であり、少なくとも1,500名の軽中度新冠肺炎を有するハイリスク非入院患者のベネフォブビル(550 mg、2回5日)の治療効果を評価した。
“日の出-3”の主な設計目的はbemnifosbuvirを単一療法として評価することであるが(初歩的な分析)、bemnifosbuvirと適合する抗ウイルス薬を受けた比較的に小さい一部の患者における連合治療の効果(二次分析)を探索することも目的である。われわれは,併用療法による新冠肺炎治療としてのベネホブビルの効果を評価するために,併用治療を受けた比較的一部の患者のデータを用いて開発計画に情報を提供する予定である。
日の出−3号試験を行うとともに,第二世代蛋白加水分解酵素阻害剤を探すことに重点を置いて内部発見計画を進めており,ベンニホブビルと併用して新冠肺炎を治療する可能性がある。高効率、耐性が良く、DDISが限られ、薬物動態(PK)促進剤(例えばリトナビル)を必要としない蛋白加水分解酵素阻害剤を探している。パイロット化合物の最適化が進んでおり,2023年末に選定された臨床候補にINDを提出することを目標としている。
我々がこの潜在的な組み合わせを進めた理由は,ウイルス複製周期の異なる点に対する異なる作用機序を有する薬剤と,ウイルス複製周期の異なる点に対する異なる作用機序を有する薬剤を含む組み合わせ療法を歴史的に重篤なウイルス疾患を治療した前例に基づいている体外培養HCoV−229 E代理モデルで検討した。ここにあります体外培養本研究では,ベネホブビルの遊離塩基AT−511とプロテアーゼ阻害剤であるネマレビルを併用した抗ウイルス活性を評価した結果,相乗的な抗ウイルス作用を示した。
13
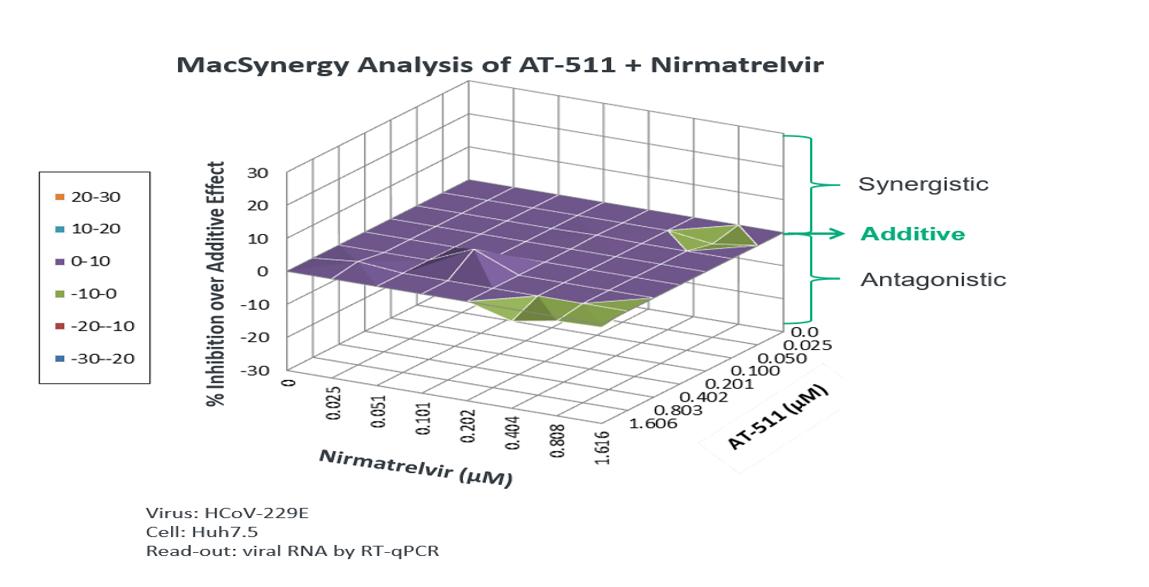
これらのデータは、フェニトフォブビルとプロテアーゼ阻害剤の併用によるSARS-CoV-2感染の治療は潜在的なメリットがあることを示していると考えられる。
標的SARS−COV−2 Niran/RdRpによる新冠肺炎の治療
SARS−CoVとSARS−CoV−2のRNAポリメラーゼ複合体は、その約30,000ヌクレオチドのウイルスRNAゲノムの転写および複製を支持する。それはRNAウイルスの中で最大、最も複雑なRNA合成機序である。SARS−CoV多サブユニットポリメラーゼ複合体は,ウイルスRdRp(“Nsp 12”),加工因子(“Nsp 7”,“Nsp 8”),校正エキソヌクレアーゼ,N 7−メチルトランスフェラーゼ(“Nsp 14”)およびヘリカーゼ(“Nsp 13”)を含む複数のNSPからなる。Nsp 12タンパク質は2つのドメインを含み、1つはRdRpコアであり、リボヌクレオチドをRNA鋳型に統合する触媒サブユニットであり、もう1つはN末端Niranドメインであり、その機能はまだ不明である。
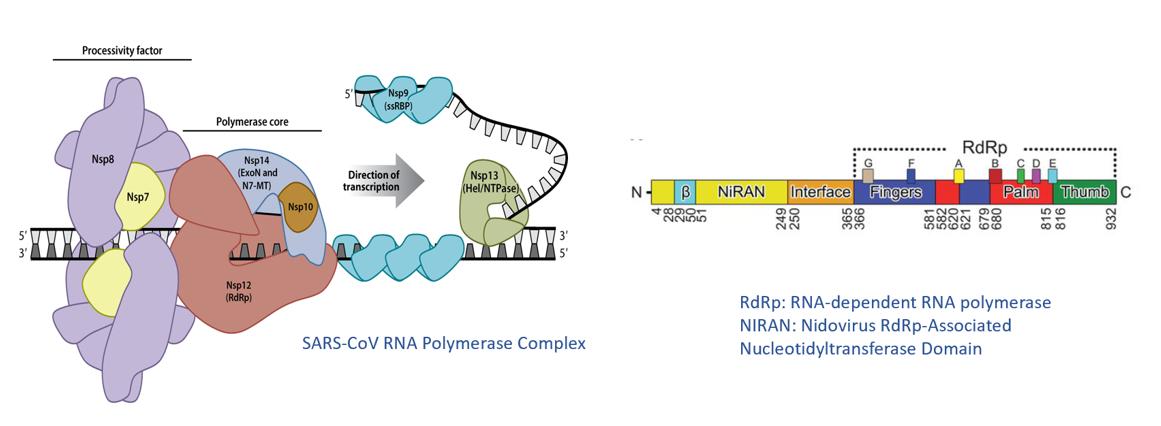
SARSコロナウイルスRNAポリメラーゼ
著者らはSARS-CoVがウイルスRNA合成を開始する機序を研究し、2つの異なる経路があることを発見した:1つはNSP 8のUMP化を通じて、Niran蛋白から起動し、媒介する;もう1つはNiran非依存方式を通じてジヌクレオチドプライマーを最初から合成する。重要なことは,両機能ともフェニニホブビルの活性三リン酸代謝物AT−9010によって抑制されることである。また,Nsp 12/7/8/RNA/AT−9100の2.98オスミウム凍結−EM四元構造が得られ,AT−9010はNiran活性中心に結合するだけでなく,RdRpにも結合され,鎖停止剤の役割を果たしていることが明らかになった。私たちはこの独特な二重身分が
14
Bemnifosbuvirの作用機序は、他の直接作用の抗ウイルス阻害剤と比較して、潜在的に高い薬剤耐性バリアを産生する。
BemnifosbuvirはウイルスRNAポリメラーゼに対してウイルス複製と転写に重要な高度に保存された酵素であるため、最近出現した変異株に対しても、そのスパイク(S)蛋白変異は受容体認識と宿主細胞膜融合過程を担当する抗ウイルス活性を維持することが予想される。現在の新冠肺炎変異はワクチンの有効性を低下させ、ウイルススパイク蛋白の突然変異によりモノクロナル抗体の有効性を除去した。将来の変異もワクチンやモノクロナル抗体の有効性に影響する可能性が予想される。
SARS-CoV-2変異体複製を効果的に抑制する
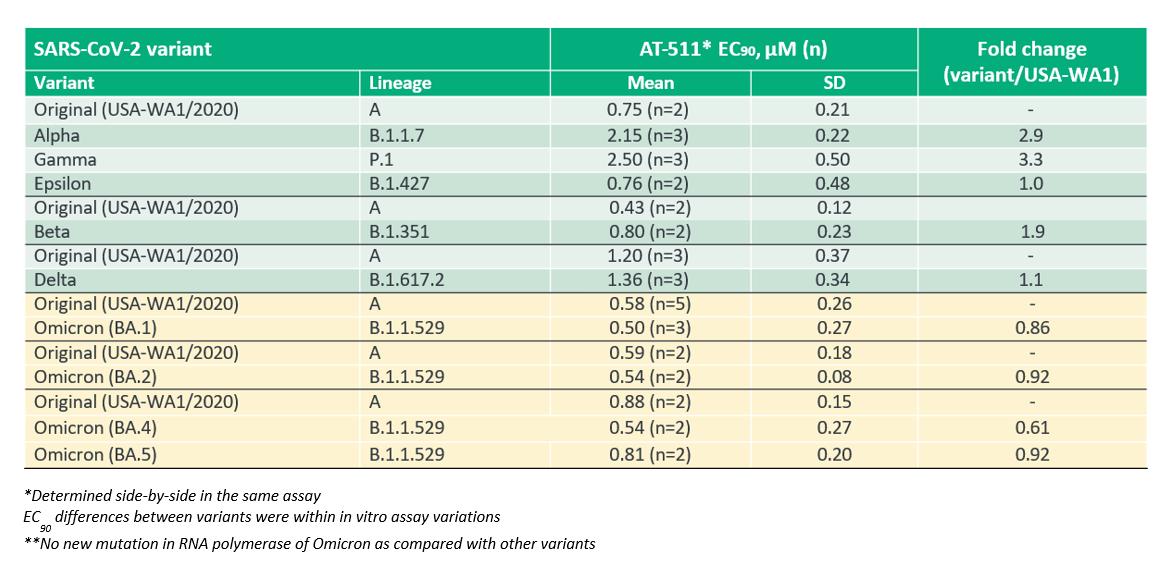
私たちは評価しました体外培養アニミホビル遊離塩基AT−511によるSARS−CoV−2 VOCとVOIの抑制作用これらの研究のデータを次の表にまとめると,AT−511はすべての主要VOCとVOIテストに対する効力を保持していることが示された。これらのデータは、高度に保存されたウイルスRNAポリメラーゼに対する化合物の重要な機構優位性を支持する。
非変異原性
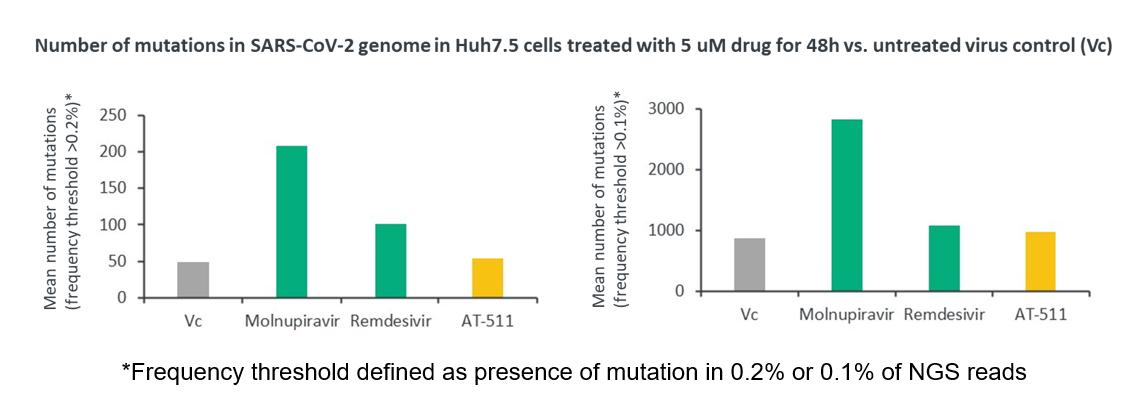
非臨床研究結果により、フェニニホブビルは突然変異を引き起こし、奇形作用がなく、しかも生殖毒性がないことを表明した。
より具体的には,SARS−CoV−2に感染したHuh 7.5細胞をAT−511(フェニルフォブビルの遊離塩基)で処理した次世代シークエンシング(NGS)分析により,ベンフォブビルは変異誘発剤ではない(これは臨床前に観察された遺伝毒性欠乏と一致する体外培養そして体内にある研究)を行い,ウイルスゲノムに変異を導入しなかった。
15
標準的な臨床前安全性、薬理学と重複用量毒性研究以外に、完成した臨床前研究により、ラットと非ヒト霊長類動物では、ベンノブビルを毎日650と1000 mg/kgの用量で13週間服用し、副作用がなかった;ベニホビルは被験ラットの雄或いは雌の生育能力に影響がなく、被験ラット或いはウサギの早期胚胎発育に影響を与えず、交尾前と交尾期間(雄)及び妊娠と哺乳期の交尾前治療によるラット(雌)の子孫発育、生殖能力或いは行動評価にも影響がないことが証明された。
臨床発展史
要約.要約
新冠肺炎の大流行開始時にわれわれの新冠肺炎計画を開始し,入院患者においてベネホブビルの全世界第二段階臨床試験を行った。その後,われわれの以前の協力者F.Hoffmann−LaRoche Ltd.とGenentech,Inc.(総称して“羅氏”と呼ぶ)とともにMOONSONGを開始し,2期外来臨床試験,MORNINGSKY,1つの3期外来臨床試験,およびMEADOWSPRING,MORNINGSKYに登録されている患者に対する3期6カ月のフォローアップ研究である。
著者らは羅氏とともに2021年10月に第二段階外来MOONSONG臨床試験を完了し、羅氏許可協定が2021年11月に終了することに伴い、著者らはそれぞれ2021年12月と2022年3月に第三段階MORNINGSKYとMEADOWSPRING臨床試験を早期に終了した。我々は,これらの患者研究から得られた重要な臨床データ,MORNINGSKYの臨床治療効果データ,および第一段階を追加的に支持する臨床薬理学研究,および健康被験者における臨床薬理学研究を用いて,新冠肺炎治療のためのベネフォブビルの設計を支援する日の出−3期3期臨床試験を提供した。
日の出−3−世界3期臨床試験
日の出-3は世界的、多中心、ランダム、二重盲検、プラセボ対照の第三段階臨床試験であり、少なくとも1,500名の軽中度新冠肺炎を有する高リスク非入院患者のベネフォブビル(550 mg、毎日2回、治療コース5日)を評価した。この試験は米国,ヨーロッパ,日本,世界の他地域の臨床試験地点で行われる。患者群は疾患進展リスクが最も高い群から構成され、80歳の患者80歳、1つ以上の主要なリスク因子を有する65歳患者および18歳の免疫機能低下患者を含み、これらはすべて新冠肺炎接種状態と関係がない。
この試験は単一療法(一次分析)としてBemnifosbuvirを評価することを目的としているが、抗ウイルス薬とBemnifosbuvir(二次分析)に適合する比較的に小さい患者のサブセットにおける併用療法の影響も探索する。この試験には、1)“支持性看護集団”(承認された抗ウイルス治療に適合しない患者、または現地に抗ウイルス薬を有さない患者)の2つの患者が含まれ、単一療法としてベンゼンフォブビルに投与された患者(予備分析)および2)“併用抗ウイルス集団”が評価され、SOCが他の適合性抗ウイルス薬との新冠肺炎に対する治療を含む場合、併用療法(二次分析)が評価される。患者は1:1のランダムな割合でBemnifosbuvir 550 mgを1日2回、ローカル使用可能SOCまたはプラセボBIDとローカル利用可能SOCを加えた治療を5日間受ける。
16
日の出-3研究の主な終点は、支持性看護集団中の少なくとも1300人の患者のすべての原因が入院または死亡して29日目までであり、この集団においてプラセボと比較して臨床的意義のある入院/死亡の減少を検出する能力があることである。試験参加患者の中で最も疾患進展リスクの高い患者を増加させることにより,入院率/死亡率は~4−6%を目標とした。60%の患者が研究ARMに登録し,支持性看護群に組み入れた後,DSMBによる中期分析を行った。各支持性看護患者群と連合抗ウイルス群中の副次的な終点は新冠肺炎合併症、受診、症状リバウンド/再発とウイルス負荷リバウンドを含む。

MORNINGSKY-世界第3段階試験
3期MORNINGSKY研究は無作為、プラセボ対照研究であり、対象は軽中度新冠肺炎を有する非入院成人と青少年患者であり、これらの患者はワクチンを接種するか否かにかかわらず、疾病進展の高リスク或いは標準リスクにある。この研究は羅氏社と協力して始まったもので、羅氏社との協力を終了したため、完成まで2021年12月に停止した。患者はランダム(2:1)に550 mgのフェニフブビルまたはプラセボ治療を受けた。主な終点は新冠肺炎症状の緩和·改善時間である。副次的な終点は入院、全因死亡率とウイルス負荷量の変化を含む。投与中止時に、216人の患者がランダム(2:1;有効:プラセボ)され、207人の患者が治療効果を評価可能な集団を構成した。この研究は47%のハイリスク群を含む広範な外来群に組み込まれ、28%の人がワクチンを接種し、56%の人がベースライン時に血清陽性であった。この研究は早期に終了したため,正式な統計比較は行われていない.
MORNINGSKY研究の主な終点である症状緩和時間は達していないが,MORNINGSKYの結果,プラセボ(70)と比較してフェニホブビルを服用した患者の入院期間は71%(2.9%対10%)減少した(p=0.047,無調整,探索性;副次的終点)。40歳以上の患者に対する亜群分析では,MORNINGSKY試験でbemnifosbuvirを服用した患者の入院期間は82%減少した。
17
この研究では死亡例はなかった。Bemnifosbuvirを服用した患者はプラセボを服用した患者と比較してウイルス負荷量の変化に有意差はなかった。プラセボと比較して、550 mgのBID用量は全体的に耐性が良好であった。薬物に関する副作用は報告されておらず,有害事象により薬物中止を検討している患者の割合は低い(bemnifosbuvirを服用している患者では2.8%,プラセボを服用している患者では7.0%)。
MEADOWSPRING試験は最初にMORNINGSKYに登録されていた患者に対して6カ月間のフォローアップ研究を行うように設計されており,MORNINGSKYに参加した患者72名のみを募集した後も2022年3月に終了し,患者数は計画より少なかった。したがって,この研究では新冠肺炎の長期症状について確実な結論を得ることができない。
新冠肺炎入院患者の世界第2段階研究
この研究は無作為、二重盲検、プラセボ対照の研究であり、中度新冠肺炎患者とプラセボ患者におけるフェニルフォブビルの治療効果を評価した。この研究は最初はBemnifosbuvir(550ミリグラムBid;A部分)の進行性呼吸不全(PRI)への影響を評価するためであったが、疾病進展の低背景率は研究を最初の設計で完成できなかった。この方案はより大量のフェニニホブビル(1100 mg、毎日2回;B部分)を探索するために修正したが、新冠肺炎治療環境の変化のため、この研究は2022年1月に早期に終了した。B部には2名の被験者(いずれもプラセボ治療を受けた)のみが参加した。
550 mg BID患者のPRI減少率は低く、治療群間に差はなかった(治療意向治療[ITT]人群:3/417.3%のフェニルニホブビル患者と4/4010.0%のプラセボ患者。A部分550ミリグラムBID被験者の全因死亡率はベニフォブウェル群で0/41,プラセボ群で5.0%(2/40)であった。また,B部1100 mg Bid群では1人のプラセボ患者が死亡した。
Bemnifosbuvir 550 mg Bid投与5日後にウイルス負荷レベルの急速な低下が認められた。翌日,bemnifosbuvir治療を受けた患者は0.6ログを経験した10ベースラインウイルス負荷の平均低下幅はプラセボに比べて大きかった。ウイルス負荷減少の持続差は8日目には不変であった。
18
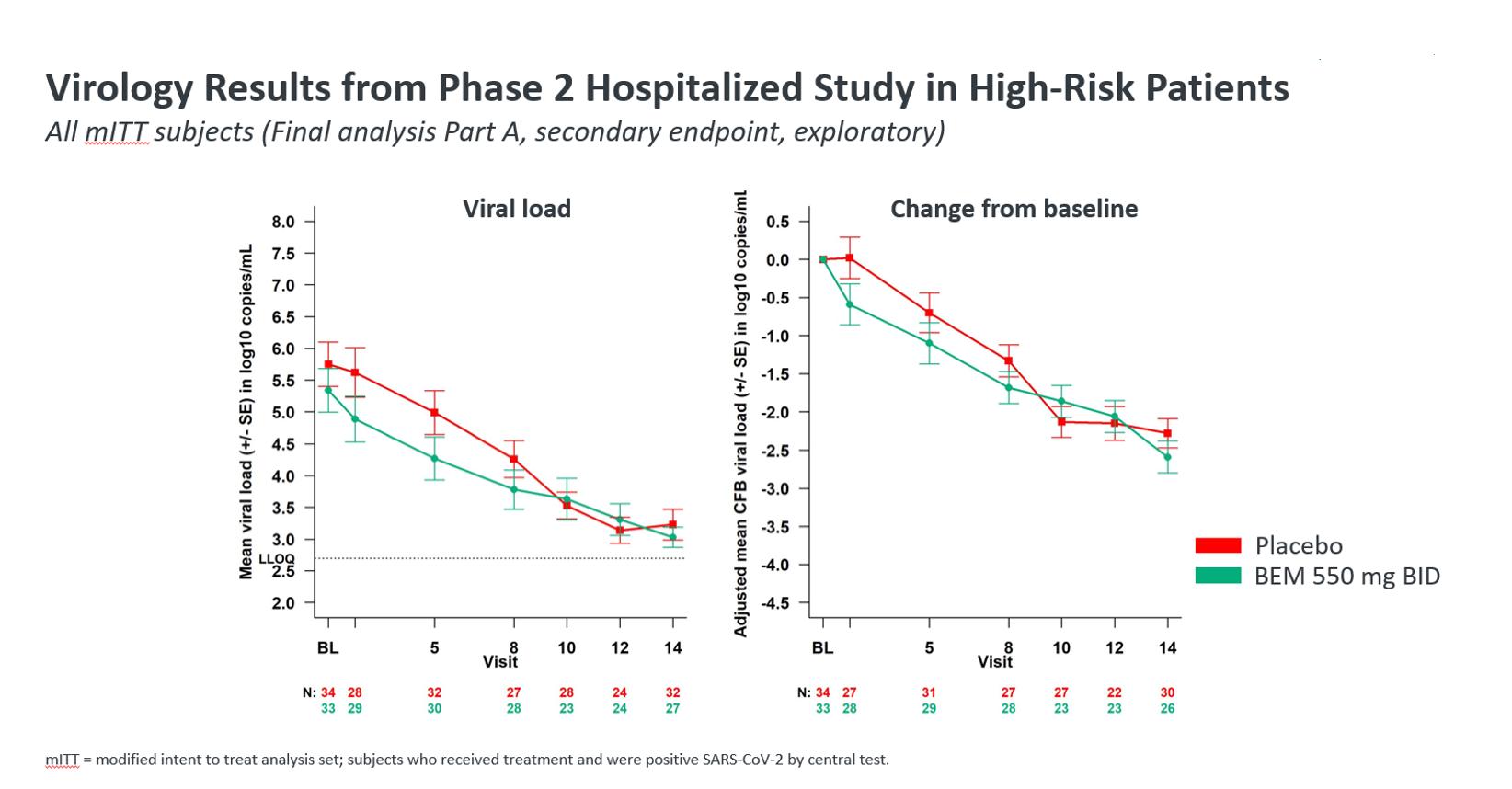
BemnifosbuvirのSARS−CoV−2抗ウイルス活性もベースラインウイルス負荷の中央値が5.35 logを超える患者で観察された10プラセボと比較して。このサブセットでは,フェニニホブビルを服用した患者は2日目(6%の患者),8日目(12%の患者),10日目(33%の患者),12日目(31%の患者)でSARS−CoV−2ウイルスの除去が達成されたが,プラセボを服用した患者の同時点での割合は0%であった。14日目(最終ウイルスサンプリング研究日)までには,フェニニホブビルを服用した患者の50%でRNAウイルスが検出されず,プラセボを服用した患者では23%であった。
550ミリグラムBID投与5日後,ベネフォブビルは全体的に耐性が良好であり,薬物に関する重篤な副作用は認められなかった。非深刻な不良事件は治療群に平均的に分布している。多くの患者の重症度は軽度から中等度であり,ベネホブビルとは無関係と評価された。
蒙松 - 世界第2段階試験
この研究は無作為、二重盲検、多中心、プラセボ対照試験であり、成人軽中度新冠肺炎外来患者(A群、n=40)とプラセボ(n=40)の成人外来患者において、逐次投与量のベンフォブビル550 mg(A群、n=30)と1,100 mg(B群、n=30)の抗ウイルス活性、安全性と薬物動態を評価する。プラセボと比較して、この研究でフェニニホブビル治療を受けた患者は、SARS−CoV−2ウイルス負荷量の減少を示す主要な終点に達しておらず、プラセボ患者の約3分の2は低リスクかつ症状の軽微な患者である。しかし,潜在的な健康状態があるハイリスク患者では,ウイルス負荷量は約0.5 log減少している107日目に、550ミリグラムBID投与とプラセボ(予め指定されたサブグループ分析キューA n=7;プラセボn=10)およびBID 1,100 mg投与と混合プラセボ(探索的サブグループ分析キューB;n=14;プラセボn=7)との比較を観察した。
この研究では,Bemnifosbuvirは全体的に耐性が良好であった。プラセボを服用した患者に何らかの有害事象(“AE”)が発生した割合は28%,550ミリグラムのBIDを服用した患者は20%,BIDを服用したBIDは1100 mgのBID患者の割合は33%であった。各治療群は3つの非薬物関連の深刻な副作用(“SAE”)があり、すべての他の副作用はすべて1級或いは2級である。胃腸(GI)に関連する副作用は最もよく見られる副作用である:プラセボ群は8%、bd群は7%である;bemnifosbuvir 1100 mg Bid群は20%である;軽度から中度の吐き気/嘔吐による研究薬物の早期中止:プラセボ群は3%、bemnifbuvir 550 mg bd群は0%、bemnifosbuvir 1100 mgBid群は17%であった。プラセボと比較して,治療群とプラセボ群の実験室異常は臨床的に有意差はなかった。
19
他の研究
MORNINGSKYの3期臨床試験と2期臨床試験を除いて、著者らは新冠肺炎計画を開始して以来、すでに支持性の1期と臨床薬理学研究を行い、1つの気管支肺胞洗浄研究、複数のDDI研究と品質バランス研究を含む。これらの研究では,Bemnifosbuvirの安全性とPKはすでに健常被験者で評価されており,用量は1100 mgと高く,1日2回,治療コースは5日間であった。
健常被験者の気管支肺胞洗浄研究の結果,フェニレホブビルは有効に肺(上皮ライナー層)に輸送され,SARS−CoV−2感染の主要部位であることが示唆された。5つの臨床DDI研究を完成し、TOPLINEの結果により、ベネホブビルと関連するDDI潜在力は全体的に低いことを示した。
ステップ1−DDI研究
一連の第一段階の研究により、薬物の相互作用状況は良好であり、CYP 3 A基質である薬物或いは外排出と肝臓摂取トランスポーター感受性基質である薬物との併用投与を含む場合、投与量を調整する必要がない。チトクロームP 3 Aは多種の薬物とサプリメントを代謝する酵素であり、この輸送体は新冠肺炎ハイリスク患者の通常処方された薬物の細胞輸送を調節している。
これらの研究では,bemnifosbuvirはCYP 3 A 4(ミダゾラム),P−糖タンパク質(ジゴキシン,シクロスポリン,カルバマゼピン),乳癌耐性蛋白と有機陰イオントランスポーターポリペプチド1 b 1(レシュバスタチン)の指標薬物とともに使用されている。薬物相互作用による可能性は低く,ベネフォブビルは通常処方されている療法と併用可能であり,これらの療法は通常脆弱な患者群が他の状況に対して服用しており,重篤な新冠肺炎に進展するリスクが高いと考えられる。
Bemnifosbuvirは健康被験者における普遍的な耐性は良好であった。MOONSONG第2期外来臨床試験の結果と一致し,健常被験者では550 mg BIDを超える用量で胃腸に関連する軽度から中等度の有害事象の発生率の増加,特に嘔気と嘔吐が認められた。550 mgのBIDはすでに耐性が良好であるため、10日まで、550 mgのBID用量は、3期日の出-3研究の5日間に選択された。
また,特殊群(例えば肝腎障害患者)において臨床薬理学的研究を支援する取り組みが行われている。
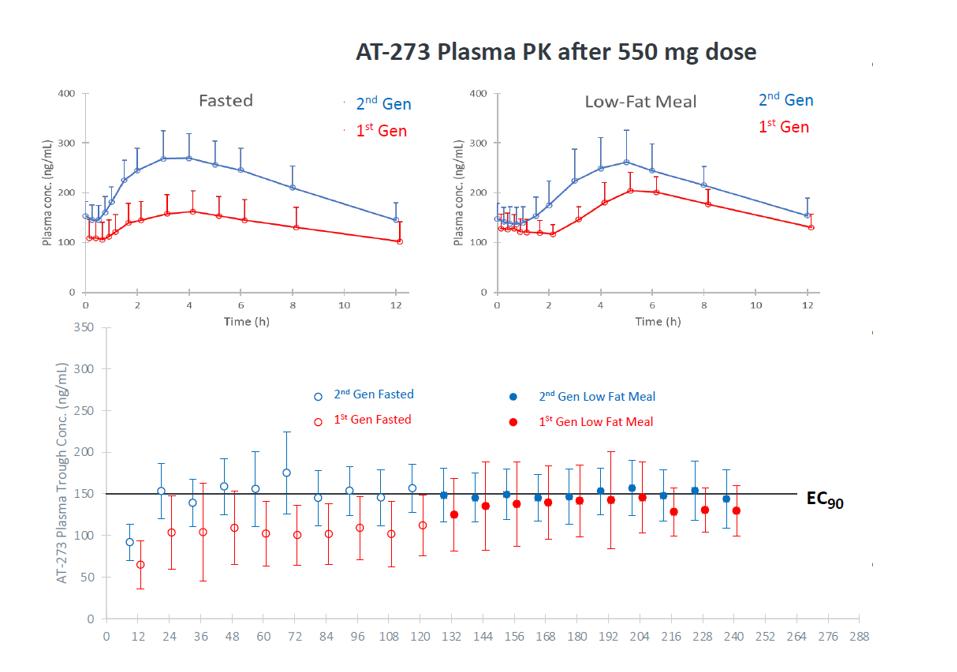
第一段階−PK研究−第二世代ベニフォブウェイ錠剤
日の出−3号臨床試験では,第二世代製剤275 mgのベニフォブウェイ錠剤を用いた。この処方を健康な被験者の第1段階研究で評価し、これらの被験者は、フェニホブビル(絶食および低脂肪食)を10日間服用した後、1日550 mg(2 X 275 Mg)を服用した。この研究の結果,第二世代錠剤はMORNINGSKY研究で用いられている第一世代錠剤よりも高いフェニフブビル活性代謝物AT−273の血漿曝露を有することが示唆された。そのほか、第二世代錠剤はもっと高いAT-273(>EC)血漿谷濃度を獲得した90SARS−CoV−2複製抑制におけるBemnifosbuvirの作用)は食物に影響を与えず、脂肪含有量とも無関係である。この研究では,健康被験者におけるBemnifosbuvirの耐性は一般に良好であった。
20
ペニホブビルとルザスビルの併用によるC型肝炎の治療
C型肝炎ウイルス
背景
C型肝炎ウイルスは血液を通じて伝播する陽性一本鎖RNAウイルスであり、主に肝臓細胞に感染する。C型肝炎ウイルスは慢性肝疾患と肝移植の主要な原因であり、輸血、血液透析と針刺を通じて伝播する。アメリカでは、麻薬注射はすべての新規C型肝炎症例の約60%を占めている。C型肝炎ウイルスの診断は、分子試験を含む血液試験に合格し、ウイルスゲノムの検出、定量化および分析を可能にし、感染を特定のウイルス遺伝子タイプに分類する。75%から85%の急性症例では,C型肝炎は慢性C型肝炎となり,潜伏期は2週間から26週まで様々であった。
C型肝炎ウイルスは7種類の遺伝子タイプと67種類の亜型に分けられ、その中で遺伝子1はアメリカのC型肝炎ウイルス症例の70%以上を占める。C型肝炎患者はまた肝機能状況による分類を行う:代償性肝硬変(肝瘢痕形成)は肝機能障害が出現していない患者であり、非代償期肝硬変は肝機能中度から深刻な損傷患者を指す。
世界保健機関のデータによると、全世界で5800万人が慢性C型肝炎ウイルス感染を患っていると推定され、毎年約150万の新しい感染病例がある。疾病管理センターが最新に発表したC型肝炎ウイルスモニタリング報告によると、アメリカのC型肝炎ウイルス感染者数は持続的に増加している。毎年約29万人がC型肝炎ウイルスに関連する肝疾患で死亡し、その中の大部分の死亡は肝硬変と肝細胞癌と関係がある。
2013年以来、急性C型肝炎の発病率は倍以上増加した(124%増加した)。しかし,報告症例数と推定症例数との間には大きな差がある。C型肝炎ウイルスに感染している人の多くは、病状が症候性疾患に発展したり、特定の臨床試験を行って診断を確認する前に、C型肝炎ウイルスは検出されない可能性があるため、自分が感染していることを知らない。したがって,症例は報告されておらず,実際の疾患流行率を歪めている。高額な医療支出(共通の治療費,肝臓移植)や末期慢性肝疾患の死亡率が報告の罹患率に比例しない場合には,報告不足の負担が生じる。
21
アメリカではC型肝炎ウイルスの発症率が上昇しています
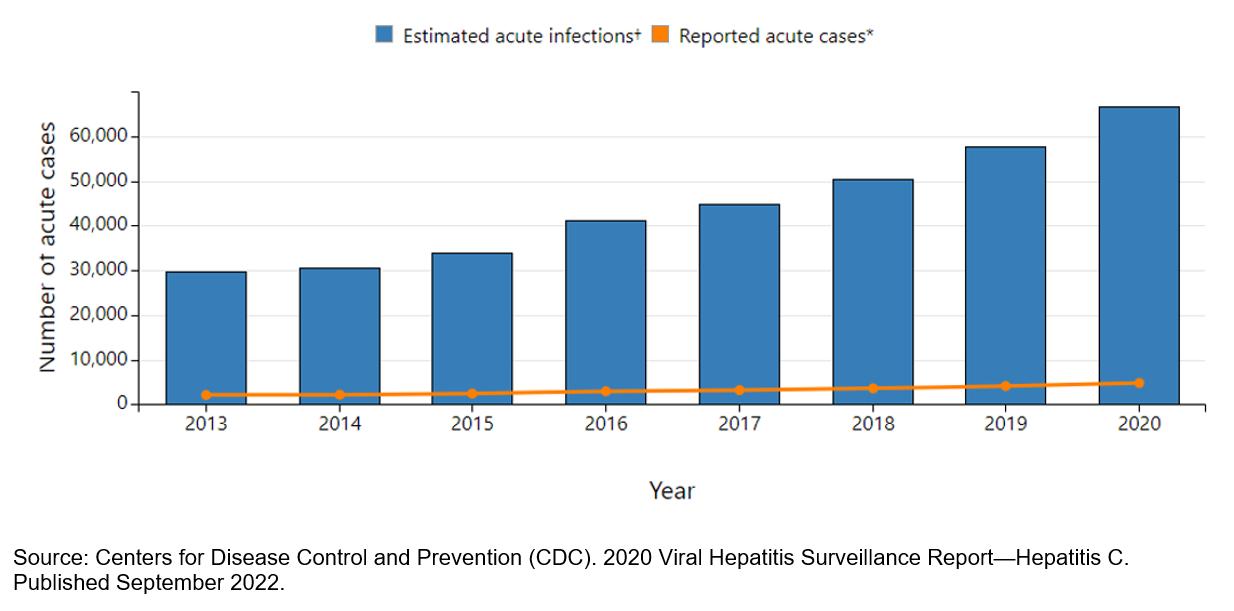
2013年から治療面で大きな進展が得られたにもかかわらず,米国では大量のサービス不足のC型肝炎患者人口が大幅に増加し続けている。発症率上昇の一部の原因は,2013年に疾病管理センターが1945年から1945年までの間に生まれたすべてのアメリカ人をスクリーニングするガイドラインを発表してから増加したC型肝炎診断であるが,発症率上昇の大きな原因はオピオイド危機,静注薬物使用,C型肝炎ウイルス再感染である。
C型肝炎の発病率の上昇に伴い、治療を受けた新しい患者の数量を相殺し、今後数年のアメリカのC型肝炎の流行率は引き続き安定を維持することが予想される。 C型肝炎治療薬の巨大な全世界市場は2050年以降まで存在すると推定されている。推定によると、2022年の全世界の売上高は40億ドルに近く、その約50%はアメリカから来ており、C型肝炎ウイルス市場は依然として大きい。
現在の治療景観
現在、C型肝炎ウイルスを予防するワクチンはまだないが、2013年から、いくつかの相次いで発売と改善された経口抗ウイルス療法はすでに大多数の患者のSVR率を95%以上に向上させ、治療持続時間は8~12週間であり、具体的には方案と患者数に依存する。先行するC型肝炎ウイルス製品は、異なる作用機序と治療標的を有する薬物からなる併用療法である:NS 3/4 Aプロテアーゼ阻害剤、NS 5 A阻害剤およびNS 5 Bヌクレオチド(T)ポリメラーゼ阻害剤。患者の遺伝子型,肝硬変状態と先の治療失敗は,治療に用いる適切な抗ウイルス治療法を決定した。アメリカでは現在慢性C型肝炎を治療する2つの主な治療法は
Epclusa(ソブビル/ウィパシビル):NS 5 B阻害剤とNS 5 A阻害剤からなる連合方案は2016年に初めてアメリカ食品と薬物管理局の許可を得た。肝硬変あるいは代償性肝硬変のない成人と小児C型肝炎ウイルス1~6型慢性感染患者の3年間の治療に適している。非代償性肝硬変患者では,Epclusaはリバビリン(プリンヌクレオシド類似体)との併用が許可されている。Epclusaの患者は12週間の治療が必要である。
Mavyret(グリパビル/ピブレビル):NS 3/4 Aプロテアーゼ阻害剤とNS 5 A阻害剤からなる連合方案は2017年に初めてアメリカ食品と薬物管理局の許可を得た。慢性感染、肝硬変或いは代償性肝硬変のない成人と児童C型肝炎患者に適している。Mavyretは、NS 5 A阻害剤またはNS 3/4 Aプロテアーゼ阻害剤(両方があるわけではないが、両方があるわけではない)を含む治療を受けた1型C型肝炎ウイルスに感染する患者のための使用も許可されている。Mavyretは1-6型C型肝炎ウイルスの8週間の治療のために最初に承認された、非肝硬変および代償性肝硬変に適した成人患者であり、これらの患者はまだ存在していない
22
以前治療したことがあります。いくつかの治療経験のある人たちの場合、治療持続時間はもっと長い(最長16週間まで)。Mavyretは非代償期肝硬変患者への使用は許可されていない。
私たちの方針は看護基準の向上を求めています
我々はベノホブビルとルザスビルの併用によるC型肝炎ウイルスの開発を行っている。BemnifosbuvirはC型肝炎ウイルス非構造蛋白5 B(“NS 5 B”)RdRpの有効な阻害剤である。Ruzasvirは1種の研究中の経口、有効、汎遺伝子型非構造蛋白5 A(NS 5 A)阻害剤であり、慢性C型肝炎ウイルス感染の治療に用いられ、著者らは2021年12月にメルク社から許可を得た。われわれのこれまでの臨床前および臨床データによると,承認されれば,この組み合わせは以下のような潜在的な利点を提供する可能性が考えられる
臨床発展
これまで、著者らは慢性C型肝炎ウイルス感染の治療を支持するために、2つのフェニニホブビルの臨床試験を完成した。
ベネホブビル単独の1期臨床試験
著者らは7日間にわたる第1段階試験を行い、健康およびC型肝炎ウイルス感染者における単一薬物としての単剤量と多用量のフェニニホブビルの作用を評価した。すべてのC型肝炎ウイルス感染者は単純なC型肝炎ウイルスリボ核酸治療を受けた≥5 log 10 IU/mL。試験の目的は安全性,耐性,PK,抗ウイルス活性を評価することである。
この試験は、健常被験者(A部分)経口単剤ベニフォブビル最大400ミリグラム塩形態(369ミリグラムラジカル)、非肝硬化性C型肝炎ウイルス感染者(B部分)単剤最大600ミリグラム塩形態(553 mgラジカル)、および非肝硬化型1 b(“GT 1”)、C型肝炎ウイルス感染者(C部分)の毎日多剤最大600ミリグラム塩形態(553 mgラジカル)の7日間の経口投与を評価した。非肝硬変型3(“GT 3”),(D部分)と肝硬変(GT 1,2,3)とC型肝炎ウイルス感染の被験者(E部分)では,他の列は毎日600ミリグラム塩形式(553 mg遊離塩基)を評価し,7日間連続した。次の表に各治療キューの用量と平均最大C型肝炎ウイルスリボ核酸減少量を示す。
88人の被験者が試験のすべての部分で用量を受け、そのうち72人の被験者が活性薬物治療を受け、16人の被験者がプラセボ治療を受けた。この試験では,bemnifosbuvirは肝硬化性と非肝硬化性C型肝炎感染患者で同等の汎遺伝子抗ウイルス活性を示した。単剤(B部分)後の平均最大C型肝炎ウイルス減少率は2.3 logであった10遊離塩基553 mg投与7日後のC型肝炎ウイルスRNA平均最大減少率は4.6 logであった10Iu/m Lです。データによると、平均最大C型肝炎ウイルスRNA減少量は4.4 log10非肝硬変型1 b(“GT 1 b”)、C型肝炎ウイルス感染者のフェニホブビル服用7日後、遊離塩基量553 mgで平均4.5 log減少した10非肝硬化性GT 3型C型肝炎ウイルス感染者の服薬7日後の投与量は1 U/mlであった。肝硬変群のPKデータは非肝硬変群と類似していた。EMaxモデルは,1日1回553 mgの遊離フェニルフェニホブビルが最大限のウイルス負荷量を減少させると予測している。
B部分の最大C型肝炎ウイルスRNA変化(非肝硬変、GT 1 C型肝炎ウイルス感染者単剤)
最大減少量(ログ)10Iu/m L) |
|
100 mg (92 mg) |
|
300 mg (277 mg) |
|
400 mg (369 mg) |
|
600 mg (553 mg) |
23
平均値±SD* |
|
0.8 |
|
1.7 |
|
2.2 |
|
2.3 |
個体 |
|
0.6, 0.8, 0.9 |
|
1.1, 1.8, 2.2 |
|
1.8, 2.2, 2.5 |
|
2.1, 2.3, 2.6 |
C部分の最大C型肝炎ウイルスRNA変化(非肝硬変、GT 1 C型肝炎ウイルス感染者の複数回の服薬)
最大減少量(ログ)10Iu/m L) |
|
プラセボ |
|
150 mg (138 mg) |
|
300 mg (277 mg) |
|
600 mg (553 mg) |
平均値±標準偏差 |
|
0.4±0.109 |
|
2.6±1.073 |
|
4.0±0.415 |
|
4.4±0.712 |
個体 |
|
0.3, 0.3, 0.4, 0.4, 0.5, 0.6 |
|
1.7, 1.8, 1.8, 2.7, 3.0, 4.5 |
|
3.4, 3.7, 3.9, 4.2, 4.2, 4.5 |
|
3.5, 4.0, 4.1, 4.3, 5.2, 5.3 |
D部分(非肝硬化性GT 3型C型肝炎ウイルス感染者の複数回注射)とE部分(肝硬化性C型肝炎ウイルス感染者の複数回注射)の最大C型肝炎ウイルスRNA変化
最大下げ幅 |
|
D-GT 3部分 |
|
E部分--肝硬変 |
|
600 mg (553 mg) |
|
600 mg (553 mg) |
|
平均値±標準偏差 |
|
4.5±0.262 |
|
4.6±0.485 |
個体 |
|
4.2, 4,4, 4.4, 4.5, 4.5, 5.0 |
|
GT1b: 4.0, 4.0, 4.5 |
*SD=標準偏差
**Qd=1日1回
ベネホブビルとダラタビルの併用2期臨床試験
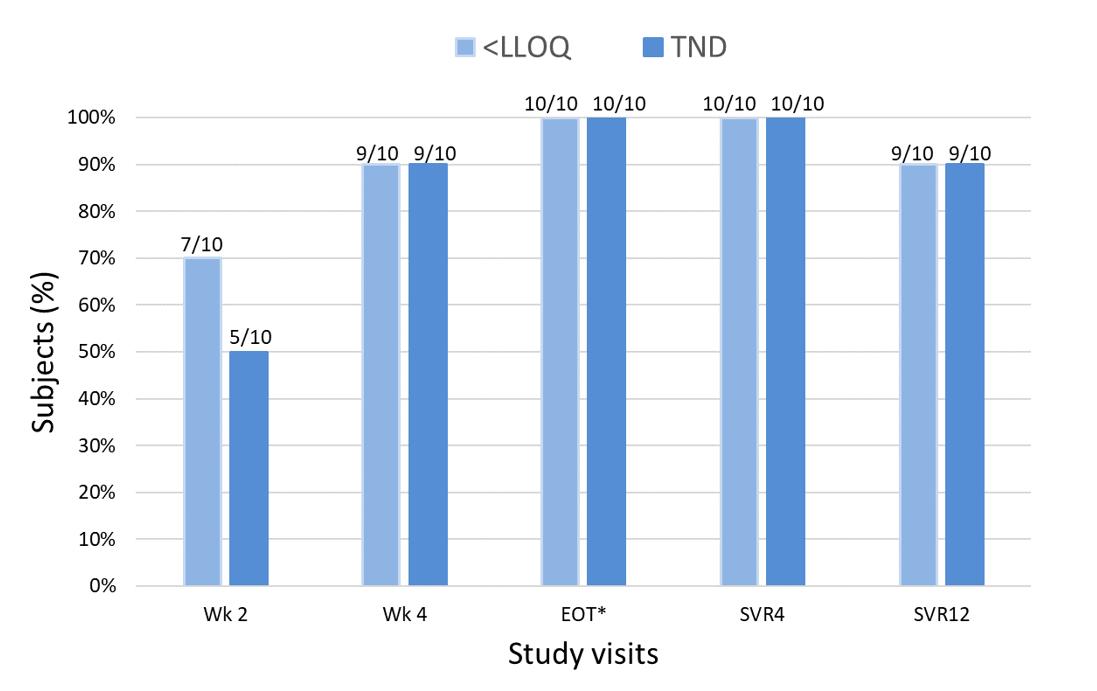
第2段階のオープンラベル臨床試験を行い、C型肝炎ウイルス感染者におけるフェニニフブビルとダラタビルの併用を評価した。ダラタビルは、許可された商業化可能なC型肝炎ウイルスNS 5 A阻害剤である。未治療の非肝硬化性GT 1 C型肝炎ウイルス感染者10人は、1日1回553 mgの遊離フェニルフェニホブビルおよび60 mgのダラタビル治療を受け、治療コースは8週間または12週間であった。この研究の主要な治療効果の終点はSVR 12(持続的なウイルス応答であり、C型肝炎ウイルスRNAと定義されている
効力の弱い第1世代C型肝炎ウイルスNS 5 A阻害剤ダラタビルを使用したにもかかわらず,すべての被験者がC型肝炎ウイルスRNAを獲得した50 参考文献と比較する).ソブビルと比較してヨーロッパ共同体は50ECと90Bemnifosbuvirの価値は約10倍低い.したがって,この場合のRAVの意味は不明である.ベースライン時には,あらかじめ存在するNS 5 A RAVは他の被験者にはなかった。
以下の図に示すように,薬物の検討開始後,ウイルス負荷は急速に低下し,70%の被験者が血漿中のC型肝炎ウイルスrnaを獲得した
C型肝炎ウイルスリボ核酸を得た被験者の割合(%)
24
ベニホブビルC型肝炎ウイルスの安全性
われわれのC型肝炎ウイルス第1期あるいは第2期臨床試験では,重篤な有害事象,用量制限毒性あるいは試験中止をきたす有害事象は認められなかった。最もよく見られる副作用は頭痛と血中脂質レベルの軽微な上昇であり,他の報告の副作用は一致したパターンはなかった。副作用の多くは重篤ではなく,ベンノフォブビルに関与しているとも考えられない。
ルザスビル
Ruzasvirは経口、汎遺伝子型別のNS 5 A阻害剤の研究であり、著者らは2021年12月にメルク社から許可を得た。メルク社が行った研究でルザスビルは体外培養強力な抗ウイルス活性を有するEC50亜~低皮モルの範囲ですべてのC型肝炎ウイルス遺伝子(3対数)10GT 1,GT 2,GT 3に感染した患者は単一治療後に認められた。この臨床抗ウイルス活性は,MavyretとEpclusaのNS 5 A阻害剤成分であるピブトリビルとウィパタビルが単一薬物として得られた効果に相当する。これらのPOCデータは、メルク社以前に行われたより大規模な2期多剤連合研究(2種類と3種類の薬物方案を含む)におけるRuzasvirの評価をサポートしている。これらの研究には,代償性肝硬変の単純治療やインターフェロン経験のある患者が含まれている。全体的に言えば、メルク社はGT 1、GT 2、GT 4とGT 6感染患者(C-Breeze 1と2)による2薬の連合研究(Ruzasvirガピリミジンヌクレオチド前駆薬uprifosbuvir、12週間)に高いSVR 12率(>90%)を観察した。GT−3代償期肝硬変患者のSVR 12発生率は低かった(40%SVR 12;C−Breeze 1)。この低い発生率はGT−3肝硬変患者でヌクレオチドuprfosbuvirに関連する抗ウイルス活性が低下したためと考えられ,Ruzasvir投与量が180 mgに増加したことによりこの群におけるSVR 12の発生率(68%SVR 12;C−Breeze 2)が大きく増加し,GT−3肝硬変患者におけるRuzasvirと温存用量に関する臨床抗ウイルス活性が強調された。
C型肝炎ウイルスに感染した1200人以上の参加者は、リバビリンの使用または使用しない2-薬剤および3-薬物レジメンの一部として、Ruzasvirの1日当たり180 mgまでの用量を受け、24週間まで持続した。全体的な安全データにより、Ruzasvirは全体的に耐性が良好であり、実験室、バイタルサイン或いは心電パラメータ値は治療と一致する変化がなかった。メルク社が行ったすべての研究では,深刻な有害事象や有害事象による中止はまれである。
25
BemnifosbuvirとRuzasvirの併用によるC型肝炎治療の理論的基礎
ベネホブビルの抗ウイルス効力,特により治療が困難な3型感染患者において,ルザスビルとベネホブビルの併用がメルク社以前に行われた研究で観察されたSVR 12率を改善する可能性があると信じられている。
本ニホブビルとルザスビルの患者への併用をさらに支援するために行った体外培養C型肝炎ウイルスGT 1 bレプリコン分析(Huh-Luc/neo-ET)における協同実験では、C型肝炎ウイルス複製子細胞は異なる濃度のAT-511、ビンニフォブビルの遊離塩基とルザスビルによって単独または連合して処理された。以下の図に示すように,これらの実験は,併用によるC型肝炎ウイルス複製抑制作用がいずれの薬剤単独よりもはるかに大きいことを示しており,両阻害剤間に相乗的抗ウイルス作用があることが示唆された。
離体する協同作用:C型肝炎ウイルスGT 1 b複製子(Huh−Luc/neo−ET)を用いて測定した
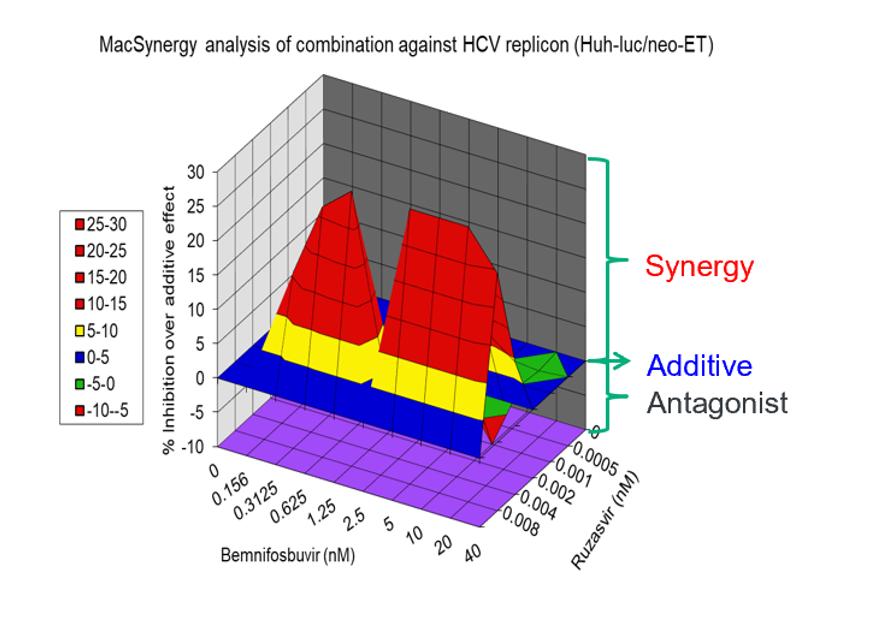
13週間のラット連合毒性研究において、BemnifosbuvirとRuzasvir単独或いは連合投与500 mg/kg/dの時の耐性は良好であった。3つの用量群のいずれも、試験品に関連する副作用に気づかなかった。単独あるいは併用投与では,フェニニホブビル,その代謝産物はルザスビルの全身曝露と類似しており,両薬剤間に有意な薬物相互作用がないことが示唆された。
結論的に、これらのデータはbemnifosbuvirとruzasvirの併用による慢性C型肝炎ウイルス感染治療の臨床発展を支持する。
計画的臨床発展
2023年第2四半期に、著者らはbemnifosbuvirとruzasvirの連合治療による単純C型肝炎ウイルス感染患者の第二段階試験を開始し、肝硬変がなくても代償性肝硬変であっても。本研究では,汎遺伝子組合せ治療8週間後の安全性と有効性を評価することを目的とし,550 mg/dのベネホブビルと180 mgのruzasvirからなる。C型肝炎ウイルスの直接作用に感染した抗ウイルス新薬患者約280名は,約60名の患者を含み,この第2段階の研究に組み込まれる予定である。研究の主な終点は安全性と治療後12週間のSVRである。他のウイルス学的終点はウイルス学的失敗、治療後24週間のSVRと薬剤耐性を含む。
26
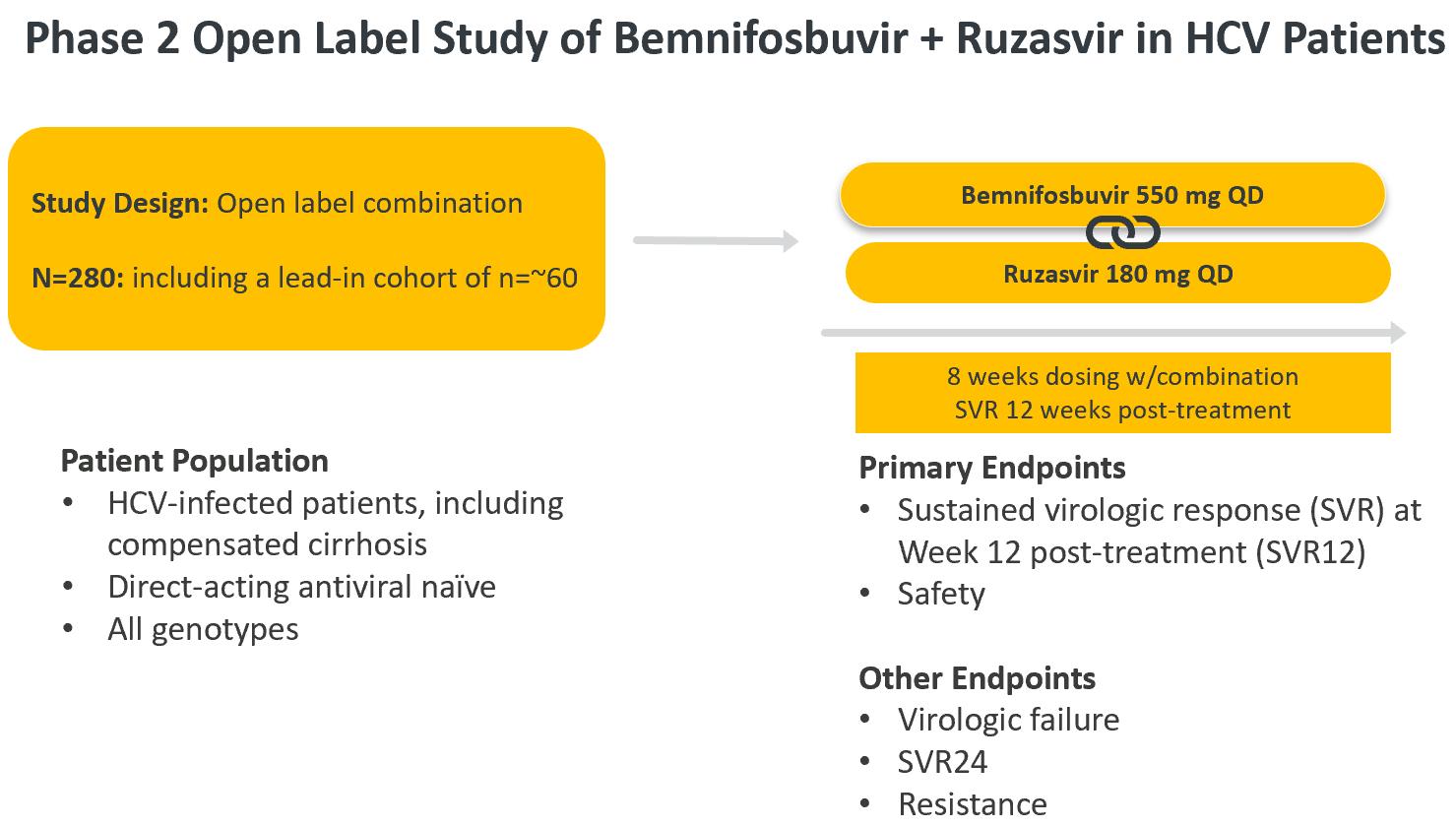
この研究の結果が陽性であれば,将来多くの患者群でBemnifosbuvirとruzasvirの併用治療8週間あるいはそれ以上(6週間)のより大規模な研究,リバビリン治療12週間の非代償性肝硬変患者での研究を支持する可能性がある。
現在,我々は,ビンネフォブビルとルザスビルの間の潜在的なDDIおよびこれら2つの薬物のPKに対する食物の影響を評価するための健康被験者の第1段階臨床研究を行っている。
羅氏許可協定
2020年10月,我々はF.Hoffmann−La Roche LtdとGenentech,Inc.とBemnifosbuvir,AT−511とその予備化合物(AT−752を含む)(“許可化合物”),任意の許可化合物を含む製品(“ライセンス製品”)と関連診断薬(“Companion Diagnostics”)の世界開発,製造,商業化に関するライセンス契約(“ロー氏ライセンス契約”)を締結した。
羅氏許可協定に基づいて羅氏に付与された権利の一部の対価格として、羅氏は2020年11月に3億5千万ドルを前払いした。また、2021年6月に発展マイルストーンを実現した後、私たちは羅氏から5000万ドルを追加した。
羅氏許可協定の期限内に,羅氏と我々は世界規模で新冠肺炎のためのベノホビルを共同開発し,このような開発活動に関するコストを折半した。
我々が2021年11月に羅氏の終了通知を受けた後、羅氏許可協定は2022年2月10日に終了した。終了日までに,羅氏とのコスト分担協定に基づいて負担するベネホブビルの開発に関する義務も終了した。
羅氏許可協定の終了に伴い、著者らは羅氏のすべての使用分野における研究、開発、製造と商業化許可化合物、許可製品と随伴診断薬物の全世界独占権利を再獲得した。
メルク社とのライセンス契約
2021年12月、マーク社とRuzasvirの開発、製造、商業化についてライセンス合意(“Merckライセンス契約”)を達成しました。Ruzasvirは我々が開発しているNS 5 A阻害剤であり,ベネホブビルと併用してC型肝炎ウイルスの治療に用いられている。
27
メルク許可協定の条項によれば、私たちは、メルクの独占(特定の保持権利の制約の下での内部研究)、再許可および世界的許可を取得し、特定のメルク特許および技術の下で研究、開発、製造、製造、使用、輸入、輸出、販売、要約または他の方法でRuzasvir(“化合物”)またはその化合物を含む製品(各製品“製品”)をヒトのすべての治療または予防用途(“分野”)に使用する。
私たちがメルク許可協定によって得た権利を考慮すると、私たちはメルクに2500万ドルの前金を支払い、いくつかの開発と規制のマイルストーンを達成した時にメルクマイルストーンに合計1.35億ドルを支払うことを要求され、私たちがいくつかの販売ベースのマイルストーンを実現した時のメルクマイルストーンへの支払いは合計3億ドルに達する。また、製品の年間純売上高に応じて階層的特許権使用料をメルクに支払うことに同意しており、高い1桁パーセントから10代程度までの範囲であるが、何らかの調整が行われる可能性がある。私たちの特許使用料支払い義務は、(I)製品(または製品に含まれる化合物)の許可されたメルク特許の有効な権利要件の最後の満了を要求し、(Ii)製品が同国で初めて商業販売された後の期間まで、国/地域および製品に基づいて継続される。
メルク許可協定の条項によると、いくつかの国/地域で少なくとも1つの製品を開発し、商業化するために、商業的に合理的な努力をする義務がある。
メルク許可協定の期限は、メルク許可協定の下で生じるすべての印税支払義務が満了するまで、製品および国/地域に基づいて継続される。便宜上、私たちは90日前にメルク許可協定を終了することを書面で通知することができます。他方がメルク許可協定の条項に実質的に違反した場合、いずれもメルク許可協定を終了する権利があるが、60日間の治療期間を遵守し、他方が破産または債務を相殺しない場合には終了しなければならない。もし私たちがメルク許可協定に従って私たちに許可された任意のメルク特許の有効性または実行可能性にいかなる妨害または反対手続きまたは他の挑戦を提起し始めた場合、または私たちが他の方法でこのようなメルク特許のいかなる延期または任意の補充保護証明書の付与に反対する場合、メルク社はメルク許可協定を直ちに終了する権利がある。
メルク許可協定が終了すると、メルクが私たちに与えた許可は終わります。私たちが便宜上“メルク許可プロトコル”を終了するとき、またはメルクが“合意”を終了するとき、メルクは、終了時に存在するため、ルザスビルを唯一の有効な製剤として含む製品を開発、製造、または商業化するために、我々のいくつかの特許およびノウハウの独占的、全額支払い、永久的、再許可可能な許可を有するであろう。また、メルク社が要求を出した場合、メルク社または私たちが安全問題以外の便利な理由でメルク社の許可協定の通知を終了した後のしばらくの間、メルク社と交渉し、ruzasvirおよびbemnifosbuvirからなる製品の開発、製造または商業化に必要ないくつかの特許および独自技術の非独占使用料許可をメルク社に付与する義務がある。製品が終了時に存在するので、いくつかの許可条項はメルク許可協定に予め規定されている。
製造業
私たちは現在、臨床前や臨床候補製品を生産するための製造施設を持ったり運営したりしていませんし、将来的に自分たちの製造業務を開発したり運営したりする計画もありません。私たちは現在第三者契約製造組織(“CMO”)によって私たちの候補製品を生産し、臨床前と臨床用途に使用している。私たちはCMOに依存していますが、私たちも豊富な製造経験を持っており、私たちの製造パートナーとの関係を監督することができます。私たちは私たちの候補製品を作るために必要などんな材料も一つ以上の供給源から得られると信じている。
競争
臨床段階のバイオ製薬会社として,製薬やバイオテクノロジー業界からの様々な会社の競争に直面している。これらの会社には小さな会社もあれば、大企業も含まれており、彼らは私たち自身よりも多くの財政と技術資源を持っており、より長い運営歴史を持っている。私たちはまた、学術、政府、民間研究機関の知的財産権、技術、製品開発と競争する可能性がある。
28
私たちの競争相手は私たちよりもっと多くの財務資源、成熟した市場地位、研究開発、製造、臨床前と臨床テスト、監督管理の承認と精算及びマーケティング許可を得た製品に関する専門知識を持っているかもしれない。これらの競争相手はまた、合格した科学、販売、マーケティングと管理者を募集し、維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。
もし私たちが開発した任意の候補製品が承認されれば、その成功に影響を与える重要な競争要素は、それらの有効性、安全性、利便性、価格、および政府および他の第三者支払者から補償を受ける可能性がある。もし私たちの競争相手が私たちが開発する可能性のある任意の製品よりも効果的で、副作用が少なく、より深刻ではなく、より便利で、あるいはより安い製品を開発し、商業化すれば、私たちの任意の候補製品に対するビジネス機会は減少または消失する可能性がある。私たちの競争相手も私たちよりも早く彼らの製品のためにFDAや他の規制部門の承認を得て、私たちよりも早く製品を商業化することができるかもしれない。
私たちの現在の目標分野では、私たちは次のような競争相手を知っている
SARS-CoV-2
多くの治療法およびワクチンはそれぞれ米国および他の多くの国で緊急治療と新冠肺炎の予防のために承認または許可されている。承認または許可された製品のほかに、新冠肺炎の治療に使用されている他の薬剤も開発されている。
現在承認または許可されている新冠肺炎治療の直接作用抗ウイルス療法は以下の通りである
現在開発中の新冠肺炎を治療する他の経口研究薬は、
探究的治療 |
会社 |
行動メカニズム |
発展段階 |
GS-5245 |
ジリッド科学会社は |
ヌクレオシド類似体 |
第3段階 |
VV 116 |
君士生物科学 |
ヌクレオシド類似体 |
第3段階 |
エンシリビル(S-217622) |
ヒノキ |
プロテアーゼ抑制物 |
第3段階 |
29
シンノリビル(SIM 0417) |
江蘇ヒムセル製薬会社 |
プロテアーゼ抑制物 |
ステップ2/3 |
EDP-235 |
エナンタ製薬会社 |
プロテアーゼ抑制物 |
第二段階 |
PBI-0451 |
パルデス生物科学 |
プロテアーゼ抑制物 |
第二段階 |
パンタランディール |
SyneuRx |
プロテアーゼ抑制物 |
第二段階 |
上記に記載された抗ウイルス薬に加えて、いくつかのモノクロナル抗体は、新規肺炎の予防または治療のために緊急に使用されることが以前に許可されている。これらの許可は現在米国で撤回されているが、将来のSARS-CoV-2変種に有効なモノクロナル抗体を開発し、新冠肺炎の治療に許可または承認する可能性がある。
新冠肺炎を予防するために緊急に使用されるワクチンおよび関連ワクチン増強剤を承認または許可することを含む、
新冠肺炎の潜在的な治療方法とワクチンは依然として絶えず発展している。以上のリストは,本年度報告10−K表までに,米国で緊急用途または臨床開発が行われている製品または候補製品の承認または認可がなされており,これらの製品または候補製品は,フェニニホブビル療法との競争が最も激しい可能性があると考えられるが,新冠肺炎が開発されている各療法の完全なリストではない。
C型肝炎ウイルス
FDAが承認した慢性C型肝炎患者の治療にはEpclusaが含まれている®経口固定用量NS 5 B阻害剤ソモブビルとNS 5 A阻害剤Velpatasvirとの組み合わせ、ハヴォニ®固定用量組合せソブビルおよびNS 5 A阻害剤リディパビル,Vosevi®固定用量の3倍の組み合わせです
30
Sofosbuvir,velpatasvirとvoxilaprevir,NS 3/4 Aプロテアーゼ阻害剤,およびSovaldi,GIlead Sciences,Inc.,Mavyretにより販売されているNS 5 B阻害剤®ブランド製品のほかに、ジリッドはその子会社のアゼルグア治療会社を通じて、エバーヴィ社によって販売されているNS 3/4 A酵素阻害剤グリカビルとNS 5 A阻害剤ピブトリビルの組み合わせ、およびメルク社によって販売されているNS 3/4 A酵素阻害剤エルバスビルとNS 3/4 A酵素阻害剤の組み合わせZepatier Gepatierを発売した。有限責任会社です。米国には,世界の他の地域では臨床開発の異なる段階にあるC型肝炎ウイルス研究試薬が他にある可能性があるにもかかわらず,後期開発段階にある研究試薬は何も知られていない。
商業化する
私たちは現在、私たちの候補製品の世界開発権を保留することによって、私たちの製品の組み合わせの価値を最大限に高めることができると信じています。しかし、許可または承認された候補製品の価値をさらに最大化するために、ある市場におけるパートナーの商業化された専門知識やリソースにアクセスして利用できるように協力を求めることができるかもしれない。私たちが開発に成功した任意の候補製品の米国での商業化を助けるために、既存の商業インフラを持つ第三者と共同普及計画を達成する可能性があります。アメリカ以外では、私たちは商業許可協定を締結するかもしれない。現在、私たちは販売、マーケティング、商業製品流通インフラを何も持っておらず、第三者と既存の手配を達成しておらず、私たちの候補製品をアメリカや他の場所で商業化しています。
知的財産権
私たちのビジネス成功は、SARS-CoV-2およびC型肝炎ウイルスに対するプリンヌクレオチド化合物を含む、ウイルス疾患を治療するためのヌクレオチド治療製品のための特許保護を取得し、維持する能力にある程度依存する。我々は、剤形、用量レジメンおよび投与処方に加えて、私たちのヌクレオチド化合物を単独で使用し、他の治療薬と組み合わせて使用するために、私たちの固有化合物を保護し、私たちのヌクレオチド化合物を用いてウイルス疾患を治療する方法を求めている。私たちはまた私たちのヌクレオチド化合物を製造する製造プロセスの保護を求めている。私たちの成功はまた、他人の固有の権利を侵害、流用、または他の方法で侵害することなく運営し、他人の侵害、流用、または他の方法で私たちの固有の権利を侵害することを防止する能力があるかどうかにかかっている。
私たちの政策は、米国および外国の特許出願を提出することによって、私たちの独自の地位を保護することであり、これらは、私たちのノウハウ、発明、および改善をカバーしており、これらは、私たちの業務の発展と実施に非常に重要である。さらに、我々は現在、米国、ヨーロッパ、および他の管轄地域に適用される場合に、特許期限の調整、回復、および/または特許期間の延長を求める予定である。私たちはまた、ビジネス秘密、技術ノウハウ、持続的な技術革新、潜在的な許可内の機会に依存して、私たちの独自の地位を発展させ、維持しています。また、適切な場合には、規制機関が私たちの薬物製品を承認するのに要する時間を補うために、規制データ独占期間を提供する米国、ヨーロッパ、他の国の法的枠組みから利益を得ることが予想される。
2023年2月1日現在、私たちは15個の特許ファミリーの唯一の所有者であり、これらの特許シリーズは、以下に説明するように、物質組成、薬物組成、使用方法、および製造プロセスを含む、私たちの候補製品および固有ヌクレオチド化合物を含む。2023年2月1日現在、我々が世界で所有している特許資産には、250件以上の未解決、付与または許可された特許出願が含まれており、そのうち14件が米国特許が発行されており、8つの係属中の米国非仮出願、4つの係属中の米国仮出願、5つの特許協力条約(PCT)に基づいて提出された未解決の国際特許出願、および米国以外の国で国家起訴段階に入った200件以上の未解決または承認された特許出願が含まれている。
2023年2月1日現在,我々はMSD International GmbH(Merck,Sharp&Dohme Corp.)の3つの特許シリーズの独占ライセンス者である.NS 5 A阻害剤ruzasvir(MK-8408)を含む物質組成、製造プロセス、および処方は、フランス、イギリス、およびドイツでそれぞれ発行された2つの特許、および欧州特許庁で出願されている米国特許および出願中の特許の2つを含む。
私たちの特許の排他的条項はそれらを獲得する国/地域の法律にかかっている。われわれが現在出願を提出している国では,特許期間は最初の提出日から20年である
31
非臨時特許出願。米国特許の期間は、規制部門の薬物の販売承認を得るのに要した時間(特許期限延長)を補償するために延長することができ、または米国特許商標局が特許訴訟中に遭遇した遅延(特許期限調整と呼ばれる)を延長することができる。例えば、1984年の“薬品価格競争と特許期限回復法”、すなわちハッジ·ワックスマン法案は、FDAが承認した新しい化学実体薬物の特許期間を特許満了後最大5年間延長することを許可している。特許期間の延長の長さは,薬物が審査過程で規制審査と職務調査にある時間の長さと関係がある。米国では、特許期間の延長は、製品が承認された日から合計14年間の特許期間を超えることはできず、承認された薬物またはその使用方法に関する特許のみを延長することができ、承認された薬物またはその使用方法に関する特許請求を延長することしかできない。連合はまた補完保護証明書と呼ばれる似たような特許延期を持っている。特定の他の管轄区域でも、特許の有効期間を延長するための法的枠組みがある。私たちは現在、私たちが合格特許を持っていて、延長期間を得ることができる任意の司法管轄区域内で、私たちが発行した任意の特許のために特許期間の延長を求めるつもりです。しかし、米国のFDAを含めて適用可能な規制機関は保証されません。このような延長を承認すべきかどうか、延長期間が承認されても評価することに同意します。さらに、私たちの特許が延長されても、その特許は、特許の延長部分を含めて、米国または外国の最終管轄権裁判所によって無効または強制執行不可能と判断される可能性がある。
現在発行されている特許および特許出願は、現在の臨床候補AT−511、bemnifosbuvir、AT−281(AT−752の無料ベース)およびAT−752の物質組成をカバーしており、出願が最終司法管轄権裁判所によって発行され、疑問を受けたときに有効である場合、これらの特許および特許出願は、可能な特許期限の調整または延長を考慮することなく、2036~2038年に満了する。現在、AT−511およびbemnifosbuvirを使用してSARS−CoV−2を治療することに関連する特許出願は、2040年から2041年まで満了するであろう。出願が最終管轄権裁判所によって発行され、疑問が提起されたときに有効である場合、可能な特許期間の調整または延長は考慮されない。AT-511およびbemnifosbuvirを使用してC型肝炎を治療するために現在発行されている特許および特許出願は、出願が疑問視されたときに最終管轄権裁判所によって発行され、有効性を維持し、可能な特許期間の調整または延長を考慮しない場合、2036年から2042年の間に満了する。現在、AT−281およびAT−752を使用してデング熱を治療することに関連する特許出願は、2036年から2043年の間に満了し、出願が最終管轄権裁判所によって発行され、疑問が提起されたときに有効である場合、可能な特許期間の調整または延長は考慮されない。
しかし、私たちの承認された製品の市場を保護するために私たちが依存する可能性がある特許を含む私たちの任意の特許は、最終管轄権裁判所によって無効または強制的に実行できないと判断されるかもしれない。あるいは、私たちは私たちの特許の期限に影響を与えるか、実行可能な方法で訴訟を解決することが私たちの利益に合致すると決定することができる。米国および他の管轄区域の特許法または特許法解釈の変化は、私たちの発明を保護し、私たちの知的財産権を実行する能力を弱める可能性がある。したがって、私たちは、私たちの特許または第三者特許の特許請求の範囲の広さまたは実行可能性を予測することができない。製薬とバイオテクノロジー産業の特徴は特許と他の知的財産権に関する広範な訴訟だ。我々が我々のヌクレオチド化合物の特許地位およびこれらの化合物の使用を獲得し保持する能力があるかどうかは、付与されたまたは付与可能な特許主張を成功的に実行できるかどうかに依存する。私たちは、私たちが提出したか、または提出可能な任意の未解決特許出願または第三者からの許可が、任意の追加特許の発行をもたらすかどうかを知らない。私たちが所有または将来獲得可能な公開特許は、挑戦、無効、または回避される可能性があり、任意の発行特許に基づいて付与される権利は、同様の技術を有する競合他社に対して十分な保護または競争優位性を提供することができない可能性がある。さらに、我々の競争相手は、同様の作用機序を有する薬剤を独立して開発し、それを商業化し、および/または特許を侵害することなく、私たちの治療方法または戦略を複製することができるかもしれない。私たちが開発する可能性のある薬物の臨床開発と規制審査には多くの時間がかかるため、私たちのどの薬も商業化される前に, 任意の関連特許は、商業化後、任意のそのような特許の任意の利点を弱めるために、非常に短い期間でのみ失効または有効に維持される可能性がある。知的財産権に関するリスクに関するより多くの情報は、第1部第1 A項を参照されたい。“リスク要因--知的財産権関連のリスク”
32
我々の特許シリーズは,2023年2月1日まで,以下にさらに紹介する.
AT-511とフェニホブビル
AT−511またはその薬学的に許容される塩(例えば、フェニレホブビル)、AT−511またはその薬学的に許容される塩の医薬組成物、およびAT−511またはその塩を用いてC型肝炎を治療する方法が記載されている第1の特許ファミリーを有する。このシリーズは7つのすでに発行されたアメリカ特許(US PAT.番号9,828,410;10,000,523;10,005,811;10,239,911;10,815,266;10,870,672;10,870,673)およびAT−511またはその薬学的に許容される塩、関連化合物、およびそれらの医薬組成物に関する係属中の米国特許出願。この特許シリーズは現在も国家起訴段階にあり、またはアフリカ地域知的所有権機関、オーストラリア、ブラジル、カナダ、中国、コロンビア、ユーラシア特許庁、エジプト、欧州特許庁、グルジア、香港、インドネシア、イスラエル、インド、日本、韓国、メキシコ、マカオ、マレーシア、ナイジェリア、ニュージーランド、フィリピン、ロシア、サウジアラビア、シンガポール、タイ、ベトナム、ウクライナ、南アフリカ、アラブ首長国連邦で承認されている。私たちは20件以上の外国特許が付与または許可され、20件以上の特許出願が出願されている。発行、有効、および実行可能な場合、この特許シリーズの予想満了年は、国の法律によって得られる可能性のある任意の延長、調整、または回復期間を考慮することなく、2036年である。
私たちはまた、フェニニホブビル、医薬組成物、およびフェニトフォブビルを用いたC型肝炎の治療方法を専門とする第2の特許ファミリーを有している。このシリーズは2つの発行された米国特許(US PAT)を含む。10,519,186号および米国特許第10,906,938号)およびフェニニホブビルに関する係属中の米国特許出願。この家族は現在アルゼンチン、アポロ、オーストラリア、ブラジル、カナダ、中国、コロンビア、欧州特許庁、欧州特許庁、グルジア、香港、インドネシア、イスラエル、インド、日本、韓国、メキシコ、マレーシア、ナイジェリア、ニュージーランド、ロシア、シンガポール、台湾、タイ、ベトナム、ウクライナ、ウズベキスタン、南アフリカで国家起訴段階にある。私たちは外国特許10件以上を付与し、25件以上を申請している。この特許シリーズの予期される有効期間は2038年であり、発行され、有効かつ強制的に実行可能である場合、米国または他の国の法律に従って可能な任意の延長、調整、または回復期間は考慮されない。
我々は2つの特許ファミリーを有し、AT−511またはフェニフブビルを用いてSARS−CoV−2を治療する方法を開示している。これらの家族は、許可された米国特許(米国特許番号10,874,687)を含み、3つの係属中の米国出願と、アルゼンチン、ARIPO、オーストラリア、バーレーン、ブラジル、カナダ、チリ、中国、コロンビア、エクアドル、エジプト、欧州特許庁、欧州特許庁、グルジア、インド、イスラエル、日本、ヨルダン、クウェート、リビア、マレーシア、メキシコ、モロッコ、ニュージーランド、ニカラグア、ナイジェリア、オマーン、フィリピン、ロシア、サウジアラビア、シンガポール、南アフリカ、韓国、台湾、タイ、チュニジア、ウズベキスタン、ベトナムで決定された出願を含む。これらのファミリーによって発行された特許は、有効かつ強制的に実行可能である場合、米国または他の国の法律によって提供される可能性のあるいかなる延期、調整、または回復期限も考慮することなく、2040年または2041年と予想される。
正鎖RNAウイルス感染の治療または予防のためのAT-511または薬学的に許容されるその塩の使用を開示する第5の特許ファミリーを有する黄ウイルス科デング熱、西ナイル、黄熱病などのウイルス感染。このシリーズは、出願中の出願と、許可された特許(米国特許番号:10,946,033)とを含み、現在、オーストラリア、ブラジル、カナダ、中国、欧州特許庁、欧州特許庁、香港、インドネシア、日本、韓国、マレーシア、ナイジェリア、ロシア、シンガポール、タイ、ベトナム、南アフリカで出願または承認されている。私たちは30件以上の外国特許を取得し、20件以上の特許出願が出願されている。この特許シリーズの予期される有効期間は2037年であり、発行され、有効かつ強制的に実行可能である場合、米国または他の国の法律に従って可能な任意の延長、調整、または回復期間は考慮されない。
6つ目の特許ファミリーを有し、肝炎後肝硬変患者のC型肝炎ウイルスの治療のためのAT−511およびフェニホブビルを開示した。この家族には審査待ちのアメリカ申請が含まれている。この家庭は現在、中国、欧州特許庁、香港、日本、韓国、ロシア、台湾地区で全国起訴段階にある。この特許シリーズの予期される有効期間は2039年であり、発行され、有効かつ強制的に実行可能である場合、米国または他の国の法律に従って可能な任意の延長、調整、または回復期間は考慮されない。
私たちは7番目の特許ファミリーを持ち、SARS-CoV-2ウイルス変異または薬剤耐性形態を治療する方法を説明した。この一連のものはPCTに基づいて提出された国際出願と
33
アルゼンチンで申請し、台湾で申請します。本特許出願に基づいて出願された非仮特許出願から発行された特許は、有効かつ強制的に実行可能であれば、米国又は他の国の法律に基づいて可能な期限調整を考慮することなく、満期年は2041年と予想される。
8つ目の特許ファミリーも有し、AT−511およびベノブビルを製造する方法が開示されている。この家族には審査待ちのアメリカ申請が含まれている。これらの仮特許出願に基づいて出願された非仮特許出願によって発行された特許は、有効かつ実行可能であれば、米国または他の国の法律に基づいて可能な期限調整を考慮することなく、満期年は2041年と予想される。
私たちはまた、AT-511およびベノシブビルを製造するための追加プロセスを開示する第9の特許シリーズを持っている。この一連はPCTの次の国際出願分野を含む。これらの非仮特許出願から発行される特許は、有効かつ実行可能であれば、米国又は他の国の法律に基づいて行われる可能性のあるいかなる期限調整も考慮することなく、満期年は2041年と予想される。
私たちはまた10番目の特許家族を持ち、ベノホブビルの新しい形態を公開した。この一連のものは、PCTに従って提出された国際出願と、カナダでの係属中の出願を含む。本特許出願に基づいて出願された非仮特許出願から発行された特許は、有効かつ強制的に実行可能であれば、米国又は他の国の法律に基づいて可能な期限調整を考慮することなく、満期年は2042年と予想される。
AT−281とAT−752
上述した第1の特許シリーズには、許可された米国特許第10,875,885号を含むAT−281、その薬学的に許容される塩(例えば、AT−752)およびAT−281またはその薬学的に許容される塩の医薬組成物およびC型肝炎ウイルス感染を治療するためのそれらの使用も記載されている。
上述した第2の特許シリーズには、米国特許第10,906,928号に含まれるAT−752およびAT−752の医薬組成物も記載されている。この特許シリーズにおける係属中の米国特許出願は、AT−752およびAT−752の医薬組成物をカバーする。
上述した第5の特許シリーズはまた、デング熱(米国特許番号10,946,033)、黄熱病およびジカウイルスを含むRNAウイルス感染の治療または予防のためのAT-281またはその薬学的に許容される塩を開示している。したがって、3つの特許ファミリーは、AT−281またはAT−752と、AT−281またはAT−752を使用してウイルス感染を治療する方法とを記載している。
我々が所有する別の特許シリーズは、AT−752の有利な剤形、AT−752の投与レジメン、およびAT−752を含むデング熱治療の併用療法を開示する3つの米国仮出願からなる。これらの非仮特許出願から発行される特許は、有効かつ実行可能であれば、米国または他の国の法律に基づいて行われる可能性のあるいかなる期限調整も考慮することなく、満期年は2043年と予想される。
ルザスビル
私たちはMSD International GmbH(メルク、シャープ、ドルム社)の3つの特許シリーズを独占的に許可した。Ruzasvir(MK-8408)の物質組成、製造方法と調合を紹介し、RuzasvirはC型肝炎ウイルスを治療する汎遺伝子NS 5 A阻害剤である。物質組成をカバーする家族は、許可された米国特許(米国特許番号9,555,038)と、フランス、イギリス、ドイツで取得された特許とを含む。期日は2034年を予定している。製造プロセスを記述する家族は、期限が2036年と予想される付与された米国特許(米国特許番号10,457,690)を含む。処方を記述する家族は、係属中の米国特許出願と欧州特許庁の未解決特許出願とを含み、承認された場合、2039年に満了する予定である。
私たちはまた、bemnifosbuvirとruzasvirの組み合わせをカバーするPCTに従って提出された国際出願を独占的に所有しており、承認された場合、その有効期間は2042年になるだろう。
34
政府規制と製品審査
アメリカ連邦、州と地方の各級政府当局及びその他の国の政府当局は私たちが開発している製品の研究、開発、テスト、製造、品質管理、承認、ラベル、包装、貯蔵、記録保存、販売促進、広告、流通、マーケティングと輸出入などの方面に対して広範な監督管理を行った。新薬は新薬申請(“NDA”)プログラムを通じてFDAの承認を得なければならず,その後米国で合法的に発売されることができる。
アメリカの薬物開発プロセスは
米国では,FDAは“連邦食品,薬物と化粧品法”(FDCA)とその実施条例に基づいて薬物を規制している。規制の承認を得て、その後、適切な連邦、州、地方、外国の法規と条例を遵守する過程には、多くの時間と財政資源が必要だ。
FDAが米国で発売される前に必要とされるプログラムは、一般に以下のような態様を含む
米国で候補製品の第1回臨床試験が開始される前に,スポンサーはINDをFDAに提出しなければならない。INDは人間がIND製品を使用することをFDAが許可する要求である。IND提出の中心焦点は臨床研究の全体的な研究計画と方案である。INDには動物や体外培養研究製品の毒理学、薬物動態学、薬理学と薬効学特徴を評価する研究;化学、製造と制御情報;及び任意の利用可能なヒトデータ或いは文献を用いて研究製品の使用を支持する。INDはヒト臨床試験が始まる前に発効しなければならない。提出されると、INDはFDAが30日以内に提案された臨床試験に対して安全懸念または問題を提起しない限り、FDAが受信した30日後に自動的に有効になる。この場合,INDは臨床的に放置される可能性があり,INDスポンサーやFDAは臨床試験が開始される前に未解決の問題や問題を解決しなければならない。したがって,INDの提出はFDA認可による臨床試験の開始につながる可能性があり,そうでない可能性もある。
臨床試験は、GCPに従って合格した研究者の監督の下でヒト被験者に研究製品を服用することを含み、すべての研究被験者に任意の臨床研究への参加についてインフォームドコンセントを提供することを含む。臨床試験
35
その他にも,研究目標を詳細に説明し,セキュリティを監視するためのパラメータ,有効性基準を評価するためのプロトコルに基づいて評価を行う.製品開発中に行われる各後続の臨床試験および後続の任意のレジメン修正は、既存のINDに個別に提出されなければならない。INDは活発であるが,前回の進展報告以来行われてきた臨床試験と非臨床研究の結果をまとめた進展報告は,他の情報を除いて少なくとも年に1回FDAに提出しなければならず,深刻かつ意外な疑わしい有害事象を知るためにFDAおよび調査者に書面IND安全報告を提出しなければならず,他の研究から発見された同一または類似の薬物に曝露されたヒトへの重大なリスク,動物または動物からの発見体外培養測定により、人類に対して重大なリスクがあり、及び任意の臨床上重要な疑わしい副作用の発生率は方案或いは研究者マニュアルに記載されているより増加した。
また,臨床試験を行うことを推奨する各地点の独立したIRBまたは倫理委員会は,その場所で臨床試験を開始する前に任意の臨床試験の計画およびそのインフォームドコンセントを審査·承認し,完成まで研究を監視しなければならない。いくつかの研究はまた、臨床研究スポンサーによって組織された独立した合格専門家グループの監視を含み、このグループは、研究のいくつかのデータへのアクセスに基づいて、研究が指定されたチェックポイントで行うことができるかどうかを許可するデータ安全監視委員会と呼ばれ、被験者に受け入れられない安全リスクまたは他の理由があると判定された場合、治療効果を示さない場合、臨床試験を停止する可能性がある。FDAあるいはスポンサーは研究対象や患者が受け入れられない健康リスクに直面していることを発見することを含む、様々な理由で臨床試験を一時停止することができる。同様に,臨床試験がIRBの要求に沿って行われていない場合,あるいは研究製品が患者の予期しない深刻な傷害に関連している場合,IRBはその機関の臨床試験の承認を一時停止または終了することができる。
人体臨床試験は通常3つの連続段階に分けて行われ、これらの段階は重複或いは合併する可能性がある
第1段階:候補製品は、最初に健康なヒト対象に導入され、場合によっては、標的疾患または状態の患者に導入される。これらの研究は、人体内での製品の安全性、用量耐性、吸収、新陳代謝と分布、および用量増加に関連する副作用をテストし、可能な場合に有効性の早期証拠を得ることを目的としている。
第二段階:候補製品を特定の疾患または状態の限られた患者集団に使用して、初歩的な治療効果、最適な用量および用量計画を評価し、可能な不良副作用および安全リスクを決定する。より大規模かつより高価な3期臨床試験を開始する前に、複数の2期臨床試験を行って情報を得ることができる。
第三段階:候補製品は拡大した患者集団に使用され、用量をさらに評価し、臨床治療効果の統計的顕著な証拠を提供し、さらに安全性をテストし、通常複数の地理的に分散した臨床試験地点で行われる。これらの臨床試験は研究製品の全体的なリスク/収益比率を確定し、製品の承認に十分な基礎を提供することを目的としている。
承認後試験は,第4段階研究と呼ばれることがあり,最初の上場承認後に行われる可能性がある。これらの試験は,治療適応が予想される患者の治療から追加的な経験を得るために用いられている。場合によっては,FDAはNDAを承認する条件として4期臨床試験を強制的に実行することができる。
新薬開発期間中、スポンサーはいつかFDAと会う機会がある。これらの要件は、INDを提出する前、第2段階の終了時、および秘密協定の提出前にある可能性がある。他の時間に会議を開催することを要求することができます。これらの会議は,スポンサーにこれまで収集してきたデータに関する情報を共有する機会を提供し,FDAにアドバイスを提供し,スポンサーとFDAに次の段階の開発について合意することができる。スポンサーは通常、第二段階試験終了時の会議を用いて第二段階臨床結果を検討し、新薬承認を支持すると考えられる重要な第三段階臨床試験計画を提出する。
臨床試験と同時に、会社は通常追加の動物研究を完成し、薬物化学と物理特性に関する追加情報を開発し、cGMPの要求に基づいて最終的に商業量産製品のプロセスを決定しなければならない。
36
製造過程は一貫して高品質の候補製品ロットを生産できる必要があり、また、メーカーは最終薬物の身分、強度、品質、純度をテストする方法を開発しなければならない。また,適切な包装を選択·テストし,候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
アメリカの審査と承認の流れ
すべての適用された法規要件に基づいて必要なすべてのテストが成功したと仮定し,製品開発,臨床前および他の非臨床研究および臨床試験の結果,製造過程の記述,薬物化学の分析テスト,提案されたラベルおよびその他の関連情報は,その製品の発売承認を要求するセキュリティプロトコルの一部としてFDAに提出される。秘密協定の提出には相当な使用料が必要であり、いくつかの限られた場合には、そのような費用を免除することができる。また,孤児薬として指定された製品については,NDAに対して使用料を評価せず,非孤児適応も含まれていない限りである。
FDAは、届出を受ける前に、提出後の最初の60日以内にすべてのNDAを予備審査して、それらが十分に完全であるかどうかを決定し、実質的な審査を行うことができる。FDAは秘密協定の申請を受け入れるのではなく、より多くの情報を提供することを要求するかもしれない。この場合、秘密協定と追加情報を再提出しなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出されると、FDAは、製品がその予期される用途に対して安全かつ有効であるかどうかを決定し、その製造がcGMPに適合するかどうかを決定して、製品の特性、強度、品質、および純度を保証および維持するためにNDAを検討する。現在発効している“処方薬使用料法案”(PDUFA)によると,FDAは標準NDA提出日から10カ月以内に新たな分子実体を審査させ,提出されたNDAに行動させることを目標としている。この審査には通常12カ月の時間が必要であり,NDAがFDAに提出された日から計算すると,FDAは申請提出後約2カ月で“届出”決定を下すためである。
FDAは新薬の申請を諮問委員会に提出するかもしれない。諮問委員会は,臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請を審査,評価し,申請を承認すべきかどうか,どのような条件でアドバイスを提供すべきかを担当する。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。
秘密協定を承認する前に、FDAは通常、製品を生産する1つまたは複数の施設を検査する。FDAは、製造プロセスおよび施設がcGMPに適合していることを決定し、要求された仕様で製品が一貫して生産されることを保証するのに十分でない限り、申請を承認しないであろう。さらに、NDAを承認する前に、FDAは、GCPに適合することを確実にするために、1つまたは複数の臨床場所を検査することができる。
FDAはセキュリティプロトコルを評価した後,承認状または完全な返信(“CRL”)を発行する.この薬剤の商業マーケティングを承認し、特定の適応の処方情報を提供する。CRLは,申請の審査周期が完了したことを示しており,申請は現在の形で承認されない.CRLは、一般に、FDAによって決定されたNDAにおける特定の欠陥を記述し、追加の臨床試験、または臨床試験、非臨床研究、または生産に関連する他の重要で時間のかかる要件のような追加の臨床データを必要とする可能性がある。CRLを発行すると,スポンサーはNDAを再提出し,手紙で発見されたすべての不足点を解決したり,申請を撤回したりしなければならない.このようなデータや情報を提出しても,FDAはNDAが承認基準を満たしていないと認定する可能性がある。
1つの製品が規制によって承認された場合、このような承認は、特定の適応が付与され、製品が発売される可能性のある指定された用途を制限または制限することができる。例えば、FDAは、製品の利点がそのリスクよりも大きいことを確実にするために、リスク評価および緩和戦略(“REMS”)を有するNDAを承認する可能性がある。REMSは、既知または潜在的な薬物に関連する深刻なリスクを管理し、これらの薬物の安全な使用を管理することによって、薬物ガイドライン、医師のコミュニケーション計画、または制限された投与方法、患者登録および患者登録などの安全な使用を確保する要素を含むことができる安全戦略である
37
他のリスク最小化ツールですFDAはまた,提案されたラベルを変更したり,適切な制御や仕様を作成したりすることを条件に承認することも可能である.承認されると、発売前と上場後の要求に対する遵守が保たれていない場合、あるいは製品が市場に進出した後に問題が発生した場合、FDAは製品承認を撤回する可能性がある。FDAはまた、商業化後の製品の安全性および有効性をさらに評価および監視するために、1つまたは複数の承認された第4段階の研究および監視を要求することができ、これらの承認された研究の結果に基づいて製品のさらなる販売を制限する可能性がある。さらに、新しい立法によって生成された要求を含む新しい政府要求が確立される可能性があり、またはFDAの政策が変更される可能性があり、これは、規制承認のスケジュールまたは他の方法で行われている開発計画に影響を与える可能性がある。
そのほか、“小児科研究公平法”はスポンサーに大多数の薬物、新しい有効成分、新しい適応、新しい剤形、新しい投与方案或いは新しい投与経路に対して小児科臨床試験を行うことを要求している。PREAによれば、元のNDAおよびサプリメントは、スポンサーが延期または免除を受けていない限り、小児科評価を含まなければならない。要求された評価は、すべての関連する小児科亜集団において主張される製品の適応の安全性および有効性を評価し、各安全で有効な小児科亜群に対する製品の用量および投与をサポートしなければならない。スポンサーまたはFDAは、小児科亜群の一部または全部の小児科臨床試験の延期を要求することができる。延期は、小児科臨床試験が完了する前に、成人で使用を許可する準備ができていることを発見するか、または小児科臨床試験が開始される前に追加の安全性または有効性データを収集する必要があることを含むいくつかの理由があるかもしれない。FDAは、必要な評価を提出できなかった、延期された最新の状況を維持し、または小児科処方承認要求を提出できなかった任意のスポンサーに、規定に適合しない手紙を送信しなければならない。
緊急使用許可
FDA局長は、衛生公衆サービス部長(“HHS”)の許可に基づいて、発表された公衆衛生緊急事態に関連する場合には、FDAの規定に適合しない製品のためにEUAを発行することによって、製品の販売を許可することができる。HHSがEUAを発表する前に、大臣は、国家安全に重大な潜在的影響を与えるか、または重大な潜在的影響を有する公衆衛生緊急事態の存在を決定し、特定の生物学的、化学的、放射線または核剤(“CBRN”)に関連するか、またはCBRNの特定の疾患または状態に起因して緊急状態を宣言しなければならない。2020年2月4日、衛生と衛生局局長はこのような突発的な公衆衛生事件が存在することを確定し、新冠肺炎感染を招くSARS-CoV-2ウイルスに関連した。脅威または緊急事態が決定されると、衛生·公衆サービス部部長は、いくつかのタイプの製品のためにEUA(EUA声明と呼ばれる)を発行する理由がある緊急事態の存在を宣言しなければならない。2020年3月27日、米国衛生·公衆サービス部長官は、SARS-CoV-2-新冠肺炎の大流行中に薬物および生物製品を許可する理由がある場合があるが、発表された任意のEU合意の条項を遵守しなければならないと、国家安全または海外在住米国市民の健康および安全に影響を及ぼす可能性がある公衆衛生緊急事態を決定したことを発表した。
EUA宣言が発行されると、FDAは、その宣言範囲に属する製品に対してEUAを発行することができる。EUAを発行するためには、FDA専門家は、(1)EUA宣言で示されているCBRNが、深刻なまたは生命を脅かす疾患または状態を引き起こす可能性がある、(2)既存の全ての科学的証拠に基づいて、CBRNによって引き起こされる疾患または状態を効果的に診断、治療または予防する可能性があり、その既知および潜在的利益がその既知および潜在的リスクよりも大きいと信じる理由があり、(3)十分で承認され、利用可能な製品代替品がないと結論しなければならない。EUAに制約された製品は、ラベルおよびマーケティング要件を含むEUAの条件に依然として適合しなければならない。さらに、EUAで製品を販売する許可は、EUA宣言が発効する期間に限定され、FDAは、場合によってはEUAを取り消すことができる。
開発と審査計画を加速する
FDAは合格した候補製品に一連の迅速な開発と審査計画を提供した。例えば、FDAの高速チャネル計画は、特定の基準に適合する候補製品の審査プロセスを加速または促進することを目的としている。具体的に、もし新薬が深刻或いは生命に危害を及ぼす疾病或いは状況を治療することを目的とし、そしてこの疾病或いは状況が満たされていない医療需要を解決する潜在力を示した場合、迅速な通路指定を受ける資格がある。高速道路について
38
候補製品の場合、FDAは、完全な出願を提出する前にNDAを考慮する審査部分をスクロールすることができ、スポンサーがNDA部分を提出するスケジュールを提供した場合、FDAはNDAの部分を受け入れることに同意し、スケジュールが許容可能であると判断し、スポンサーはNDAの第1の部分を提出する際に任意の必要な使用料を支払うことができる。
重篤または生命に危険な疾患や状況を治療しようとする候補品も,その開発や審査を加速するための画期的な療法指定を受ける資格がある可能性がある。予備臨床証拠が、候補製品が単独または1つまたは複数の他の薬物または生物製品と組み合わせて使用される可能性があることを示す場合、候補製品は、1つまたは複数の臨床重要終点において、既存の治療法よりも実質的な改善を示す可能性があり、例えば、臨床開発早期に観察された実質的な治療効果を示す場合、候補製品は突破的治療称号を得ることができる。この指定には、Fast Track計画のすべての機能と、第1段階で開始されたより密集したFDA相互作用および指導と、高度な管理者の参加を含む候補製品開発および審査を加速する組織約束とが含まれる。
迅速なチャネル指定または突破療法指定を有する製品を含むFDA承認を提出する任意の薬物のマーケティング申請は、優先的な審査および承認の加速など、FDAが開発および審査を加速することを意図している他のタイプの計画の資格に適合する可能性もある。候補製品が重篤な疾患の治療のために設計され、承認された場合、市販製品と比較して安全性または有効性の面で有意な改善が提供される場合、NDAは優先審査を受ける資格がある。FDAは、審査を促進するために、優先審査として指定されたNDAを評価するために追加のリソースを使用しようと試みるだろう。FDAは,提出日後6カ月以内に優先審査指定を有する出願を審査するように努力しているが,その現在のPDUFA審査目標に基づいて,新分子実体NDAを審査する期間は10カ月である。
しかも、候補製品は加速された承認を受ける資格があるかもしれない。深刻または生命を脅かす疾患または疾患を治療するための候補製品は、候補製品が臨床利益を合理的に予測する可能性のある代替終点、または不可逆的な発病率または死亡率よりも早く測定可能な臨床終点、不可逆的に不可逆的な発病率または死亡率または他の臨床的利益を合理的に予測する臨床終点に有効であることを決定する際に、病状の重症度、希少性または流行率、および代替治療の利用可能性または不足を考慮しながら、加速承認を得る資格がある可能性がある。承認の条件として、FDAは、一般に、予測の臨床的利益を検証および説明するために、承認を加速させる薬剤のスポンサーに、十分かつ制御された検証臨床試験を行うことを要求する。スポンサーが必要な臨床試験をタイムリーに行うことができなかった場合、またはそのような試験が予期される臨床的利益を検証できなかった場合、加速承認された製品は、加速脱退プログラムの影響を受ける可能性がある。さらに、FDAは現在、販売促進材料の事前承認を承認を加速する条件とすることを要求しており、製品の商業発売時間に悪影響を及ぼす可能性がある。
高速チャネル指定、画期的な治療指定、優先審査、承認の加速は承認基準を変更することはありませんが、開発や承認過程を加速する可能性があります。1つの製品がこれらの計画のうちの1つまたは複数に適合していても、FDAは、製品がもはや資格条件に適合していないことを後で決定することができ、またはFDAの審査または承認を決定する期間が短縮されないことができる。
承認後に要求する
FDAによって製造または流通が許可された任意の製品は、記録保存、有害事象報告、定期報告、製品サンプリングおよび流通、ならびに製品広告および販売促進に関連する要件を含むFDAによって普遍的かつ持続的に規制される。承認後、承認された製品の大多数の変更は、新たな適応または他のラベル宣言を追加するなど、FDAの事前審査および承認を経なければならない。どんな発売された製品についても、継続的な年間計画費用があります。
薬品メーカー及びその下請け業者はFDAとある州機関に彼らの機関を登録することを要求され、FDAとある州機関の定期的な抜き打ち検査を受け、cGMPに符合するかどうかを見て、これは薬品メーカーにあるプログラムと書類の要求を加えた。製造プロセスの変更は厳しく規制されており,変更の重要性により,FDAの承認を得ておく必要がある可能性がある
39
実施する。FDAの規定はまた、cGMPとのいかなる偏差も調査·是正し、NDA保有者およびNDA保有者が使用を決定する可能性のある任意の第三者製造業者に報告することを要求する。そのため、メーカーは生産と品質管理の分野で時間、お金、精力をかけ続け、cGMPやその他の法規遵守性を維持しなければならない。
規制要件や基準が守られていない場合、あるいは製品発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。その後、製品には、予期されない深刻度または頻度の不良事象、または生産プロセス、または法規要件を遵守できなかったことを含む以前に未知の問題が存在し、新しいセキュリティ情報を追加するために承認されたラベルの改訂を招く可能性があり、新しい安全リスクを評価するために発売後研究または臨床研究を実施するか、またはREMS計画に従って流通制限または他の制限を実施する可能性がある。他の他の潜在的な結果には
FDAは薬品のマーケティング、ラベル、広告、販売促進を密接に規制している。1社はFDAが承認したラベルの規定に基づいて、安全性と有効性、純度、効力に関する声明しか提出できない。FDAと他の機関は非ラベル用途の普及を禁止する法律法規を積極的に施行している。これらの要求を守らないことは、否定的な宣伝、警告状、改正広告、および潜在的な民事と刑事罰を招く可能性がある。医師はその独立した専門医学判断に基づいて、製品ラベルに記載されていない用途、およびFDAテストと承認された用途とは異なる合法的に利用可能な製品のために処方することができる。医師は,異なる場合,このような非ラベル使用が多くの患者の最適な治療法であると考えるかもしれない。FDAは医者が治療を選択する時の行動を規範化しない。しかしながら、FDAは、製品ラベルの外で使用される問題におけるマーケティング担当者のコミュニケーションを制限している。連邦政府は、ラベル外使用の不当な普及の疑いがある会社に巨額の民事と刑事罰金を科し、会社がラベル外販売促進に従事することを禁止している。FDAおよび他の規制機関はまた、企業に同意法令または永久禁止を締結し、これらの法令または永久禁止に基づいて、特定の販売促進行為を変更または制限することを要求する。しかしながら、会社は、FDAによって承認された製品ラベルと一致する真で誤解されない情報を共有するかもしれない。
マーケティング排他性
FDCAによって許可されたマーケティング排他的条項は、いくつかのマーケティング申請の提出または承認を延期する可能性がある。FDCAは新しい化学実体秘密協定の承認を得た最初の申請者に5年間のアメリカ国内の非特許データ排他期を提供した。FDAが以前に同じ活性部分を含む他の新薬を承認していなければ,薬物は新しい化学実体であり,活性部分は薬物物質の作用を担う分子やイオンである。排他的な期間内にFDAは承認しないかもしれませんし、略称新薬の審査さえ受けないかもしれません
40
出願(“ANDA”)または第505条(B)(2)条に従って提出されたセキュリティ協定(“505(B)(2)セキュリティ協定”)は、出願人が承認に必要なすべてのデータを参照するための合法的な権利を有していない場合、同じ活性部分に基づく別の薬剤のために別の会社によって提出され、元の革新薬と同じ適応であるか、別の適応のためのものであるかにかかわらず、同じ活性部分に基づく別の薬剤のために別の会社によって提出される。しかしながら、出願がイノベーターNDA所有者がFDAに記載された特許のうちの1つを含む特許が無効または未侵害証明である場合、4年後に出願することができる。
FDAが、出願人が行っているまたは後援する新しい臨床研究(バイオアベイラビリティ研究を除く)が、既存の薬剤の新しい適応、用量または強度のような承認申請に不可欠であると考えている場合、FDCAは、NDAに3年間の市場排他性、または既存のNDAの補充を提供することもできる。この3年間の排他性は、この薬剤が新しい臨床研究に基づいて承認された修正のみを含み、FDAがANDAまたは505(B)(2)NDAを許可することは禁止されておらず、元の適応または使用条件の有効成分を含む薬剤のために使用される。5年と3年の排他性は完全な秘密協定の提出や承認を延期したり承認したりしないだろう。しかしながら、完全なセキュリティプロトコルを提出する出願人は、安全かつ有効であることを証明するために、任意の臨床前研究および十分かつ良好に制御された臨床試験を参照するために必要な権利を行うか、または得ることを要求されるであろう。
小児科専門権はアメリカで利用可能なもう一つのマーケティング専門権だ。スポンサーがFDAの書面要求に応じて児童に臨床試験を行う場合、小児科排他性規定は別の排他期に加えて6ケ月のマーケティング排他性を追加する。書面出願の発表はスポンサーに述べた臨床試験を要求しない。
他の医療保険法
製薬会社は連邦政府およびそれらが業務を展開している州と外国司法管轄区当局の追加医療監督と法執行を受けている。このような法律には、米国連邦および州の反リベート、詐欺および乱用、虚偽声明、定価報告および医師の支払いの透明性に関する法律および法規が含まれているが、これらの法律および法規は、医薬品の価格設定および医師および他の登録医療専門家への支払いまたは他の価値移転、および米国以外の管轄地域の同様の外国の法律に関するものである このような外国の法律や条例の範囲は上記の規定よりも広い可能性があり,支払者にかかわらず適用可能である。これらの法律と法規は大きく異なる可能性があり、コンプライアンス作業をさらに複雑化させる可能性がある。例えば、EUでは、多くのEU加盟国が具体的な反贈与法規を採択し、医療製品の商業行為、特に医療専門家や組織に対する商業行為をさらに制限している。また、最近では、医療専門家や実体への支払いや価値移転の規制を強化する傾向が見られ、多くのEU加盟国は、米国の製薬会社に対する要求と同様に、製薬会社に対する報告や透明性要件(通常は年に1回)を実施する全国的な“サンシャイン法案”を採択した。いくつかの国はまた、商業コンプライアンス計画を実施することを要求するか、またはマーケティング支出および価格設定情報の開示を要求する。このような任意の法律または任意の他の適用可能な政府法規に違反することは、行政民事および刑事罰、損害賠償、罰金の返還、追加の報告要件および監督義務、契約損害賠償、業務の縮小または再構成、政府医療計画から除外され、および/または監禁を含む重大な処罰をもたらす可能性がある。
保証と精算を請け負う
私たちが規制承認を求める可能性のある任意の候補製品のカバー範囲と精算状態には、重大な不確実性がある。米国および他の管轄地域での販売は、連邦医療保険、医療補助、TRICARE、退役軍人管理局などの政府医療計画、管理型医療組織、個人医療保険会社を含む第三者支払者が十分な保険と十分な補償を提供するかどうかにある程度依存する。私たちあるいは私たちの顧客が私たちの候補製品のために精算を求める価格は第三者支払人の疑問、値下げ、あるいは拒否されるかもしれません。
第三者支払い者が製品に保険を提供するかどうかを決定するプロセスは、一般に、支払者が製品に支払う支払率を設定するプロセスとは分離されている。アメリカでは、保険や補償について、支払者間に統一された政策はありません。以下の方面の決定について
41
いずれかの製品、保険範囲、精算金額を保証するかどうかは、一つずつ計画した上で策定されています。第三者支払者が自己の保険·精算政策を設定する際には,通常連邦医療保険引受政策や支払制限に依存するが,独自の方法や承認の流れもある。そのため、製品の保証範囲と精算範囲は支払人によって異なる。したがって、保証範囲の決定プロセスは、しばしば時間がかかり、高価なプロセスであり、製造業者が製品の使用のために個々の支払人に科学的および臨床的支援を提供する必要がある可能性があり、保証範囲および適切な補償を一貫的に適用すること、または最初に十分な補償を得ることを保証することはできない。
第三者決済者は、価格、医療製品およびサービスの医療必要性および費用効果、ならびにそれらの安全性および有効性を検討することにますます挑戦している。価格制御及びコスト制御措置を講じ、既存の制御及び措置を有する司法管区においてより限定的な政策をとることにより、承認された製品の販売をさらに制限することが可能となる。第三者支払者は、私たちの候補製品が他の利用可能な療法と比較して医療的に必要であるか、または費用対効果があると思わないかもしれないし、有利な保証に必要なリターン率を確保することは、十分なコスト利益率を生成できないかもしれないし、または薬物開発への私たちの投資の適切なリターンを達成するために十分な価格レベルを維持することができないかもしれない。また、任意の製品の第三者精算または第三者支払人が製品を保証しないと決定することは、医師の使用や患者のその製品に対する需要を減少させる可能性がある。
医療改革
米国や外国の司法管轄地域では、医療保健システムに関するいくつかの立法と規制改革および提案された改革が継続されており、これらの改革は候補製品の上場承認を阻止または延期し、承認後の活動を制限または規制し、候補製品の利益のある販売に影響を与える可能性がある。
米国の政策立案者や支払者の中では,医療システムの変革を推進することに大きな興味があり,医療コストの抑制,質の向上および/または参入拡大を既定の目標としている。米国では、製薬業はこれらの努力の重点であり、重大な立法計画の大きな影響を受けてきた。2010年3月、“保健·教育和解法案”(“ACA”と総称する)により改正された“患者保護·平価医療法案”が可決され、政府や民間保険会社が医療保健に資金を提供する方式を大きく変え、製薬業に大きな影響を与えた。ACAはブランド薬品メーカーが支払うべき医療補助リベートの最低レベルを15.1%から23.1%に引き上げた;医療補助管理保健組織が支払った薬品に対してリベートを要求した;メーカーにカバーギャップ割引計画に参加することを要求した。この計画では、メーカーはそのカバーギャップ期間中に条件を満たす受益者にブランド薬品交渉価格を適用する販売時点割引を提供することに同意しなければならず、メーカーの外来薬物として連邦医療保険D部分の条件に組み入れられた。連邦政府の特定プロジェクトにある“ブランド処方薬”を販売する薬品メーカー或いは輸入業者に対して差し引くことのできない年会費を徴収する;新しい方法を実施し、吸入、注入、点滴、移植或いは注射した薬品に対してメーカーの医療補助薬品還付計画下のリベートを計算する;医療補助計画の資格基準を拡大する;新しい患者を中心とした結果研究所を創立し、優先事項を監督、確定し、臨床治療効果の比較研究を行う, そして、このような研究に資金を提供し、連邦医療保険と医療補助サービスセンター(CMS)に医療保険と医療補助革新センターを設立し、革新的な支払いとサービス交付モデルをテストして、連邦医療保険と医療補助支出を低減し、処方薬支出を含む可能性がある。
ACAのいくつかの側面は公布以来、司法と政治的な挑戦を受けてきた。2021年6月17日、米国最高裁はACAに対する最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。最高裁が裁決を下す前に,総裁·バイデンはACA市場による医療保険の取得を目的とした2021年2月15日から2021年8月15日までの特殊保険期間を開始する行政命令を発表した。行政命令はまた、作業要求を含む医療補助モデル項目および免除計画の再検討、医療補助またはACAによる医療保険カバー範囲の獲得による不必要な障害をもたらす政策の再検討、医療補助またはACAによる医療保険の取得を制限する既存の政策および規則の見直しを指示する。
42
さらに、ACAが公布されて以来、他の立法改正も提案され、採択された。これらの変化には、提供者の連邦医療保険支払総額の減少が含まれ、2013年4月1日に施行され、その後の法規の立法改正により、国会が追加的な行動を取らない限り、2020年5月1日から2022年3月31日まで支払いを停止することが2032年まで有効となる。また、2013年1月2日、2012年の“米国納税者救済法”が法律に署名し、病院を含むいくつかの医療サービス提供者に支払う医療保険を減らし、政府が提供者に多額の金を取り戻す訴訟時効を3年から5年に延長したまた、2021年3月11日、“2021年米国救援計画法案”が法律に調印され、2024年1月1日から法定の医療補助薬品還付上限が撤廃され、現在の上限は薬品メーカーの平均価格の100%となっている。
また、政府は最近、メーカーがその販売する製品に価格を設定する方式をより厳格に審査し、国会で数回の調査を行い、連邦と州立法を提出し、製品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、政府の薬品に対する補償方法を改革することを提出し、公布した。2022年8月16日、2022年の“インフレ低減法案”または“アイルランド共和軍”が法律となった。他の事項を除いて、アイルランド共和軍はある薬品のメーカーに連邦医療保険との価格交渉を要求し(2026年から)、連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを徴収し、インフレを超える価格上昇を罰し(初めて2023年に満期)、D部分のカバーギャップ割引計画の代わりに新しい割引計画を採用する(2025年から)。アイルランド共和軍は衛生·公衆サービス部長官が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。このような理由と他の理由で、アイルランド共和軍がどのように実施されるのかは不明だ。
アメリカの個別州もますます積極的に薬品の価格を制御するための法規を実施しており、価格或いは患者の精算制限、割引、ある製品への参入とマーケティングコスト開示の制限及び透明性措置を含み、場合によっては、他の国からの輸入と大量調達を奨励することを目的としている。また、ある州および地域医療当局と個別病院はますます入札プログラムを使用して、どの薬品とサプライヤーがその医療計画に組み入れられるかを決定する。ルイジアナ州とワシントン州は2019年に入札プログラムを使用し、最近ミネソタ州は2021年に入札プログラムを使用して、連邦医療保険でカバーされている人々および処罰機関の人々を含む特定の集団のC型肝炎抗ウイルス療法サプライヤーとの契約を確実にする。他の州でも現在似たような討論が行われている。また、第三者支払者と政府当局は、参考定価システムや公表割引や値札にますます興味を持っている。
2021年12月13日,EUは衛生技術評価(“HTA”)に関する2021/2282号条例を採択した。この規定は2022年1月に施行されたが、2025年1月から適用され、その間に実施に関する準備と手順が取られるだけである。この条例は施行されると、関連製品を対象に段階的に実施されるだろう。この規定は、新医薬製品を含む衛生技術の評価におけるEU加盟国の協力を促進し、これらの分野で共同臨床評価を行うEUレベルの協力に基礎を提供することを目的としている。この法規は、EU加盟国がEU範囲内で汎用的なHTAツール、方法、およびプログラムを使用することを可能にし、患者に最大の潜在的影響を有する革新的な衛生技術の共同臨床評価を含む4つの主要分野で協力することを可能にし、科学コンサルティングを連合し、開発者はHTA当局にアドバイスを求め、新興衛生技術および将来性のある技術を決定し、他の分野で自発的な協力を継続することができる。個別EU加盟国は、衛生技術の非臨床(例えば、経済、社会、倫理)の評価を引き続き担当し、定価と精算について決定する。
将来的には、より多くの州、連邦、外国の医療改革措置が取られることが予想され、いずれも連邦と州政府が薬品や医療サービスのために支払う金額を制限する可能性があり、これは承認されると、私たちの候補製品に対する需要の減少または追加の価格設定圧力を招く可能性がある。
43
データのプライバシーとセキュリティ
多くの州と連邦の法律、法規、標準は、健康に関連する個人情報および他の個人情報の収集、使用、アクセス、秘密およびセキュリティを規定しており、現在または将来的には、私たちの運営または私たちのパートナーの運営に適用される可能性がある。アメリカでは、多くの連邦と州の法律法規は、データ漏洩通知法、健康情報プライバシーと安全法及び消費者保護法律法規を含み、健康に関連する個人情報とその他の個人情報の収集、使用、開示と保護を規範化している。プライバシーとセキュリティ法律、法規、その他の義務は絶えず変化し、互いに衝突し、コンプライアンス作業を複雑化させ、調査、訴訟あるいは行動を招き、重大な民事および/または刑事罰およびデータ処理の制限を招く可能性がある。
また、いくつかの外国法は、健康に関するデータを含む個人データのプライバシーおよびセキュリティを管理する。例えば,EU一般データ保護条例(GDPR)は,ヨーロッパ経済圏内の個人の個人データの処理に厳しい要求をしている。GDPRを遵守しなければならない企業は、より強力なデータ保護要件の規制法執行、および不正があれば2000万ユーロまたは不適合会社の世界年収の4%までの罰金が科される可能性があるコンプライアンス義務およびリスクに直面しなければならない。また,2021年1月1日から会社はGDPRとイギリスGDPRを遵守しなければならず,後者は改正された2018年のイギリスデータ保護法とともにイギリス国内法にGDPRを保持している。イギリスのGDPRはGDPRでの罰金、すなわち最高2000万ユーロ(1750万ポンド)または世界売上高の4%の罰金を反映している。
アメリカ以外の政府規制
アメリカの法規以外に、私たちはEUなどの他の司法管轄区の各種法規の制約を受けて、その中には臨床試験、マーケティング許可及び私たちの製品が一旦承認された後の任意の商業販売と流通を含む。私たちがFDAの候補製品の承認を得るかどうかにかかわらず、私たちは外国の規制機関が臨床試験を開始したり、これらの国でその製品を販売する前に必要な承認を得なければならない。臨床試験、審査過程、製品許可、定価と精算を指導する要求と手続きは国によって異なる。適用される外国の監督管理要求を守らなければ、他のほかに、罰金、監督管理の一時停止または撤回、製品のリコール、製品の差し押さえ、経営制限、刑事起訴などの処罰を受ける可能性がある。
非臨床研究と臨床試験
アメリカと類似して、EUの非臨床と臨床研究の各段階は重要な監督管理によって制御されている。
非臨床研究を行うのは,新たな化学や生物物質の健康や環境安全性を証明するためである。非臨床(薬物-毒性)研究は、EU指令2004/10/ECに規定されている良好な実験室操作原則(“GLP”)を遵守しなければならない(特定の医薬製品、例えば放射性ラベルのための放射性薬物前駆体については、別の正当な理由がない限り)。特に、体外と体内の非臨床研究は必ずGLP原則に従って計画、実行、モニタリング、記録、報告とアーカイブを行い、GLP原則は組織過程の品質体系と非臨床研究の条件として一連の規則と標準を定義した。このようなプロス基準は経済協力と開発組織の要求を反映する。
EUでは,医療製品の臨床試験はEUと国家法規,国際協調会議(“ICH”)の良好な臨床実践に関するガイドライン(“GCP”)および“ヘルシンキ宣言”からの適用法規要求と倫理原則に適合しなければならない。臨床試験の発起人がEU内で成立していない場合、それはEU実体をその法定代表者として指定しなければならない。スポンサーは臨床試験保険証書を購入しなければならず、大多数のEU加盟国では、スポンサーは臨床試験で負傷したいかなる研究対象にも“非のない”賠償を提供する責任がある。
EUの臨床試験に関連する規制構造は最近変化した。EU臨床試験条例(CTR)は2014年4月に採択され,EU臨床試験条例が廃止された
44
その指示は2022年1月31日に施行された。指示とは異なり、CTRはすべてのEU加盟国に直接適用され、加盟国がそれをさらに国家法律として実施する必要はない。CTRは臨床試験情報システムを通じてEU全体の臨床試験の評価と監督過程を著しく調整し、このシステムは集中したEU門戸とデータベースを含む。
臨床試験指令は,臨床試験を行う各加盟国で主管する国家衛生当局と独立した倫理委員会に単独の臨床試験申請(CTA)を提出することを要求しているが,FDAやIRBのように,CTRは集中的な手続きを導入し,多センター試験の単一申請の提出のみを要求している。CTRは、スポンサーが各会員国の主管当局と道徳委員会に文書を提出することを可能にし、各会員国が決定を下すことを可能にする。他の事項以外に、CTAは試験方案のコピーと被調査薬品の生産と品質情報を含む調査薬品ファイルを含まなければならない。
CTAの評価手続きも統一されており、すべての関連加盟国による共同評価を含み、道徳基準を含む各加盟国が個別にその領土に関する具体的な要求を評価する。各会員国の決定は集中されたEUポータルサイトを通じてスポンサーに伝達される。CTAが承認されると,臨床研究開発は継続可能である。
CTRは3年間の過渡期が予想される。進行中の臨床試験と新たな臨床試験がCTRによってどの程度制御されるかはそれぞれ異なる。2022年1月31日までに“臨床試験指令”に基づいて申請を提出した臨床試験、または(Ii)は2022年1月31日から2023年1月31日までの間であり、スポンサーはEU臨床試験指令を適用する臨床試験を選択しており、2025年1月31日まではこの指令の管轄を受けている。この日以降,すべての臨床試験(行われている臨床試験を含む)はCTR条項に拘束される。
臨床試験で使用される薬品は良好な生産規範(GMP)に従って生産しなければならない。他の国と連合の範囲の規制要件も適用される可能性がある。
マーケティング許可
私たちの未来の候補製品をEUと他の多くの外国司法管轄区に推進するために、私たちは単独の規制承認を受けなければならない。より具体的には、EUでは、候補医薬製品はマーケティング許可(MA)を得た後にのみ商業化することができる。EU規制制度によると、規制機関による候補製品の承認を得るためには、M&A申請(MAA)を提出しなければならない。このようにする過程は,他を除いて医薬製品の性質に依存する。2つのタイプのMAがあります
45
集中プログラムにより,環境評価機関が重大な影響評価を評価する最長期限は210日であった。
EUでは、満たされていない医療需要に対して、国民の健康に大きな影響を与えることが期待される革新的な製品は、米国の2016年3月の画期的な治療指定と同様の優先薬品(PRIME)計画のような一連の迅速な開発·審査計画を得る資格がある可能性がある。EMAは、満たされていない医療ニーズに対する薬物開発に対するEMAの支援を強化するための自発的計画であるPRIME計画を開始する。その基礎は、有望な薬剤を開発している会社との相互作用と早期対話を増加させ、彼らの製品開発計画を最適化し、より早期に患者に接触するのを助けるために、彼らの評価を加速させることである。Prime指定を受けた製品開発者は加速評価を受ける資格が期待されるが,これは保証ではない。Primeの称号を持つ候補製品のスポンサーは多くのメリットを得ることができ、しかし限らず、早期にEMAと積極的な監督対話を行い、臨床試験設計とその他の開発計画要素を頻繁に討論し、及びファイルを提出した後にMAA評価を加速する。CHMPの専任連絡先と調査委員は、EMA委員会レベルで製品のより多くの理解を促進するために、Prime計画の早期に任命されたことが重要である。最初の会議はこれらの関係を開始し、EMAの多学科専門家チームを含み、全体的な発展と監督戦略に関する指導を提供した。
また,EUでは,必要なすべての安全性や有効性データが得られていない場合には,“条件付き”MAが付与される可能性がある.条件付きMAは,失われたデータの生成やセキュリティ対策の増加を確保する条件を満たさなければならない.有効期間は1年で、すべての条件が満たされるまで年に1回更新しなければならない。未完成の研究が提供されると、“標準”のMAとなることができる。しかし,EMAが設定した時間範囲でこれらの条件を満たしていなければ,MAは更新を停止する.また、“特別な場合”では、出願人が、製品が許可され、特定の手順に従った後であっても、正常な使用条件下での有効性および安全性に関する包括的なデータを提供できないことを証明することができれば、MAも承認することができる。特に期待される適応は非常にまれであり,現在の科学的知識状態では網羅的な情報を提供することが不可能である場合や,データを生成することが一般的に受け入れられている倫理原則に違反する可能性がある場合がある.このMAは、重篤な疾患または満たされていない医療需要のために承認されるべき医薬製品を保持し、出願人は、MAに付与するために必要な合法的な完全データセットを有さないので、条件付きMAに近い。しかし,条件付きMAと異なり,申請者は失われたデータを提供する必要もなく,提供する必要もない.“特殊な場合”のMAは最終的に承認されているが,毎年薬品のリスク−収益バランスが審査されており,リスク−収益比が有利でなければMAは撤回される。
上記の手順により、MAを付与するために、欧州市場管理局又はEU加盟国主管当局は、製品の品質、安全性及び有効性に関する科学的基準に基づいて、製品のリスク効果バランスを評価する。MAの初期期限は5年である.この5年後、許可はリスク-収益バランスを再評価した上で無期限に更新することができる。
46
データとマーケティングの排他性
連合はまたデータと市場排他性に機会を提供する。MAを受信した後、参照製品は、通常、8年間のデータ独占権と他の2年間の市場独占権とを得る。承認された場合、データ固有期間は、模倣薬または生物類似薬の申請者がEUで模倣薬または生物類似薬を申請することを防止することになり、参照製品がEUで初めて許可された日から8年以内に、参照製品プロファイルに含まれる臨床前および臨床試験データに依存する。市場排他期は成功した模倣薬或いは生物類似申請者がEUでその製品を商業化することを阻止し、参考製品がEUの初めてのMAから10年後まで。MA保持者が10年の最初の8年間に1つまたは複数の新しい治療適応の許可を得た場合、10年間の市場専門期間は最大11年に延長されることができる。許可前の科学的評価では、これらの適応は既存の治療法と比較して有意な臨床的利益をもたらすと考えられる。しかし、一つの製品がEU規制機関によって新しい化学実体とみなされることは保証されず、製品はデータ排他性を得る資格がない可能性がある。
小児科発展
EUでは,新医薬製品のMAAはEMAの小児科委員会(PDCO)と合意した小児科調査計画(PIP)に適合するために小児科群で行われた研究結果を含まなければならない。PIPは,MAが求められている薬物の小児科適応を支援するために,データ発生時間とアドバイスの措置を規定している。PDCOは、成人における製品の有効性および安全性を証明するのに十分なデータがあるまで、PIPの一部または全ての措置の実施を延期する義務を許可することができる。さらに、小児科臨床試験データが不要または不適切に提供される場合、PDCOは、子供に無効または安全でない可能性があるので、これらのデータを提供する義務を免除することができ、この製品は、治療のために使用される疾患または状態が成人集団でのみ発生することが予想される、または小児科患者の既存の治療に対して有意な治療利益がない場合である。すべてのEU加盟国でMAを取得し、研究結果を製品情報に含めると、否定的な場合であっても、その製品は、6ヶ月間の補充保護証明書の取得延期(任意の補充保護証明書が承認時に有効である場合)、または孤児医薬製品について、孤児市場の独占経営権を2年間延長することを許可する資格がある。
上記のEU規則は、27のEU加盟国にノルウェー、リヒテンシュタイン、アイスランドを加えた欧州経済圏(EEA)に一般的に適用されている。
EU及び加盟国が臨床試験の進行、生産承認、医薬製品のM&A及びそのような製品のマーケティングに適用される法律を遵守せず、M&Aを付与する前及び後、医薬製品の製造、法定医療保険、賄賂及び反腐敗、又は他の適用される法規の要求は、行政、民事又は刑事罰を招く可能性がある。これらの処罰は、臨床試験の遅延または許可を拒否すること、またはMAの承認、製品の撤回およびリコール、製品の差し押さえ、一時停止、MAの撤回または変更、生産の完全または部分的な一時停止、流通、製造または臨床試験、経営制限、禁止、免許停止、罰金、および刑事罰を含むことができる。
イギリスの離脱とイギリスの規制枠組み
2021年1月1日の離脱移行期間が終了して以来、イギリス(イングランド、スコットランド、ウェールズ)はEU法律の直接的な制約を受けていないが、アイルランド/北アイルランド議定書の条項によると、EU法律は北アイルランドに一般的に適用されている。二次立法で連合王国法律に転換されたEU法律は依然としてイギリスに適用されている。しかし、現在イギリス議会に提出されている“2022年保留EU法律(撤回と改革)法案”によると、いかなる保留EU法律も明確に保留され、国内法によって国内法として吸収されていない場合、または部級法規によって延長され(2026年6月23日より遅くない)、自動的に失効し、2023年12月31日までに撤回される。しかし、(EU)CTRなどの新しい立法はイギリス(GB)には適用されない。“2021年医薬品·医療機器法”によると、国務長官または“適切な当局”は、医療製品や医療機器分野の既存の法規を改正または補充する権利がある。これにより将来的には副次的な方法で新しい規則を導入することができる
47
立法は、人類薬物、臨床試験と医療機器領域の監督管理格差と未来の変化を処理する上で柔軟性を許容することを目的としている。
2021年1月1日以来、薬品と医療製品規制機関(MHRA)はイギリスの独立した薬品と医療機器監督機関であった。北アイルランド議定書の結果として、北アイルランドはイングランド、ウェールズ、スコットランドとは異なる規則、GBが適用されるだろう;全体的に、北アイルランドはEUの規制制度に従うが、その国家主管機関はMHRAであるだろう。
臨床試験に関するイギリスの規制枠組みは、既存のEU立法(二次立法によってイギリス法律に定着された)に由来する。2022年1月17日,MHRAはイギリスの臨床試験立法の見直しについて8週間の諮問を開始した。相談は2022年3月14日に終了し、臨床試験の審査を簡略化し、革新を促進し、臨床試験の透明性を高め、リスク比率を高め、そして患者と公衆の臨床試験への参加を促進することを目的とした。協議の結果は密接に注目されており、イギリスが(EU)CTRと一致するか、規制の柔軟性を維持するために規則から逸脱するかを決定するだろう。
MHRAは、150日間の評価およびスクロール審査プログラムを含む、患者に利益を得る新薬を優先的に得るプログラムを含む、国家許可プログラムを変更している。中央許可製品のためのすべての既存のEU MAは、MA所有者が脱退を選択しない限り、GBでのみ有効であり、無料で、2021年1月1日にイギリスMAに自動的に変換またはキャンセルされる。集中プログラムを用いて欧州経済区全体で効果的なM&Aを獲得するためには,欧州経済区に会社を設立しなければならない。したがって,イギリスが離脱して以来,イギリスで設立された会社はEU集中化プログラムを使用することはできず,ヨーロッパ経済区実体はいかなる集中式MAを持たなければならない。英国で製品を商業化するためにイギリスMAを得るためには、申請者はイギリスで設立されなければならず、イギリスで製品を商業化するためには、イギリスの国家認可プログラムのうちの1つまたはイギリス離脱後に残りの国際協力プログラムのうちの1つに従ってMAを取得しなければならない。
MAの元孤児の称号はないだろう。逆に、MHRAは、対応するMA申請を審査しながら孤児指定申請を審査する。これらの基準は基本的に同じであるが,すべて市場のためにオーダーメイドされている,すなわちEUではなくイギリスのこのような疾患の流行率は万分の5を超えてはならない。孤児の称号が与えられた場合、市場独占期間はこの製品がGBで初めて承認された日から設定される。
人的資本資源
2023年2月20日までに、私たちは70人の常勤従業員がいて、そのうち23人の従業員は医学、博士、あるいは薬学博士の学位を持っています。これらの常勤従業員のうち、49人の従業員が研究開発活動に従事している。私たちの従業員の中の一人も労働組合代表でもなく、集団交渉協定のカバー範囲もない。私たちは私たちが従業員と仲がいいと思う。
私たちの人的資本資源は、私たちの既存と新しい従業員の誘致、採用、維持、激励、統合に重点を置いている。私たちの競争的株式、現金給与、そして福祉計画の主な目的はこのような優先順位を促進して支持することだ。我々の人的資本資源戦略は全面的であり、協力、創業と結果を重視する方式が科学的厳格な原則に基づく核心的な仕事方式を促進することを目的としていると考えられる。私たちの発展に伴い、私たちは私たちの人的資本資源キットを評価し続ける予定だ。
組織する
ATEA製薬会社は2012年7月に設立され、2014年3月に主要業務を開始した。同社はマサチューセッツ州ボストンにあります。ATEA製薬証券会社はマサチューセッツ州の会社で、2016年に設立され、ATEA製薬会社の完全子会社である。
利用可能な情報
我々は,我々のForm 10−K年次報告,Form 10−Q四半期報告,Form 8−K現在の報告,委託書およびその他の情報,およびこれらの報告の修正を米国証券取引委員会(“米国証券取引委員会”)に提出または電子的に提出した。公衆は、インターネットを介して米国証券取引委員会のウェブサイト上で、これらおよび他の米国証券取引委員会の届出文書を取得することができる。私たちのウェブサイトでは
48
米国証券取引委員会にこれらの報告書を提出または提出した後、できるだけ早く合理的で実行可能な範囲内で、“投資家”の欄でこれらの報告書の写しを無料で提供してください。
私たちの執行担当者と役員に関する情報
次の表は,本年度報告10−K表までの日,各役員と役員の名前,年齢,ポストを示している。
|
|
|
||||
名前.名前 |
|
年ごろ |
|
|
ポスト |
|
行政員 |
|
|
|
|
|
|
ジャン·Pierre Sommadossi博士 |
|
|
66 |
|
|
社長と最高経営責任者兼取締役会長 |
アンドレア·コクラン |
|
|
60 |
|
|
首席財務官、執行副総裁、法律及び秘書 |
ジャネット·ハモンド医学博士 |
|
|
62 |
|
|
首席発展官 |
マリア·アラントサ·ホルガー医学博士 |
|
|
54 |
|
|
首席医療官 |
ジョン·ワフリカ |
|
|
59 |
|
|
首席商務官 |
ウェイン·フォスター |
|
|
54 |
|
|
常務副総裁兼首席会計官 |
役員.取締役 |
|
|
|
|
|
|
フランクリン·バージャー(1)(2) |
|
|
73 |
|
|
重役(筆頭取締役) |
ジェローム·アダムス、医学博士(3)(4) |
|
|
48 |
|
|
役員.取締役 |
バーバラ·ダンカン(1)(3) |
|
|
58 |
|
|
役員.取締役 |
ブルーノ·ルシディ(1)(2) |
|
|
63 |
|
|
役員.取締役 |
Polly A.Murphy,D.V.M.,Ph.D.(3)(4) |
|
|
58 |
|
|
役員.取締役 |
ブルース·ボルスキー、医学博士(2)(4) |
|
|
68 |
|
|
役員.取締役 |
(1)監査委員会のメンバー。
(2)賠償委員会メンバー。
(3)指名及び企業管理委員会委員。
(4)戦略と公共政策委員会委員。
行政員
ジャン·Pierre Sommadossi博士当社の創業者であり、2012年7月から総裁兼CEO、取締役会長を務めてきました。これまで、彼は1998年から2010年まで生物製薬会社Idenix PharmPharmticals,Inc.で共同創立し、複数のポストを担当し、CEO兼最高経営責任者と会長を含む。Sommadossi博士は1998年に生物製薬会社PharmAsset,Inc.を他者と共同で設立した。Sommadossi博士は2021年2月からABG Acquisition Corporationの取締役会メンバーを務め、2013年以来Panchrest,Inc.の取締役会議長を務め、2013年以来医療保健分野のマーケティング許可代表を務め、2021年以来生物技術会社Biothea Pharma,Inc.の取締役会議長を務めている。Sommadossi博士は2004年以来BioExec研究所の取締役会メンバーを務めてきた。これまで、Sommadossi博士は2015年6月から2022年5月まで生物製薬会社Kezar Life Science、Inc.取締役会主席を務め、2016年10月から2020年11月まで生物製薬会社Rafael PharmPharmticals、Inc.取締役会副主席を務め、2020年9月から2021年1月まで生物製薬会社PegaOne,Inc.取締役会議長を務めた。2010年から2021年にかけて、ソマドーシーはハーバード医学院発見委員会のメンバーも務めた。フランスのマルセイユ大学で博士号と薬学博士号を取得した。私たちはSommadossi博士が生物技術業界の広範な科学、運営、戦略と管理経験で彼を私たちの取締役会に在任する資格があると信じている。
アンドレア·コクラン2020年10月から私たちの最高財務官を務め、2014年9月から私たちの秘書を務め、2013年12月から執行副総裁、法律顧問を務めています。コクランさんはまた、2014年9月から2020年10月までの間に総裁行政執行副総裁を務めたことがある。私たちに加入する前に、Corcoranさんは2011年から2012年までバイオテクノロジー会社で戦略と財務総監の上級副総裁を務め、2007年から2011年まで生物製薬会社トレックス社で総法律顧問兼秘書を務め、2007年から2011年までIdix執行副総裁を務めた
49
1998年から2007年まで製薬会社に勤務した。コクランさんはボストンカレッジ法学部で法学博士号を取得し、プロヴィデンズ学院で学士号を取得した。
ジャネット·ハモンド医学博士2020年8月から私たちの首席開発官を務めてきた。著者らに加入する前に、ハモンド博士は2016年11月から2020年8月まで生物製薬会社AbbVie,Inc.で総裁副主任兼一般医学と伝染病開発部治療区域主管を務め、2011年3月から2016年11月までF.Hoffmann-La Rocheで伝染病全世界主管高級副総裁と薬物研究と早期開発主管中国を務めた。ハモンド博士は南アフリカのケープタウン大学で医学博士と博士号を取得し、ジョン·ホプキンス大学衛生·公衆衛生学院で臨床研究理学修士号を取得した。
マリア·アラントサ·ホルガー医学博士2021年1月から我々の首席医療官を務め,2020年10月から代理首席医療官を務め,2020年8月以来臨床科学部執行副総裁を務めている。我々に参加する前に,ホルガー博士は2019年10月から2020年8月まで生物港製薬会社で副総裁を務め,薬物警戒や医療を担当していた。これまで、ホルガー博士は2017年7月から2019年8月まで羅氏ニューヨーク革新センター臨床プロジェクト実行全世界責任者総裁副主任を務め、2012年から2016年まで羅氏F.Hoffmann-La羅氏伝染病転化医学の全世界責任者を務めた。ホルガー博士はサンタンダー医学院で医学博士号を取得し、西奈山医学院で小児科入院医師と小児科伝染病研究員の学位を完成した。
ジョン·ワフリカ2018年10月以来、私たちの首席商務官を務めてきました。私たちに参加する前に、Vavrickaさんは、2018年3月~2021年6月に共同で設立され、バイオテクノロジー会社Biothea Pharma,Inc.のCEOを務めています。これまで、さん·ワヴリカは2007年~2015年に創業し、世界的な製薬会社イローコ製薬会社のCEO、CEOを務めています。ヴァブリカさんは、西北大学で学士号を取得した。
ウェイン·フォスター2022年1月から弊社常務副財務兼首席会計官総裁を務め、これまで2019年12月から2022年1月まで上級副総裁財務行政を務めてきた。私たちに加入する前に、フォスターさんは、バイオ製薬会社メルサナ社で2012年1月から2019年9月まで財務副社長を務めていました。フォスターさんは、マサチューセッツ大学アーマースター校の学士号を取得した。
役員.取締役
フランクリン·バージャー2019年9月から取締役会メンバーと牽引機関を務めてきた。2005年6月以来、伯傑はコンサルティング会社FMB Research LLC取締役の創業者兼管理職を務めてきた。Bergerさんはまた、2010年5月からBellus Health,Inc.の取締役、2015年3月からESSA Pharma Inc.の取締役、2016年1月からKezar生命科学の取締役、2014年10月からAtreca Inc.の取締役、2020年5月からRain Treateutics Inc.の取締役、2021年4月からRain Treateutics Inc.の取締役CEOを務める。バージャーさんは、2014年10月から2020年12月までの間にトーカゲンの取締役会メンバーを務め、2016年2月から2020年12月までのProteostantage Treateutics社の取締役会メンバーを務め、2014年10月から2021年4月までFive Prime Treeuticsの取締役会メンバーを務めていました。バージャーさんはジョン·ホプキンス大学で学士号と修士号を取得し、ハーバードビジネススクールで工商管理修士号を取得した。私たちは、バージャーさんはバイオテクノロジー業界の株アナリストの財務的背景と経験に加えて、複数の上場企業の取締役会で働いている経験に加えて、私たちの取締役会に在籍する資格があると信じています。
ジェローム·アダムス医学博士2021年5月以来、私たちの取締役会のメンバーを務めてきた。アダムス博士は2021年10月以来、普渡大学健康公平イニシアチブの役員責任者を務めてきた。アダムス博士は20人を務めていますこれは…。2017年9月から2021年1月まで米国衛生局局長を務め,その間にオピオイドの流行に専念し,新冠肺炎タスクフォースのメンバーであった。これまでアダムス博士は2014年11月から2017年9月までインディアナ州衛生専門員を務めており,そこでインディアナ州全域で前例のないHIV爆発への取り組みを主宰していた。2014年に公共サービスを開始するまで,アダムス博士はインディアナ大学麻酔系の麻酔科医と助教授であり,2008年1月から2017年9月までであった。彼のキャリアの初期には、アダムス博士は礼来会社の臨床研究アシスタントだった。彼は米国医学会を含む複数の専門組織で指導職を務めていた
50
インディアナ州医学会とインディアナ州麻酔学者協会ですアダムス博士はボルチモア県メリーランド大学で生化学学士と心理学学士号を取得し、インディアナ大学医学院で医学博士号を取得し、カリフォルニア大学バークレー校で公衆衛生修士号を取得した。アダムス博士の公共部門での豊富な経験は、彼の新冠肺炎タスクフォースでの仕事を含めて、彼が私たちの取締役会のメンバーになる資格があると信じています。
バーバラ·ダンカン2020年10月以来当社の取締役会メンバーを務めてきました。2009年5月から2016年6月まで、ダンカンさんはIntercept PharmPharmticals,Inc.で首席財務官兼財務担当者を務めた。ダンカンさんは2020年11月以来Fusion PharmPharmticals Inc.の取締役会長を務め、2016年6月からJounce治療会社の取締役会メンバーを務め、2016年6月からAdaptimmune治療会社の取締役会メンバーを務め、2017年6月以来Ovid治療会社の取締役会議長を務め、2023年2月以来Halozyme社の取締役会メンバーを務めている。これまで、ダンカンさんは2019年3月から2020年10月まで免疫医学会社の取締役会メンバーを務め、2016年11月から2018年4月までInnoviva、Inc.取締役会メンバーを務め、2015年6月から2020年1月までAEVI遺伝子医薬会社の取締役会メンバーを務め、2016年11月から2021年5月までObsEva S.A.取締役会メンバーを務めた。ダンカンさんはルイジアナ州立大学で学士号を取得し、ペンシルベニア大学ウォートンビジネススクールで工商管理修士号を取得した。私たちは彼女がバイオテクノロジー業界と上場会社での経験から、ダンカンさんが私たちの取締役会に就く資格があると信じている。
ブルーノ·ルシディ2014年9月から当社の取締役会メンバーを務めてきました。ルシーディは2013年7月以来、バイオテクノロジー会社の独立コンサルタントを務めてきた。2017年1月から2020年6月まで、ルシーディは経済発展機関バローニャ貿易·外国投資局で生命科学の専門家を務めている。ルシーディさんは2017年10月から2019年9月まで、臨床前段階のバイオ製薬会社AgenTus TreeuticsのCEOを務めています。ルシーディさんは製薬業界で35年以上の経験を持っている。彼は百時美施貴宝、ジョンソン、グラクソ·スミスクラインで高級管理職を務め、ヨーロッパとアメリカのいくつかの生物製薬会社の最高経営責任者と取締役会議長を務めたことがある。LucidiさんはフランスのVillejuifのGustave Roussy研究所で腫瘍学のトレーニングを受け、フランスのパリ高等商業大学で会社のマーケティングと戦略管理トレーニングを受け、ニューヨーク投資銀行アカデミーで金融、M&A、買収に関するトレーニングを受けました。私たちはLucidiさんが生命科学の分野で豊富な経験を持っているので、私たちの取締役会で働く資格があると信じています。
ボリー·A·マーフィーD.V.M博士2020年8月から私たちの取締役会のメンバーを務めてきた。2020年8月以来、マーフィー博士はUroGen製薬会社の首席商務官を務めてきた。これまで、マーフィー博士は2008年9月から2020年8月までファイザー社で多数の指導職を務め、2019年1月から2020年8月まで総裁兼ファイザー腫瘍業務部門商業発展主管を務め、2017年6月から2018年12月まで総裁兼ファイザーグローバルマーケティング及び商業発展業務部門の主管を務め、2013年11月から2018年5月までファイザー中国副総裁兼戦略及び業務発展主管を務めた。マーフィー博士は2022年9月からCelcuity Inc.の取締役会メンバーを務めてきた。マーフィー博士はアイオワ州立大学で医学博士と医学博士号を取得した。私たちはマーフィー博士が私たちの取締役会に勤めている資格があると信じています。彼女は製薬業の業務発展と商業化の面で豊富な経験を持っているからです。
ブルース·ボルスキー医学博士2014年11月以来、当社の取締役会メンバーを務めてきました。ボルスキー博士はニューヨークのミニョラにある長島ニューヨーク大学ランゲニ病院医学部主任で、2015年5月からずっとそこで医者をしています。2019年2月以来、ニューヨーク大学ロングアイランド医学院医学部教授と主任を務め、ニューヨーク大学ロングアイランド医学院副院長も務めている。ボルスキー博士は有力な臨床ウイルス学者であり、HIV/エイズ、B型肝炎ウイルス、C型肝炎ウイルスとその他のウイルス感染の臨床研究において積極的な役割を果たしている。1998年12月から2015年5月まで,ポールスキー博士はシナイ山サンルーク山病院や西奈山ルーズベルト病院に勤務し,そこで医学部主任や感染症科主任などを務めた。ボルスキー博士はウェイン州立大学で医学博士号を取得しました。ボルスキー博士は生命科学業界で豊富な臨床経験を持っているので、私たちの取締役会に就く資格があると信じています。
51
第1 A項。RISK因子です。
以下に説明するリスクおよび不確定要因、ならびに当社の合併財務諸表および関連説明、ならびに“経営結果および財務状況の検討および分析”を含む今年度の報告書Form 10-Kの他の情報を慎重に考慮しなければなりません。上記のいずれかのリスクが発生した場合、私たちの業務、財務状況、運営結果、あるいは見通しは重大な悪影響を受ける可能性がありますので、私たちの普通株の市場価格は下落する可能性があり、あなたはすべてまたは一部の投資を損失する可能性があります。いくつかの要因により、以下に説明する要因を含むため、我々の実際の結果は、これらの前向き陳述において予想される結果とは大きく異なる可能性がある。
新冠肺炎関連リスク
われわれはベニホブビルを新冠肺炎治療の潜在薬として開発することに大きな不確実性がある。
われわれは新冠肺炎の治療のためのベニフォブビルを開発してまだ早期段階であり,新冠肺炎を治療する潜在薬としてベネフォブビルの開発に成功しない可能性がある。2020年10月、私たちはF.Hoffmann-LaRoche Ltd.とGenentech,Inc.(総称して“羅氏”)と許可協定(改訂された“羅氏許可協定”)を締結し、この協定に基づいて、Bemnifosbuvir、米国国外での開発と商業化権利(特定のC型肝炎ウイルス用途を除く)を含む私たちのいくつかの化合物に関する独占的な許可を羅氏に授与した。2021年4月、著者らは羅氏と一緒に無作為、二重盲検、多中心、プラセボ対照の3期MORNINGSKY臨床試験を開始し、外来環境下で軽中度新冠肺炎を有する成人と青少年患者のフェニルフォブビルを研究し、その後、著者らはMEADOWSPRINGを開始し、これは3期6ケ月のフォローアップ研究であり、以前MORNINGSKYに登録した患者の中で、フェニトフォブビル治療が新冠肺炎の長期後遺症に与える影響を評価する。第三段階臨床試験はすでに開始されており,新冠肺炎患者のベネホブビルを評価する2つの第2段階臨床試験が行われている。そのうちの1つは第2段階臨床試験で入院患者を募集し、もう1つの第2段階MOONSONG臨床試験は外来患者を募集する。2021年10月、我々は羅氏と共にMOONSONGを完成し、全体的な研究群でプラセボを服用した患者と比較して、ベースラインレベルから軽中度新冠肺炎患者のSARS-CoV-2ウイルス数を低下させる主要な終点には達しておらず、そのうちの約3分の2の登録患者が低リスクかつ症状の軽微な患者であることを発表した。2021年11月、羅氏は羅氏許可協定を終了し、2022年2月10日から発効すると通知した。2021年12月,新冠肺炎による治療パターンの変化, 新しい経口抗ウイルス治療レジメンの有用性を含めて,3期ごとのMORNINGSKYおよびMEADOWSPRING臨床試験を中止することにした。われわれは両3期研究とも十分な数の患者を募集せずにあらかじめ指定された統計分析を行った。2022年1月,入院患者で行われた第2段階臨床試験を終了することにした。
それにもかかわらず,われわれはベニフォブビルの開発を進めており,ハイリスク非入院患者の新冠肺炎の治療に用いるとともに,単一と連合戦略を求めている。私たちは単一療法や併用療法が成功するかどうか分からない。2022年11月、我々は世界的、多中心、無作為、二重盲検、プラセボ対照の3期臨床試験であるSunISE-3を開始し、少なくとも1,500名の軽中度新冠肺炎を有するハイリスク非入院患者のベネホブビル(550 mg、2回/5日)を評価した。
日の出−3号試験を行うとともに,ベンニホブビルと併用して新冠肺炎を治療する可能性のある独自のプロテアーゼ阻害剤の発見に努めている。私たちはbemnifosbuvirと組み合わせて評価するための潜在的なタンパク質加水分解酵素阻害剤を内部で発見し、開発する努力はまだ非常に早期の段階にあり、この努力が成功するかどうか、あるいは成功すれば、私たちが発見したプロテアーゼ阻害剤製品候補製品がいつ臨床開発に許可される可能性があるかどうかを知らない。新たに発見された候補プロテアーゼ阻害剤製品または任意の他の候補製品の臨床開発を開始する前に、INDをFDAまたはCTAに提出し、米国以外の同様の規制機関に提出することを支援するために、広範な臨床前研究を完了する必要がある。
任意の候補製品の臨床前試験および臨床試験に使用可能な十分な数の材料の製造は、複雑で、長く、時間がかかり、
52
この過程はコストが高い。したがって,我々が予想していたスケジュールにこのようなINDやCTAを提出できるかどうかは確認できず,規制機関が新たに発見されたプロテアーゼ阻害剤の臨床試験開始を許可するかどうかも決定できない。
代替的に、候補プロテアーゼ阻害剤薬剤を開発および商業化する権利を第三者から許可または得ることができる。提案、交渉、および実施獲得は、新規冠肺炎の潜在的治療のためのプロテアーゼ阻害剤またはフェニニホブビルと組み合わせて可能性のある任意の他の候補製品を得ることができるかもしれないが、長く複雑なプロセスである可能性がある。他の会社には、より多くの財務、マーケティング、販売資源を持つ会社が含まれており、これらの候補製品の買収を競争しているかもしれません。私たちは私たちが受け入れられると思う条項で他の候補製品の権利を得ることができないかもしれない(もしあれば)。
さらに、私たちが提案したbemnifosbuvirとプロテアーゼ阻害剤を併用する方案のような連合方案の臨床試験を評価することは、連合方案の各構成要素の効果を十分に証明し、FDAまたは他の監督管理機関を満足させる必要があることを含む追加のコスト、時間およびリスクに直面する。例えば,早期臨床試験を行い,任意の新たに発見された酵素阻害剤候補薬の安全性を評価し,後期臨床試験を行い,ベネホブビルとこの酵素阻害剤の組み合わせを評価することが求められると予想される。
我々はすでに約束し,引き続き大量の財力と人的資源を投入し,ベネホブビルを単一療法として開発し,ベネホブビルと併用可能なプロテアーゼ阻害剤候補薬を発見し,ベネフォブビルと新冠肺炎治療のための酵素阻害剤を併用した(いずれの薬剤も候補であるベネホブビルCOV 19製品)。もし私たちが1つ以上のBemnifosbuvir COV 19候補製品の開発に成功できなければ、私たちは他の開発計画の資源を占有し、Bemnifosbuvir COV 19製品の候補を開発するために専用の資源を回復できない可能性があり、これは私たちの業務に大きな悪影響を及ぼすかもしれない。予想される時間内に日の出-3臨床試験を達成できない場合、または私たちの第3段階日の出-3臨床試験および他の臨床試験のデータが1つまたは複数の候補Bemnifosbuvir COV 19製品のさらなる開発または商業化をサポートしていない場合、あるいは投資家が私たちの臨床試験の任意の設計に負の反応を示し、臨床試験または臨床試験データを完成させる時間が予想され、私たちの普通株に対する需要が大幅に低下する可能性があり、私たちの普通株価格が大幅に低下する可能性があり、これは私たちの株主に大きな損失をもたらす可能性がある。
そのほか、著者らは現在新冠肺炎に対する経口抗ウィルス治療が切実に必要であると考えているが、行われている新冠肺炎の大流行の持続時間と程度はまだ確定しておらず、SARS-CoV-2が1種の地方性人類コロナウイルスになるかどうかも不明であり、現在の大流行消退後に人の群れの中で伝播する可能性がある。大流行が散逸した場合、新しい感染の数または重症度、ワクチンの有効性、他の治療選択の有効性、または他の態様の著しい低下によっても、治療に対する需要は大幅に減少する可能性がある。Bemnifosbuvir COV 19候補製品の商業化前または直後に新しい療法への需要が減少すれば,開発に成功し承認されれば,我々の業務は悪影響を受けるであろう。
Bemnifosbuvir COV 19候補製品は,開発と承認に成功しても,新冠肺炎からの他の療法やワクチンの激しい競争に直面することが予想され,これらの療法やワクチンの使用が許可または承認されているか,あるいは開発中である。
多くのバイオテクノロジーや製薬会社は,新冠肺炎の治療法や新冠肺炎を引き起こすSARS−CoV−2ウイルスに対するワクチンの開発を継続している。その中の多くの会社は、大型製薬会社を含み、より多くの発展資源と成熟した商業化能力を持っている。
現在、いくつかのワクチンと関連するワクチンブースターが新冠肺炎のために承認または許可されており、2つの経口抗ウイルス療法を含む新しい冠肺炎を治療する治療法もあり、ファイザーによって開発されたパシクロビルとムック社とリチベック治療会社によって開発されたラグフリオは、いずれもハイリスク新冠肺炎患者の緊急使用が許可されている。
さらに、ギレド科学社が開発したヌクレオチドプロドラッグVekluryは、軽度の患者を有する成人および入院または未入院の小児患者の新規冠肺炎の治療に許可されている
53
中度新冠肺炎と高リスクは入院と死亡を含む重度新冠肺炎に進展した。開発中の新冠肺炎療法にはGS−5245,経口ヌクレオシド前駆体薬がジリッド科学社によって開発されており,現在行われている無作為,二重盲検,プラセボ対照の3期臨床試験は,高入院リスクのある非入院COVID患者を対象とした。
治療薬に加えて、新冠肺炎を予防するための能動免疫ワクチンと、初期ワクチン接種後に起動される免疫効果を延長するためのワクチン“ブースター”は、米国食品·薬物管理局によって緊急使用が許可または許可されている。ファイザー社、バイオテクノロジー社、Moderna社を含むワクチンメーカーは、現在と未来のSARS-CoV-2変種に対してより大きく持続的な免疫効果を有する可能性がある新しいワクチンと増強剤の開発を継続している。
他の製薬と生物製薬会社はより多くのワクチンと治療法を開発している。他の新冠肺炎治療のための経口直接作用抗ウイルス薬を開発した会社はEnanta製薬会社、Shionogi&Co.,Ltd.,Pardes Biosciences,Inc.,君実生物科学会社、江蘇Simcere製薬会社とSyneuRx社を含む。
現在承認または許可されている製品や他社が開発している製品を考慮すると,我々が開発する可能性のあるどの治療法も激しい競争に直面している可能性がある。治療効果、安全性、コストまたは他の要素によってますます多くの新冠肺炎治療薬と区別された治療方法を開発できない場合、または何らかの治療方法が治療基準になった場合、より便利またはより低コストで使用することができ、または任意のエンティティが許可または承認された治療方法をより成功的に商業化することに成功し、候補薬物テノホビルCOV 19が承認されても、新しい冠肺炎の治療に使用されるこのような製品の商業化に成功したり、他の治療方法やワクチンと競合したりすることに成功し、私たちの業務や運営に悪影響を及ぼす可能性がある。
新冠肺炎の流行は私たちの業務と財務業績に実質的な悪影響を及ぼすかもしれない。
SARS-CoV-2変種と亜変種の全世界範囲での出現はますます多くの感染を招き、この感染ワクチンを接種した人の中で突破的な感染を発生することを含む。各国政府が新冠肺炎の伝播を減少させるために実施した旅行禁止令、在宅命令、その他の措置は、2020年の新冠肺炎の初発生に対応し、全世界の商業運営と経済活動の広範な中断を招いた。今後の事件の再出現は、新冠肺炎の伝播を減少させるための措置の更新を招く可能性があり、その多くは現在緩和されている。公衆衛生指令に応答し、従業員の新冠肺炎のリスクを最小限に抑えることを支援するために、すべての従業員の在宅勤務を許可することを含め、予防措置を継続して維持していく。私たちの多くの第三者パートナー、例えば、私たちのCMO、臨床研究機関(CRO)、サプライヤー、その他の機関は、類似した予防措置を取り、維持し続けている。大流行の間、これらの措置は時々私たちの業務を乱し、私たちのいくつかの臨床計画とスケジュールを延期します。例えば,2020年には,われわれのC型肝炎ウイルス臨床開発プロジェクトは臨床試験所の国や医療機関による新冠肺炎予防措置により閉鎖され,われわれのC型肝炎ウイルス臨床開発プロジェクトは一時停止した。2023年、私たちはC型肝炎ウイルス臨床試験を再開する予定です。
新冠肺炎疫病の延長或いは灰再発が著者らの運営に与える影響は深刻である可能性があり、そして著者らの業務、財務状況と運営結果に負の影響を与える可能性がある。例えば、最近中国政府が封鎖措置を緩和し、新冠肺炎が広範囲に発生した。もし持続的あるいは新一波の新冠肺炎感染が中国の業務活動を乱した場合、私たちは臨床試験材料の肝心な成分を調達し、製造する能力は不利な影響を受ける可能性がある。新冠肺炎疫病が私たちの業務と財務業績に不利な影響を与える程度で、それはまた本“リスク要素”の節で述べた多くの他のリスク要素の増加を招く可能性があり、例えば私たちの臨床試験スケジュールに関連する要素、私たちは臨床試験対象を募集し、私たちの候補製品を生産するために必要な材料を獲得する能力、そして私たちの資金を調達する能力。
54
新冠肺炎の大流行の変化或いは任意の他の公衆衛生危機の発生は著者らの臨床試験に実質的な不利な影響を与える可能性がある。
新冠肺炎の大流行の変化あるいは他の公衆衛生危機の発生により、私たちはもっと多くの中断を経験するかもしれません。これは私たちの臨床試験に深刻な影響を与えるかもしれません
特定の変種或いは亜変種に感染した新冠肺炎の症状、進展と伝播は多くの方面で異なり、症状の重症度と伝播率を含む。変異の迅速と持続出現及び疾病表現の変化は著者らが新冠肺炎患者の中で臨床試験を行うために追加的な挑戦を提出した。例えば,新冠肺炎患者には様々な症状や副作用が出現しており,臨床試験研究者がわれわれの臨床試験で観察されたいかなる有害事象がフェニルフォブビルに関与しているのか潜在疾患と一致しているのかを決定することは困難である可能性がある。深刻さの増加や
55
有害事象の発生は、bemnifosbuvirや私たちが開発を求めている任意の併用療法に関連していると考えられ、その規制承認を延期または阻止する可能性があり、これは、私たちの業務、財務状況、および運営結果に実質的な悪影響を及ぼす可能性がある。そのほか、新冠肺炎の臨床試験の治療効果と抗ウィルス結果は感染を招く1つ或いは多数の変種、疾病の伝播性と重症度及び関連する入院と死亡などの要素の影響を受ける可能性がある。したがって,大流行の進展に伴い,臨床試験期間全体で発生する応答率は時間とともに変化する可能性がある。
私たちの財務状況と資本要求に関連するリスク
私たちの経営の歴史は限られており、承認された抗ウイルス製品の開発や商業化に成功したり商業化したりしていないことは、私たちの業務のこれまでの成功を評価することが難しくなり、私たちの将来の生存の将来性を評価することも困難になるかもしれない。
私たちは臨床段階の生物製薬会社です。これまで、私たちの業務は、会社に資金と人員を提供し、私たちの技術を開発し、私たちの候補製品を決定し、開発することに限られています。我々の将来性は,生物製薬会社が運営初期によく遭遇する不確実性,リスク,費用,困難を考慮しなければならない。我々は、いかなる後期または重要な臨床試験を完成させ、市場の承認を得たり、商業規模の製品を生産したり、成功した製品の商業化に必要な販売やマーケティング活動を行うことができるか、または第三者代表にこれらの活動を行わせることができることを証明していない。したがって,我々がより長い運営歴史や開発成功,市場承認と商業化抗ウイルス療法の歴史を持っていれば,我々の将来の成功や生存能力の予測は当然のように正確ではないかもしれない。
さらに、私たちは予測できない費用、困難、合併症、遅延、および他の既知と未知の障害に直面するかもしれない。例えば,新冠肺炎領域の変化により,2021年12月にわれわれの3期MORNINGSKY試験を中止し,最近になってその後の3期臨床試験SUNISE−3を開始し,フェニニホブビルの軽中度新冠肺炎ハイリスク外来患者に対する治療作用を評価した。日の出−3試験の主な終点は,現地標準看護療法を併用せずにbemnifosbuvirまたはプラセボ治療のみを受けた登録患者において,29日目にすべての原因が入院または死亡したことである。我々の現在の研究設計とサンプル量は,プラセボキュー中の入院率は少なくとも4%と予想されている。プラセボ患者のキュー内の実際の入院率が低い場合、サンプルサイズを増加させ、試験に組み込まれる全体的な患者数を増加させる必要がある可能性がある。これは裁判をもっと難しくし、裁判を完了する時間と費用が増加すると予想される。新冠肺炎の構造の変化に伴い、新冠肺炎の検出率、診断率、入院率は引き続き大きな差がある。多くの地理的地域報告の入院率は、報告された特定の地域の患者総人数が4%未満であることを含む大幅な変動を継続する可能性がある。
もし私たちが開発に成功し、任意の候補製品の承認を得たら、研究開発に集中した会社からビジネス活動を支援できる会社に移行する必要があります。私たちはこの転換で成功しないかもしれない。
私たちが引き続き業務を発展させることに伴い、私たちは様々な要素によって、私たちの財務状況と経営業績は様々な要素によって異なる四半期と毎年大幅に変動する可能性があり、その中の多くの要素は私たちがコントロールできないと予想している。例えば,羅氏が羅氏許可協定を中止する決定も,羅氏が2022年2月10日,すなわち羅氏許可協定終了の発効日後に,新たな冠肺炎治療のためのフェニホブビルの開発に関する費用の義務を分担している。
また、羅氏許可協定を終了したさらなる結果として、私たちは羅氏から2020年に受信した前金と、2021年に私たちが受け取った記念碑的支払い以外のいかなる収入も得ないだろう。したがって、今後の経営業績の指標として、本報告または任意の他の特定の四半期または年次報告書に含まれる結果に依存してはなりません。
設立以来、多くの運営費が発生しており、予測可能な将来に多くの追加運用費用が発生すると予想されている。私たちはどんな製品もありません
56
どんなビジネス収入も生まれ、私たちは2022年と予測可能な未来に利益を維持しないと予想している。
設立以来、私たちは多くの運営費用を発生させた。2022年12月31日までの年度と2021年12月31日現在の年度では,それぞれ1兆307億ドルと2.13億ドルである。2021年,羅氏は羅氏許可協定を終了したため,前払金と羅氏から受け取ったマイルストーン支払いに関する繰延収入残高に関する収入を会計上確認し,2021年12月31日までの年度の営業収入を記録した。2022年12月31日までの年度では、営業収入は何も記録されておらず、2023年や予想される将来も営業収入は実現しないと予想されています。
私たちはどんな製品も商業化していないし、製品販売から何の収入も得られなかった。私たちのほとんどの財政資源は、私たちの臨床試験と臨床前開発活動を含む研究と開発に投入されている。予測可能な未来には、臨床開発を通じて候補製品を推進し、臨床前開発を継続し、私たちの研究と開発活動を拡大し、ベンフォブビルと連携して潜在的な肺炎治療のための任意の候補製品を求める可能性があり、臨床前研究と臨床試験を完成させ、監督部門の承認を求め、監督部門の承認を得た後、私たちの製品を商業化することができるので、巨大な追加運営費用と運営損失を招くことが予想される。
FDAまたは外国規制機関の承認を得て、米国または海外で任意の候補製品をそれぞれ販売するためには、FDAまたは外国規制機関がその期待用途に対して安全かつ有効であることを満足的に証明するために、NDAまたは同様の申請をFDAに提出しなければならない。この論証には大量の研究と動物試験からの広範なデータが必要であり、これらの試験は非臨床或いは臨床前研究、及び人体試験と呼ばれ、臨床試験と呼ばれる。
また,時間の経過とともに候補製品を後続の臨床段階に進めるコストが大幅に増加することが多い。司法管轄区域であっても、私たちのすべての候補製品を市場に承認する総コストは巨大であり、正確に予測することは難しいだろう。医薬品開発に関連する多くのリスクおよび不確実性のため、費用を増加させる時間または金額を正確に予測することができないか、または製品の商業化から収入を発生させ、または再び利益を達成することができるかどうかを正確に予測することができるかどうか。われわれの臨床開発活動の推進に伴い,特に軽中度新冠肺炎高リスク外来患者の第三段階日の出−3臨床試験の治療,およびベネホブビルとルザスビルの併用によるC型肝炎の第二段階臨床試験の評価に伴い,われわれの費用は大幅に増加することが予想される。さらに、以下のような場合、私たちの費用も大幅に増加するだろう
57
また、私たちが任意の製品の開発、商業化、許可に成功し、製品収入を創出する能力は、大量の追加リスクと不確実性の影響を受ける。私たちが製品販売から任意の収入を得る前に、私たちの各候補製品、および私たちが発見、許可、または他の方法で得られる可能性のある任意の未来の候補製品は、追加の臨床前および/または臨床開発、少なくとも1つの司法管轄区域の規制承認、製造供給、生産能力、流通ルートおよび専門知識の確保、外部サプライヤーの使用、商業組織の確立、大量の投資、および重大なマーケティング努力を必要とするだろう。そのため,将来的には,現金を用いた運営活動を継続し,運営費用や運営損失が生じることが予想される。現金の使用や運営費や運営損失の発生はすでに我々の運営資本に悪影響を与えており,引き続き悪影響を及ぼすことが予想される。
将来の費用や損失額と、私たちが今後数年で利益を達成したり維持したりする能力は不確実だ。私たちはどんな商業収入も生まれていない製品は、予測可能な未来に製品の商業販売から収入を生み出すことは期待されておらず、製品の販売から収入が生じることは決してないかもしれない。私たちが製品収入を創出し、利益を維持する能力は、候補製品の臨床開発の成功、FDAと外国規制機関の必要な規制承認の獲得、製造と販売能力の確立、私たちの製品に対する市場の受容度(承認されれば)、マーケティングインフラの構築、または他の方法で私たちが承認された候補製品を商業化すること、および十分な資金を集めて私たちの活動に資金を提供することに依存するだろう。私たちはこのような事業の中のどれも成功しないかもしれない。もし私たちの業務の一部または全部が成功しなければ、私たちの業務、将来性、および経営結果は実質的な悪影響を受ける可能性がある。
私たちは受け入れ可能な条件で提供できないか、または根本的にできないかもしれない大量の追加資金が必要になるだろう。必要なときに必要な資本を得ることができなければ、私たちの製品開発や商業化努力を延期、制限、減少、または中止させる可能性があります。
設立以来、私たちは多くの運営費用を発生させた。われわれの現在と計画中の業務活動に関連する巨額の費用,特に軽中度新冠肺炎高リスク外来患者の第3段階日の出−3臨床試験,およびフェニホブビルとルザスビルの併用によるC型肝炎治療の第2段階臨床試験の評価を予定している。また,bemnifosbuvir COV 19製品の開発や他の候補製品の発見,買収,開発に関する巨額の費用が発生することが予想される。
私たちは今後の臨床試験と臨床前開発を支援するために追加の資本を引き続き必要とし、私たちは株式発行、債務融資、マーケティングと流通手配、その他の協力、戦略連合と許可手配、または他の出所を通じて資金を調達することができる。他の融資源は割引の条件で提供されない可能性があり、もしあれば。もし私たちが受け入れ可能な条件下で追加の資金を調達することに成功できなければ、計画中の臨床試験を開始または完了することができないかもしれないし、FDAやいかなる外国の規制機関も私たちの任意の候補製品に対する規制承認を求めることができず、製品開発の中止を余儀なくされる可能性がある。また、追加融資を得ようとすると、私たちの経営陣の日常活動からの時間や注意力を分散させ、候補製品開発作業を損なう可能性があります。
58
私たちの現在の運営計画によると、2022年12月31日まで、私たちの現金と現金等価物は、2026年までの運営費用と資本支出要求を満たすのに十分になると信じている。この推定は、間違っていることが証明される可能性があるという仮定に基づいており、私たちは現在予想されているよりも早く私たちが利用できる資本資源を使用することができる。私たちは、このような発売と商業化がパートナーの責任ではないことを前提として、現在および任意の未来の候補製品を発売し、商業化するために多くの追加資金を必要とするだろう。また,我々の開発努力の過程で他の予期しないコストが発生する可能性がある.我々の計画と期待される臨床試験の設計と結果は高度に不確定であるため、私たちが開発した任意の候補製品の開発と商業化に成功するのに必要な実際の金額を合理的に見積もることはできない。
私たちの未来の資本需要は多くの要素に依存していますが、これらに限定されません
現在、私たちは外部資金源や他の支援を約束していないし、私たちは受け入れ可能な条件で追加資金を提供するかどうか、または全くできないかどうかを決定することもできない。新冠肺炎の大流行は、新しいと現有の新冠肺炎変異体の変化、及び国内或いは政治的動揺(例えばウクライナとロシアの間の持続的な戦争)を含む地政学的事件を含み、すでに全世界の金融市場の深刻な混乱を招いた。また、最近または未来の市場変動、インフレ激化、金利上昇は、持続すれば、私たちの融資コストを増加させ、私たちが獲得することを制限する可能性がある
59
未来の流動性の潜在的な源。もし私たちが受け入れられる条項やタイムリーな基礎の上で十分な追加資本を調達できなければ、私たちは候補製品の開発や商業化、または他の研究開発計画を大幅に延期、削減、または停止しなければならないかもしれない。
私たちは製品販売から何の収入も得ていないので、利益を得ることができないかもしれない。
終了した羅氏許可契約により受け取った何らかの支払いにより会計上収入を確認したため、2021年12月31日までの年度の営業収入を確認した。しかし、羅氏許可協定が終了した後、私たちは羅氏または他のいかなる第三者からも収入であることが確認された増額支払いを受けなかった。そのため,2022年12月31日までの1年間に1兆307億ドルの運営損失が発生した。私たちが将来の利益を達成し維持する能力は私たちが製品販売から収入を創出する能力にかかっている。ロ氏許可協定以外に、私たちは何の収入も生まれておらず、私たちが臨床開発に成功し、監督部門の承認を得て、私たちの少なくとも1つの候補製品を商業化することに成功しない限り、製品収入を生成することも望んでいない。私たちの候補製品は異なる開発段階にあり、ある場合には追加の臨床前研究を行う必要があると予想され、すべての場合には追加の臨床開発及び監督審査と承認、大量投資、十分な商業製造能力と重大なマーケティング努力を得て、製品販売から任意の収入を得ることができる。現在、私たちは少なくとも今後数年以内に製品販売から収入を得ないと予想している。私たちの創造能力は複数の要因に依存していますがこれらに限定されません
60
上に列挙された多くの要素は私たちがコントロールできないことであり、私たちに重大な遅延を招いたり、規制部門の許可を得たり、私たちの候補製品を商業化することを阻止するかもしれない。私たちの候補製品を商業化することができても、製品販売後に収益性を維持することができず、収益性に対する外部の期待を満たすことができないかもしれません。もし私たちが収益性を達成したり維持したりすることができなければ、私たちの収益性に対する外部の期待に達しなければ、私たちの普通株の価値は大きな悪影響を受けるだろう。また、私たちがどんな製品を販売することで十分な収入を得ることができなければ、私たちは運営を続けることができないかもしれない。
純営業損失の繰越や他の税務属性を使って課税収入を相殺する能力は一定の制限を受ける可能性があります。
2022年12月31日現在、米国連邦純営業損失繰越(NOL)1,940万ドルがあり、将来の課税収入(あれば)を相殺することができ、そのうち40万ドルは2034年に満期になり、そのうち1,900万ドルは満期になっていないが、その使用は制限されており(2022年12月31日以降の課税年度)、毎年年間課税所得額の80%を差し引くことができる。また、2022年12月31日現在、3,000万ドルの州NOLがあり、あれば、将来の課税収入を相殺し、2042年に満期を迎えることができます。2021年12月31日までの年間で、連邦と州NOL約5280万ドルと5260万ドルをそれぞれ使用し、連邦と州研究開発信用繰越はそれぞれ70万ドルと30万ドルだった。
一般的に、改正された1986年の“国内税法”(以下、“税法”と呼ぶ)第382条と383条によると、会社は“所有権変更”が発生し、通常その持分所有権は3年以内に価値によって50%を超えると定義され、その利用変更前のNOLとその研究開発信用は将来の課税所得額を相殺する能力が制限される。私たちのNOLと研究開発信用の転換は以前の所有権変更による制限を受ける可能性があり、もし私たちが所有権変更が発生すれば、私たちがNOL(以前に使用していない)と研究開発信用の転換を利用する能力は規則382と383節の更なる制限を受ける可能性がある。
2021年12月31日までに第382条の研究を完了し,その間に所有権移転は発生していないことを示した。しかし、この結論は税務機関の疑問を受けるかもしれない。また、規則382及び383条の規定によれば、将来、私たちの株式の変化、その中のいくつかは私たちの制御範囲を超える可能性があり、所有権の変化を招く可能性があります。これらの原因により、著者らは現有のNOL或いは研究開発信用の繰り越し或いは未来に発生する可能性のある純運営損失と研究開発信用を利用できないかもしれない。私たちは2022年12月31日までの年度の第382条の研究を完了している。
知覚された市場またはビジネス機会が、さらなる投資または他の戦略業務、財務または他の理由で、私たちの業務に実質的な損害を与え、私たちの株価に悪影響を及ぼす可能性があると考えるならば、候補製品の開発をいつでも延期、一時停止、または終了させることができる。
私たちがすでに行っているか、または将来可能な臨床前研究および臨床試験の結果が、戦略、業務、
61
財務的または他の理由は、候補製品の新興特徴が重要な市場で規制機関の承認を得ず、意味のある市場承認を得ることができないか、またはその予想される指示または市場において任意の競争優位性を提供するか、または株主に著しい見返りをもたらす可能性があると判断または信じることを含む。2023年2月,AT−752を第2段階臨床試験に進めた後,AT−752の治療とデング熱予防の臨床開発を休止した。この行動をとるのは,期待されるより長い時間と,デング熱治療のための抗ウイルス薬の臨床開発に関連する他の挑戦である。われわれは最近,呼吸器合胞体ウイルス治療の候補製品を探す努力も中止した。この行動をとるのは,我々の管理チームの重点強化を促進し,われわれの計画のより先進的な治療適応に資源を提供するためである。これらの行動および他の臨床計画または候補製品の任意の他の同様の遅延、一時停止または終了は、私たちの業務、運営結果、または財務状態に実質的な損害を与える可能性がある。
私たちの候補製品の発見、開発、臨床前と臨床テスト、製造と規制承認に関連するリスク
私たちのビジネスは、Bemnifosbuvir COV 19候補製品を1つ以上開発する能力に強く依存しています。もし私たちがBemnifosbuvir COV 19候補製品を開発することに成功できなければ、私たちの業務は損害を受けるだろう。私たちの業務はまた、C型肝炎の治療のためのbemnifosbuvirとruzasvirの組み合わせを含む他の最先端の候補製品の成功に非常に依存しており、これは大量の追加の臨床テストを必要とし、その後、私たちは監督部門の承認を求め、商業販売を開始することができるかもしれない。これらの候補製品が臨床開発に失敗し、規制部門の承認を得なかったり、商業化に成功しなかったり、商業化の面で深刻に遅延された場合、私たちの業務は損なわれるだろう。
私たちの業務と未来の成功の大部分は、私たちが開発し、監督機関の承認を得て、1つ以上の候補bemnifosbuvir COV 19製品と、C型肝炎を治療するためのbemnifosbuvirとruzasvirの組み合わせを商業化する能力にかかっている。私たちは現在商業販売が許可されていない製品も、候補製品の開発も完了していません。私たちは永遠に適切な製品を開発できないかもしれません。短期的に、私たちの大部分の努力と支出は潜在的なBemnifosbuvir COV 19候補製品の開発に引き続き使用されると予想される。他の事項に加えて、この仕事は、軽中度の新冠肺炎を有する高リスク外来患者の治療、および追加の非臨床および臨床開発、ならびにフェニニホブビルと組み合わせて新冠肺炎を治療する薬剤または候補薬剤の発見、取得または許可の使用に関連する費用のための、我々の第3段階日の出-3期臨床試験の成功を達成する必要があるであろう。新冠肺炎は現在全世界の大流行であるが、それは予測不可能であり、治療方法は任意の候補ビンニフォブビルCOV 19製品を含み、新変種或いは亜変種の出現及び新しいワクチン、ワクチン促進剤と治療薬物の開発と許可と承認によって時代遅れまで不利な影響を受ける可能性がある。さらに、今後数年以内に、私たちの大部分の努力と支出は、bemnifosbuvirとruzasvirの併用治療C型肝炎ウイルスの開発に力を入れることになると予想され、これは追加の臨床開発、臨床、医療事務、製造活動の管理、複数の司法管轄区域で規制の承認を受け、製造供給を確保し、商業組織を構築する必要がある, 巨大な投資と巨大なマーケティング努力。私たちが規制部門の承認を得ても、私たちは現在または未来の候補製品の中でどんな製品が臨床試験で成功し、監督部門の承認を得るか、商業化に成功するかどうかを決定することはできない。また、任意の候補製品の開発が延期または一時停止される可能性があり、これは、候補製品を商業化することに成功する能力に影響を与える可能性がある。
もし私たちの競争相手が病気を治療する製品を開発し、適用すれば、私たちが候補製品の開発に成功する前に、私たちが現在または未来の候補製品を治療のために開発している特定の患者集団、あるいは私たちの競争相手が私たちの候補製品よりも優れた製品を開発していれば、私たちの潜在的な市場シェアはもっと小さくなるか、全く存在しないかもしれない。私たちがマーケティングの任意の候補製品の承認を得ても、私たちの候補製品が他の商業化に成功したり、市場に広く受け入れられている商業代替製品と同じように効果的で効果的かどうかを判断することはできません。承認されれば,どの製品の安全性や有効性の特徴も臨床試験で観察された結果と一致するか,あるいはbemnifosbuvir COV 19候補製品に対して持続的変異に対する有効性を示すことは確認できない
62
新冠肺炎の病原体であるSARS−COV−2ウイルス。もし私たちがBemnifosbuvir COV 19候補製品または私たちの他の最先端の候補製品の臨床開発に成功しなかった場合、これらの候補製品は必要な監督管理の承認を得ておらず、これらの候補製品の開発または承認に重大な遅延が発生した場合、あるいはいかなる承認された製品も商業的に成功せず、私たちの業務、財務状況、運営結果は重大な損害を受ける可能性がある。
FDAと類似の外国規制機関の規制承認過程は長く、高価で、時間がかかり、本質的に予測できない。もし私たちが最終的に規制部門の候補製品の承認を得ることができなければ、私たちは製品収入を生むことができなくなり、私たちの業務は深刻な損害を受けるだろう。
FDAの承認なしに、私たちはアメリカで商業化、マーケティング、普及、またはどんな候補製品も販売してはいけない。外国の規制機関もまた似たような要求を実施した。米国食品医薬品局および同様の外国規制機関の承認を得るのに要する時間は予測不可能であり、通常、臨床試験開始後数年後に承認される必要があり、これは、関連する候補製品のタイプ、複雑性および新規性、および候補製品が対象とする疾患または条件、特に急速に進化し続ける新しい冠肺炎を含む多くの要素に依存する。さらに、候補製品の臨床開発過程において、承認政策、法規、または承認を得るために必要な臨床データのタイプおよび数が変化する可能性があり、司法管轄区域によって異なる可能性があり、承認の遅延または承認申請の不承認の決定を招く可能性がある。
監督管理機関は審査過程中にかなりの自由裁量権を持っており、いかなる申請を受け入れることを拒否することができ、著者らのデータが承認を得るのに十分ではなく、追加の臨床前、臨床或いはその他の研究を行う必要があることを決定することもできる。私たちはまだ候補品のための秘密協定を提出していないし、規制部門の承認も得ていない。私たちは、私たちの候補製品の人体上の安全性と有効性を証明し、規制機関を満足させるために、追加の臨床前または非臨床研究と臨床試験を完成させなければならない。そして、私たちはこれらの承認を得ることができ、そして私たちの既存の候補製品または私たちが将来開発を求める可能性のあるどの候補製品も、規制部門の承認を得ることができないかもしれない。私たちの候補製品の申請は多くの理由で規制部門の承認を得ることができないかもしれませんが、以下の理由に限定されません
この長い承認過程と,臨床試験結果の予測不可能性は,規制部門の承認を得ることができず,任意の候補製品を市場に出すことができず,業務を大きく損なう可能性がある。しかも、私たちが承認されても、規制部門は承認するかもしれない
63
私たちの任意の候補製品の適応は、私たちが要求するものよりも少ないか、または限られており、狭い適応、警告またはリスク評価および緩和策(“REMS”)または同様のリスク管理措置の形態で重大な制限を加える可能性がある。規制当局は、私たちが開発する可能性のある製品に請求しようとしている価格を承認しないかもしれないし、高価な発売後の臨床試験の表現によって承認されるかもしれないし、候補製品のラベルには、候補製品の商業化に必要または必要なラベル宣言が含まれていないことが承認されるかもしれない。上記のいずれの状況も私たちの業務を深刻に損なう可能性がある。
臨床開発は、患者を募集して臨床試験に参加することを含み、高価で、長く、不確定な過程である。われわれの臨床試験は大きな遅延やコストに遭遇する可能性があり,あるいは予想されるタイムライン上で臨床試験を行うことや完成できない可能性があり,まったくなければ。
FDAや他の同様の外国規制機関が私たちの候補製品の販売を許可する前に、私たちの候補製品の安全性と有効性を証明するために、臨床前開発と広範な臨床試験を完了しなければならない。臨床テストは高価で時間がかかり、不確実性も存在する。1つまたは複数の臨床試験の失敗は、例えば、2021年10月に第2段階MOONSONG臨床試験における総患者集団の主要な終点に到達できなかったようなプロセスの任意の段階で発生する可能性がある。臨床前研究と早期臨床試験の結果は後の臨床試験の成功を予測できないかもしれない。新冠肺炎に対する治療法の開発では特に,我々が開発したBemnifosbuvir COV 19候補製品を含め,ウイルスや疾患の進行速度が非常に速く,開発中の候補製品が臨床開発が完了する前に淘汰される可能性がある。そのほか、臨床前と臨床データ、特に探索性終点の分析と患者グループからのデータの分析、例えば著者らは臨床開発に依存するMORNINGSKY研究のデータを継続し、よく異なる解釈の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると考えているが、彼らの薬物はまだ発売許可を得ていない。今まで、私たちは私たちのどんな候補製品の後期または重要な臨床試験も完成していません。私たちの計画や進行中の臨床試験が計画通りにスタートしたり、計画通りに完成したりする保証はありません, もし本当にあれば。将来のINDまたは同様の出願を提出することが、適用される場合、FDAまたは他の規制機関が将来の臨床試験をタイムリーに開始することを可能にするかどうかも決定できません。また,これらの試験が開始されても,規制当局のこのような臨床試験の一時停止や終了を招く可能性がある。1つまたは複数の臨床試験の失敗は試験の任意の段階で起こる可能性があり、私たちの将来の臨床試験は成功しないかもしれない。成功またはタイムリーな臨床試験の開始または完了を妨げる可能性があるイベントは、これらに限定されない
64
また,新冠肺炎の持続的な変化による事件,イベント,中断は,我々が開始,登録,われわれの計画中と進行中の臨床試験を開始,あるいは完了する際に困難や遅延に遭遇する可能性を増加させる可能性がある。特に、新冠肺炎の発病率、検査と診断率、ウイルス突然変異による迅速に変化した新冠肺炎治療看護標準の変化、医療保健提供者が獲得した急速に増加した知識、ワクチンの比率と持続性及びますます多くの治療選択
65
これはわれわれの日の出−3期臨床試験およびわれわれが開始する可能性のある他の新冠肺炎に関連する臨床試験の順調な完成に影響する可能性がある。
私たちの第3段階日の出-3臨床試験、または私たちが開始する可能性のある任意の他の計画における臨床試験を成功させることができない場合は、私たちの追加コストをもたらしたり、製品販売から収入を得る能力を損なう可能性があります。さらに、私たちが私たちの候補製品を製造したり、調合を変更したりすれば、私たちは修正された候補製品を以前のバージョンに関連付けるために追加的な研究を行うことを要求されるかもしれません。臨床試験の遅延は、承認された製品が特許保護を受ける時間を短縮することも可能であり、私たちの競争相手が私たちよりも先に製品を市場に出すことが可能であり、候補製品を商業化する能力を弱める可能性があり、私たちの業務を深刻に損なう可能性がある。
臨床試験が、我々、dsmbによって一時停止または終了された場合、またはFDAまたは任意の他の規制機関によって臨床試験が一時停止または終了された場合、またはそのような試験を行っている機関のIRBsは、その臨床研究者およびその審査を受けた場所の参加を一時停止または終了する場合、遅延に遭遇する可能性もある。このような機関は、様々な要素のために臨床試験を一時停止または終了する可能性があり、これらの要素は、監督管理要求または著者らの臨床規程に従って臨床試験を行うことができなかったこと、FDAまたは他の監督機関の臨床試験操作または試験場所の検査の実施による臨床休止、予見できない安全問題または副作用、候補製品の使用のメリット、政府法規または行政措置の変化、または十分な資金が不足して臨床試験を継続することを含む。
また,海外での臨床試験は,我々が現在行っているように,そうでなければ他の候補製品の試験を継続することが予想され,追加のリスクをもたらし,臨床試験の完了を遅らせる可能性がある。これらのリスクには,外国に登録された患者が医療サービスや文化的慣習の違いにより臨床案を遵守できなかったこと,外国規制計画に関する追加行政負担を管理すること,およびこのような外国に関連する政治的·経済的リスクがある。
また,われわれの臨床試験の首席研究員は時々私たちの科学コンサルタントやコンサルタントを務め,このようなサービスに関する報酬を得る可能性がある。場合によっては、私たちはFDAまたは同様の外国規制機関にいくつかの関係を報告することを要求されるかもしれない。FDAや同様の外国の規制機関は結論を出す可能性があり、私たちと主要な研究者との財務関係は利益の衝突をもたらしたり、他の方法でこの研究の解釈に影響を与えたりする。したがって,FDAや同様の外国の規制機関は,適用された臨床試験地点で発生するデータの完全性を疑問視する可能性があり,臨床試験自体の効用が脅かされる可能性がある。これは、FDAまたは同様の外国規制機関が私たちの上場申請の承認を遅延または拒否することを招き、最終的に私たちの1つ以上の候補製品が上場承認を拒否することにつながる可能性がある。
私たちの候補製品のすべての臨床試験の完成を遅延することは私たちのコストを増加させ、私たちの候補製品の開発と承認過程を遅くし、そして私たちの製品販売と製品収入を創造する能力を遅延或いは危険にさらす可能性がある。さらに、臨床試験の開始または完了遅延をもたらす多くの要因は、最終的には、私たちの候補製品が規制部門の承認を得ることを拒否される可能性もある。したがって、私たちの臨床試験に生じるどんな遅延も、候補製品を商業化する独占的な権利を持つ任意の期限を短縮することができ、私たちの競争相手は私たちの前に製品を市場に出すかもしれません。これは私たちの候補製品の商業的可能性を著しく低下させるかもしれません。このようなどんな状況でも、私たちの業務、財政状況、そして見通しに大きな被害を及ぼす可能性がある。
また,FDAや他の規制機関の臨床試験に関する政策が変わる可能性があり,追加の政府法規が公布される可能性がある。例えば,欧州連合(“EU”)の臨床試験に関する規制構造が最近変化している。EU CTRは2014年4月に採択され,EU臨床試験指令(“臨床試験指令”)が廃止され,2022年1月31日に適用が開始された。臨床試験指令は、各加盟国で国家衛生当局および独立した倫理委員会に単独のCTAを提出することを要求しているが、CTRは集中的なプロセスを導入し、すべての関連加盟国に申請を提出することのみを要求している。CTRは、スポンサーが各会員国の主管当局と道徳委員会に文書を提出することを可能にし、各会員国が決定を下すことを可能にする。♪the the the
66
CTAの評価手続きも統一されており、すべての関連加盟国による共同評価を含み、道徳基準を含む各加盟国が個別にその領土に関する具体的な要求を評価する。各会員国の決定は集中されたEUポータルサイトを通じてスポンサーに伝達される。CTAが承認されると,臨床研究開発は継続可能である。CTRは3年間の過渡期が予想される。進行中の臨床試験と新たな臨床試験がCTRによってどの程度制御されるかはそれぞれ異なる。2022年1月31日までに“臨床試験指令”によるCTAの臨床試験については,“臨床試験指令”は過渡期に基づいて3年間適用される。(I)2022年1月31日までに“臨床試験指令”に基づいて申請を提出した臨床試験、または(Ii)2022年1月31日から2023年1月31日までの間、かつスポンサーが“臨床試験指令”を適用する臨床試験を選択し、2025年1月31日までこの指令によって管轄されている。この日以降,すべての臨床試験(行われている臨床試験を含む)はCTR条項に拘束される。2023年1月31日現在,われわれが提出した日の出−3期3期臨床試験とわれわれのC型肝炎ウイルス2期臨床試験に関するCTAは臨床試験指令に基づいて提出されている。私たちはCTRによるCTAの提出を2023年2月1日から始めた。私たちがCTRで資料を提出する経験は限られている。我々と我々の第三者サービスプロバイダ(例えば臨床研究機関(“CRO”)がCTR要求を遵守することは、我々の開発計画に影響を与える可能性がある。
イギリスがどの程度その規制をEUと統合することを求めているのかは不明だ。臨床試験に関するイギリスの規制枠組みは、既存のEU立法(二次立法によってイギリス法律に定着された)に由来する。2022年1月17日、イギリス薬品と保健品監督局(MHRA)はイギリスの臨床試験立法の再制定について8週間の相談を展開した。相談は2022年3月14日に終了し、臨床試験の審査を簡略化し、革新を促進し、臨床試験の透明性を高め、リスク比率を高め、そして患者と公衆の臨床試験への参加を促進することを目的とした。協議の結果は密接に注目されており、イギリスがCTRと一致するか、CTRから乖離しているかを決定して、規制の柔軟性を維持するだろう。イギリスはその法規をEUが採用している新しい方法と緊密に結合しないことを決定し,他の国ではなく,イギリスでの臨床試験のコストに影響を与える可能性がある。
もし私たちが既存の要求の変化にゆっくりあるいは適応できない場合、あるいは新しい要求を採用したり、臨床試験を管理する政策を採用すれば、私たちの発展計画も影響を受ける可能性がある。
私たちは私たちのいくつかの候補製品を他の治療法と組み合わせて開発するつもりで、これは私たちを追加的なリスクに直面させるだろう。
併用療法は通常ウイルス感染の治療に用いられる。われわれは現在,新冠肺炎とC型肝炎に対する併用療法の開発を計画している。併用療法の開発は,我々が開発可能な任意の併用療法における個々の有効成分の安全性と有効性を証明するなど,より多くの臨床リスクに直面している。
新冠肺炎患者の治療には,ベネホブビルを単一療法として開発したほか,ベネホブビルと研究中の蛋白加水分解酵素阻害剤からなる併用製品が開発される予定であり,現在内部発見が行われている。C型肝炎ウイルスの治療に対して、著者らは現在ルザスビルと連合したbemnifosbuvirを開発しており、これはFDA或いは類似外国の監督管理機関の許可を得ていない候補製品である。私たちは私たちが開発した任意の候補製品をマーケティングして販売することができないかもしれません。これらの候補製品は、最終的に市場の承認を得ていない任意の承認されていない療法と組み合わせています。
FDAまたは同様の外国の規制機関がこれらの他の組み合わせ薬を承認しない場合、または私たちの候補製品と共同評価された薬物または生物製品に安全性、有効性、製造または供給の問題が生じることを選択した場合、私たちは共同治療案の承認を得たり、候補製品をマーケティングしたりすることができないかもしれない。
さらに、第三者開発療法や候補製品と組み合わせて使用される療法が十分な数の臨床試験や私たちの候補製品の商業化を生産できない場合、または併用療法のコストが目を引くほど高い場合、私たちの開発および商業化努力が損なわれることは、私たちの業務、財務状況、運営結果、および成長の見通しに悪影響を及ぼすだろう。
67
著者らの候補製品は深刻な不良事件、不良副作用或いはその臨床開発を阻止し、その監督管理の承認を阻止し、その商業潜在力を制限し、或いは重大な負の結果を招く可能性のある他の特性と関係があるかもしれない。
私たちの候補製品によって引き起こされる有害事象または他の副作用は、私たち、私たちの協力者、試験の任意のDSMBまたは規制機関の臨床試験の中断、延期または停止を招く可能性があり、より厳格なラベルまたはFDAまたは他の同様の外国規制機関の規制承認遅延または拒否を招く可能性がある。
臨床試験を行っている間、患者は彼らの研究医に疾病、傷害、不快感を含む彼らの健康変化を報告した。一般に、研究されている候補製品がこれらの状況をもたらしているかどうかを決定することは不可能だ。私たちがより大きく、より長く、より広い臨床試験で私たちの候補製品を試験するとき、またはこれらの候補製品の使用がより広くなるにつれて(規制部門の許可を得た場合)、患者は、以前の試験で観察された疾患、傷害、不快感、および他の有害事象、および以前の試験で発生しなかったか、または検出されなかったことを報告する。多くの場合,研究製品が大規模臨床試験で試験を行った後,あるいは承認後に患者にビジネス規模の製品を提供した後にのみ,副作用を検出することができる場合がある。
もしどんな深刻な不良事件が発生した場合、私たちが開発した任意の候補製品または製品の臨床試験または商業流通は一時停止または終了される可能性があり、私たちの業務は深刻な損害を受ける可能性がある。治療に関連する副作用はまた、患者の募集と患者の試験完了或いは潜在的責任クレームを招く能力に影響を与える可能性がある。規制当局は、私たちに、任意のまたはすべての目標適応の任意の候補製品または製品の販売を停止することを要求することを、さらなる開発を停止し、承認を拒否するか、または要求することができる。もし私たちが任意の臨床試験または商業化努力を延期、一時停止または終了することを要求された場合、これらの候補製品または製品の商業的見通しが損なわれる可能性があり、それらまたは私たちが開発した他の候補製品から製品収入を創出する能力が延期またはキャンセルされる可能性がある。さらに、もし私たちの1つまたは複数の候補製品が発売承認され、私たちまたは他の人が後にこのような製品によって引き起こされる不良副作用または有害事象を発見した場合、多くの潜在的な重大な負の結果をもたらす可能性があるが、これらに限定されない
これらの事件のいずれも、特定の候補製品に対する市場の受け入れ度を達成または維持することを阻止することができ、承認されれば、私たちの業務、財務状況、および運営結果を深刻に損なう可能性がある。
68
われわれが臨床試験で患者を募集する際に困難に遭遇すると,われわれの臨床開発活動は延期されたり,他の悪影響を受けたりする可能性がある。
様々な理由から,臨床試験では患者登録の困難に遭遇する可能性がある。臨床試験方案に基づいて適時に臨床試験を完成し、他の事項以外に、試験が終了するまで十分な数の患者を募集する能力があるかどうかに依存する。患者の入選は多くの要素に依存するが、これらに限定されない
さらに、我々の臨床試験は、我々の候補製品と同じ治療分野または同様の分野にある製品を他の臨床試験と争い、この競争は、私たちの試験に参加することを選択する可能性のある患者のうちの1つによる試験に参加することを選択する可能性があるので、使用可能な患者数およびタイプを減少させる可能性がある。例えば,われわれの潜在的競争相手によってスポンサーされたいくつかの臨床試験は,軽中度新冠肺炎を有するハイリスク外来患者に対する経口抗ウイルス薬の治療作用を評価していることが知られており,日の出−3への参加を求めている患者群と同様である。合格臨床研究者の数が限られているため、著者らはいくつかの競争相手が使用した同じ臨床試験地点で著者らのいくつかの臨床試験を行う可能性があり、これは著者らがこれらの臨床試験地点で臨床試験を行うことができる患者数を減少させる。
競合、特に新冠肺炎の場合、疾患および治療パターンの変化、検出および診断率または他の原因による患者募集遅延は、コスト増加を招く可能性があり、または計画中の臨床試験の時間または結果に影響を与える可能性があり、これらの試験の完了または開始を阻止し、候補製品開発を推進する能力に悪影響を及ぼす可能性がある。
FDAの迅速なチャネル指定は、より速い開発や規制審査や承認過程を招くことがなく、我々の候補製品が上場承認される可能性も増加しない可能性がある。
候補製品が深刻または生命に危険な疾患を治療するために使用され、候補製品がそのような疾患が満たされていない医療需要を解決する潜在力を示す場合、候補製品スポンサーは迅速なチャネル認証を申請することができる。Fast Track認証を取得した候補製品のスポンサーは,製品開発期間中にFDA審査チームとより頻繁にインタラクションする機会がある可能性があり,新薬申請(“NDA”)が提出されると,NDAは優先審査を受ける資格がある可能性がある。Fast Track候補製品の秘密保持プロトコルも
69
スクロール審査を行う資格があり,完全なセキュリティプロトコルを提出する前に,FDAは機密プロトコルの各部をスクロール審査することが考えられる.
FDAは幅広い自由裁量権を有しており、任意の特定の候補製品迅速チャネル称号が付与されているかどうか。したがって、Bemnifosbuvir COV 19候補製品を含む他の候補製品のためのこのような迅速なチャネル認証を求めることができますが、FDAが承認することを保証することはできません。私たちが高速チャネル認証を受けたとしても、私たちは従来のFDAプログラムよりも速い開発過程、審査、または承認を経験しないかもしれない。FDAが我々の臨床開発プロジェクトのデータが高速チャネルの指定をサポートしなくなったと考える場合,その指定を撤回する可能性がある。しかし、高速チャネル認証を受けた多くの候補製品は承認されなかった。
われわれは現在米国以外の地点で我々の候補製品の臨床試験を行っており,将来的にはより多くの臨床試験を行うことが選択される可能性があり,FDAは外国地点での試験データを受け入れない可能性がある。
我々は現在進行中であり,将来的には米国以外の臨床試験,我々の候補製品を行う予定である。FDAや同様の外国規制機関が米国や他の管轄地域以外で行われている臨床試験を受けた研究データは,何らかの条件によって制限される可能性があり,まったく受け入れられない可能性もある。外国の臨床試験からのデータが米国での上場承認の唯一の根拠となることを意図している場合、FDAは通常、(I)データが米国の人口および米国の医療実践に適用されない限り、(I)データが米国の人口および米国の医療実践に適用されない限り、(Ii)試験は公認能力を有する臨床研究者によってGCP規定に従って行われる;および(Iii)データはFDAによる現場検査を必要とせず、またはFDAがそのような検査が必要であると考える場合、FDAは現場検査または他の適切な方法でデータを検証することができる。また,海外の研究データが承認の唯一の根拠としようとしなくても,その研究がINDの制限を受けなければ,FDAはGCP要求に基づいて行われない限り,そのデータを上場承認申請の支援として受け入れず,FDAはその研究からのデータを現場検査で検証することができる(必要であれば)。多くの外国の規制機関もまた似たような承認要求を持っている。また、このような外国裁判は、裁判を行う外国司法管轄区域に適用される現地法によって管轄される。FDAや同様の外国規制機関が米国や適用司法管轄区域以外で行われた試験データを受け入れることは保証されない。FDAや同様の外国の規制機関がこのようなデータを受け入れなければ, これは高価で時間がかかる可能性がある追加の試験を必要とし、私たちが開発する可能性のある現在または未来の候補製品が適用される司法管轄区域で商業化承認を得られない可能性がある。
また,米国国内外の複数の管轄地域での臨床試験には固有のリスクがある
著者らが時々発表或いは公表した臨床試験の一時、“背線”と初歩的なデータはより多くの患者データの獲得に従って変化する可能性があり、そして監査と検証手続きの制限を受け、これは最終データの重大な変化を招く可能性がある。
私たちは時々私たちの臨床前研究と臨床試験の初歩的或いは主要なデータを公開する可能性があり、これらのデータは当時利用可能なデータの初歩的な分析に基づいて、結果及び関連する発見と結論は特定の研究或いは試験の関連データをより全面的に審査した後に変化する可能性がある。仮説、推定、計算、
70
結論は私たちがデータの一部を分析し、私たちがすべてのデータを全面的かつ慎重に評価する機会がないか、または機会がないかもしれないということだ。したがって、私たちの報告書のトップラインまたは予備データは、同じ研究報告の最終結果とは異なる可能性があり、または他のデータが受信されて十分に評価されると、異なる結論または考慮要因が、これらの予備またはバックラインデータを合格させる可能性がある。トップラインと予備データも依然として監査とチェック手続きの制約を受けており、これは最終結果が私たちが以前に公表した予備またはバックラインデータと大きく異なる可能性がある。したがって、最終データが利用可能になる前に、トップラインデータは慎重に表示されなければならない。
私たちはまた時々私たちの臨床前研究と臨床試験の中期データを開示することができる。私たちがその後完了する可能性のある臨床試験の中期データは、患者登録の継続およびより多くの患者データの獲得に伴い、あるいは私たちの臨床試験の患者が彼らの疾患の他の治療を継続するにつれて、1つまたは複数の臨床結果が実質的に変化する可能性があるというリスクを受ける可能性がある。初期または中期データと最終結果との間の不利な差は、私たちのビジネスの将来性を深刻に損なう可能性があります。しかも、私たちまたは私たちの競争相手が中間データを開示することは私たちの普通株の価格変動を招くかもしれない。
さらに、規制機関を含む他の人は、私たちの仮定、推定、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品または製品の承認または商業化、およびわが社の全体的な状況に影響を与える可能性がある。
さらに、開示された特定の研究または臨床試験に関する情報を選択することは、通常、広範な情報に基づいており、あなたまたは他の人は、私たちが決定した重要な情報または他の適切な情報が私たちの開示に含まれることに同意しない可能性がある。もし私たちが報告した中期、営業または予備データが実際の結果と異なる場合、または規制機関を含む他の人が結論に同意しない場合、私たちが承認を得て私たちの候補製品を商業化する能力は損なわれる可能性があり、これは私たちの業務、経営業績、将来性、または財務状況を損なう可能性がある。
私たちは他の候補製品を決定して成功させる努力で成功しないかもしれない。
私たちの戦略の一部は新しい候補製品を決定することを含む。例えば,タンパク質加水分解酵素阻害剤製品を発見するために内部努力を行っており,フェニニホブビルと併用して新冠肺炎を治療できる可能性がある。これらの努力および他の候補新製品を決定するために開始された任意の後続の発見努力は、これらのリスク要因で議論されているものを含む、臨床開発のための候補製品を生成できない可能性がある
71
私たちがより多くの適切な製品を決定して成功させて商業化することができなければ、私たちの業務戦略や私たちの財務状況に悪影響を及ぼすだろう。
私たちは成功しない可能性のある潜在的な候補製品に集中する可能性があり、私たちはより成功する可能性のある他の候補製品を開発する機会を放棄しなければならないかもしれない。
私たちは、最終的に成功しないことが証明された潜在的な製品候補に、私たちの努力と資源を集中させるか、または私たちの財務的予想に合わないマーケティング製品を許可または購入することを選択することができます。したがって、可能な商業製品または利益の市場機会を利用することができず、他の候補製品または他の疾患を追求する機会を放棄または延期することが要求される可能性があり、これらの疾患は、後により大きな商業潜在力を有することが証明される可能性がある。
また、私たちの財力と人的資源は限られており、私たちの主要な候補製品の開発に重点を置いており、特にbemnifosbuvir COV 19とbemnifosbuvirはruzasvirと併用してC型肝炎を治療しているため、私たちは他の未来により大きな商業潜在力が証明された候補製品を追求する機会を放棄または延期する可能性がある。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。現在および将来の研究開発計画および他の特定の適応の将来の候補製品への支出は、いかなる商業的に実行可能な将来の候補製品も生じない可能性がある。もし私たちが特定の未来の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは協力、許可、または他の特許権使用料の手配によって、これらの未来の候補製品に貴重な権利を放棄するかもしれないが、この場合、私たちはこれらの未来の候補製品の独占開発権と商業化権利を維持することがより有利になるだろう。
承認を加速する経路や米国国外の同様の迅速な承認経路を使用することで、FDAがいくつかの候補製品を承認することを保証しようと試みるかもしれません。もし私たちがそのような承認を得ることができなければ、私たちは私たちの予想を超える追加の臨床前研究や臨床試験を要求される可能性があり、これは必要な市場承認を得る費用を増加させ、必要な市場承認を受けることを延期するかもしれない。FDAの加速承認や外国規制機関の同様の迅速な承認を得ても、我々の検証的試験が臨床的利益を確認していない場合、または厳格な上場後の要求を遵守していない場合、FDAまたは外国規制機関は、承認の加速または同様の条件付き承認の撤回を求める可能性がある。
我々は,重篤で生命に危険な疾患を治療するためのいくつかの候補製品を開発しているため,FDAの加速承認経路に基づいてこのような候補製品の承認を求めることにする可能性がある。候補製品が深刻または生命に危険な疾患または状態を治療することを意図し、一般に既存の治療法よりも有意な利点を提供し、候補製品が臨床的利益を合理的に予測する可能性のある代替終点または中間臨床終点に影響を及ぼすと決定された場合、候補製品は加速承認を得る資格がある可能性がある。FDAは臨床利益は特定の疾病の背景下で臨床意義のある積極的な治療効果であり、例えば不可逆的な発病率或いは死亡率であると考えている。承認を加速するために、代替終点は1つの標識であり、例えば実験室測定、放射画像、バイタルサイン或いは他の臨床利益を予測できると考えられる指標であるが、それ自体は臨床利益の測定基準ではない。中間臨床終点は不可逆的発病率或いは死亡率への影響の前に測定できる臨床終点であり、それは不可逆的な発病率或いは死亡率或いは他の臨床利益に対する影響を合理的に予測する可能性がある。
加速承認経路は既存療法に対する新薬の優位性は直接の治療優位ではないかもしれないが、患者と公衆衛生の観点から見ると臨床上重要な改善状況である。承認された場合、承認を加速することは、一般に、薬剤の臨床的利益を検証および説明するために、勤勉な方法で検証的研究を行うことにスポンサーが同意するかどうかに依存する。スポンサーがこのような研究をタイムリーに行わなかった場合、またはそのような検証的研究が薬剤の予期される臨床的利益を検証できなかった場合、FDAは、薬剤の承認を迅速に撤回することができる。また、2022年12月、総裁は2023年度まで米国政府に資金を提供する総合支出法案に署名した。総合法案には“2022年食品·薬物総合改革法案”が含まれており、この法案は他にも導入されている
72
FDAによる検証性試験の監視を強化することを含む、承認を加速された製品に対するFDAの監督能力を拡大するが、これらの改革の最終的な影響はまだ不明である。
EUでは、必要なすべてのセキュリティおよび有効性データが取得されていない場合には、“条件付き”上場許可が付与される可能性がある。条件付きマーケティング許可は、欠落データを生成すること、またはセキュリティ対策を増加させるために必要な条件を保証することに依存する。条件付きマーケティング許可の有効期間は1年であり、すべての関連条件が満たされるまで年に1回更新しなければならない。適用可能な未定研究が提供されると、条件付きマーケティング許可は“標準”マーケティング許可となることができる。しかしながら、条件が欧州医薬品局(“EMA”)が設定された時間枠内で満たされていない場合、マーケティング許可は更新を停止する。また、“特殊な場合”は、出願人が正常な使用条件であることを証明することができれば、製品が許可され、特定のプログラムを採用しなければならない後であっても、その有効性や安全性に関する包括的なデータを提供することができなければ、上場を承認することができる。予想される適応が非常にまれであり,現在の科学的知識状態では網羅的な情報を提供することが不可能である場合や,データを生成することが一般的に受け入れられている倫理原則に違反する可能性がある場合がある。このようなタイプのマーケティング許可は、深刻な疾患または満たされていない医療需要のために許可される医薬品のために保持されるので、条件付きマーケティング許可に近いが、出願人は、マーケティング許可を付与するために必要な法定完全データセットを有していない。しかしながら、条件付きマーケティング許可とは異なり、出願人は、失われたデータを提供する必要はなく、提供する必要はない。“特別な場合”のマーケティング許可は最終的に付与されたにもかかわらず, 毎年薬品のリスク−収益バランスを審査し,リスク−収益比が有利でなければ上場許可を撤回する可能性がある。
EUの主管監督機関は広範な自由裁量権を有しており、特別な場合にこのような条件付きマーケティング許可またはマーケティング許可が付与されているかどうかは、このような評価または許可が承認されても、従来のプログラムよりも速い開発過程、審査、または許可を経験しない可能性がある。さらに、このようなマーケティング許可の廃止または脅しは、候補製品の臨床開発に不確実性または遅延をもたらす可能性があり、承認されれば、私たちの製品および候補製品の商業化の将来性を脅かす可能性がある。このような事件は私たちの業務、財務状況、そして運営結果に大きな影響を及ぼすかもしれない。
私たちが私たちの候補製品に加速承認を求めること、または迅速な規制指定を得ることを求める秘密協定を提出することを決定した場合、そのような提出または申請が受け入れられるか、または開発加速、審査または承認がタイムリーに承認されるか、または全く保証されない保証はない。候補製品が加速承認または任意の他の形態の加速開発、審査または承認を得ることができない場合、候補製品の商業化までの時間が長くなり、候補製品の開発コストが増加する可能性があり、市場での競争地位を損なう可能性がある。
私たちが必要な臨床前研究と臨床試験を完成しても、マーケティング承認過程は高価で、時間と不確定であり、私たちあるいは任意の未来のパートナーが私たちが開発した任意の候補製品の商業化承認を得ることを阻止するかもしれない。
我々が開発する可能性のある任意の候補製品およびその開発および商業化に関連する活動は、設計、テスト、製造、安全、効果、記録保存、ラベル、貯蔵、承認、広告、販売促進、販売、流通を含み、FDAと米国の他の規制機関、および他の国の同様の機関によって全面的に規制されている。私たちが開発した任意の候補製品は効果がないかもしれません。中程度の効果しかないかもしれません。あるいは不良または意外な副作用、毒性、あるいは他の特徴があることが証明される可能性があり、上場承認を得ることを阻止したり、商業使用を阻止したり制限したりする可能性があります。候補製品のマーケティング承認が得られなければ、指定された司法管轄区域で候補製品を商業化することはできないだろう。私たちはまだどのような管轄区域の規制機関からもどんな候補製品のマーケティングの承認を得ていない。私たちが開発している候補製品や私たちが将来開発を求めるかもしれない製品は規制部門の承認を得られないかもしれません。会社として私たちは届出の面で経験がない
73
マーケティング承認を得るために必要なアプリケーションをサポートし、この過程で第三者CRO、サプライヤー、サプライヤー、または規制コンサルタントに依存することが予想されます。監督部門の承認を得るためには、各監督機関に広範な臨床前と臨床データ及び支持情報を提出し、候補製品の安全性と有効性を確定する必要がある。監督管理の承認を得るためには、製品の製造過程に関する情報を関連監督機関に提出し、関連監督機関が製造施設を検査する必要がある。
米国や海外では,上場承認を得る過程は高価であり,大量の臨床試験が必要であれば,長年の時間を要する可能性があり,本当に承認された場合,関連する候補製品のタイプ,複雑性,新規性を含む様々な要因によって大きく異なる可能性がある。開発期間中の市場承認政策の変化、追加法規又は法規の変化、又は各提出製品申請に対する規制審査の変化は、出願の承認又は拒絶の遅延を招く可能性がある. 例えば、欧州委員会が2020年11月に開始した欧州薬品戦略イニシアティブを背景に、EU薬品立法は現在全面的な審査が行われている。医薬製品に関するいくつかの立法文書の改正に関する提案(規制排他的期限,迅速チャネルの資格などが改訂される可能性がある)。2023年第1四半期に欧州委員会によって採択される予定だ。提案された改正は,欧州議会と欧州理事会の同意と採択(2024年末または2025年初めまではないと予想される)を得ると,長期的には製薬業に大きな影響を与える可能性がある。
FDAと他国の類似機関は承認過程においてかなりの自由裁量権を有しており、いかなる申請も拒否することができ、私たちのデータが承認を得るのに十分ではなく、追加の臨床前、臨床、あるいは他の研究を行う必要があると決定することもできる。そのほか、臨床前と臨床試験から得られたデータの異なる解釈は候補製品の上場承認を延期、制限或いは阻止する可能性がある。私たちが最終的に得たどのマーケティング承認も限られているかもしれないし、制限されたり、承認された後の約束は、承認された製品が商業的に実行できないようにすることができる。
もし私たちが承認を得る上で遅延に遭遇した場合、あるいは私たちが開発する可能性のある任意の候補製品の承認を得ることができなければ、これらの候補製品のビジネス見通しが損なわれる可能性があり、私たちが製品収入を作る能力は実質的に損なわれるだろう。
私たちが開発中の新冠肺炎を治療するbemnifosbuvir COV 19製品についてfdaにEUAを求めたり、外国の規制機関から類似の緊急使用許可を求めたりするかもしれません。もし私たちがこのような許可を得たり維持できなかったら、私たちは予想よりも長い臨床開発過程を行う必要があるかもしれません。私たちの業務は損害を受ける可能性があります.
もし私たちが当時私たちの3期日の出-3臨床試験と他の新冠肺炎の臨床試験から十分なデータを得たら、私たちは私たちの候補製品Bemnifosbuvir COV 19についてFDAのEUAあるいは他の外国の監督機関の類似の緊急使用許可を求めるかもしれない。FDAは、ある場合にのみEUAを発行する権利があり、例えば、公衆衛生緊急時には、衛生·公衆サービス部(HHS)秘書の声明に基づいて、緊急事態が存在し、あるタイプの製品がEUA(EUA声明と呼ばれる)を発行する理由がある。2020年3月27日、衛生·公衆サービス部部長は、当時、新冠肺炎の大流行期間中に薬品と生物製品を許可する理由がある場合があったが、その後特定の製品に対して発表された任意のEU合意の条項を遵守しなければならないと発表した。EUA宣言が発行され、有効性が維持されると、FDAは、その宣言範囲に属する製品に対してEUAを発行することができる。EUAを発行するためには、FDA専門家は、(1)EUA宣言で示されている化学、生物、放射性または核剤またはCBRNが、深刻または生命に危険な疾患または状態をもたらすことができ、(2)既存の全ての科学的証拠に基づいて、製品がCBRNに起因する疾患または状態を効果的に診断、治療または予防する可能性があり、製品の既知および潜在的利益がその既知および潜在的リスクよりも大きいと信じる理由があり、(3)十分な、承認され、利用可能な製品代替品がないと結論しなければならない。現在2種類の経口直接作用の抗ウイルス薬パシクロビルがある(ネマレビルとリトナビル)とLagevrio(モヌビラビル) 新冠肺炎ハイリスク患者の緊急使用を許可する。
74
FDAがEUAを承認する基準は、従来の審査手順に従ってNDAを承認する基準よりも低く、私たちの1つ以上の候補製品のためにEUAを求めて取得しても、承認が必要であれば、FDAはそのような候補製品のNDAを承認することを保証することはできません。したがって、Bemnifosbuvir COV 19候補製品のEUAを取得したとしても、追加の臨床試験を行う必要があり、その後、その候補製品にNDAまたは同様のマーケティング申請を提出する必要があるかもしれない。
EUAによる製品販売の許可は、EUA宣言が発効する期間に限定され、FDAは、場合によってはEUAを取り消すことができる。EUAに対するFDAの政策は意外に変わるかもしれない。私たちはどんな許可も得られなければ、どのくらい維持されるのか予測できない。FDAが新しいと絶えず変化する公共衛生情報と臨床証拠に応答することに伴い、FDAの新冠肺炎の診断、治療或いは緩和のためのワクチンとその他の製品に関する政策は依然として絶えず変化している。2023年1月31日、バイデン政府は2023年5月11日から新冠肺炎突発公衆衛生事件を中止する計画を発表した。 公共部門の会計基準が発表されていない場合、衛生·公衆サービス部長官は、将来のEU会計基準を発行する理由がある場合も失効したと認定することができる。この場合、私たちは任意のBemnifosbuvir COV 19候補製品を商業化するために秘密協定を求めることが要求されるだろう。
したがって、私たちの3期日の出-3臨床試験と私たちが開始する可能性のある任意の他の新冠肺炎臨床試験のデータが陽性であると信じても、私たちはBemnifosbuvir COV 19候補製品のEUAまたは他の緊急許可を得ることができないかもしれない。さらに、私たちがEUAまたは同様の緊急許可を得たとしても、このようなEUAまたは他の許可は撤回される可能性があり、私たちはそのような製品のNDA承認を得るまで、任意の商業化活動を停止することを要求されるかもしれない。どんな商業化活動の停止も、私たちの業務、財務状況、そして運営結果に悪影響を及ぼすだろう。
たとえ私たちの候補製品がFDAの承認を得ても、私たちは決してアメリカ以外で承認されたり、そのような製品を商業化したりすることはありません。これは私たちがそのすべての市場潜在力を達成する能力を制限するだろう。
米国以外の市場で任意の製品を販売するためには、安全性と有効性に関する他国の多くの規制要件を確立し、遵守しなければならない。一国で行われる臨床試験は、他の国の規制部門に受け入れられない可能性があり、一国の規制承認は、他のどの国でも規制承認を受けることを意味するものではない。承認手続きは国によって異なり、追加の製品テストと検証、および追加の行政審査期限が含まれる可能性があります。外国の監督管理機関の承認を求めることは著者らの重大な遅延、困難とコストを招く可能性があり、追加の臨床前研究或いは臨床試験を必要とする可能性があり、これは高価で時間がかかるだろう。各国の規制要求には大きな違いがある可能性があり、私たちの製品がこれらの国で発売されることを延期または阻止する可能性がある。これらと他の規制要求を満たすことは高価で、時間がかかり、不確定であり、予期しない遅延が生じる可能性がある。また、私たちはどの国でも規制承認を得ることができず、他の国の規制承認過程を延期したり、マイナス影響を与える可能性がある。私たちは国際市場を含めてどの司法管轄区域での販売も承認されていない候補製品は、組織として、国際市場で規制承認を受けた経験がありません。もし私たちが国際市場の規制要求を守らない場合、あるいは必要な承認を得て維持できなければ、私たちが製品全体の市場潜在力を実現する能力は損なわれるだろう。
現在あるいは未来の候補製品が市場の承認を得ても、それは医者、患者、第三者支払人と医学界の他の人が商業成功に必要な市場受容度に達することができないかもしれない。
もし私たちが開発した任意の現在または未来の候補製品が市場の承認を得たら、単一薬物としても他の治療法と併用しても、医師、患者、第三者支払人、および医学界の他の人の十分な市場受容度を得ることができないかもしれない。例えば,現在承認されている抗ウイルス製品は医学界でC型肝炎ウイルスの治療に用いられており,米国では2種類の経口抗ウイルス薬PaxlovidとLagevrioが現在新冠肺炎の治療に許可されており,医師はこれらの療法に依存し続ける可能性がある。もし私たちが開発した候補製品が十分な受容度に達していなければ、著しい製品収入が生じないかもしれません
75
利益を実現する。任意の候補製品が商業販売のために承認された場合、市場の受容度は多くの要因に依存するが、これらに限定されない
資金不足や世界的な健康問題によるFDAや外国規制機関の中断は、重要な指導部や他の人員の採用、保留、配置の能力を阻害する可能性があり、あるいは新製品や修正された製品のタイムリーまたは開発、承認、商業化を他の方法で阻止することは、私たちの業務にマイナスの影響を与える可能性がある。
FDAと外国監督機関が新製品を審査または承認する能力は、政府予算と資金レベル、法定、監督と政策の変化、FDAまたは外国監督機関のキーパーソンの雇用と保留、ユーザー費用の支払いを受け入れる能力、および大量のスポンサーが迅速に新冠肺炎に流入する事件を含む様々な要素の影響を受ける可能性がある。したがって、FDAと外国規制機関の平均審査時間は近年変動している。また,他の研究開発活動を援助する政府機関への政府の援助は政治過程の影響を受けており,この過程は本質的に不安定で予測不可能である。FDAや他の機関の中断も、新薬が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。例えば、ここ数年間、米国政府は何度か閉鎖されており、FDAのようないくつかの規制機関は、FDAのキー従業員を休暇にし、キー活動を停止しなければならない。
また、新冠肺炎の流行に対応するため、アメリカ食品薬品監督管理局は国内外の異なる場所の製造施設の大部分の検査を延期した。FDAはすでに実行可能な情況下で国内施設に対する標準検査操作を回復したが、FDAは依然としてその検査活動の変化を監視と実施し、その従業員及び監督会社の安全を確保し、絶えず変化する新冠肺炎疫病に適応し、ウイルスの灰再発或いは新変種の出現は更なる検査遅延を招く可能性がある。新冠肺炎の流行に対応するために、米国以外の規制機関も同様の制限措置や他の政策措置をとっている。政府が長期的に停止している場合、または世界的な健康問題がFDAまたは他の規制機関の定期検査、審査または他の規制活動を阻害し続けている場合、FDAまたは他の監督管理機関が私たちの規制提出を適時に審査し、処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。
私たちの保険証書は高くて、いくつかの商業リスクから私たちだけを保護して、これは私たちを重大な未保険債務に直面させます。
臨床試験製品責任保険がありますが、私たちの業務が遭遇する可能性のあるすべての種類のリスクに保険をかけていません。私たちが現在維持しているいくつかの保険は一般責任、財産、自動車、労働者補償、傘と役員と高級管理者保険を含みます。
臨床試験監督提出過程の一部として、多くの国で、私たちは現地の保険を提供することを要求されています。もし彼らが臨床試験に参加したり接触したりしたためであると信じている場合、臨床試験に関連する人員を保証します
76
臨床試験で研究されている研究候補製品。これらの現地保険証書の取得に時間がかかる可能性があり、特定の国での臨床試験の予想開始が遅れる可能性がある。さらに、これらのローカル保険シートは、被害者側が提起する可能性のあるすべてのクレームをカバーしていない可能性があり、私たちのクレーム弁護に関連する損失および私たちに不利ないかなる判決もカバーするのに十分ではない可能性があります。
私たちが将来得た任意の追加の製品責任保険は、私たちが受ける可能性のあるいかなる費用や損失を補償するのに十分ではないかもしれない。また、保険範囲はますます高くなっており、将来的には、責任による損失から私たちを保障するために、合理的なコストや十分な金額で保険範囲を維持することができないかもしれない。もし私たちの候補製品が市場の承認を得たら、私たちは商業製品販売を含む保険を購入するつもりです;しかし、私たちは商業的に合理的な条項や十分な金額で製品責任保険を得ることができないかもしれません。私たちに提出された成功した製品責任クレームや一連のクレームは、私たちの株価を下落させる可能性があり、判決のクレームが私たちの保険範囲内または金額が私たちの保険範囲を超えなければ、私たちが開発した任意の候補製品の開発と商業化を阻止または制限することを含む、私たちの運営や業務結果に悪影響を及ぼす可能性があります。私たちは特定の生物或いは危険廃棄物保険を受けていません。私たちの財産、意外と一般責任保険は生物或いは危険廃棄物の暴露或いは汚染による損害と罰金は明確に含まれていません。したがって、汚染や傷害が発生した場合、損害賠償責任を請求されたり、私たちの資源を超えた罰金が科されたりする可能性があり、私たちの臨床試験や規制承認は一時停止される可能性があります。
上場企業として、将来的に取締役や上級者責任保険をより難しく、より高価にする可能性があり、低減された保険限度額や保証範囲を受け入れること、または同じまたは同様の保証範囲を得るためにより高いコストを発生させることが求められる可能性がある。したがって、私たちは合格した人を私たちの取締役会、取締役会委員会に参加したり、役員にしたりすることがもっと難しいかもしれない。しかし、私たちは私たちが既存の保険を維持し、十分な保険を提供できるかどうか分からない。いかなる重大な未保険債務も私たちが大量の金額を支払う必要があるかもしれません。これは私たちの現金と現金等価物の状況と運営結果に悪影響を及ぼすでしょう。
情報技術システムの故障,ネットワーク攻撃や欠陥が発生すると,我々の業務や運営が影響を受ける可能性があり,我々の業績に大きな影響を与える可能性がある.
我々の情報技術システム、および我々のCROおよび他の請負者およびコンサルタントのシステムは、コンピュータウイルスおよび他のマルウェア(例えば、恐喝ソフトウェア)、許可されていないアクセスまたは他のネットワークセキュリティ攻撃、外部または内部当事者の不適切な行為、人為的エラー(例えば、社会工学、ネットワーク釣り)、自然災害(ハリケーンを含む)、テロ、戦争、火災および電気通信または電気故障の攻撃、故障、および破損を受けやすい。私たちの通常の業務プロセスでは、知的財産権、機密情報、臨床前および臨床試験データ、独自の業務情報、個人データ、ならびに私たちの臨床試験対象および従業員の個人識別健康情報を含む敏感なデータを直接または間接的に収集、記憶および送信し、我々のデータセンターおよびネットワーク、または第三者のデータセンターおよびネットワーク上にある。このような情報の安全な処理、維持、そして伝達は私たちの行動に必須的だ。
私たちは、私たちの重要な第三者(例えば、協力者)とセキュリティ対策を実施しているにもかかわらず、私たちの情報技術システムは、ハッカーまたは内部非行者の攻撃を受けやすいか、または人為的ミス、技術的脆弱性、汚職、または他の中断によって破壊される可能性がある。世界各地からの未遂攻撃と侵入の数、持続性、強度と複雑性の増加に伴い、セキュリティホールや破壊のリスクは普遍的に増加し、特にコンピュータハッカー、外国政府とネットワークテロリストを含むネットワーク攻撃或いはネットワーク侵入を介している。新冠肺炎の大流行により、私たちはまたより多くのネットワーク安全リスクに直面している。私たちのインターネット技術への依存と私たちの遠隔作業の従業員の数のため、これはネット犯罪者の抜け穴を利用するためにもっと多くの機会を作るかもしれない。私たちはすべての種類の安全脅威を予見できないかもしれないし、これらすべての安全脅威に対して効果的な予防措置を取ることができないかもしれない。サイバー犯罪者が使用する技術は常に変化し、起動前に識別されない可能性があり、外部サービスプロバイダ、組織犯罪支店、テロ組織または敵意外国組織などの外部団体を含む様々なソースから来る可能性がある
77
政府や機関ですそのような理由で、私たちはまたセキュリティホールに遭遇する可能性があり、このような脆弱性は長い間発見されないかもしれない。発見されても、攻撃による事件または違反を十分に調査または修復することができない可能性があり、これらの攻撃は、制御を回避し、法医学的証拠を検出および除去または混同するためのツールおよび技術をますます使用している。私たちのデータ保護の仕事と情報技術への私たちの投資は、私たちのシステムまたは私たちのCROと他の請負業者やコンサルタントのシステムの重大な故障、データ漏洩、または破壊を防止することを保証することはできません。
私たちと私たちの特定のサービス提供者たちは時々ネットワーク攻撃とセキュリティ事件の影響を受ける。これまで重大なシステム故障,事故,セキュリティホールを経験したことはないと考えられるが,このような事件が発生して我々の運営中断を招くと,我々の候補製品開発計画に重大な中断を招く可能性がある.例えば、完成した、行われている、または計画されている臨床前研究または臨床試験のデータ損失は、私たちの規制承認作業を遅延させ、データを回復または複製するコストを著しく増加させる可能性がある。任意の中断またはセキュリティホールが、私たちのデータやアプリケーションを紛失したり、破損したり、個人、機密、または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの候補製品のさらなる開発が延期される可能性があります。
セキュリティホールまたは他のイベントが、不正な取得または無許可使用、開示、配布、または他の方法で個人情報を処理する場合、プライバシー法およびセキュリティ法に従って個人、政府当局、監督機関、メディア、および他の当事者に通知する必要がある場合があります。このようなアクセス、開示、または他の情報の損失は、法的クレームや訴訟、個人情報のプライバシーを保護する法的責任と重大な規制処罰を招く可能性があり、このような事件は私たちの運営を乱し、私たちの名声を損なう可能性があり、人々が私たちと私たちの臨床試験を行う能力に自信を失わせる可能性があり、これは私たちの名声に悪影響を与え、私たちの候補製品の臨床開発を延期する可能性がある。さらに、私たちの保険カバー範囲は、私たちのシステムの中断または破壊によって引き起こされる可能性のある財務、法律、商業、または名声損失をカバーするのに十分ではないかもしれません。
医療保健法やその他の法律コンプライアンス事項に関するリスク
私たちは広範囲で費用の高い政府によって規制されるだろう。
私たちの候補製品はFDA、医療保険と医療補助サービスセンター(CMS)、アメリカ衛生と公衆サービス部の他の部門、アメリカ司法省、州と地方政府及びアメリカ海外の相応機関の監督と監督を含む広範かつ厳格な国内政府の監督を受ける。FDAは薬品の研究、開発、臨床前と臨床試験、製造、安全性、有効性、記録保存、報告、ラベル、包装、貯蔵、承認、広告、販売、販売、流通、輸出入を監督する。もし私たちの製品が海外に販売されれば、それらはFDAの承認を得たかどうかにかかわらず、外国政府の広範な規制を受けるだろう。そのような外国の規制は米国の相応の規制要求と同じように高く、さらに高いかもしれない。
政府の規制は私たちの製品の研究、開発、製造、販売のコストとリスクを大幅に増加させた。各候補製品の臨床前試験と臨床試験を含む監督管理審査と承認過程は、長く、高価であり、不確定である。私たちは臨床試験の規制許可を得て維持しなければならない。私たちが販売しようとしているすべての製品は規制部門の許可を得なければなりません。製品が使用する製造施設は検査され、法律の要求に適合しなければなりません。監督部門の承認を得るためには、各期待用途に対する製品の安全性と有効性、効力および純度を決定するために、各提案された治療適応のために、広範な臨床前および臨床データおよび他の支持情報を提出する必要がある。開発と承認過程には長年の時間がかかり、大量の資源が必要であり、製品の承認を招くことは決してないかもしれない。
特定の製品の規制承認を得ることができても、承認はその製品の指定された医療用途を制限する可能性があり、そうでなければ、製品の普及、販売、流通能力を制限する可能性があり、コストの高い上場後監督を要求することができ、および/または継続的な発売後研究を要求する可能性がある。承認された製品の材料変更、例えば変更または修正されたラベルの製造には、さらなる規制審査および承認が必要となる可能性がある。一度
78
承認された後、任意の承認が撤回される可能性があり、例えば、製品が以前に未知のセキュリティ問題のような以前に未知の問題が存在することが後に発見された場合。
私たち、私たちのコンサルタント、CMO、CRO、または他のサプライヤーが規制プロセスの任意の段階で適用される規制要件を遵守できなかった場合、このような不遵守が承認申請または承認申請の補充剤の遅延を招く可能性がある;FDAを含む規制機関が、未解決の市場承認申請または承認された申請の補充剤の審査を拒否する;警告状、罰金、輸入および/または輸出制限、製品のリコールまたは差し押さえ、禁止、完全または部分的な生産停止、民事処罰、以前に承認されたマーケティング申請または許可証の撤回、FDAまたは他の規制機関が政府契約に対する提案;および/または刑事起訴。
今後の医療法や政策の公布は、候補製品の商業化の難しさやコストを増加させ、私たちの業務に悪影響を及ぼす可能性があります。
米国、EU、その他の司法管轄地域では、医療システムは、複数の立法や法規の変化、提案された変化が引き続き発生することが予想され、これらの変化は、私たちの候補製品の発売承認を阻止または延期する可能性があり、マーケティングの承認を得た任意の候補製品の承認後の活動を制限または規制し、定価と精算に影響を与え、このような製品を販売する利益能力に影響を与える可能性がある。特に,米国連邦や州レベルでは,医療コストの低減と医療の質の向上を図る取り組みが継続されている。また,新たな条例や既存の医療法規や条例の解釈もしばしば採択されている。
2010年3月、“患者保護·平価医療法案”(ACA)が公布され、政府や民間保険会社が医療に資金を提供する方法が大きく変更された。ACAの条項の中で、製薬とバイオテクノロジー産業にとって最も重要な条項は:
ACAのいくつかの側面は公布以来、司法、国会、行政面で挑戦されてきた。2021年6月17日、米国最高裁はいくつかの州がACAに対する最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。
また、ACAが公布されて以来、米国は他の立法改正を提案し、採択した。2011年8月、“2011年予算統制法案”は医療保険総金額を強制的に削減
79
サプライヤーへの支払いは、2013年4月1日から施行され、その後の立法改正により2031年まで有効となる。また、2021年3月11日には、2024年1月1日から法定の医療補助薬品還付上限が撤廃され、現在の上限は薬品メーカーの平均価格の100%となる“2021年米国救援計画法案”が署名された。
また、支払い方法は、医療立法や規制措置の他の変化の影響を受ける可能性がある。例えば、協力医療は、結果に基づく精算など、新たな支払い·交付モデルを開発することができる。また、最近政府はメーカーが製品を販売するための価格設定方式の審査を強化し、アメリカ議会がいくつかの調査を行い、薬品定価の透明性を高め、連邦医療保険下の処方薬コストの低減及び定価とメーカー患者計画との関係を審査するための連邦立法を提出し、公布した。最近は2022年8月16日に、“インフレ低減法案”(“IRA”)が法律に署名された。他の事項を除いて、アイルランド共和軍はある薬品のメーカーに連邦医療保険との価格交渉(2026年から)を要求し、価格は交渉できるが上限があり、連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを実施し、インフレを超える価格上昇を処罰し(2023年に初めて満了)、D部分のカバーギャップ割引計画の代わりに新しい割引計画を用いる(2025年から)。アイルランド共和軍は,衛生·公衆サービス部(“HHS”)秘書が最初の数年間,規制ではなく指導によって多くの規定を実施することを許可した。このような理由と他の理由で、アイルランド共和軍がどのように実施されるのかは不明だ。私たちは将来的により多くのアメリカ連邦医療改革措置を取ることが予想され、そのいずれも米国連邦政府が医療製品やサービスのために支払う金額を制限する可能性があり、これは私たちの候補製品に対する需要の減少や追加の価格設定圧力を招く可能性がある。
アメリカ各州もますます立法を通じて、価格或いは患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示と透明性措置を含む薬品と生物製品の定価を制御するための法規を実施しており、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。法律で規定されている第三者支払者の支払金額の価格制御またはその他の制限は、私たちの業務、運営結果、財務状況、見通しを損なう可能性があります。また,地域医療当局や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。これは私たちの候補製品に対する最終的な需要を減らすか、あるいは私たちの製品の価格設定に圧力を与えるかもしれない。
EUでは、承認されれば、同様の政治、経済、規制発展が候補製品を利益的に商業化する能力に影響を与える可能性がある。価格およびコスト制御措置に対する持続的な圧力に加えて、EUまたは加盟国レベルの立法発展は、著しい追加的な要求や障害を招く可能性があり、これは私たちの運営コストを増加させる可能性がある。EUで医療サービスを提供することは、医療サービスの確立と運営、薬品の定価と精算を含み、ほぼ完全に国家の法律と政策の問題であり、EUの法律や政策ではない。この点で,各国政府や保健サービス提供者は,保健サービスの提供や製品の定価や精算に異なる優先順位や方法を持っている。しかし、全体的に、大多数のEU加盟国の医療予算制限は、関連医療サービス提供者の薬品定価と精算の制限を招いた。加えて、製品の開発·マーケティングを希望するEUや国の規制負担が増加しており、これは、候補製品の上場承認を阻止または延期し、承認後の活動を制限したり、規制したりし、候補製品を商業化する能力(承認されれば)に影響を与える可能性がある。
米国やEU以外の市場では,精算や医療支払い制度は国によって異なり,多くの国で特定製品や療法に価格上限が設定されている。
今後の法律や政策の制定は、候補製品の上場承認を得る難しさやコストを増加させ、私たちの業務に悪影響を及ぼす可能性があります。
米国では、承認後の要求を拡大し、薬品の販売や販売促進活動を制限するための立法·規制提案がなされている。私たちはより多くの立法改正、あるいはFDAの法規、ガイドラインを公布するかどうかを決定することができない
80
説明が変わるかどうか、またはこれらの変更が私たちの候補製品のマーケティング承認(あれば)に与える影響は何かもしれません。また、FDA承認過程に対する国会のより厳格な審査は、上場承認を著しく延期または阻止し、より厳しい製品ラベルと上場後のテストとその他の要求の制約を受ける可能性がある。
私たちはアメリカ、EU、または任意の他の司法管轄区域の将来の立法または行政行動によって生じる可能性のある政府規制の可能性、性質、または程度を予測することができない。もし私たちまたは私たちが接触する可能性のある任意の第三者が既存の要求の変化に緩やかに適応できない場合、または新しい要求や政策を採用することができない場合、または私たちまたはそのような第三者が規制コンプライアンスを維持できない場合、私たちは法執行行動の影響を受ける可能性があり、利益を達成したり維持することができないかもしれない。
私たちの候補製品が規制部門の承認を得ても、私たちの製品は監督部門の審査と発売後の要求を受けるだろう。
私たちが入手可能な任意の候補製品の規制承認は、候補製品の安全性および有効性を監視するために、規制機関および監督機関に報告書を提出することを要求し、年齢または医療条件、警告、予防措置または禁忌症などの指定された集団の使用制限に関連する重大な制限を含む可能性があり、重い承認後の研究またはリスク管理要件を含む可能性がある。例えば、FDAは、私たちの候補製品を承認するためにREMSを必要とすることができ、これは、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師トレーニングおよびコミュニケーション計画、または安全な使用を保証する他の要素を必要とする可能性がある。さらに、もし私たちの候補製品が承認されたら、それは製造、ラベル、包装、貯蔵、広告、販売促進、サンプリング、記録保存、発売後の研究と提出安全、治療効果と他の発売後の情報の持続的な規制要求を受け、アメリカの連邦と州要求、比較可能な外国監督機関の要求を含む。製造業者および製造業者の工場は、品質管理および製造手順がcGMPおよび類似法規に適合することを確保することを含む、FDAおよび同様の外国規制機関の広範な要求を遵守しなければならない。したがって、私たちと私たちの契約製造業者は、cGMPおよび同様の要求に対する遵守状況、および任意の承認されたマーケティング申請で行われた約束の遵守状況を評価するために、持続的な審査および検査を受ける。したがって、私たちと私たちと協力している他の人たちは、製造、生産、品質管理を含むすべての規制コンプライアンス分野で時間、お金、エネルギーをかけ続けなければならない。
FDAまたは他の規制機関が、意外な深刻または頻度の不良事象のような以前に未知の問題があることを発見した場合、または製品を生産する施設に問題がある場合、または製品の販売促進、マーケティングまたはラベルに同意しない場合、規制機関は、製品の市場からの撤退を要求することを含む、製品または私たちに制限を加える可能性がある。もし私たちが適用される規制要求を遵守できなければ、規制当局や法執行当局は他の措置を取るかもしれない
政府は違法の疑いのあるいかなる調査にも対応するために多くの時間と資源を必要とする可能性があり、否定的な宣伝が生じる可能性がある。現行の規制要件を遵守しないいかなる行為も、私たちの商業化と創造能力に悪影響を及ぼす可能性がある
81
製品です。もし規制制裁が施行されたり、規制承認が撤回されたら、私たちの業務は深刻な損害を受けるだろう。
さらに、FDAおよび他の規制機関の政策は変化する可能性があり、私たちの候補製品に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布される可能性がある。米国や海外の将来の立法や行政や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちが既存の要求の変化に緩やかに適応できない場合、あるいは新しい要求や政策を採用することができない場合、あるいは規制コンプライアンスを維持できない場合、私たちは法執行行動の影響を受ける可能性があり、利益を達成したり維持したりすることができないかもしれない。
FDAと他の規制機関は非ラベル使用の普及を禁止する法律法規を積極的に実行している。
もし私たちの候補製品が承認され、私たちがこれらの製品のラベル外用途を不正に普及させたことが発見されれば、私たちは重大な責任を負うかもしれない。FDAや他の規制機関は、承認されれば、処方製品(例えば、私たちの候補製品)に対する販売促進主張を厳格に規制することができる。特に、製品は、製品が承認されたラベルに反映されるように、FDAまたは他の規制機関によって承認されていない使用に使用されてはならない。もし私たちが候補製品のマーケティング承認を得たら、医者は承認されたラベルと一致しない方法で患者に処方するかもしれない。もし私たちがこのようなラベル外の使用を普及させることを発見されたら、私たちは重大な責任を負うかもしれない。米連邦政府は、ラベル外使用の不当な普及の疑いがある会社に巨額の民事と刑事罰金を科し、いくつかの会社がラベル外普及に従事することを禁止している。FDAはまた、企業に同意法令または永久禁止を締結し、これらの法令または永久禁止に基づいて、特定の販売促進行為を変更または制限することを要求する。もし私たちが私たちの候補製品の普及を成功的に管理できなければ、承認されれば、私たちは重大な責任を負う可能性があり、これは私たちの業務や財務状況に実質的な悪影響を及ぼすだろう。
我々の業務運営および調査者,医療専門家,コンサルタント,第三者支払者,患者組織と顧客との現在と将来の関係は,適用される医療規制法の制約を受け,処罰される可能性がある。
私たちの業務運営および調査者、医療専門家、コンサルタント、第三者支払人、患者組織と顧客との現在および将来の手配は、広範に適用される詐欺と乱用、および他の医療に関する法律と法規に直面する可能性があります。これらの法律は、私たちがどのように研究し、承認されれば、マーケティング、販売、販売、私たちの候補製品をどのように研究するかを含めて、私たちが業務を展開する業務または財務配置と関係を制限するかもしれません。これらの法律には限定されません
82
83
我々の内部運営と将来の第三者の業務配置が適用される医療法律や法規に適合することを確保することは、多くのコストに及ぶ。政府当局は、私たちのビジネス行為は、私たちと医師や他の医療保健提供者との関係を含むかもしれませんが、その中の一部の人は、私たちに提供されたサービスによって株式または株式オプションの形で補償され、承認されれば、候補製品の注文や使用に影響を与える可能性があり、現在または未来に適用される詐欺および乱用または他の医療保健法律および法規に関する現行または将来の法規、法規、機関指導または定例法に適合しない可能性があります。私たちの業務が上記の任意の法律または私たちに適用される可能性のある他の任意の政府の法律および法規に違反していることが発見された場合、私たちは、MedicareおよびMedicaidまたは他の国または司法管轄区域の同様の計画、違反、返却、個人監禁、契約損害、名声損害、利益減少、および私たちの業務の削減または再編に関する告発を解決するために、民事、刑事および行政処罰、損害賠償、罰金、MedicareおよびMedicaidまたは他の国または司法管轄区域の同様の計画のような重大な処罰を受ける可能性がある。もし私たちがそれと業務を行うことを期待している任意の医師や他の提供者や実体が適用されない法律を遵守していないことが発見された場合、彼らは政府の援助された医療計画や監禁から除外されることを含む刑事、民事または行政制裁を受ける可能性があり、これは私たちの業務運営能力に影響を及ぼす可能性がある。さらに、このような行動を防御するには費用がかかり、時間がかかる可能性があり、多くの人的資源が必要となる可能性がある。だから…, たとえ私たちが私たちに提起される可能性のあるどんな訴訟も防ぐことに成功したとしても、私たちの業務は損害を受ける可能性がある。
私たちは政府の規制や他の法的義務の制約、特にプライバシー、データ保護、情報セキュリティに関連する義務を受けており、消費者保護法の制約も受けており、これらの法律は私たちのマーケティング行為を規範化し、不公平または詐欺的な行為ややり方を禁止している。私たちは実際にこのような義務を守らないと私たちの業務を損なう可能性があると思う。
私たちはデータのプライバシーと安全に関連した様々な法律と法規によって制限されている。米国と世界は新しいプライバシールールを制定しており、既存のルールは更新され強化されている。例えば、カリフォルニア消費者プライバシー法(CCPA)は2020年1月1日に施行され、カリフォルニアの消費者のためのプライバシー権を創出し、ある個人情報を処理するエンティティのプライバシーとセキュリティ義務を増加させ、カリフォルニアの個人に新しい情報を開示することを要求し、これらの個人に新たな能力を与え、特定の個人情報を売却しないことを選択し、違反行為に対する民事処罰と、データ漏洩訴訟のデータ漏洩を増加させることが予想される個人訴訟権を規定する。さらに、2023年1月1日に全面的に施行されたカリフォルニアプライバシー権法案(“CPRA”)は、CCPAを大幅に改正し、カバーする企業に追加の消費者権利手続き、データ使用の制限、より高いリスクデータの新しい監査要求、および特定の敏感なデータ使用からの選択を含む追加のデータ保護義務を課した。それはまた、実質的な法規の発行を許可し、プライバシーおよび情報セキュリティの法執行を強化し、追加のコンプライアンス投資および潜在的なビジネスプロセスの変化を必要とする可能性がある新しいカリフォルニア州データ保護機関を作成した。バージニア州、コロラド州、コネチカット州、ユタ州でも同様の法律が可決され、他の州や連邦政府も同様の法律を提出し、米国がより厳しいプライバシー立法に傾いている傾向を反映している。このような法律の公布は相互衝突の要求があり、コンプライアンスを挑戦的にする可能性がある。CCPA、CPRA、または他の国内プライバシーおよびデータ保護法律の制約または影響を受ける場合, このような法的要求を遵守できなかったどんな責任も私たちの財政状況に悪影響を及ぼすかもしれない。
これらの多く、複雑でよく変化する法規を遵守することは高価で困難であり、いかなるプライバシー法またはデータセキュリティ法に準拠しない場合、または流用、紛失または他の不正処理、敏感または機密患者、消費者または他の個人情報を使用または開示することに関連する任意のセキュリティイベントまたは違反行為が、私たち、CROまたはビジネスパートナーまたは他の第三者にかかわらず、私たちの業務、財務状態、および運営結果に悪影響を及ぼす可能性がある。このような悪影響は、調査コスト、重大な罰金および処罰、補償、特殊、懲罰および法定損害賠償、訴訟、私たちのプライバシーおよび安全慣行に関する同意令、影響を受けた個人に通知、信用監視サービスおよび/または信用回復サービスまたは他の関連サービスの提供を要求すること、私たちの営業許可証に対する不利な行動、名声損害、および禁止救済を含むことができるが、これらに限定されない。
84
私たちの海外での業務はまたデータ保護部門のより厳しい審査や注目を受ける可能性がある。例えば,EU GDPRは2018年5月に発効し,欧州経済圏内の個人データの処理に厳しい要求をしている ヨーロッパ経済圏での私たちの活動を背景にしていますGDPRとEU個別加盟国の関連実施法律は、EU以外の会社がEU内の個人に商品またはサービスを提供するために、または彼らの行動(臨床試験を含む)を監視するために処理された個人データを含むEU健康データおよび他の個人データの収集および使用を管理している。GDPRを遵守しなければならない企業は、より強力なデータ保護要件の規制法執行、および不正があれば2000万ユーロまたは不適合会社の世界年収の4%までの罰金が科される可能性があるコンプライアンス義務およびリスクに直面しなければならない。上記に加えて、GDPR違反は、規制調査、名声被害、コマンド停止/変更、当社のデータの処理、実行通知、および/または評価通知(強制監査のための)をもたらす可能性があります。会社はまた、代表訴訟および他の集団訴訟タイプの訴訟(個人が傷害された)を含む民事クレームに直面する可能性があり、巨額の賠償または損害賠償責任、および関連コスト、内部資源移転および名声損害をもたらす可能性がある。
その他の要求では、“個人資料保護法”規定は、“個人資料保護法”によって規定された個人資料を、米国を含むそのような個人資料を十分に保護することが発見されていない第三国に移転し、2020年7月、EU裁判所(“EU裁判所”)は、組織がいかに合法的に個人資料を欧州経済区から米国に移転するかを制限し、国際移転の目的のためにプライバシーシールドを無効にし、標準契約条項(“SCC”)の使用にさらに制限を加えることである。2022年3月、米国とEUは無効な法規に代わる新たな規制制度を発表したが、バイデン総裁が2022年10月7日に署名した米国の信号情報活動の保障措置の強化に関する行政命令を除いて、この新たなEU-米国データプライバシーの枠組みは実施されていない。2020年7月16日のCJEU裁決の後、欧州裁判所と規制機関の裁決は国際データ転送に制限的な方法をとった。規制当局が個人資料出力メカニズムについてさらなる指針を出すのに伴い、SCCが使用できない場合、および/または法執行行動を開始すると、追加コスト、クレームおよび/または規制調査または罰金を受ける可能性があり、および/または他の方法で私たちが業務を展開している国と地域の間で個人資料を移転できない場合、これは私たちがサービスを提供する方法、地理的位置、または私たちの関連システムと業務の隔離に影響を与え、私たちの財務業績に悪影響を及ぼす可能性がある。
また、2021年1月1日から、イギリスのEU離脱の過渡期が終了した後、GDPRや同様の処罰とは異なるが、1,750万GBまでの罰金や違反会社の前財政年度の世界年収の4%を含む、1,750万GBまでの罰金や違反会社の前会計年度の世界年収の4%を含む英国のデータ保護制度(イギリスGDPRとイギリス2018年データ保護法)を遵守しなければならない。
私たちのCROまたは他の第三者サービスプロバイダが、私たちまたは私たちのサプライヤー、試験患者、調査者、および臨床サイトの従業員の個人識別および他の敏感なまたは機密情報にアクセスできることを保証することはできません。私たちはこれに責任があります。彼らは私たちが加えた契約義務に違反しないか、またはデータセキュリティの脆弱性や試みに遭遇しないことを保証できません。これは、プライバシー法律法規下での私たちの義務違反および/または逆に私たちの業務、運営結果、財務状況に悪影響を及ぼす可能性があります。私たちの契約措置と私たち自身のプライバシーと安全保障に関する措置が、第三者の処理、使用、保存、転送に関連するリスクから私たちを保護することを保証することはできません。上記のいずれも、我々の業務、財務状況、経営結果及び見通しに実質的な悪影響を及ぼす可能性がある。
私たちは私たちが後援している臨床試験から得られた健康情報のプライバシーに関する責任に直面するかもしれない。
アメリカの大多数の医療保健提供者は、私たちがそれから患者の健康情報を取得する研究機関を含み、すべてHIPAAが公布したプライバシーと安全法規の制約を受け、この法規は“経済と臨床健康情報技術法案”によって改正された。我々は現在HIPAA下の保証実体やビジネスパートナーとして行動しているとは思わないため,HIPAAの要求や処罰を直接受けることはない.しかし、HIPAAの刑事規定によると、誰でも起訴される可能性がある
85
直接または協力教唆または共謀原則の規定に基づいている。したがって、事実および状況に基づいて、私たちが知らずにHIPAAがカバーするヘルスケア提供者または研究機関から個人識別可能な健康情報を受信し、その医療提供者または研究機関が個人識別可能な健康情報の開示に関するHIPAAの要求を満たしていない場合、重大な刑事罰に直面する可能性がある。連邦貿易委員会(“FTC”)によると、HIPAAが適用されなくても、消費者の個人情報安全を保護する適切な手順をとることができず、連邦貿易委員会法に違反した不公平な行為ややり方を構成したり、商業に影響を与えたりする。連邦貿易委員会は、ある企業のデータセキュリティ対策が、消費者情報の感度および数、その業務の規模および複雑さ、および安全性の向上および脆弱性を低減するために使用できるツールのコストのために合理的かつ適切になると予想している。個人が識別可能な健康情報は敏感なデータと考えられ,より強力に保護されるべきである。
私たち、CROおよび第三者サービスプロバイダは、健康に関する情報を含む敏感な情報を受信し、維持し、これらの情報は、私たちが臨床試験全体にわたって、私たちの研究協力の過程で受け取ったものである。したがって,州法の制約を受け,個人情報が漏洩した場合に影響を受けた個人や州規制機関に通知することが要求される可能性があり,HIPAAが保護している健康情報よりも広い情報種別である。我々の米国以外の臨床試験計画は,イギリスのGDPR,GDPR,その計画を実施するEU加盟国の立法を含む国際データ保護法に関与する可能性がある。また、EUは2022年1月31日に施行されたEU臨床試験条例を採択した。この規定は臨床試験によるデータの使用に新たな義務を課し,ヨーロッパ患者に臨床試験を背景に処理された個人データにアクセスする機会を与えている。
私たちは米国以外の活動に追加的なコンプライアンス要求を加え、規定を守らないために追加的な強制執行リスクを発生させた。もし我々のCROや他の第三者請負業者が個人データをEUから米国に移す厳しいルールを遵守できなかった場合、このような協力者に対する刑事·行政制裁を招く可能性があり、これは私たちの業務に悪影響を及ぼす可能性がある。さらに、いくつかの健康プライバシー法、データ漏洩通知法、消費者保護法、および遺伝子試験法は、私たちおよび/または私たちの協力者の運営に直接適用される可能性があり、個人の健康情報を収集、使用、および伝播することに制限を加えることができる。
さらに、私たちまたは私たちの協力者が健康情報を取得した患者、および私たちとこの情報を共有する提供者は、私たちが情報を使用して開示する能力を制限する法定または契約権利を持っている可能性がある。私たちは適用されるプライバシーとデータセキュリティ法律を持続的に遵守することを保証するために、多くの資本と他の資源を費やす必要があるかもしれない。私たちがプライバシー権を侵害したり、私たちの契約義務に違反していると主張して、私たちが責任がないことが発見されても、弁護は高価で時間がかかる可能性があり、否定的な宣伝を招く可能性があり、私たちの業務を損なう可能性があります。
もし私たちまたは第三者CMO、CROまたは他の請負業者またはコンサルタントが適用される連邦、州、または外国法規のプライバシー要件を遵守できない場合、私たちは一連の規制行動の影響を受ける可能性があり、これらの行動は、私たちまたは私たちの請負業者が私たちの候補製品を開発および商業化する能力に影響を与える可能性があり、商業化できる任意の影響を受ける製品の販売を損害または阻止する可能性があり、あるいは私たちの製品の開発、商業化、マーケティングのコストと支出を大幅に増加させる可能性がある。いかなる脅威や実際の政府の法執行行動も否定的な宣伝を生む可能性があり、そうでなければ、これらの資源は私たちの業務の他の側面に使用できるように大量の資源を投入することが要求される。ソーシャルメディアをますます使用することは、責任、データセキュリティが破壊され、または名声を損なう可能性がある。上記のいずれも、当社の業務、財務状況、経営結果、見通しに重大な悪影響を及ぼす可能性があります。
私たちは環境、健康、安全法律法規の制約を受けており、私たちは環境コンプライアンスや救済活動に関連する責任と巨額の費用に直面する可能性がある。
私たちの業務は、私たちの開発、テスト、製造活動を含めて、多くの環境、健康と安全法律法規の制約を受けています。その他の事項以外に、これらの法律及び条例は、化学溶媒、ヒト細胞、発癌化合物、変異原性化合物及び生殖、実験室に毒性作用を有する化合物の制御使用、処理、放出及び処置及び登録の維持などの危険材料及び生物材料を管理する
86
手術や血液が伝播する病原体に接触する。もし私たちがこのような法律法規を守らなければ、私たちは罰金や他の制裁を受けるかもしれない。また、ある環境法は、行為が発生した場合の過ちや合法性を考慮することなく、責任を負うことができる。
我々と同様の活動をしている他社と同様に,危険や生体材料の放出や接触に関する責任を含む現在および歴史的活動に固有の環境責任リスクに直面している。環境、健康、そして安全の法律法規はもっと厳しくなっている。私たちは、将来の環境コンプライアンスや救済活動に多くの費用を発生させることを要求されるかもしれませんが、この場合、私たちの第三者メーカーの生産努力や私たちの開発努力は中断または遅延する可能性があります。
私たちと私たちの職員たちは内部と外部のコミュニケーションの手段としてソーシャルメディアツールをますます利用している。
変化するソーシャルメディア伝播ガイドを監視し、適用されるルールを遵守しようと努力していますが、私たちまたは私たちの従業員がソーシャルメディアを使用して私たちの候補製品やビジネスを伝播することは、適用要件に違反していることが発見される可能性があります。さらに、私たちの従業員は、法律法規、私たちの政策、および他の法律または契約要件に適合しない方法でソーシャルメディアを意図的または意図的に使用することができ、これは、規制法執行行動、責任、商業秘密または他の知的財産権の損失を招き、または私たち従業員、臨床試験患者、顧客、および他の人の個人情報公開をもたらす可能性がある。さらに、ソーシャルメディア上の私たちまたは私たちの候補製品に関する否定的な投稿やコメントは、私たちの名声、ブランドイメージ、および名声を深刻に損なう可能性があります。これらの事件はすべて私たちの業務、将来性、経営業績、財務状況に重大な悪影響を及ぼす可能性があり、私たちの普通株の価格に悪影響を及ぼす可能性があります。
商業化に関連するリスク
競争相手の発展は私たちの製品や技術を時代遅れにしたり、競争力に欠けたり、私たちの市場規模を縮小させるかもしれません。
私たちの業界の特徴は広範な研究と開発作業、技術の急速な発展、激しい競争と独自製品に対する高度な重視である。新製品の関連市場への参入と先進技術の出現に伴い、私たちの候補製品は激しい競争とますます激しい競争に直面すると予想される。私たちは製薬、バイオテクノロジー、専門製薬会社を含む多くの異なる源からの潜在的な競争に直面している。学術研究機関、政府機関および公共·民間機関も競争力のある製品と技術の潜在的な源である。私たちの競争相手はすでに先進的な技術や方法を持っているか、または開発する可能性があり、これは彼らに競争優位を提供するかもしれない。これらの競争相手の多くは、承認または開発されている製品を有している可能性があり、これらの製品は、候補製品に対する私たちの治療カテゴリに属する。また、これらの競争相手の多くの会社は、単独で運営しても、そのパートナーと一緒により大きな研究開発プロジェクトを運営しても、あるいは私たちよりもはるかに多くの財務資源を持っていて、以下の点でより多くの経験を持っている
もしこれらの競争相手が私たちがより安全で、より効果的で、あるいはより安い治療法やワクチンを発売する前に市場に進出すれば、私たちの候補製品が商業化が承認されれば、利益にならないかもしれないし、開発を続ける価値もないかもしれない。製薬産業の技術は急速で大きな変化を経験しており、私たちはそれが引き続きそうすることを予想している。私たちがその開発に関連したいかなる費用を回収する前に、私たちが開発したどんな化合物、製品、またはプロセスは時代遅れになったり、経済的になったりする可能性がある。私たちの候補製品の成功は
87
製品の有効性、安全性、信頼性、獲得性、タイミング、監督管理許可範囲、受容度と価格などの要素。著者らが成功した他の重要な要素は候補製品の開発速度、臨床開発と実験室テストの完成、監督管理の許可及び潜在製品の製造と販売の商業ロットを含む。
承認された治療法には大きな競争があり,新冠肺炎については,われわれの目標疾患に対する認可治療法や開発中の治療法にも競争がある。多くの承認された薬物は有名な治療法や製品であり,医師,患者,第三者支払者に広く受け入れられている。製薬とバイオテクノロジー会社は,新冠肺炎のワクチンや治療法の開発と承認,われわれの目標疾患の適応ごとの治療法の異なる段階にある。現在、いくつかのワクチンと関連するワクチン推進剤が新冠肺炎のために承認または許可されており、2つの経口抗ウイルス療法、パシクロビルおよびラグフリオを含む新冠肺炎を治療する治療法も使用可能であり、いずれもハイリスク新冠肺炎患者の緊急使用が許可されている。また、Vekluryは静脈内投与されたヌクレオチドプロドラッグであり、新冠肺炎の治療に許可された成人および小児患者であり、これらの患者は入院しているか、または入院していない場合、軽度から中等度の新冠肺炎を有し、入院および死亡を含む重篤な新冠肺炎に発展するリスクが高い。開発中の新冠肺炎療法にはGS−5245,経口ヌクレオシド前駆体薬があり,ジリッド科学社がハイリスク非入院COVID患者の無作為二重盲検プラセボ対照試験それは.EpclusaやMavyretなどの経口抗ウイルス薬を含むいくつかの薬剤がC型肝炎の治療に承認されている。私たちの候補製品は、既存の製品や現在開発されている製品と直接または間接的に競争することを目的としている。たとえ私たちの製品が承認されて商業化されても、私たちの候補製品は病院、医者、あるいは患者の市場で受け入れられないかもしれない。病院、医者、あるいは患者は結論を出すかもしれないが、私たちの製品は既存の薬物よりも安全ではなく、より効果的で、あるいはもっと魅力的である。もし私たちの候補製品がどんな理由でも市場の承認を得られなければ、私たちの収入潜在力は減少し、これは私たちの収益性に実質的な悪影響を及ぼすだろう。
私たちの多くの競争相手は私たちより多くの資本資源、強力な候補製品ルート、成熟した市場地位及び研究開発、製造、臨床前と臨床テスト、監督管理の承認及び清算とマーケティングの許可を得た製品に関する専門知識を持っている。したがって、私たちの競争相手は私たちよりも早く製品の商業化や特許や他の知的財産権保護を達成するかもしれない。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの競争相手はまた合格した臨床、監督、科学、販売、マーケティングと管理人員を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、そして著者らの計画と相互補完或いは必要な技術を獲得する上で私たちと競争している。もし私たちの競争相手が開発し商業化した製品が、私たちが開発する可能性のあるいかなる製品よりも安全で、より効果的で、副作用が少なく、より深刻ではなく、より便利で、あるいは私たちが開発する可能性のある任意の製品を時代遅れにしたり、競争力を持たなくしたりすれば、これは私たちの業務と運営に実質的な悪影響を与え、私たちのビジネス機会は減少または消滅するかもしれない。
私たちの候補製品の商業化の成功は、政府当局と健康保険会社が設立した保険範囲、十分な精算レベル、価格設定政策にある程度依存するだろう。もし私たちの候補製品が保証範囲を獲得または維持し、十分な補償を得ることができなければ、承認されれば、これらの製品を販売する能力を制限し、製品収入を創出する能力を低下させる可能性がある。
新たに承認された製品の保険カバーと精算に関する不確実性が大きい。米国では,連邦医療保険や医療補助計画のような第三者支払者,個人や政府支払者を含め,新薬や生物製品のカバー範囲を決定する上で重要な役割を果たしている。私たちが私たちの候補製品を商業化することに成功できるかどうかは、政府衛生行政部門、個人健康保険会社、その他の組織がこれらの製品と関連治療に提供する保険範囲と十分な補償にある程度依存する。政府当局と他の第三者支払人、例えば個人健康保険会社や健康維持組織は、彼らがどのような薬を支払うかを決定します
88
精算レベル。政府や個人支払者が提供する保険範囲と精算範囲は,多くの患者が治療費を負担できる鍵となっている。
第三者支払者は、医薬品およびサービスの価格にますます挑戦しており、同等の模倣薬、生物学的に似ている、またはより安い治療法が利用可能な場合、多くの第三者支払者は、特定の薬物および生物製品への保険および精算を拒否する可能性がある。第三者支払者は,我々の製品が代替可能であると考え,価格の低い製品のみを患者に精算することを提案するかもしれない。医師の監督下で管理されている製品については、このような薬物が高い価格に関連することが多いため、保険および適切な補償を得ることは特に困難である可能性がある。私たちの候補製品に対してより良い治療効果あるいはより良い管理利便性を示しても、既存の第三者療法の価格は私たちの候補製品に対する費用を制限するかもしれません。これらの支払者は、特定の製品の精算状態を拒否または撤回したり、新製品や既存市場製品の価格を低すぎるレベルに設定したりして、候補製品への投資から適切なリターンを実現できなくなる可能性がある。精算が得られない場合や限定精算だけでは、候補製品の商業化に成功できない可能性があり、候補製品に満足な財務リターンを得ることができない可能性もある。
米国では,連邦医療保険や医療補助計画のような第三者支払者,個人や政府支払者を含め,新薬や生物製品の保険や精算範囲を決定する上で重要な役割を果たしている。連邦医療保険計画は、個人や他の政府支払人がどのように新薬保険と精算政策を制定するかのモデルとしてますます多く使われている。しかし、米国の第三者支払者では、製品の保証や精算に統一された政策はない。そのため、製品の保証範囲と精算範囲は支払人によって異なる。したがって、保証範囲の決定プロセスは通常、時間がかかり、高価なプロセスであり、私たちの候補製品を使用するために単独で各支払人に科学的かつ臨床的な支援を提供する必要があるが、保証範囲と十分な補償が一致するか、または最初に得られる保証はない。一部の第三者支払者は,新たなあるいは革新的な薬物療法の保証範囲をあらかじめ承認しておく必要がある可能性があり,このような治療法を用いた医療提供者に精算する。また、精算に関する規則や条例は常に変化しており、場合によっては短時間で通知されており、これらの規則や条例が変わる可能性があると考えられる。第三者決済者が私たちの候補製品の保証範囲と精算についてどのように決定するか予測できません。
アメリカ以外では、国際業務は一般的に広範な政府価格制御と他の市場監督管理を受けており、EUと他の司法管轄区のコスト制御措置に対する日々の重視はすでに私たちの候補製品の価格設定と使用に圧力を与え続けると信じている。多くの国では、国家衛生システムの一部として、医療製品の価格は異なる価格制御メカニズムの制約を受けている。他の国は会社が自ら医療製品を価格設定することを許可しているが、会社の利益を監督してコントロールしている。追加の外国価格規制や定価規制の他の変化は、私たちの候補製品に受け取ることができる費用を制限するかもしれません。そのため、米国以外の市場では、米国に比べて候補製品の精算が減少する可能性があり、商業的に合理的な収入や利益を生み出すには不十分である可能性がある。
また,米国や海外の政府や第三者支払者が医療コストの制限や低減に力を入れており,これらの組織が新承認製品のカバー範囲や精算レベルを制限する可能性があるため,候補製品のカバーや十分な支払いを提供できない可能性がある。私たちは、管理的医療の傾向、医療機関のますます増加する影響力、立法行動(最近公布されたアイルランド共和軍を含む)による追加立法の変化により、私たちの候補製品の販売に関連した価格設定圧力に直面することを予想している。全体的に,医療コストの低下圧力は大きくなり,特に処方薬や生物製品,外科手術やその他の治療が行われている。そのため、新製品の参入にはますます高い壁が設けられている。
また、衛生部部長が2020年3月に新冠肺炎の流行開始時に公衆衛生緊急状態に入ることを発表して以来、アメリカ連邦政府は新冠肺炎の治療法の主要な買い手であった。2023年1月31日、バイデン政府は新冠肺炎PHEの中止計画を発表し、2023年5月11日から発効した。 私たちが開発に成功したとしても
89
Bemnifosbuvir COV 19候補製品については、もしその製品が政府調達要求を満たすことができなければ、有効な競争ができない可能性があり、もし政府のこのような製品に対する調達需要が供給が飽和しすぎ、患者の需要が減少し、あるいはこのような調達のための支出が減少した場合、私たちの将来の運営結果は不利な影響を受ける可能性がある。また,米国連邦政府がどのくらいの期間にわたって新冠肺炎療法の主要な買い手であるかは不明である。最近、米国連邦政府は新冠肺炎療法、ワクチン、診断検査の調達と流通を商業市場に移行する意向があるとの情報がある。この移行の時間はまだ確定しておらず、多くの条件の制約を受けており、その中には大量の増額支出があるかどうかを含み、アメリカ連邦政府の新冠肺炎対応計画に更なる資金を提供する。このようなアメリカ連邦政府調達と流通ルートから商業市場ルートへの移行の影響は多方面であり、アメリカ人の無料或いは巨額の補助金を受ける新冠肺炎療法、ワクチンと診断サービスを停止することが含まれる可能性がある。患者参入におけるこのような変化は、製品の使用に影響を与える可能性があり、私たちの候補製品を商業化することに成功し、治療分野で私たちの候補製品から満足できる財務的リターンを得る能力を制限する可能性がある。
もし私たちが単独でまたは第三者と協力して販売、マーケティング、流通能力を確立できなければ、私たちは私たちの任意の候補製品を商業化することに成功できないかもしれません。承認されれば、どんな製品収入も発生できないかもしれません。
私たちは製品を販売、マーケティング、流通する人員やインフラが限られていて、会社として候補製品を商業化した経験がありません。そのような組織を設立して維持する費用はそうする費用効果を超えるかもしれない。
承認されれば、アメリカや世界各地の他の市場で私たちの候補製品をマーケティングするために、自分の重点販売、流通、マーケティングインフラを構築することができるかもしれません。私たち自身の販売、マーケティングと流通能力を確立することは、私たちが合格者を募集し、維持し、適切に激励し、十分な販売手がかりを生成し、販売とマーケティング人員に十分な訓練を提供し、異なる地理的位置に分散した販売とマーケティングチームを効果的に管理する能力を含む大量の費用とリスクに関連する。もし私たちの内部販売、マーケティング、流通能力の開発にどんな失敗や遅延が生じた場合、どの製品の発表を延期する可能性があり、承認されれば、私たちの候補製品の商業化に悪影響を及ぼすだろう。また、販売チームを募集してマーケティング能力を確立する候補製品のビジネス発表が何らかの理由で延期または発生していない場合には、これらの商業化費用を早期または不必要に発生させる。これは費用が高いかもしれません。もし私たちが私たちの販売とマーケティング担当者を維持したり再配置できなければ、私たちの投資は損失します。
私たちの候補製品の商業化を阻害するかもしれませんがこれらに限定されません
内部販売、マーケティング、流通能力を確立しないことや、特定の国/地域でそうしないことを決定することができない場合、協力手配を求めることができるかもしれません。もし私たちが
90
協力手配は、私たちの販売は、協力者の製品に対する戦略的興味と、そのような協力者が製品を商業化することに成功する能力に大きく依存するだろう。
もし私たちが自分の販売チームを構築したり、協力関係を構築して私たちの任意の候補製品を商業化することができなければ、候補製品の潜在的な商業化を延期したり、これらの候補製品に対する販売やマーケティング活動の範囲を縮小させられたりする可能性があります。もし私たちが私たちの支出を増やして商業化活動に資金を提供することを選択すれば、私たちは追加的な資本を得る必要があり、これらの資本は受け入れ可能な条件で私たちに提供できないかもしれないし、全く得られないかもしれない。私たちは理想的な場合よりも早い段階でパートナーと手配することができ、私たちは私たちの任意の候補製品に対する権利を放棄すること、または他の方法で私たちに不利な条項に同意することを要求される可能性があり、いずれも私たちの業務、運営結果、および見通しに悪影響を及ぼす可能性がある。
もし私たちが十分な販売、マーケティング、流通能力を確立できなければ、私たち自身も第三者と協力しても、私たちの候補製品を商業化することに成功せず、利益を上げることができず、重大な追加損失が生じる可能性がある。私たちは大手製薬会社を含む多くの会社と競争し、これらの会社は広範かつ資金的で十分なマーケティングと販売業務を持っている。マーケティングおよび販売機能を実行するための内部チームまたは第三者の支援がなければ、これらのより成熟した会社との競争に成功することができないかもしれない。
また、十分な販売、マーケティング、流通能力が確立されていても、定価モデル、サプライチェーン、配信メカニズムなどにおける全体的な業界動向の進展は、私たちの予想から外れる可能性がある。もしこれらや他の業界の傾向が予想されていない方法で変化した場合、私たちは私たちの候補製品を商業化したり、利益を達成することに成功できないかもしれません。
私たちの将来の成長は私たちが外国市場に進出する能力にある程度依存するかもしれないが、そこでは追加の規制負担と他のリスクと不確定要素の影響を受けるだろう。
私たちの将来の収益性は候補品を海外市場で商業化する能力にある程度依存するかもしれませんが、私たちは第三者との協力に依存するかもしれません。私たちは現在、私たちの候補製品を海外市場で商業化するパートナーはいない。私たちは私たちの候補製品の海外市場での開発と商業化の機会を評価している。外国市場関連規制機関の規制承認を得るまで、私たちは私たちの候補製品のマーケティングや普及は許可されておらず、私たちはいかなる候補製品の規制承認も得られないかもしれない。他の国で単独の規制承認を得るためには、これらの国/地域の私たちの候補製品の安全性と有効性に関する多くの異なる規制要求、および私たちの候補製品の臨床試験や商業販売、定価、流通などの規制要求を遵守する必要があり、これらの管轄区域で成功するかどうかを予測することはできない。もし私たちの候補製品が承認され、最終的に私たちの候補製品を海外市場で商業化すれば、私たちは追加のリスクと不確実性に直面するだろう
91
私たちが承認されたどの製品の海外販売も、政府の規制、政治的、経済的不安定、貿易制限、関税変化の悪影響を受ける可能性がある。
もし私たちの候補製品が商業化されることが承認されれば、私たちは第三者と選択的に協力して、アメリカ以外のいくつかの司法管轄区域でマーケティングを行うことができるかもしれない。私たちは国際製薬業務に関連する追加のリスクに直面すると予想されていますが、これらに限定されません
組織として、私たちは以前このような分野で経験がなかった。また、EUや多くのEU加盟国や南米や東南アジアなどの他の世界地域で加えられている複雑な規制、税収、労働者、その他の法律要件を遵守する必要があるだろう。米国に本社を置くバイオテクノロジー会社の多くは、北米以外で製品をマーケティングする過程は非常に挑戦的であることを発見した。
特定の法律と政治的危険もまた外国の行動に固有のものだ。私たちが事業を展開する可能性のある国では、外国政府が民間企業を国有化する可能性があり、これにはリスクがある。特定の国や地域では、テロやこのような活動に対する反応は、アメリカでの私たちの行動よりも大きな脅威になる可能性がある。特定の国/地域の社会および文化規範は、実質的な法律および法規の遵守を要求する政策を含む、わが社の政策を遵守することを支持しない可能性がある。また、我々が業務を展開することが可能な国の全体的な経済·政治状況の変化は、私たちの財務業績や将来の成長にリスクとなる。また、商業化を実現するためには、米国以外で財務的かつ商業的に強力なパートナーを探す必要があり、これらのパートナーは、私たちが要求する高製造および法律および規制コンプライアンス基準を遵守することになり、これは私たちの財務業績にとってリスクである。私たちが世界で業務を経営するにつれて、私たちの成功は私たちがこれらのリスクと他の関連リスクを予測し、効果的に管理する能力にある程度依存するだろう。これらの要素や他の国際業務に関連する要因の結果が、私たちの業務、財務状況、または経営結果に悪影響を与えないことは保証されません。
いくつかの国、特にEUでは、処方薬の価格設定は政府によって統制されている。これらの国では,薬品発売許可を受けた後,政府当局との定価交渉にはかなりの時間がかかる可能性がある。一部の国で精算や定価の承認を得るためには、臨床試験を行う必要があるかもしれません。私たちの候補製品のコスト効果を他の利用可能な治療法と比較します。もし私たちの製品が精算を受けられない場合、あるいは範囲や金額が制限されている場合、あるいは定価が満足できないレベルに設定されている場合、私たちの業務は損害を受ける可能性があり、実質的かもしれません。
私たちの潜在的な製品に対する責任訴訟は、私たちが重大な責任を招き、私たちが開発する可能性のある任意の製品の商業化を制限するかもしれません。
92
臨床試験で私たちの候補製品を使用して市場の承認を得たどの製品を販売しても製品責任クレームのリスクに直面させます。消費者、医療提供者、製薬会社、または他の販売または他の方法で私たちの製品に接触した人は、私たちに製品責任を請求するかもしれません。予期せぬ悪影響を持つ製品に基づく集団訴訟では,多額の判決が下されることがある.もし私たちが製品責任クレームを正当化することに成功できなければ、私たちは大量の責任とコストを発生するかもしれないが、これらは保険範囲内ではないかもしれない。さらに、製品責任クレームは、是非曲直や最終結果にかかわらず、以下のようになる可能性がある
潜在的な製品責任クレームを防止するために、許容可能なコストで十分な製品責任保険を獲得または保持することができず、私たち単独または会社パートナーと開発した製品の商業化を阻止または阻害する可能性がある。臨床試験保険がありますが、私たちの保険書も各種の排除があります。私たちは製品責任クレームの影響を受けるかもしれませんが、私たちは保険範囲を受けていません。私たちは私たちの保険範囲の制限を超えたり、私たちの保険カバー範囲内でない裁判所の裁決または和解合意で達成された任意の金額を支払う必要があるかもしれません。私たちはこれらの金額を支払うために十分な資本を持っていないか、または得ることができません。私たちが未来の会社のパートナーと合意しても、私たちは損害賠償を受ける権利があります。何かクレームがあれば、この賠償は利用できないか十分かもしれません。
製造に関するリスクと第三者への依存は
私たちは引き続き第三者に依存して私たちの研究計画、臨床前研究、臨床試験のための材料を製造する予定で、私たちはこれらの当事者のいずれとも長期契約をしていません。このような第三者への依存は、私たちが十分な数のそのような材料や、私たちが開発して商業化する可能性のある任意の候補製品を持っていない、またはそのような供給が許容可能なコストで私たちに提供できなくなり、これが私たちの開発や商業化努力を延期、阻止、または損害する可能性があるというリスクを増加させる。
我々は第三者が我々の臨床試験と研究活動、臨床前と臨床開発のための材料を開発することに依存し、引き続き依存することが予想される。もし私たちのすべての候補製品が発売承認されれば、私たちは第三者に依存して商業生産を行う予定だ。私たちは現在、臨床前および臨床試験材料を提供するために使用されているいかなる第三者メーカーと長期合意を達成しておらず、私たちは注文した方法で必要な材料を購入している。その中のいくつかのメーカーは私たちの生産に重要であり、これらのメーカーを失ったり、受け入れられるコスト或いは品質で数量を得ることができず、著者らの臨床前研究或いは臨床試験を適時に行う能力を遅延、阻止或いは弱める可能性があり、そして著者らの開発と商業化努力に実質的な悪影響を与える。
93
私たちは第三者メーカーに依存して上場承認された候補製品に商業供給を提供したい(あれば)。
私たちは第三者製造業者と必要な合意を維持または確立することができないかもしれないし、受け入れ可能な条項でそうすることができないかもしれない。第三者製造業者と合意できても、第三者製造業者に依存することは、限定されないが、追加のリスクをもたらす
Ruzasvirについては、私たちの活性薬物成分の独占サプライヤーは中国に位置し、私たちのすべての候補製品については、ベノブビルを含み、それぞれの製造技術の規制出発材料のすべてのサプライヤーは中国に位置している。私たちはそのような第三者製造業者を引き続き使用する予定だ。私たちの中国のメーカーのいかなる生産中断または私たちの需要を満たすのに十分な数量の製品を生産することができなくても、現在の新冠肺炎の疫病、自然災害、その他の流行病、貿易中断、あるいはその他の原因によっても、私たちの日常的な業務を運営する能力を弱める可能性があり、そして私たちの候補製品を研究開発し続ける能力を弱める可能性がある。例えば、“ウイグル族強制労働保護法”は、貨物が強制労働生産を使用していないことが証明できない限り、中国のある新疆地域からの輸入を禁止しており、この立法はグローバルサプライチェーンに悪影響を与え、私たちの業務や運営結果に悪影響を及ぼす可能性がある。
私たちは生産過程のすべての側面を完全にコントロールすることができず、私たちの契約製造パートナーに依存してcGMP法規と同様の法規要件を遵守して、活性医薬物質と完成品を生産する。第三者メーカーは、米国以外のcGMP法規や同様の規制要件を遵守できない可能性がある。もし私たちの契約製造業者が私たちの規格およびFDAまたは他の機関の厳格な規制要件に適合した材料を生産することに成功しなければ、彼らはその製造施設の許可を確保および/または維持することができないだろう。しかも、私たちの契約製造業者が十分な品質管理、品質保証と
94
合格者。FDAまたは同様の外国規制機関が、これらの施設が私たちの候補製品を生産するために許可されていない場合、または将来的にそのような許可を撤回する場合、私たちは代替製造施設を探す必要があるかもしれません。これは、私たちの候補製品を開発、獲得、またはマーケティングする能力に深刻な影響を与えます。私たちまたは私たちの第三者製造業者が適用された法規に従わないことは、罰金、禁止、民事処罰、遅延、承認の一時停止または撤回、許可証の取り消し、候補製品または薬物の差し押さえまたはリコール、運営制限、および刑事起訴を引き起こす可能性があり、いずれも候補製品または薬物の供給に重大な悪影響を与え、私たちの業務および運営結果を損なう可能性があります。
私たちの第三者メーカーは十分な品質と数量で私たちの候補製品の生産規模を拡大することに成功できないかもしれません。これは私たちの候補製品の臨床進歩と商業化を損なう可能性があります。
私たちの候補製品を臨床試験し、任意の承認された製品を商業化するために、私たちのメーカーはこれらの製品を大量に生産する必要があります。しかし、彼らは、私たちの任意の候補製品または製品の製造能力をタイムリーに、または費用効果のある方法で成功的に向上させることができないかもしれない、または全くできないかもしれない。また,拡張活動中に品質の問題が生じる可能性がある.もし私たちまたはどのメーカーも十分な品質と数量で私たちの候補製品の生産規模を成功的に拡大することができなければ、これらの候補製品の開発、テスト、臨床試験は延期または不可能になる可能性があり、いかなる最終製品の規制承認や商業発売は延期または獲得できない可能性があり、これは私たちの業務を深刻に損なう可能性がある。供給源は時々中断する可能性があり、中断すれば、私たちのメーカーが必要な規模で私たちの候補製品を生産する能力を破壊するだろう。もし私たちの候補製品の臨床的または商業的供給需要を満たすことができなければ、あるいは商業的に合理的な条項でそうすれば、私たちの候補製品を開発し、私たちの製品を商業化することに成功することができないかもしれない。
私たちは候補製品で使用されるすべてのコンポーネントに複数の供給源を提供していません。長期供給契約もありません。私たちのいくつかのサプライヤーは私たちの生産に重要です。もし私たちがキーサプライヤーを失ったら、私たちが候補製品開発を完成させる能力に実質的な悪影響を及ぼすかもしれない。もし私たちの候補製品が規制部門の承認を得たら、私たちはそれを商業化するためにその部品の供給を拡大する必要があるだろう。
私たちはベノホブビルまたはルザスビルまたは私たちの任意の他の候補製品を生産するために使用されるすべてのコンポーネントに複数のソースを提供していません。Ruzasvirについては、私たちの活性薬物成分の独占サプライヤーは中国に位置し、私たちのすべての候補製品については、ベノブビルを含み、それぞれの製造技術の規制出発材料のすべてのサプライヤーは中国に位置している。私たちは私たちのどの部品供給業者とも長期供給協定を持っていない。私たちは私たちの候補製品のための追加的な供給源を作ることができないかもしれないし、受け入れられる条項でそうすることができないかもしれない。製造サプライヤーはcGMP品質と監督管理要求及び類似の監督管理要求を遵守しなければならず、著者らの候補製品に関連する製造、テスト、品質管理と記録保存を含み、そして適用監督機関の持続的な検査を受ける必要がある。製造業供給者たちはまた地域、州、連邦法規と許可要求によって制限されている。もし私たちのどのサプライヤーもすべての適用された法規と要求を遵守できなかった場合、長時間の遅延と供給中断を招く可能性があります。
私たちの候補製品の部品サプライヤーの数は限られています。必要であれば、他のサプライヤーから供給を受ける必要があれば、私たちは本当にあれば、商業的に合理的な条件でこれらの供給を得ることができないかもしれない。他の会社と協力するために私たちの製造プロセスを再設計するには多くの時間と費用が必要かもしれませんが、再設計プロセスは比較可能な研究や接続研究のようなより多くの研究の必要性を引き起こす可能性があります。さらに、私たちのいくつかのサプライヤーは私たちの生産に重要であり、もしこれらのサプライヤーが私たちの競争相手に奪われたり、他の方法で流失したりすれば、私たちの開発と商業化努力に実質的な悪影響を及ぼすだろう。
どの上場承認の一部としても、規制当局は製品を承認する前に成功しなければならない検査を行う。もし製造業者がこのような規制検査を成功的に完了できなかったら、遅延を招くだろう。承認されたサプライヤーの供給が中断されれば、商業供給が深刻に中断される可能性がある。代替サプライヤーは以下の方法で資格を得る必要がある
95
秘密協定修正案や追加文書は、これ以上の遅延を招く可能性がある。新たなサプライヤーに依存して商業生産を行う場合、FDAや米国以外の他の規制機関も追加的な研究を行う必要がある可能性がある。サプライヤーの交換は大量のコストを伴う可能性があり、私たちに必要な臨床およびビジネススケジュールの遅延を招く可能性がある。
もし私たちが合理的な価格で必要な供給を得ることができない場合、候補製品開発を達成する能力に実質的な悪影響を与える可能性があり、あるいは規制部門の候補製品の承認を受けた場合、私たちがそれを商業化する能力に大きな悪影響を及ぼす可能性がある。
私たちは第三者に依存して臨床前研究と臨床試験を行う。もし第三者がGCPによって適時に臨床試験を行うことができなかった場合、私たちが監督部門の私たちの候補製品に対する承認或いは商業化を求めたり、獲得したりすることを遅延或いは妨害する可能性がある。
我々は第三者に依存して我々の第三段階日の出-3臨床試験、著者らの臨床前研究とその他の臨床試験の肝心な方面を行い、私たちは第三者に依存して私たちの候補製品の未来の臨床試験と臨床前研究を行う予定である。具体的には,我々は使用·依存しており,医療機関,臨床研究者,CRO,コンサルタントを継続して使用·依存し,われわれの臨床案や法規の要求に基づいて臨床試験を行う予定である。これらのCRO、調査者、その他の第三者はこれらの実験の進行とスケジュール、その後のデータ収集と分析に重要な役割を果たしている。我々は我々の第三者請負業者の活動を管理する合意があるが,彼らの実際の表現への影響は限られている.しかし、私たちは私たちのすべての臨床試験が適用された方案と法律、法規、科学的基準に従って行われていることを確実にする責任があり、私たちのCROと他の第三者への依存は私たちの規制責任を免除しない。私たちと私たちのCROはGCP要求を遵守しなければならない。これらの要求は、FDAと同様の外国規制機関が私たちの臨床開発におけるすべての候補製品に対して実行する法規とガイドラインである。規制当局は,試験スポンサー,主要調査者,試験地点を定期的に検査することでこれらのGCPを実行している。もし私たちまたは私たちの任意のCROまたは試験サイトが適用されたGCPに従わなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられるかもしれませんが、FDAまたは同様の外国の規制機関は、私たちのマーケティング申請を承認する前に追加の臨床試験を行うことを要求するかもしれません。特定の規制機関で検査した後、保証することはできません, このような監督機関は私たちの任意の臨床試験或いは研究活動がGCP規定に符合することを確定する。また,われわれの臨床試験では米国以外のcGMPや類似法規の下で生産された製品を使用しなければならない。私たちがこれらの規定を遵守しないことは、私たちが臨床試験を繰り返す必要があるかもしれないが、これは規制部門の承認過程を遅らせるだろう。
我々の臨床試験や臨床前研究を行ういかなる第三者も我々の従業員ではなく,また,我々がそのような第三者と合意して我々に提供してくれた救済措置を除いて,いかなるCRO,研究者,あるいは他の第三者がこのような試験を行うのに十分な時間と資源を投入したり,契約に応じて履行するかを保証することはできない。もしこれらの第三者のいずれかが予想される最終期限内に達成できなかった場合、私たちの臨床方案を遵守したり、規制要求を満たしたり、あるいは他の方法で不合格を示した場合、私たちの臨床試験は延長、延期、または終了される可能性がある。また、私たちと契約した多くの第三者も、私たちの競争相手を含む他の商業実体と関係があるかもしれません。彼らはまた、これらの実体のための臨床試験や他の薬物開発活動を行っているかもしれません。これは、私たちの競争地位を損なう可能性があります。さらに、私たちの臨床試験の首席研究者は時々私たちの科学顧問または顧問を務めることができ、そのようなサービスに関連する現金および現金等価物または株式補償を得ることができる。これらの関係および任意の関連賠償が知覚的または実際的な利益の衝突をもたらし、またはFDAまたは外国の規制当局が、財務関係が試験の解釈に影響を及ぼす可能性があると結論した場合、適用される臨床試験場所で生成されたデータの完全性が問われる可能性があり、臨床試験自体の効用が脅かされる可能性があり、これにより、FDAまたは外国規制機関に提出された任意のNDAまたは同様の申請が遅延または拒否される可能性がある。そのような遅延や拒否は私たちが候補製品を商業化することを防ぐことができる。
もし治癒されていない重大な違約が発生したら、私たちのCROは私たちとの合意を終わらせる権利がある。また,われわれの臨床試験に参加した被験者の安全性を合理的に証明できれば,われわれのいくつかのCROやほとんどの臨床試験地点ではわれわれとの合意を終了する権利がある。
96
もし私たちがこのような第三者との任意の関係が終わったら、私たちは他の第三者と合意したり、商業的に合理的な条項でそうすることができないかもしれない。CRO、調査者、および他の第三者の交換または追加は、管理時間および重点を必要とする追加のコストに関連しています。しかも、新しいCROが仕事を始める時、自然な過渡期がある。したがって,遅延が生じ,必要な臨床開発スケジュールを満たす能力に実質的な影響を与える可能性がある。CRO、調査者、その他の第三者との関係を慎重に処理しているにもかかわらず、将来的に挑戦や遅延に遭遇しない保証はなく、これらの遅延や挑戦が私たちの業務、財務状況、および見通しに実質的な悪影響を与えないことを保証することはできない。
私たちは第三者と協力して私たちの候補者を開発して商業化するかもしれない。私たちは協力関係を構築し、維持することに成功できないかもしれません。これは、私たちの候補製品の開発と商業化に成功する能力を大きく制限するかもしれません。
私たちは私たちの候補製品の開発と商業化のために協力関係を求めるかもしれない。もし私たちがどの第三者と任意の協力計画を達成すれば、私たちの協力者の投入を共有または制限して、彼らと開発する可能性のある任意の候補製品の開発または潜在的に商業化された資源の数と時間を求めることができるかもしれない。私たちがどんな協力からも製品収入を作る能力は、私たちの協力者が彼らに割り当てられた機能を成功的に実行する能力に依存するだろう。私たちは私たちが参加したどんな協力も成功するかどうかを予測できない。
私たちの候補製品の協力には多くのリスクが含まれている
97
例えば、2022年2月に羅氏許可プロトコルは終了し、この合意に基づいて、米国以外でベンフォブビルを開発するいくつかの権利と、ベンフォブビルを商業化するいくつかの権利とを羅氏に独占的に付与した。そのため,ロ氏社がベノジブビルの開発と生産において行っているすべての活動は,臨床試験材料の世界的な生産と臨床試験に必要な何らかの操作を含めて,ほとんど停止している。基本的に、これらのすべての活動は、Bemnifosbuvir COV 19候補製品を世界的に開発し続けるために必要であり、私たちは今、このような活動の進行と費用を完全に担当しています。
適切な協力を求めることで、私たちは激しい競争に直面するかもしれない。バイオテクノロジーと製薬会社間の業務合併は潜在的な協力者の数を減少させる。また、交渉過程は時間も複雑であり、私たちはタイムリーで、受け入れ可能な条件で協力を交渉することができず、交渉することさえできないかもしれない。それができなければ、私たちが協力を求めている候補製品の開発を削減したり、潜在的な商業化を延期したり、販売やマーケティング活動の範囲を縮小したり、私たちの支出を増やしたり、自分で開発や商業化活動を行ったりしなければならないかもしれません
98
料金です。もし私たちが私たちの支出を増やし、私たち自身の開発や商業化活動に資金を提供することを選択すれば、私たちは追加の資本を得る必要があるかもしれないし、これらの資本は受け入れ可能な条件で私たちに提供できないかもしれないし、全く得られないかもしれない。もし私たちが十分な資金がなければ、私たちは候補製品をさらに開発したり、それらを市場に出して製品収入を生成することができないかもしれない。
もし私たちが任意の候補製品を開発して商業化するために協力すれば、私たちまたは私たちの協力者が合意によって付与された権利を行使しないことを選択した場合、または私たちまたは私たちの協力者が候補製品を既存の運営にうまく統合できない場合、私たちはこのような取引の利点を達成できないかもしれない。さらに、もし私たちと私たちの任意の協力者との合意が終了すれば、私たちの協力者が私たちに権限を与えた技術と知的財産権へのアクセスが制限されたり、完全に終了したりする可能性があり、これは、私たちが協力者の技術や知的財産権を利用して私たちの候補製品を開発し続けることを延期するか、あるいはこれらの候補製品の開発を完全に停止することを要求するかもしれない。例えば、メルク社とのライセンス契約が終了すれば、RuzasvirとC型肝炎を治療する私たちの主要な候補製品bemnifosbuvirとの共同開発、製造、商業化を停止することを要求されます。私たちがメルク社と別の合意を締結することができなければ、条項は私たちに不利になるかもしれません。私たちはまた、適切な代替協力者を見つけたり、新しい協力者を誘致することが難しいことを発見するかもしれませんし、私たちの開発計画が延期されるかもしれませんし、商業·金融界での私たちのイメージが悪影響を受ける可能性があります。いずれの協力者も、製品開発、規制承認、商業化に関連する多くのリスクに直面する可能性があり、これらのリスクは本“リスク要因”の節で説明されているが、私たちの協力者にどんな負の影響も私たちに悪影響を及ぼす可能性がある。
協力者および私たちが依存する他の人によって提供される独立して確認されていないデータは、虚偽、誤った、または不完全であることが証明される可能性がある。
私たちはCRO、科学者と協力者のような第三者サプライヤーに依存して、私たちの計画、臨床前研究或いは臨床試験と私たちの業務に関連する重要なデータとその他の情報を提供してくれます。これらの第三者が不正確、誤ったデータ、または不完全なデータを提供する場合、私たちのビジネス、将来性、および運営結果は、重大な悪影響を受ける可能性がある。
私たちの従業員と独立請負業者は、首席調査者、CRO、コンサルタント、サプライヤー、および私たちが従事する可能性のある研究、開発、監督、製造、品質保証および他の薬品機能および商業化に関連する任意の第三者を含み、規制基準および要求を遵守しないことを含む不適切な行為または他の不適切な活動に従事する可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。
最高調査者、CRO、コンサルタント、サプライヤー、および研究、開発、規制、製造、品質保証および他の医薬品機能および商業化に関連する任意の第三者の不正行為を含む可能性のある研究、開発、規制、および他の医薬品機能および商業化に関連する任意の第三者の不正行為を含む可能性があり、これらの行為または不正な活動は、(I)FDAおよび他の同様の規制機関の法律および法規、これらの機関に真実、完全かつ正確な情報を報告することを要求する法律、(Ii)製造基準;(Iii)データプライバシー、セキュリティ、腐敗防止、詐欺および乱用、および他の医療に関する法律、または(Iv)真の、完全かつ正確な財務情報およびデータを報告することを要求する法律。具体的には、医療業界の販売、マーケティング、および商業配置は、詐欺、不正行為、リベート、自己取引、および他の乱用行為を防止するための広範な法律法規によって制限されている。これらの法律法規は、広範な価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネス計画を制限または禁止する可能性があります。これらの法律制約を受けた活動はまた、臨床前研究或いは臨床試験過程で得られた情報の不適切な使用或いは虚偽陳述に関連する可能性があり、臨床前研究或いは臨床試験中に詐欺性データを作成し、或いは薬品を不法に流用し、これは規制制裁を招き、そして著者らの名声に深刻な損害を与える可能性がある。従業員や他の第三者の不正行為を識別して阻止することは常に可能ではない, このような活動を検出し防止するための予防措置は、未知または未管理のリスクまたは損失を効果的に制御することができないか、またはそのような法律または法規を遵守できないことによる政府の調査または他の行動または訴訟から私たちを保護することができない可能性がある。さらに、誰かや政府がこのような詐欺や他の不正行為を告発する可能性があり、起こらなくてもリスクに直面している。もし私たちにこのような行動を取ったら私たちは
99
私たちの権利を自己弁護または維持することに成功し、これらの行動は、重大な民事、刑事および行政処罰、損害賠償、金銭、罰金、引き渡し、Medicare、Medicaid、他の米国連邦医療計画または他の司法管轄区域の医療計画に参加することから除外される可能性があり、違反容疑、個人監禁、他の制裁、契約損害、名声損害、利益減少および将来収益の減少、および私たちの業務を削減するための重大な民事、刑事と行政処罰、損害賠償、罰金、引き渡しを含む、私たちの業務および運営結果に重大な影響を与える可能性がある。
もし私たちのCMOとCROが傷害や適用法違反の方法で危険や生体材料を使用すれば、損害賠償責任を負う可能性があります。
我々の研究と開発活動は,我々のCMOが使用する化学材料および我々のCROが使用する医療や生体材料を含む潜在的危険物質の制御使用に関するものである。我々のCMOとCROはアメリカとその運営所の国の連邦、州と地方の法律法規の制約を受け、医療と危険材料の使用、製造、貯蔵、処理と処置を管理する。我々のCMOとCROがこれらの材料を使用,処理,貯蔵,処分する手順は法律で規定された基準に適合していると信じているが,医療や危険材料による汚染や傷害のリスクを完全に除去することはできない。このような汚染や傷害のため、私たちは責任を負うかもしれないし、地方、都市、州、または連邦当局はこれらの材料の使用を制限し、私たちの業務運営を中断するかもしれない。事故が発生すると、私たちは損害賠償責任や罰金を要求されるかもしれません。責任は私たちの資源範囲を超えるかもしれません。一般に,医療,生物,危険材料の不適切な処理による責任を保険にかけることはない。適用される環境法律法規の遵守はコストが高く、現在または未来の環境法規は私たちの研究、開発、生産努力を損なう可能性があり、それによって私たちの業務、将来性、財務状況、または運営結果を損なう可能性がある。
知的財産権に関するリスク
もし私たちの技術や候補製品について私たちの知的財産権を獲得、維持、実行、十分に保護できない場合、あるいは取得した特許や他の知的財産権保護の範囲が広くなければ、私たちの競争相手は私たちと似ているか同じ技術や製品を開発し、商業化する可能性があり、私たちの技術や候補製品の開発と商業化に成功する能力は不利な影響を受ける可能性がある。
私たちは、特許、商業秘密保護、および秘密協定の組み合わせによって私たちの知的財産権を保護し、他社がAT-511(bemnifosbuvirの無料基地)、bemnifosbuvir、および私たちが許可した化合物ruzasvirおよびそれらの使用または製造、または私たちの任意の他の候補パイプライン製品および任意の未来の候補製品の複製を防止する。私たちの成功は私たちがアメリカや他の国でこのような候補製品の特許保護を獲得して維持する能力があるかどうかに大きくかかっている。
特許訴訟プロセスは高価で、時間がかかり、複雑であり、私たちは必要または望ましいすべての特許または特許出願を合理的なコストまたはタイムリーに提出、起訴、維持、強制執行または許可することができないかもしれない。私たちは、私たちの従業員、CRO、コンサルタント、科学コンサルタント、および他の請負業者などと、私たちの研究開発成果の秘密または特許可能な当事者と秘密協定を締結する権利がありますが、これらの当事者のいずれかは、合意に違反し、特許出願を提出する前にこのような成果を開示し、特許保護を求める能力を危険にさらす可能性があります。また、科学文献で発見された出版物は、実際の発見よりも遅れがちであり、米国および他の管轄区域の特許出願は、通常、出願後18ヶ月後に発表され、一部の特許出願は、発表前にそうであった。したがって、私たちは、私たちが私たちの特許または係属中の特許出願で主張された最初の発明であるか、または発明または候補製品に関連する任意の特許出願を最初に提出することを決定することができない。また、規制承認に遅延が生じた場合、特許保護の下で候補製品を販売する期間が短縮される可能性があります。
製薬分野の特許実力は複雑な法律,事実,科学的問題に関連しており,不確実である可能性がある。特許保護を受ける前に、私たちの研究開発成果で特許を申請できることを確認できない可能性があります。私たちが持っている特許出願
100
発行された特許声明は、米国または他の国/地域での私たちの候補製品をカバーすることにならないかもしれない。我々の特許および特許出願に関連するすべての潜在的に関連する既存技術が発見されていることは保証されず、これらの技術は、特許を無効にするか、または係属中の特許出願から特許が発行されることを阻止する可能性がある。特許が確実に発行されても、これらの特許が私たちの候補製品をカバーしていても、第三者は、そのような特許の発明性、所有権、有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、そのような特許を縮小または失効させるか、または実行不可能と認定される可能性がある。私たちの未解決および将来の特許出願は、私たちの技術または候補製品を保護する特許の発行、または他社が競争技術および候補製品を商業化することを効果的に阻止する特許をもたらすことができないかもしれません。また、私たちが提出したいかなる米国臨時特許出願も、関連する臨時特許出願を提出してから12ヶ月以内に非臨時特許出願を提出しない限り、発行された特許になる資格がない。もし私たちがいかなる非臨時特許出願を直ちに提出しない場合、私たちは臨時特許出願の優先日と、臨時特許出願に開示された発明に関する任意の特許保護を失う可能性がある。
また,それらが挑戦されていなくても,我々の特許や特許出願は,我々の知的財産権を十分に保護し,我々の候補製品に排他性を提供したり,他の人が我々の主張をめぐる設計を阻止したりすることができない可能性がある.さらに、第三者が我々の特許を侵害することなく、類似または代替製品または方法を作成して同様の結果を達成しないことは保証されない。これらの結果のいずれも、第三者の競争を阻止する能力を弱める可能性があり、第三者競争は私たちの業務に悪影響を及ぼす可能性がある。
もし私たちが持っている私たちの計画または候補製品に関連する特許出願が発表されなかった場合、もし私たちが現在または未来に発表された特許の保護の広さや強度が脅かされている場合、またはそれらが私たちの候補製品に意味のある排他性を提供できなかった場合、会社が私たちと協力して候補製品を開発することを阻止したり、現在または未来の候補製品を商業化する能力を脅かすかもしれない。私たちは最近私たちの候補製品に関するいくつかの特許出願を提出した。私たちは、もしあれば、発行された特許、任意のそのような特許の広さ、または発行された特許が無効であることが発見されるかどうか、強制的に実行できないか、または第三者によって脅かされるかどうかをもたらす保証を提供することはできない。これらの特許または私たちが持っている任意の他の特許への成功に対するいかなる反対も、私たちが開発する可能性のある任意の候補製品を商業化することに成功するために必要な権利を奪う可能性がある。
特許の発行は,その発明性,所有権,範囲,有効性または実行可能性に関する決定的な要素ではなく,我々の特許は米国や海外の裁判所または特許庁で挑戦される可能性がある。さらに、特許の発行は、第三者が(承認された場合)私たちの候補製品の販売を阻止するか、または私たち自身の特許技術を実践することを阻止する可能性があるので、特許発明を実践する権利を与えてくれない。
2011年の“Leahy-Smith America発明法”(“Leahy-Smith Act”)を含む米国の広範囲な特許改革立法は、私たちの知的財産権の実力や実行可能な不確実性を増加させ、それを保護するコストを増加させる可能性がある。Leahy-Smith法案は、特許出願起訴方式および特許訴訟に影響を与える条項を含む米国特許法の複数の重大な改正を含む。“ライシー·スミス法案”(Leahy-Smith Act)によると、同一発明を主張する異なる方が2つ以上の特許出願を提出した場合、米国は、どちらが特許を付与すべきかを決定するために“先発明”から“先出願”制度に移行する。これは、私たちが発明から特許出願を提出するまでの間に迅速に進み、勤勉に特許出願を提出することを要求するだろうが、場合によっては、私たちが発明に関する特許出願を迅速に提出または起訴することを阻止する可能性がある。ライシー·スミス法案はまた、既存技術に適合した開示範囲を拡大した。さらに、第三者が2013年3月16日に“Leahy-Smith Act”適用条項が発効する前に特許出願を提出した場合、第三者は、それが私たちの特許請求の範囲に含まれる任意の主題の第1の発明であるかどうかを決定するために、米国で介入手続きを開始することができる。第三者が事前に発行した既存技術を米国特許商標局(“USPTO”)に提出する必要もあるかもしれない。
ライシー·スミス法案は、米国が発行した特許に挑戦するための新たなプログラムを初めて作成し、付与後審査、当事者間の審査、派生プログラムを含み、これらのプログラムはUSPTOで行われた対抗プログラムであり、一部の第三者はこれらのプログラムを利用してきた
101
競争相手が発行した特許の一部または全部の権利要件を取り消します。優先日が2013年3月16日以降の特許については,我々のすべての特許出願があり,第三者は特許発表後9ヶ月の窓口でライセンス後審査出願を提出することができる。特許が2013年3月16日までに提出された場合は,特許発表後ただちに当事者間の審査出願を提出することができる。優先権日が2013年3月16日以降の特許については、ライセンス提出後の審査出願の9ヶ月の期限が満了した後に当事者間審査出願を提出することができる。贈与後の再審手続きは任意の疑問理由に基づいて提起することができるが、当事者間の再審手続きは公表された既存技術に基づいて疑問を提起することしかできない。米国特許商標局のこれらの対抗性訴訟は、米国特許の有効性を推定することなく、米国連邦裁判所の訴訟において特許主張を審査することを含む。USPTOは2018年11月13日に施行された最終規則を発表し、現在使用されている米国連邦裁判所が現在使用している同じ請求項構築基準を使用して、USPTO訴訟における特許請求の範囲を解釈することを発表した。これは、使用される言葉の簡単で一般的な意味である。もし私たちの任意の特許がこのようなUSPTO訴訟で第三者の挑戦を受けた場合、私たちがその特許を成功裏に守ることは保証されず、これは、独占特許権の喪失を含む挑戦された特許権を失うことになり、あるいは特許主張が縮小され、無効または実行できなくなり、これは、他の人が類似または商業化または同じ技術および製品を使用することを阻止する能力を制限するか、または私たちの技術および候補製品の特許保護期間を制限する可能性がある。このような手続きは大きなコストを招く可能性があり、私たちの科学者と経営陣に時間がかかる必要があります, 最終的な結果が私たちに有利であっても。
以上のような理由により、我々の特許権の発行、範囲、有効性、実行可能性、商業価値には大きな不確実性が存在し、これは私たちの業務、財務状況、経営結果、将来性に重大な悪影響を及ぼす可能性がある。
知的財産権侵害、流用、または他の侵害行為に対する第三者のクレームは、大量のコストを招いたり、私たちの開発と商業化努力を阻害したり、延期したりする可能性がある。
私たちのビジネスの成功は、私たちが実際の侵害、流用、または他の方法で第三者の特許および他の独占権を侵害することを避けることにある程度かかっている。米国国内外では、製薬業には、特許侵害訴訟、介入、異議、再審、認可後、各当事者間の審査手続き、米国特許商標局および外国司法管轄区における同様の訴訟、例えば欧州特許庁での異議など、特許および他の知的財産権に関連する訴訟が大量にある。我々が開発候補を求めている分野には,第三者が所有する米国や外国が発行した特許や係属中の特許出願が多く存在する.製薬業を含む知的財産権に依存する業界の多くは、競争相手に対する優位性を得るための手段として知的財産権訴訟を利用している。製薬業界の拡張や特許の発行、および上場企業としてより大きな知名度と市場露出率を得ることにより、当社の候補製品が第三者特許権侵害のクレームを受けるリスクが高まる可能性がある。一部のクレーム者は私たちよりもはるかに多くの資源を持っている可能性があり、私たちよりも複雑な知的財産訴訟費用をより大きく長時間受けることができるかもしれない。また,特許権の強制執行による特許料の抽出と和解にのみ重点を置いた特許持株会社が我々を対象とする可能性がある。
第三者は私たちが許可されていない状況で彼らのノウハウを使用していると主張するかもしれない。物質の組成、薬物送達、製造方法、または治療方法に関する当社の候補製品の使用または製造に関連する第三者特許または特許出願が存在する可能性がある。私たちは、私たちの技術、製品、成分、およびその使用が侵害されないか、流用しないか、または他の方法で第三者特許または他の知的財産権を侵害することを保証することはできません。特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、我々の候補製品が発行された特許を侵害する可能性がある。さらに、第三者は将来的に特許を取得し、私たちの技術を使ってこれらの特許を侵害したと主張するかもしれない。発表された係属中の特許出願は、後で特許請求を提出することができ、私たちの候補製品または私たちの候補製品の使用をカバーするか、またはいくつかの制限を受ける可能性がある。特許発行後、特許請求の範囲は、依然として法律解釈、特許における書面開示、および特許の起訴履歴に依存する。特許または保留出願の関連性または範囲の説明は正しくないかもしれませんが、これは私たちの候補製品をマーケティングする能力に悪影響を及ぼすかもしれません。成功に挑戦するために
102
連邦裁判所における米国特許の有効性、私たちは有効性の推定を克服しなければならない。これは重い負担であるため、このような米国特許主張の無効について明確で納得できる証拠を提出することを要求するため、司法管轄権を有する裁判所が、このような米国特許の主張の無効を宣言する保証はない。管轄権のある裁判所が任意の第三者特許を持ち、私たちの任意の候補製品の組成、任意の候補製品の製造プロセス、または任意の候補製品の使用方法をカバーする場合、任意のそのような特許の所有者は、適用特許に基づいてライセンスを取得し、これらの特許が商業的に合理的な条項で提供されないか、またはその特許が満了する前に、その候補製品を商業化する能力を阻止することができるかもしれない。
提訴や論争のある訴訟手続きの法的敷居が低いため,勝訴確率の低い訴訟や訴訟手続きが提起される可能性があり,弁護には大量の資源が必要である.訴訟と論争のある訴訟手続きはまた高価で時間がかかるかもしれないが、私たちのこのような訴訟手続きの中の相手は私たちよりも多くの資源を投入してこのような法的行動を起訴する能力があるかもしれない。私たちの製品候補が商業化に近づいていれば、上場企業に関するより大きな知名度を得るにつれて、このような訴訟や訴訟に巻き込まれるリスクが高まる可能性があります。第三者は、そのようなクレームの是非を考慮することなく、既存の特許または将来付与される可能性のある特許に基づいて、侵害クレームを私たちに提示することができる。我々は、我々の技術および製品候補およびその用途に関連する可能性のあるすべての知的財産権を知らないかもしれないし、第三者知的財産権が無効であると誤って結論を出すことができるか、または私たちの活動および製品候補が侵害されていない、流用していない、または他の方法でそのような知的財産権に違反している可能性がある。したがって、私たちは私たちの技術と製品候補を決定することができません。あるいは私たちの開発と商業化は、第三者の知的財産権を侵害、流用、または他の方法で侵害することもありません。
我々にクレームをつけた当事者は、禁止または他の公平な救済を受ける可能性があり、これは、私たちの1つまたは複数の候補製品のさらなる開発および商業化を効果的に阻止することができ、および/または私たちの名声および財務業績を損なうことができる。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、経営陣や従業員資源を当社の業務から大量に移転する可能性がある。また、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスだと思う場合、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。もし私たちに対する侵害クレームが成功した場合、私たちは故意に侵害した3倍の損害賠償金と弁護士費、特許権使用料の支払い、私たちの侵害製品の再設計、登録商標に関するクレームで私たちの製品候補製品を再命名すること、または第三者から1つ以上のライセンスを取得することを含む巨額の損害賠償を支払わなければならないかもしれない。これらは大量の時間とお金の支出を必要とする可能性があり、不可能であるか、技術的に不可能である可能性がある。しかも、私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。たとえ私たちが許可を得ることができても、それは私たちの競争相手と他の第三者が私たちに許可された同じ技術にアクセスできるように非排他的である可能性がある;または、また、それは商業市場での私たちの成功的な競争の能力を阻害または破壊する条項を含むことができる。上記のいずれも、我々の業務、財務状況、経営結果及び見通しに実質的な悪影響を及ぼす可能性がある。
多くの会社や大学は、私たちの製品と同じ分野で特許、すなわちヌクレオチドプロドラッグを出願し、これらの特許出願は私たちに不利になる可能性があり、これは私たちの業務に影響を与える可能性があり、成功すれば、高価な訴訟を招き、私たちの製品の収益性に影響を与え、および/または製品の販売または使用を禁止する可能性がある。
私たちの候補製品は主にヌクレオチド前駆体薬物、あるいはヌクレオチドリン酸アミドである。多くの会社や大学はこの一般分野で特許出願を持ち,ウイルス適応症,例えばgilead PharmAsset,LLC.,gilead Sciences,Inc.,Merck&Co.,Bristol Myers Squibb,F.Hoffmann−La Roche,カーディフ大学,大学学部カーディフコンサルティング会社,NuCana,plc,Alios Biophma,Medirなどを含む特許を発行している。もしこれらの会社、大学、または他の会社のいずれかが保有している特許が私たちの任意の候補製品またはその使用または製造によって侵害されたと主張した場合、私たちはコストの高い訴訟に巻き込まれる可能性があり、これは私たちの業務に影響を与え、私たちの従業員の時間を占め、彼らの注意を分散させる可能性があり、訴訟が成功すれば、私たちの製品の収益力に影響を与えたり、その販売を禁止したりする可能性がある。2019年6月3日、私たちは国際特許協力条約特許出願に関する匿名第三者観察文書を受け取りました
103
半硫酸塩形態のAT−511、またはベノブビルをカバーする第2の特許シリーズ。この観察結果は,遊離塩基AT−511における半硫酸塩フェニホブビルの特許性に全体的に挑戦した。2019年8月1日、私たちは、ビンネフォブビルが心臓ではなく肝臓に比例して集中していないので、AT-511のフェニルフォブウェル半硫酸塩がAT-511の遊離塩基を考慮して明らかではないことを記載した回復書を提出し、犬モデルに示されているように、C型肝炎が肝臓疾患であるため、より多くのC型肝炎の治療効果を提供することができ、毒性を低下させることができる。また,所見への対応は提案されていないが,サルではフェニルフォブビルの肺での半減期や濃度が肝臓よりも高いことが,われわれの新冠肺炎適応に関与していることが示唆されている。2020年8月10日、匿名者が、我々の薬剤であるフェニフブビルを用いてC型肝炎患者の代償性または非代償性肝硬変を治療する方法に関する我々の特許協力条約特許出願に対して第三者の意見を提出した。匿名側は,AT−511の半硫酸塩(フェニニホブビル)がC型肝炎ウイルス感染の肝硬変患者の治療に有効であると主張している。私たちは2020年10月2日にこの観察に対する反応を提出したが、その中で私たちはいくつかの理由で同意しなかった, われわれの肝硬変治療法申請を含めてAT−511の半硫酸塩は公開されておらず,代償性肝硬変患者の治療は非代償性肝硬変患者に直接適していないことが指摘されている。我々の特許協力条約特許出願は,フェニトフォブビルを用いた肝硬変C型肝炎ウイルス感染患者の治療効果を支援するヒトデータを提供している。匿名の第三者の意見および私たちの回答は、私たちそれぞれの特許出願のどの国でも読んで考慮するために文書に置かれている。2019年12月、米国特許庁はベニホブビルの組成物をカバーした特許を発行した。しかしながら、上述した発行された米国特許を除いて、これらの意見が、いかなる司法管轄区のような特許協力条約特許出願の国家段階届出から発行された特許を取得する能力に悪影響を与えないことは保証されない。私たちは現在または未来に行われている可能性のある特許出願がこの分野で働いている競争相手の業務に影響を与えていることを知らないかもしれない。特許出願は、通常、提出後6~18ヶ月以内に発行され、すでに提出されている出願において新たな特許請求がなされることは、一定期間にわたって公衆(我々を含む)には見えないことがあり、または公開されている場合、私たちはまだ見ることができない。私たちの一般的な技術分野で業務に従事する第三者が、本登録期間の前または期間を含めて、私たちの1つまたは複数の製品またはその使用または製造方法をカバーする特許主張を提起しないことは保証されない。もし本当にこのような状況が発生したら、私たちはこのような特許や出願を無効にしようとする措置を取らなければならないかもしれない, 私たちはそうしないことを選択するかもしれないし、成功しないかもしれない。特許または出願のライセンスは、商業的に合理的な条項で取得できないか、または全く得られない可能性がある。
私たちの製品は、米国で1984年に改正された“薬品価格競争と特許期限回復法”(“ハッジ·ワックスマン法案”とも呼ばれる)に制約されており、これは私たちの製品を販売しようとしている後発薬会社と訴訟を起こすリスクを増加させ、特許保護を失う可能性があります。
我々の臨床候補薬物はFDA薬物評価·研究センターによって審査された薬物分子であるため,商業化後,米国ではハーチ−ワックスマン法(Hatch−Waxman Act)特許訴訟手続きの制約を受け,イミテーション製薬会社がFDAにANDAを提出することを可能にし,生物学的同等性データのみを用いて我々の薬物を販売する承認を得る。“ハッジ·ワックスマン法”によると、私たちの医薬製品またはその使用方法をカバーする特許をFDAの“承認された医薬製品と治療同等性評価アセンブリ”に列挙する機会があり、FDAのオレンジブックと呼ばれることがある。
現在,米国ではFDAは我々のすべての製品に資格のある新化学物質(“NCE”)に5年間の独占経営権を付与する可能性がある。NCEは,FDAが任意の他のNDAで承認した活性部分を含まない薬物である。イミテーション製薬会社は私たちの製品が承認されて四年後にFDAにANDAを提出することができます。模造製薬会社がANDAを提出することは特許侵害の技術行為とされている。模造製薬会社は、それは私たちの上場特許が自然期限が切れる日まで待って、私たちの製品の模造薬バージョンを販売することができますか、あるいは私たちの1つ以上の上場特許が無効で、強制執行できない、あるいは侵害されていないことを証明することができます。後者であれば、私たちは45日間模造製薬会社に特許侵害訴訟を提起するだろう。これはジェネリック医薬品会社が私たちがリストした特許を考えているので、私たちがOrange Bookに列挙した1つ以上の特許に挑戦するだろう
104
無効、実行不可能、または侵害されていない。“ハッジ·ワックスマン法”によると、訴訟が提起された場合、FDAは、私たちのデータ排他期間が終了してから30ヶ月後、または裁判所が最終判決を下し、私たちが主張する特許主張が無効、実行不可能、または侵害されていないと判断するまで、後発薬の最終承認を発表してはならない。もし私たちがOrange Bookに私たちの関連特許を正確に列挙していなければ、ANDAによるジェネリック医薬品会社の認証に直ちに訴訟を提起しなければ、あるいは私たちがそこで発生した特許訴訟に勝利しなければ、私たちは私たちの独自の保護を失うかもしれないし、私たちの製品はすぐに模倣薬になるかもしれない。しかも、私たちがOrange Bookに私たちの関連特許を正確に列挙し、直ちに訴訟を提起して訴訟に勝っても、模倣訴訟は私たちに非常に大きな弁護士費と従業員の時間コストをもたらし、長い間私たちの注意を分散させるかもしれない。また、1つ以上の模造製薬会社が1種の革新的な薬物を同時に販売しようとするのはよく見られるため、私たちは多くの訴訟のコストと気晴らしに直面する可能性がある。私たちはまた、模造製薬会社が私たちの特許が満期になる前に私たちの市場に参入することを可能にする方法で、あるいは私たちの特許の強度、有効性、または実行可能性に悪影響を与える方法で訴訟を解決する必要があると判断することができるかもしれない。
FTCまたは他の国のそれぞれの機関がどのように薬品特許訴訟を行ったり、解決したりするかによると、いくつかの製薬会社はFTCまたは対応機関の厳格な審査の対象となっており、いくつかの審査は反独占違反の告発を招き、罰金や権利喪失を招くことがある。私たちは私たちがまたそのような審査を受けないか、あるいは審査の結果が私たちに有利であることを確認することができなくて、これは罰金や処罰につながるかもしれない。
過去数年、連邦貿易委員会は連邦裁判所で複数の訴訟を提起し、“ハッチ-ワックスマン法案”と革新会社と模倣薬会社との間の訴訟和解協定に挑戦し、その反競争と呼ばれた。例えば、連邦貿易委員会は、支払いの有無にかかわらず、どんな価値のあるものも支払いであるという急進的な立場を取っている。FTCのやり方によれば、特許和解の一部として、イノベーターがOrange Book上場特許に最初に挑戦した後発薬会社に付与された180日間の期間内にライセンス後発薬の発売を開始または延期しないことに同意した場合、または支払いなしに交渉が延期される場合、FTCは受け入れられない逆支払いであると考えられる可能性がある。製薬業界は,このような合意はリスクを除去する理性的な商業決定であり,和解条項が特許の排除潜在力の範囲内であれば,反独占攻撃を受けないと弁明している。2013年、米国最高裁は、FTCがActavis,Inc.を訴えた事件において、製薬業およびFTCのいわゆる逆支払いに関する論点を賛成5票、反対3票で却下し、参入を遅延させた対価格交換に関する“逆支払い”和解が反競争分析を受けるか否かは、(A)競争に真の悪影響を与える可能性、(B)支払いの理由、(C)特許権者が反競争損害をもたらす能力、の5つの要因に依存すると考えている。(D)支払われた金額が特許の弱点を代替することができるかどうか、(E)不合理な多額の支払いの反独占責任は、訴訟当事者がその訴訟を解決することを妨げるものではなく、例えば、模造薬が特許満了前に市場に参入することを許可し、特許権者が模倣薬に費用を支払わない。さらに、逆支払いが合理的かどうかはその規模にかかっている, それは特許権者が予想している将来の訴訟費用との割合、それが支払いを代表する可能性のある他のサービスの独立性、Actavis事件の場合のように、他の任意の納得できる理由が不足している。裁判所は,逆支払い和解は独占禁止法に違反する可能性があり,標準的な反独占理由規則分析の制約を受け,協定が連邦貿易委員会で不正であることを証明する責任があるとし,このような理由規則分析の構造を下位裁判所に残した。もし私たちが医薬品会社の“ハッジ·ワックスマン法案”との訴訟を含めて薬物特許訴訟に直面すれば、私たちがどのように事件を解決するかどうか、FTCの立場に強く反対しても、私たちは巨額の費用や罰金に直面する可能性があるという活動に基づくFTC挑戦に直面するかもしれません。
特許条項は製品に対する私たちの競争地位を十分な時間で保護するのに十分ではないかもしれない。
任意の個別特許の期限は、その特許が付与された国の適用法に依存する。米国では、すべての維持費が直ちに支払われる限り、特許の有効期限は、通常、その出願提出日から20年または最初に要求される非一時的出願日である。特許期間を延長することができる場合もあるが,特許の有効期限は限られているため,提供される保護も限られている。新製品候補製品の開発、テスト、および監督審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品が満期になる前または直後に満了する可能性がある
105
商業化されています特許期間を延長する資格のある特許については,米国で特許期間の延長を求めることが予想され,できれば他の国/地域でも特許期間の延長を求めるが,我々が求めている任意の特許期間の延長,またはそのような特許期限の延長がいかなる競争優位性を提供するかは保証されない.
米国の“ハッジ·ワックスマン法”では,我々の各製品に選択特許の特許期間延長を求める機会があり,特許期間延長の長さ(あれば)は米国特許商標局とFDAの審査·承認に依存すると規定されている。
米国では、“ハッジ·ワックスマン法”は、各製品の特許が正常に満了した後に最大5年間の特許期間を延長することを許可し、治療方法特許である場合は、承認された適応(または延長期間内に承認された任意の追加適応)に限定される。特許期間延長の長さは、通常、臨床試験期間の半分にFDA審査NDA期間の全時間を加えて計算され、これらの期間内の任意の遅延時間を減算する。特許期間の延長にも制限があり,薬品承認日から14年を超えない。したがって、もし私たちが最近提出·発表された特許の特許期間延長を選択して付与された場合、私たちは可能な特許期間延長からすべてのメリットを得ることができないかもしれない。例えば、適用期間内に出願できなかったか、関連特許が満了する前に出願できなかったか、または多くの適用要件のいずれかを満たすことができなかったため、特許期間の延長が全く付与されない可能性もある。また、適用当局は、米国のFDAおよびUSPTO、および他の国/地域の任意の同等の規制機関を含み、このような延期が利用可能かどうかの評価に同意しない可能性があり、私たちの特許延期の承認を拒否するか、または私たちが要求したよりも限られた延期を承認する可能性がある。このような状況が発生すれば、私たちの競争相手は私たちの特許が満期になった後、私たちの臨床と臨床前データを参考にすることで競争製品の承認を得て、他の場合よりも早く彼らの製品を発売するかもしれない。このような状況が発生すれば、私たちが製品収入を創出する能力に実質的な悪影響を及ぼす可能性がある。
1997年,食品·薬物管理局近代化法案(“FDAMA”)の一部として,国会は児童薬物研究を行う医薬品メーカーにインセンティブを提供する法律を公布した。この法律は小児科研究と引き換えに6カ月間の排他性を規定しており,小児科排他性条項と呼ばれている。FDAMAに適合した臨床研究を行えば、私たちは私たちの法規データ独占期間と私たちの特許期間延長期間(受領すれば)で6ヶ月の期限を追加的に得ることができるかもしれない。しかし,FDAMAに適合した小児科研究を行わないことや,FDAに受け入れられないことを選択した場合,我々のデータ排他性や特許期間延長の追加6カ月の排他的延長は得られないであろう。
欧州では,補充保護証明書は規制審査期間中に失われた特許期間を補償するために特許期間を最大5年に延長することができ,合意された小児科調査計画に基づいて臨床試験データを取得すれば,さらに6カ月延長することができる。ヨーロッパのすべての国は補充保護証明書を提供しなければならないが,ヨーロッパ諸国間には統一された立法がないため,国ごとに補充保護証明書を申請·発行しなければならない。これは、国によって異なるか、または全く提供されない可能性があるこれらの証明書の申請および受信のコストが高い可能性がある。
もし私たちが商業的に合理的な条項や第三者からライセンスを得ることができない場合、あるいはこのような合意の下での私たちの義務を履行できない場合、私たちの業務は損なわれる可能性があります。
第三者の特許や他のノウハウを用いて私たちの製品を商業化する必要があるかもしれませんが、この場合、これらの第三者の許可を得ることが求められます。もし私たちがこのような技術を許可できない場合、あるいは私たちが不利な条項でそのような技術を許可することを余儀なくされた場合、私たちの業務は実質的な損害を受ける可能性がある。もし私たちが必要な許可を得ることができない場合、私たちは影響を受けた候補製品を開発したり商業化することができなくなり、これは私たちの業務に実質的な損害を与える可能性があり、そのような知的財産権を所有または制御する第三者は、私たちの販売禁止または私たち側が印税および/または他の形態の賠償義務を支払うことを求めることができる。たとえ我々が許可を得ることができても,我々の競争相手や他の第三者が我々に許可されている同じ技術にアクセスできるように非排他的である可能性がある.第三者知的財産権の許可または買収は競争分野であり、より多くの老舗企業は、魅力的または必要と考えられる第三者知的財産権許可または買収戦略をとることができる。これらの老舗会社は
106
それらの規模、資本資源及びより強い臨床開発と商業化能力のため、私たちより競争優位を持っているかもしれない。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。
もし私たちが必要な第三者知的財産権を得ることができない場合、あるいは私たちの既存の知的財産権を維持することができなければ、私たちは、私たちの技術、候補製品、またはそれらを製造する方法、または代替技術を開発または許可するのに時間と資源をかけて再設計する必要があるかもしれません。これらはすべて技術的にも商業的にも不可能かもしれません。もし私たちがそれができなければ、影響を受けた技術や候補製品を開発または商業化することができないかもしれません。これは、私たちの業務、財務状況、運営結果、将来性を深刻に損なう可能性があります。
さらに、私たちが将来の任意の許可プロトコルの義務を履行できない場合、私たちの取引相手は、これらのプロトコルを終了する権利があるかもしれません。この場合、私たちは、これらのプロトコルによってカバーされる任意の製品の開発、製造、またはマーケティングを停止させることができないか、またはそのようなプロトコルの下での他の処罰に直面する可能性があります。このような状況は、任意のこのようなプロトコルに従って開発された候補製品の価値に実質的な悪影響を及ぼす可能性がある。これらの合意を終了するか、またはこれらの合意の下で私たちの権利を減少またはキャンセルするか、または私たちの業務の利益に適合した場合に、私たちのこのような合意の下での私たちの権利を自由に譲渡または再許可することを制限することは、私たちが重要な知的財産権または技術に対する私たちの権利、またはそのような合意に依存する1つまたは複数の候補製品のさらなる開発または商業化を阻害、遅延、または禁止することを含む、新たなまたは回復された合意をあまり有利ではない条項で交渉しなければならない可能性がある。
私たちは現在何の関連訴訟にも巻き込まれていないにもかかわらず、私たちは私たちの特許や他の知的財産権を保護または強制する訴訟に巻き込まれるかもしれないが、これは高価で時間がかかり、成功しないかもしれない。
競争相手および他の第三者は、私たちまたは将来の許可者の特許、商標、著作権、または他の知的財産権を侵害、流用、または他の方法で侵害する可能性があります。したがって、私たちは第三者に侵害、流用、または他の知的財産権に関するクレームを提起する必要があるかもしれない。権利侵害や他の許可されていない使用と戦うために、私たちは国/地域にクレームをつけることを要求されるかもしれません。これは高価で時間がかかり、私たちの管理者と科学者の時間と注意力を分散させているかもしれません。私たちはこのようなクレームを提出し、追及するのに十分な財政あるいは他の資源があることを保証することはできません。これらのクレームは往々にして数年続けなければなりません。
私たちが第三者に提起したいかなるクレームも、私たちが侵害、流用、あるいは他の方法で彼らの知的財産権を侵害したと主張するように、これらの当事者に反クレームを促す可能性がある。さらに、特許侵害訴訟では、このような当事者は、私たちが主張している特許が無効または実行不可能であるか、またはその両方を反訴することができる。米国の特許訴訟では,被告が無効または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由は、新規性の欠如、明らかな、または実施できないことを含む、いくつかの法定要件のいずれかを満たしていないと言われているからかもしれない。主張を実行できない理由は,特許起訴に関連する者が起訴中に米国特許商標局に関連情報を隠蔽したり,誤った声明をしたりしたためかもしれない.第三者は米国や国外の行政機関にこのようなクレームを提起することができ,訴訟範囲外でも同様である。このようなメカニズムには、再審査、認可後審査、当事者間の審査、介入手続き、派生手続き、および外国法ドメインにおける同等の手続き(例えば、反対手続き)が含まれる。法的に無効と実行不可能と断言された後の結果は予測できない。
どのような訴訟においても、裁判所は、私たちの特許または私たちが許可した特許が無効であること、強制的に実行できないこと、および/または侵害されていないことを判断することができ、またはそのような特許の権利要件を狭く解釈することができ、または私たちの特許が関連技術をカバーしないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。任意の訴訟または弁護手続における不利な結果は、私たちの1つまたは複数の特許を全部または部分的に無効、狭義の解釈または実行できないリスクに直面させる可能性があり、私たちの特許出願を発行できないリスクに直面させる可能性があり、これらの当事者または他の競争相手に対してこれらの特許を主張する能力を制限し、第三者が類似または競合製品を製造および販売する能力を制限または排除する可能性がある。同様に商標侵害請求を主張すれば
107
裁判所は、私たちが主張する商標が無効または強制的に執行できない、または私たちが主張する商標侵害者が関連する商標に対して優先的な権利を有すると判断するかもしれない。この場合、私たちは最終的にこのような商標の使用を中止することを余儀なくされる可能性があり、これは私たちの業務に実質的な損害を与え、私たちの市場地位に悪影響を及ぼす可能性がある。
私たちが知的財産権の侵害、流用、または他の侵害を決定しても、裁判所はこれ以上のこのような活動に対して禁止を発行せず、金銭損害賠償のみを決定する可能性があり、これは十分な救済措置ではないかもしれない。さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。聴聞、動議、または他の一時的手続き、または事態発展の結果を公開することもできる。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に実質的な悪影響を及ぼす可能性がある。上記のいずれも、我々の業務、財務状況、経営結果及び見通しに実質的な悪影響を及ぼす可能性がある。
2022年4月12日、インド特許庁から特許、意匠、商標総監から事前許可反対通知を受けました。反対意見はサンカルプリハビリテーション信託会社によって提出され,AT−511またはその薬学的に許容される塩に対する我々の未解決特許主張に挑戦した。AT−511とC型肝炎治療のための私たちの特許主張を強力に守るつもりですが、インド特許庁が私たちに有利な決定を下すことは保証されず、私たちの特許主張が承認されることを許可します。しかも、インドの事前の反対は非常に遅く進展しているかもしれないので、この訴訟は数年以内に解決されないかもしれない。私たちの特許出願はAT-511の特許として発行されないし、この許可前の反対意見が私たちに有利に解決されない限り、インドでC型肝炎の治療にも使用されないだろう。もし私たちに有利な解決策がなければ、私たちはインドでAT-511の特許を得ることができないかもしれない。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
米国特許商標局、欧州特許庁、および他の特許機関は、特許出願中にいくつかのプログラム、文書、費用支払い、およびその他の同様の規定を遵守することを要求する。特許および/または出願の定期維持費、継続費、年会費、および様々な他の政府費用は、特許および/または出願の有効期間内にいくつかの段階で米国特許商標局および米国以外の様々な政府特許機関に支払われる。私たちはこれらの費用を支払うようにシステム的に注意して、私たちは外部会社を招聘し、私たちの外部弁護士に依存してアメリカではない特許代理機関にこのような費用を支払う。多くの場合、適用される規則に基づいて、滞納金を支払うことによって、または他の方法で過失失効を修正することができるが、場合によっては、規則を遵守しないことは、特許または特許出願が放棄または失効され、関連する法域特許権の一部または全部を喪失させる可能性がある。この場合、私たちの競争相手または他の第三者は、同様のまたは同じ製品または技術で市場に参入する可能性があり、これは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
私たちは世界各地で私たちの知的財産権を強制的に施行できないかもしれない。
世界各国で私たちの候補製品の出願、起訴、特許保護の費用は目を引くほど高いだろう。したがって、私たちは特定の司法管轄区域で特定の知的財産権を保護または維持保護しないことを選択することができる。特許可能な要求は特定の国では、特に発展途上国では異なる可能性がある。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、他の侵害製品を私たちが特許保護を受ける可能性があるが、特許法執行力がアメリカに及ばない地域に輸出することもできる。これらの製品は、私たちが発行できるかもしれない特許を発行していない司法管轄区域で私たちの製品と競争することができるかもしれません。将来のいかなる特許主張や他の知的財産権も、それらの競争を効果的にまたは阻止するのに十分ではないかもしれません。
また、私たちが知的財産権を保護して実行する能力は、外国の知的財産権法の変化の悪影響を受ける可能性がある。また、米国やヨーロッパ以外のいくつかの国の法律は、米国や欧州の法律のように知的財産権保護を提供していない。
108
ある外国司法管轄区では、多くの会社が知的財産権の保護と保護に重大な問題に直面している。インド、中国などの発展途上国を含むいくつかの国の法律制度は、私たちの特許や他の知的財産権の執行に不利である可能性がある。
これは私たちの特許の侵害や流用や他の方法で私たちの他の知的財産権を侵害することを阻止することを難しくするかもしれない。一部の外国の国は、革新会社が保有する承認された薬物の特許に対して強制許可を発行し、政府または1つ以上の第三者会社が外国政府が公共利益に適合していると考えている場合には、革新会社の特許権者の許可なしに承認された薬物を販売することを可能にすると表明している。例えば、インドはこのようなプログラムを使用して、国内会社が革新者の承認なしに特許薬を製造·販売することを許可している。私たちのいかなる薬物をカバーする特許が外国で強制許可されないことも保証されず、私たちがこのような強制許可を与えるかどうか、あるいはどのように影響を与えるかどうかを保証することはできない。また、ブラジルは、その規制機関ANVISAがブラジルで薬物特許を付与するか否かの決定に参加することを許可しており、特許付与決定は、特許が特許要求に適合しているか否か、このような特許が国益に適合しているか否かを含むいくつかの要因に基づいて行われている。また,他のいくつかの国でも薬物特許の実行が他のタイプの技術特許よりも困難になるように法律が制定されている。また、ドーハ宣言で説明された“貿易関連知的財産権条約”(“TRIPS”)によると、麻薬生産国は、十分な製造能力の乏しい発展途上国への麻薬輸出を許可しなければならない。したがって、米国または外国の医薬品市場は、医療分野の特許発行、強制執行、または非自発的許可に関する現在の公共政策の影響を受ける可能性がある。
また、2015年11月、貿易に関する知的財産権協定の管理を担当する世界貿易機関(“WTO”)のメンバー投票で、後発開発途上国が薬品特許の免除を強制執行しないことを2033年に延長することが決定した。私たちは現在、後発開発途上国で特許出願を提出していません。私たちの現在の意図は、今後これらの国で特許出願が提出されないことであり、少なくともある程度はWTOの薬品特許免除のためです。
また、WTOは現在、新冠肺炎ワクチンの知的財産権の放棄を検討しており、米国政府は最近、免除を支持する立場を表明している。現在の提案は新冠肺炎ワクチンをカバーする知的財産権を一時的に放棄することであるが,承認されれば放棄の最終時間と範囲は不明である。状況の複雑さを考慮して、このような免除の範囲および時間は、広範な交渉を必要とする可能性があり、これは、長期的な不確実性を招き、我々の業務に悪影響を及ぼす可能性がある。もし免除が承認され、そしてフェノキシフォブビルのような新冠肺炎療法をカバーすれば、私たちはフェニルプロピホビルを商業化し、私たちの関連技術を保護する能力は不利な影響を受ける可能性がある。
私たちは私たちの能力に頼って私たちの特許を強制的に執行することで他人の競争を阻止しますが、いくつかの管轄区域は私たちに第三者に許可を与えることを要求するかもしれません。このような強制許可は私たちを含むいくつかの候補製品に拡張することができ、これは私たちの潜在的な収入機会を制限するかもしれない。
多くの外国国には強制許可法があり,これらの法律によると,特許権者は場合によっては第三者に許可を付与しなければならない。例えば、直接立法や国際計画によって、救命製品や高価な製品の強制許可や強制許可の脅威が発展途上国でますます一般的になっている。強制許可は私たちを含むいくつかの候補製品に拡張することができ、もし彼らがマーケティング承認を得たら、これは私たちの潜在的な収入機会を制限するかもしれない。したがって、私たちはアメリカとヨーロッパ以外のいくつかの国で第三者が私たちの発明を実施することを阻止できないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することもできます。また、もし私たちが特許を実行して侵害活動を阻止する能力が不足していれば、競争相手は他の侵害製品を私たちが特許保護を持っている地域に輸出するかもしれません。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。外国司法管区で私たちの特許権の訴訟を強制的に執行することは、成否にかかわらず、巨額のコストを招き、私たちの努力と資源を私たちの業務の他の側面から移転させる可能性があります。また主要市場での知的財産権を保護しようとしていますが
109
もし私たちの製品にこのような特許権が存在すれば、私たちが私たちの製品を販売したいすべての司法管轄区域で同様の努力を開始または維持できることを確実にすることはできません。したがって、このような国で知的財産権を保護するための私たちの努力は十分ではないかもしれない。
さらに、いくつかの国は、政府機関または政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許所有者は金銭救済に限られている可能性があり,政府が侵害者であれば侵害を禁止できない可能性があり,特許の価値を大幅に低下させる可能性がある。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位は損なわれるだろう。
特許提供の保護に加えて、我々は、特許を出願できない独自技術、特許を実施しにくいプロセス、および私たちの候補製品発見および開発中に特許がカバーされていないノウハウ、情報または技術に関する任意の他の要素を保護するために、商業秘密保護および秘密保護プロトコルに依存して保護する。しかし、商業秘密は保護するのが難しいかもしれない。私たちは、当社の従業員、CRO、コンサルタント、科学コンサルタント、および他の請負業者のような、これらの技術およびプロセスにアクセスする権利のある当事者と秘密協定を締結することによって、私たちのノウハウおよびプロセスを保護することを求めています。私たちは私たちが可能であるか、または私たちの商業秘密またはノウハウとプロセスに接触したすべての当事者とこのような合意に到達したことを保証することができない。また、私たちは私たちのビルの実体安全と、私たちの情報科学技術システムの実体と電子安全を維持して、私たちの資料と商業秘密の完全性とセキュリティを維持することに力を入れています。私たちはこれらの個人、組織、そしてシステムに自信がありますが、合意やセキュリティ措置は違反される可能性があり、私たちの商業機密は漏洩する可能性があり、私たちはこのような違反に対応するための十分な救済措置がないかもしれません。強制執行側が商業秘密を不正に開示したり流用したりする主張は困難であり、高価で時間がかかり、結果は予測できない。しかも、米国国内外のいくつかの裁判所は商業秘密をあまり望んでいないか、または保護したくない。私たちの商業秘密または他の機密固有情報を盗用または無許可に開示することは、私たちの商業秘密の保護を失い、私たちの競争的地位を損ない、私たちの業務に実質的な悪影響を及ぼす可能性がある。また…, 私たちの商業秘密または他の機密固有情報を保護するための措置が不十分であると考えられる場合、私たちは、第三者が商業秘密または他の機密固有情報を流用するのに十分な追跡権を持っていない可能性がある。
さらに、競争相手または他の第三者が、我々のビジネス秘密および他の機密固有情報を他の方法で取得しないこと、または実質的に同じ技術およびプロセスを独立して発見または開発することを保証することはできない。私たちの候補製品や技術に関連する商業秘密や他の非特許知的財産権を第三者に漏洩することを阻止できない場合、私たちがこのような強制的に実行可能な商業秘密保護を持つことは保証されません。私たちは私たちの市場で競争優位性を確立または維持することができないかもしれません。これは、私たちの業務、運営結果、および財務状況に大きな悪影響を及ぼす可能性があります。
私たちは、私たちの従業員、コンサルタント、または独立請負業者が第三者の機密情報を誤って使用または開示し、私たちの従業員が彼らの元雇用主のいわゆる商業秘密を誤って使用または開示した、または私たちが私たち自身の知的財産権を持っていると主張するという告発を受けるかもしれない。
私たちは将来的に、私たちの競争相手や潜在的な競争相手を含む、大学や他のバイオテクノロジーや製薬会社に雇われていた個人を雇うことができるようになった。私たちは、私たちの従業員、コンサルタント、および独立請負業者が、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを保証するために努力しているが、私たちは、商業秘密または他の固有情報を含む、私たちまたは私たちの従業員、コンサルタント、または独立請負業者が、これらの個人の任意の元雇用主または他の第三者の知的財産権を無意識にまたは他の方法で使用または開示しているというクレームを受ける可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護できなければ、金銭損害賠償の支払いに加えて、貴重な知的財産権や人員、あるいは私たちの雇用者の能力を失う可能性があり、上記のいずれの場合も、これは私たちの業務に悪影響を及ぼす可能性があります。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
110
私たちの政策は、私たちのすべての従業員およびコンサルタントが彼らの発明を私たちに譲渡することを要求することであるが、従業員またはコンサルタントが私たちのために働いているときに他人が所有している知的財産権を使用する場合、関連するまたはそれによって生じるノウハウおよび発明の権利に関する論争が生じる可能性がある。私たちはまた、実際に私たちが自分の知的財産権と見なしているすべての当事者とこのような合意を構想したり開発したりすることに成功しないかもしれない。知的財産権の譲渡は自動的に実行されない可能性があり、あるいは譲渡協定が違反される可能性があり、私たちは第三者にクレームをつけさせられたり、私たちが私たちの知的財産権の所有権だと思うことを決定するために、私たちが提起する可能性のあるクレームを弁護したりすることができます。このようなクレームは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
私たちの独自の権利は私たちの技術と候補製品を保護するのに十分ではないかもしれませんが、知的財産権は私たちの競争優位が直面しているすべての潜在的な脅威を解決できるとは限りません。
私たちの知的財産権が提供する将来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。以下の例は例示的である
これらの事件のいずれかが発生した場合、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
111
従業員事務に関するリスク、管理成長、および私たちの業務に関するその他のリスク
私たちは私たちの組織を拡大する必要があり、私たちはこのような成長を管理することに困難に直面するかもしれないし、これは私たちの運営を混乱させるかもしれない。
時間の経過とともに、私たちの従業員数と業務範囲は大幅に増加し、特に製品候補開発、監督と臨床事務、販売、マーケティング、流通分野で大幅に増加すると予想される。私たちの成長活動を管理するためには、私たちの管理、運営、財務システムを継続して実施し、改善し、私たちの施設を拡大し、より多くの合格者を募集し、訓練し続けなければならない。私たちの財務資源が限られていることと、私たちの管理チームがこのような成長を期待している会社を管理する上での限られた経験のため、私たちの業務の拡張を効果的に管理したり、より多くの合格者を募集したりすることができない可能性があります。会社の規模を拡大する過程で、私たちは新入社員を確定、採用、統合することが難しいかもしれない。今後の成長は私たちの経営陣に大きな追加責任をもたらすだろうが、これらに限定されない
また、私たちの経営陣は、私たちの日常活動から不比例な注意を移し、これらの成長活動を大量に管理する必要があるかもしれない。私たちは私たちの業務の拡張を効果的に管理できない可能性があり、これは私たちのインフラが弱く、操作ミスを招き、ビジネス機会を失い、従業員を失い、残りの従業員の生産性を低下させる可能性がある。私たちの予想成長は大量の資本支出を必要とし、開発候補製品のような他のプロジェクトから財政資源を移すことができるかもしれない。私たちの経営陣が私たちの成長を効果的に管理できなければ、私たちの支出増加は予想を超える可能性があり、私たちは製品収入を創出および/または増加させる能力が低下する可能性があり、私たちは私たちの業務戦略を実施できないかもしれない。私たちの将来の財務業績と、私たちが任意の承認された製品を商業化し、効果的に競争する能力は、私たちが将来のどんな成長の能力を効果的に管理するかにある程度依存するだろう。
現在、予測可能な未来に、私たちは臨床試験の進行と実行、製造のほとんどの側面を含むいくつかの独立した組織、コンサルタント、コンサルタントに大きく依存していくつかのサービスを提供する。必要な場合には、独立した組織、コンサルタント、コンサルタントのサービスがタイムリーに提供される保証はなく、合格した代替者を見つけることも保証されない。さらに、私たちのアウトソーシング活動を効率的に管理できない場合、あるいは独立した組織、コンサルタント、またはコンサルタントが提供するサービスの品質や正確性が何らかの理由で影響を受ける場合、私たちの臨床試験は延長、延期、または終了される可能性があり、私たちは規制機関から候補製品の承認を得られないか、あるいは他の方法で私たちの業務を推進することができないかもしれません。私たちが既存の独立組織、コンサルタント、またはコンサルタントを経済的に合理的な条件で管理すること、または外部で他の適任な独立組織、コンサルタント、コンサルタントを見つけることができる保証はありません。新入社員を雇用したり、コンサルタントやコンサルタントチームを拡大したりすることで、私たちの組織を効果的に拡張することができない場合や、このような拡張に対応するために効率的に施設を維持したり得ることができなければ、候補製品をさらに開発して商業化するために必要な任務を実行することができない可能性があり、そのため、私たちの研究、開発、商業化の目標を達成できないかもしれません。
私たちと適格な人材やコンサルタントを競争する多くのバイオテクノロジーや製薬会社は、私たちよりも多くの財務や他の資源、異なるリスク状況、そしてより長い業界の歴史を持っています。もし私たちが高い素質の人員やコンサルタントを引き付け、維持することができなければ、候補製品を発見し、開発し、私たちの業務を経営する速度と成功は限られるだろう。
112
私たちは限られた職員だけが私たちの業務を管理して運営する。
2023年2月20日現在、私たちは70人のフルタイム従業員がいます。私たちは2つの異なる疾患適応の候補製品の臨床開発に集中しており、効率的な方法で私たちの業務を管理·運営することが求められている。私たちは私たちの候補製品を開発したり、私たちの業務を運営したり、私たちが本来達成しようとしていたすべての目標を達成するために、十分な人員レベルを雇用および/または維持することができることを保証することはできません。
もし私たちが重要な管理者や科学者を失った場合、合格した従業員、役員、高級管理職、あるいは他の重要な人員を募集できない場合、あるいは私たちの報酬コストが増加し、私たちの業務は深刻な影響を受ける可能性があります。
私たちは私たちの官僚たち、役員たち、そして重要な職員たちに強く依存している。私たちの各高級管理者、取締役と肝心な従業員は私たちの候補製品と私たちの運営に対して専門知識を持っているため、もし私たちの任意の1人以上の高級管理者、取締役或いは肝心な従業員がサービスを失ったら、臨床試験とその他の重要な活動の計画と実行を深刻に遅延、損害或いは阻止する可能性があり、これは私たちの業務の成功的な発展に必要である。私たちはある幹部と雇用協定を締結しましたが、全体的に、これらの合意は私たちの幹部がいつでも私たちとの雇用関係を終了することを阻止しません。
私たちはどんな上級管理者や役員にもキーパーソン生命保険をかけません。
私たちの役人、役員、重要な従業員の継続サービスを保留する以外に、私たちの将来の成功と成長は私たちがより多くの人員を識別、採用、維持する能力に部分的に依存するだろう。
私たちがより多くの人員を識別、採用し、維持し、必要に応じて退職した幹部と肝心な従業員を交換する能力は困難あるいはコストが高い可能性があり、また、私たちの業界で候補製品を発見または他の方法で識別·開発するために必要な技能と経験を持っている人員の数は限られており、監督部門の許可を得て製品を商業化することに成功したからである。この限られた人材バンクから募集する競争は非常に激しく、多くの製薬と生物技術会社の間の類似人員に対する競争を考慮して、私たちは受け入れ可能な条件でこれらの追加のキーパーソンを採用、訓練、維持或いは有効に激励することができないかもしれない。私たちはまた、大学や研究機関から科学や臨床人を募集する競争に直面している。また、私たちは、科学と臨床コンサルタントを含むコンサルタントとコンサルタントに依存して、私たちの研究開発と商業化戦略の制定を助けてくれます。私たちのコンサルタントとコンサルタントは、私たち以外のエンティティによって採用される可能性があり、他のエンティティとの相談やコンサルティング契約に基づいて約束することができ、これは、彼らの私たちに対する利用可能性を制限するかもしれません。もし私たちが引き続き素質の高い人材を誘致し、維持することができなければ、候補製品を開発し、商業化する能力は制限されるだろう。
私たちの多くの重要な職員たちは相当な数の普通株式を与えられたり、私たちの普通株を購入するオプションを与えられた。私たちの重要な従業員を含む私たちの従業員は、彼らが持っている株が株の元の購入価格に対して著しく変動した場合、あるいは彼らが持っているオプションの行使価格が私たちの普通株の市場価格よりも著しく低いか、あるいはそれ以上であれば、彼らは私たちを離れる可能性が高いかもしれない。
不安定な市場や経済状況は、私たちの業務、財務状況、株価に深刻な悪影響を及ぼす可能性がある。
信用と金融市場を含む世界経済は極端な変動と破壊を経験し、流動性と信用供給の深刻な減少、消費者自信の低下、経済成長の低下、失業率の上昇、金利上昇、インフレ率の上昇及び経済安定の不確定性を含む。例えば、新冠肺炎の大流行は広範な失業、経済減速と資本市場の極端な変動を招く。同様に、現在ウクライナとロシアの間の衝突は資本市場の極端な変動をもたらし、さらに世界経済の結果をもたらしているが、エネルギー、農産物、その他の基本的な商品供給に関する結果を含むがこれらに限定されない。これらの条件は、任意の必要な債務または株式融資をよりタイムリーに、または有利な条件で得ることができ、コストがより高いか、または希釈度が高くなる可能性がある。現在のウクライナやロシアとの情勢のような政治的動揺は、世界経済の変動や中断を招く可能性もある。さらにリスクがあります
113
私たちの1つまたは複数のCRO、サプライヤー、CMO、または他の第三者プロバイダは、経済低迷の中で生き残ることができないかもしれない。したがって、私たちの業務、経営業績、普通株価格は不利な影響を受ける可能性があります。
イギリスの離脱の持続的な影響は私たちの業務に否定的な影響を及ぼすかもしれない。
2021年1月1日の離脱移行期間が終了して以来、イギリス(イングランド、スコットランド、ウェールズ)はEU法律の直接的な制約を受けていないが、アイルランド/北アイルランド議定書の条項によると、EU法律は北アイルランドに一般的に適用されている。二次立法でイギリス法律に転換されたEU法律は依然としてイギリスに適用される。しかし、現在イギリス議会に提出されている“2022年保留EU法律(撤回と改革)法案”によると、いかなる保留EU法律も明確に保留され、国内法によって国内法として吸収されていない場合、または部級法規によって延長され(2026年6月23日より遅くない)、自動的に失効し、2023年12月31日までに撤回される。しかも、CTRのような新しい立法はイギリスに適用されない。イギリス政府は、国務大臣または適切な機関に、医療製品および医療機器分野の既存の法規を改正または補充することを許可する“2021年医薬品·医療機器法”を採択した。これにより,人間の薬物,臨床試験,医療機器分野の規制格差や将来の変化を解決する上で柔軟性を許すことを目的として,今後二次立法で新たなルールを導入することが可能となる。
“EU-イギリス貿易·協力協定”は2021年1月1日に発効した。TCAは製薬企業に影響を与える条項(関税と関税に関する条項を含む)を含む。また、薬品に関するいくつかの具体的な規定がある。これらの措置には,GMPの薬品生産施設の検査と発表を相互に認めるGMPファイルが含まれている。しかし、TCAはイギリスとEUの薬品法規と製品標準の卸売相互承認を含まず、将来イギリスはEUとは異なる現地要求がある可能性があり、これは将来イギリスで発生する臨床と開発活動に影響を与える可能性があると予想される。同様に、イギリスでの臨床試験提出と活動データは、EMA臨床試験情報システム(“CTIS”)においてEU諸国の臨床試験提出と活動データとバンドルすることができず、イギリスの将来の臨床と開発活動の複雑性、コスト、潜在リスクをさらに増加させる。英国のEU離脱が英国とEUとの関係をどの程度変えるかは、政治的にも経済的にも大きな不確実性がある。
英国、EU、世界事業における英国離脱の長期的な影響は、TCAおよびイギリスとEU間の任意の他の関連協定の実施と応用の影響に依存する。MHRAは2021年1月1日からイギリスの独立した薬品と医療機器規制機関である。北アイルランド議定書の結果として、北アイルランドが適用される規則は、イングランド、ウェールズ、スコットランド、およびイギリスとは異なり、全体的に、北アイルランドはEUの規制制度に従うだろうが、その国家主管機関はMHRAであるだろう。
これらの事態はすでに発展し、世界経済状況と世界金融市場の安定に重大な悪影響を与え続ける可能性があり、世界市場の流動性を著しく減少させ、主要市場参加者がある金融市場で運営する能力を制限する可能性がある。特に、イギリスの金融と銀行市場、そしてイギリスとヨーロッパの規制過程のかなり不確実な時期を招く可能性もある。このような不確実性により、世界金融市場は大幅な変動を経験する可能性があり、これは私たちの普通株の市場価格に悪影響を及ぼす可能性がある。資産評価、通貨レート、信用格付けも市場変動の激化の影響を受ける可能性がある。イギリスが離脱後に金融法律と法規、税収と自由貿易協定、知的財産権、データ保護法、サプライチェーン物流、環境、健康と安全法律法規、移民法、雇用法を含むEU規則と法規を置換または複製することを決定したため、イギリスの将来の法律法規は透明性が不足し、これはイギリスの外国直接投資を減少させ、コストを増加させ、経済活動を抑制し、資本を得る機会を制限する可能性がある。英国とEUが受け入れ可能な離脱条項について交渉できない場合、または他のEU加盟国が離脱を求める場合、イギリスと他のEU加盟国との間、または欧州経済圏全体の間のバリアフリー参入が減少または撤廃される可能性がある。
114
イギリスの離脱と新しい規制制度により、私たちはまた新しい規制コストと挑戦に直面する可能性があり、これらのコストと挑戦は私たちの運営に悪影響を及ぼす可能性がある。しかも、ポンド、ユーロ、ドルの間の通貨為替レートはイギリスの離脱の影響を受け続ける可能性がある。
私たちの普通株に関するリスクは
私たちの主要株主と経営陣は私たちのかなりの割合の普通株を持っており、株主の承認が待たれる事項に大きな影響を与える可能性があります。
2022年12月31日現在、私たちの役員、役員、>5%の株主実益は私たちの普通株のかなりの割合を持っています。したがって、このような株主が力を合わせれば、このような所有権地位を通じて私たちに影響を与える能力があるかもしれない。このような株主たちは株主の承認を必要とする事項を決定することができるかもしれない。例えば、これらの株主は、取締役選挙を制御したり、我々の組織文書の修正または任意の合併、資産売却、または他の重大な会社取引の承認に影響を与えることができるかもしれない。これは、私たちの株主の一つとして、あなたの最適な利益に合致すると思うかもしれませんので、私たちの普通株に対する能動的な買収提案や要約を阻止または阻止することができます。
証券や業界アナリストが私たちの業務に関する研究や報告を発表しない場合、あるいは彼らが私たちの株に不利または誤った意見を発表すれば、私たちの業務が良好であっても、私たちの株価や取引量が低下する可能性がある。
私たちの普通株の取引市場は、業界や証券アナリストが発表した私たちまたは私たちの業務に関する研究と報告の影響を受けています。私たちは現在証券と産業アナリストに対する研究報告書が限られている。もし私たちの報道を担当したり、私たちのアナリストが私たちの普通株格付けを引き下げたり、私たち、私たちの業務モデル、私たちの知的財産権あるいは私たちの株式表現に不利または誤った意見を発表した場合、あるいは私たちの臨床前研究や臨床試験と運営結果がアナリストの期待を満たしていない場合、私たちの株価は低下するかもしれません。2021年第4四半期にアナリストが行ったように、私たちは第2段階MOONSONG臨床試験のデータに対して報告を発表したので、この報告は全体的な患者数と他の事件の主要な研究終点に達していません。これらのアナリストのうちの1人以上が私たちへの報告を停止したり、私たちに関する報告書を定期的に発表できなかったりすれば、私たちは金融市場で可視性を失う可能性があり、これは逆に私たちの株価や取引量を低下させる可能性がある。
当社の登録証明書や再記述の法律やデラウェア州の法律の条項は、わが社の買収をより困難にする可能性があり、これは私たちの株主に有利になる可能性があり、私たちの株主が現在の経営陣を交換または更迭しようとすることを阻止するかもしれません。
当社の再記載された会社登録証明書および当社の改正および再記述された定款の条項は、株主が有利と思われる可能性のある会社の合併、買収、または他の支配権変更を阻止、延期、または阻止する可能性があり、あなたの株式から割増取引を得ることができることを含む。これらの条項はまた、投資家が未来に私たちの普通株に支払いたい価格を制限し、それによって私たちの普通株の市場価格を下げる可能性がある。また、我々の取締役会が責任を持って我々の管理チームのメンバーに命じているため、これらの規定は、株主が取締役会のメンバーを交換する難しさを増やすことで、現在の経営陣の任意の試みを交換または罷免することを阻害または阻止する可能性がある。他の事項を除いて、これらの規定には規定が含まれている
115
また、私たちはデラウェア州に登録して設立されたので、私たちはデラウェア州会社法第203条の規定によって管轄されています。この条項は、規定された方法で合併または合併が承認されない限り、私たちが発行した議決権のある株を15%以上保有している人が取引日後3年以内に私たちと合併または合併することを禁止しています。
私たちの会社登録証明書は、特定の裁判所が私たちの株主が起こしうるいくつかの訴訟の独占フォーラムとして特定の裁判所を指定し、これは、私たちの株主が有利な司法フォーラムを得ることが私たちとの紛争を処理する能力を制限するかもしれない。
私たちが再声明した会社登録証明書は、私たちが書面で代替裁判所を選択することに同意しない限り、デラウェア州衡平裁判所は、株主が私たちに提起したクレームに関連する大部分の法的訴訟の唯一かつ独占的な裁判所であるが、“取引法”に規定されている任意の責任または義務または連邦裁判所の排他的管轄権を有する任意の他のクレームを強制的に執行するための訴訟を除外し、デラウェア州衡平裁判所が標的管轄権の欠如によって却下された任意の訴訟を、デラウェア州の別の州または連邦裁判所で提起することができる。私たちが再記述した会社登録証明書は、私たちが書面で代替裁判所を選択することに同意しない限り、アメリカ連邦地域裁判所は1933年に証券法に基づいて提出された訴因を解決する独占的な裁判所になると規定している。当社の株式の権益を購入又はその他の方法で取得した者又は実体は、当社の上述した再記載会社の登録証明書の規定に同意したとみなされなければならない。
この条項は、会社紛争の解決に特に経験のある総理や証券法紛争の解決に経験のある連邦裁判官のデラウェア州法律の適用における一貫性を向上させ、他のフォーラムよりも速いスケジュールで効率的に事件を管理し、多裁判所訴訟からの負担を保護するため、私たちに利益を与えると信じている。しかしながら、この条項は、司法裁判所において、任意の株主が、私たちまたは私たちの取締役、上級管理者、従業員、または代理とトラブルを発生させることに有利であると考える株主のクレームを提起する能力を制限する可能性があるので、私たちの役員、高級管理者、従業員、および代理人に対する訴訟を阻止する可能性がある。他社の会社登録証明書における同様の選択裁判所条項の実行可能性が法的手続きで疑問視されており、我々に提起された任意の適用訴訟において、裁判所は、我々が再記述した会社登録証明書に含まれる選択裁判所条項が、このような訴訟において適用されないか、または実行できないことを発見する可能性がある。もし裁判所が私たちが再記載した会社登録証明書に含まれる裁判所条項の選択が訴訟で適用されないか、または実行できないことを発見した場合、私たちは他の管轄区域でこのような訴訟の解決に関連する追加費用を発生させる可能性があり、これは私たちの業務、財務状況、または運営結果に悪影響を及ぼす可能性がある。
116
一般リスク因子
追加資本の調達は私たちの株主に追加的な希釈をもたらし、私たちの運営を制限し、私たちの技術や候補製品に対する権利を放棄することを要求し、私たちの株価を下落させる可能性があります。
これまで、製品販売から相当な収入を得ることができれば、株式発行、債務融資、マーケティング、流通手配、その他の協力、戦略連合、許可手配の組み合わせで現金需要を満たすことができるかもしれません。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画のために十分な資金があると考えても、追加の資本を求めることができる。
私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、あなたの所有権資本は希釈され、これらの証券の条項は清算または他の特典を含む可能性があり、普通株主としての権利に悪影響を及ぼす可能性がある。債務融資および優先株融資に関連する可能性のある協定には、追加債務を招く、資本支出を行う、配当金を発表する、私たちの株を償還する、特定の投資を行うこと、特定の合併、合併または資産売却取引に従事するなど、私たちの業務の契約を制限または制限する能力が含まれる。もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配によってより多くの資金を調達する場合、私たちは、私たちの技術、将来の収入源、または候補製品に対する貴重な権利を放棄するか、または私たちに不利になる可能性のある条項でライセンスを付与することを要求されるかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できない場合、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは私たちは自分で開発とマーケティングをより望んでいた候補製品の権利を与えます。
特許法、及び米国及び他の司法管区裁判所及び他の機関の法執行を弱めることは、私たちの特許保護能力に影響を与える可能性がある。
過去数年間、米国最高裁判所と下級裁判所は特許事件においていくつかの意見を発表し、多くの人はこれらの意見が米国の特許保護を弱める可能性があると考えているか、あるタイプの革新は特許を申請できないと考える場合もあれば、いくつかのタイプの革新は特許を申請できないか、全体的に法廷で特許無効を宣言しやすいと考えている。また,最近では米国や他の国の特許法をより多く改正することが提案されており,採択されれば,我々のノウハウのために特許保護を受ける能力,あるいは我々のノウハウを実行する能力に影響を与える可能性がある。米国議会、米国裁判所、米国特許商標局、および他の国の関連立法機関および他の機関の将来の行動によれば、特許を管理する法律法規は予測不可能な方法で変化する可能性があり、それにより、私たちが新しい特許を獲得したり、私たちの既存特許と将来獲得可能な特許を実行し、保護する能力を弱める可能性がある。
一部の外国司法管轄区の法律は知的財産権の保護程度はアメリカに及ばず、多くの会社は外国の司法管轄区の保護と知的財産権の保護に重大な困難に直面している。もし私たちが知的財産権を保護する上でこのような困難に遭遇したり、他の理由で外国の管轄区域で私たちの知的財産権を効果的に保護できない場合、私たちのビジネスの見通しは深刻な損害を受ける可能性があります。例えば、私たちは、インドの患者オフィスの特許、外観設計、商標総監に許可前の反対意見を提出したという通知を受けた。AT-511の特許主張を弁護しようとしていますが、私たちが成功する保証はありませんし、インド特許庁が特許主張を承認する保証もありません。そうであれば,このような訴訟手続きの開始と継続による不確実な要因は,我々が市場で競争する能力に大きな悪影響を与える可能性がある.外国対抗訴訟の費用も高い可能性があり、多くの外国司法管轄区では、敗訴側は勝訴側の弁護士費を支払わなければならない。
私たちは買収や戦略的協力を行う可能性があり、これは私たちの業務を混乱させ、株主の株式希釈を招き、私たちの財務資源を減少させ、私たちに債務を発生させたり、負債を負担したり、他のリスクに直面させたりする可能性がある。
将来的には、他の業務、製品、または技術を得るために取引を行うことができ、または許可を含む戦略的協力を達成することができる。もし私たちが適切な買収や協力候補を見つけたら、私たちは有利な条項でこのような買収や協力を達成することができず、甚だしきに至っては完成できないかもしれない。どんな買収や協力も私たちの競争地位を強化しないかもしれませんが、これらの取引は
117
お客様や投資家に否定的に見られるかもしれませんが、私たちはこのような買収や協力の期待的な利益を決して達成しないかもしれません。私たちは、買収に関連する債務を発生させるか、または被買収会社の株主に私たちの普通株式または他の株式証券を発行することを決定することができ、これは私たちの既存株主の所有権パーセンテージを減少させるだろう。私たちは買収された業務や協力が発見されなかった債務によって損失を受ける可能性があり、これらの債務は私たちが売り手や私たちのパートナーから受ける可能性のある賠償範囲内ではない。また、任意の買収した人員、技術、運営を効果的、タイムリーかつ中断なく統合することができない可能性があります。買収や協力は、管理職の日常的な役割への関心を分散させ、キーパーソンの流失を招き、私たちの費用を増加させ、運営や他の用途に利用可能な現金や現金等価物を減らすことも可能である。将来の買収や協力の数、時間や規模を予測することもできず、どのような取引が私たちの経営業績に影響を与える可能性も予測できない。
私たちまたは私たちが依存している第三者は自然災害や流行病の悪影響を受ける可能性があり、私たちの業務の連続性や災害復旧計画は深刻な災害から私たちを十分に保護できないかもしれません。
新冠肺炎以外の自然災害(地震、火災、嵐、洪水、干ばつおよび極端温度を含むがこれらに限定されない)や流行病は、私たちの運営を深刻に混乱させ、私たちの業務、運営業績、財務状況、および見通しに実質的な悪影響を及ぼす可能性がある。気候変化はこのような事件の頻度や強度を増加させるかもしれない。また,気候変化は,温度や降水パターンの変化や海面上昇など,実環境の様々な長期変化を引き起こす可能性があり,これも我々の業務に悪影響を及ぼす可能性がある。自然災害、停電、流行病(例えば、新冠肺炎疫病)または他の事件が発生した場合、本社または従業員のすべてまたは大部分の本社または従業員がキー業務記録に連携してアクセスする仮想ネットワーク能力の使用を阻止し、キーインフラ(私たちが依存する製造施設のような)を破損したり、他の方法で運営を中断したりすることは、場合によっては長い間私たちの業務を継続することができない可能性がある。深刻な災害や同様の事件が発生した場合、我々の既存の災害復旧および業務連続計画は十分ではないことが証明される可能性がある。私たちの災害復旧と業務連続計画の限られた性質のため、私たちは大量の費用を発生する可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性があります。
計画に対するより多くの関心と変化の予想は、私たちのコストを増加させ、私たちの名声を損なうか、あるいは他の方法で私たちの業務に悪影響を及ぼすかもしれない。
様々な業界の企業は、そのESGおよび持続可能な開発実践に関連する様々な利害関係者がますます多くの審査に直面している。自発的なESGイニシアティブおよび開示の予想は、コスト増加をもたらす可能性があり(コンプライアンス、利害関係者の参加、契約、および保険に関連するコストの増加を含むが、これらに限定されない)、コンプライアンスまたは開示義務を強化するか、または私たちのサービス、財務状態、または運営結果に他の悪影響を与える。
当社および/または製品のESGイメージを改善するために、自発的な行動(例えば、自発的開示、認証、または目標など)をとることがあるが、そのような行動は、コストが高く、予期される効果を達成できない可能性がある。しかも、私たちがコントロールできない要素のせいで、私たちはこのような計画を成功的に達成できないかもしれない。そうでなくても、私たちの行動はその後、各利害関係者に不十分と認定される可能性があり、このような取り組みが現在自発的であっても、投資家や監督機関からESG努力への参加を受ける可能性がある。
特定の市場参加者は、主要機関投資家および資本提供者を含み、投資または投票決定を行う際に、第三者基準およびスコアを使用して、会社のESGプロファイルを評価する。不利なESG格付けは、投資家の私たちまたは私たちの業界に対する否定的な感情を増加させる可能性があり、これは私たちの株価および私たちが資金を得る機会とコストに悪影響を及ぼすかもしれない。ESGイベントが私たちの名声に悪影響を及ぼす程度では、従業員または顧客の能力を引き付けるために効果的な競争を阻害する可能性もあり、これは私たちの運営に悪影響を及ぼす可能性がある。
さらに、ESGトランザクションにおいて、開示に関連するより多くの規制レベルがある可能性が予想される。例えば、米国証券取引委員会は、定期報告で気候関連の開示を大幅に拡大することを求める提案された規則を公表している
118
これは、過去にこのような制御を受けていなかった事項に対して重大な追加内部制御プログラムおよびプログラムを実施し、我々の管理職および取締役会により多くの監督義務を課すことを含む、多くの追加コストを発生させることを要求する可能性がある。利害関係者が予想するこれらや他の変化は,コスト増加や審査を招く可能性があり,このリスク要因の中で決定されたすべてのリスクを悪化させる可能性がある.さらに、私たちの多くの顧客とサプライヤーは同様の予想の影響を受ける可能性があり、これは私たちが知らないかもしれないリスクを含めて追加的なリスクを増加または発生させる可能性がある。
私たちに対する訴訟は高価で時間のかかる弁護になるかもしれないし、追加的な責任を招くかもしれない。
私たちは時々法的手続きや通常の業務中または他の側面で発生したクレーム、例えば私たちの顧客または第三者が商業紛争について提起したクレーム、および私たちの現職または前任従業員が提出した雇用クレームの影響を受ける可能性がある。クレームは、政府機関、患者、または私たちの顧客のサプライヤーまたは株主を含む様々な他の当事者によってまたは代表されてもよい。従来、証券集団訴訟は、ある会社の証券市場価格が下落した後に提起されることが多かった。
私たちに関連する訴訟は、巨額のコストを招き、運営上私たちの業務を制限し、経営陣の注意と資源を分散させる可能性があり、これは私たちの業務、全体の財務状況、運営結果を深刻に損なう可能性があります。保険は既存または未来のクレームを含まない可能性があり、私たちの1つ以上のそのようなクレームを完全に賠償するのに十分ではないか、または私たちが受け入れられる条項で保険を提供し続けることができる。私たちが提出した保険や保険不足のないクレームは、予期せぬコストを招き、私たちの運営結果に悪影響を与え、私たちの株式の取引価格を低下させる可能性があります。
私たちの普通株の市場価格はずっと不安定で、大きな変動があるかもしれません。
私たちの株価はずっと変動していて、変動し続ける可能性が高い。我々の株価は極端に変動し、2021年の終値は2021年2月8日の高値88.44ドルから2021年11月23日の安値7.67ドルまで、2022年の終値は2022年1月3日の高値9.19ドルから2022年12月28日の安値4.35ドルまで様々だ。
株式市場全体、特にナスダック世界の精選市場、特にバイオ製薬会社の取引量は極めて大きな変動を経験し、この変動は特定の会社の経営業績を悪化させ、比例しないよりも、場合によってはその経営業績とは関係がない。この変動のせいで、普通株を売って利益を得ることができないかもしれません。私たち普通株の市場価格は多くの要素の影響を受けるかもしれませんが、これらに限定されません
119
私たちの実際の経営業績にかかわらず、広範な市場と業界要素は私たちの普通株の市場価格にマイナス影響を与える可能性がある。従来、証券集団訴訟は、会社証券の市場価格が変動した後に会社に提起されることが多かった。このような訴訟を提起すれば、巨額の費用と経営陣の注意と資源の移転を招く可能性があり、これは私たちの業務、財務状況、運営結果を損なうことになる。
120
財務報告の効率的な内部統制と効率的な開示制御プログラムを保持できなかった場合、財務結果をタイムリーに正確に報告したり、詐欺を防止したりすることができず、投資家がわが社の信頼に悪影響を及ぼす可能性がある。
我々は、米国証券取引委員会がサバンズ-オキシリー法案第302及び404節を実施する規則を遵守しなければならない。この2節は、経営陣に、我々の四半期及び年次報告書において財務及びその他の情報を認証し、財務報告の制御の有効性に関する年間管理報告を提供することを要求する。また、 私たちは、私たちの独立公認会計士事務所が発行した財務報告内部統制の証明報告書を含む必要があり、私たちの独立公認会計士事務所は、2022年12月31日までの年度の財務報告内部統制に対する第404条に基づく当社の内部統制の有効性を証明しなければならない。もし私たちが財務報告の内部統制に重大な欠陥を発見すれば、私たちの独立公認会計士事務所は不利な報告をするかもしれない。
第404条の要求を遵守するためには、新たな内部統制及び手続きを実施し、より多くの会計又は内部監査者を雇用するなど、他の行動をとる必要がある。我々は、引き続き内部資源を提供し、外部コンサルタントを招聘し、詳細な作業計画によって財務報告内部制御の十分性を評価し、記録し、引き続き適宜ステップ改善制御プログラムを採用し、このような制御がファイルのように機能しているかどうかをテストにより検証し、財務報告内部制御の継続報告及び改善手順を実施する必要がある。内部統制のテストと維持は、私たちの業務運営にも重要な他の問題から、私たちの経営陣の注意をそらすことができます。私たちは努力したにもかかわらず、私たちは規定された時間枠内で結論を出すことができないかもしれない、すなわち私たちは財務報告書の内部統制に有効であり、404条の要求に適合する可能性がある。財務報告書に対する私たちの内部統制を評価する際に、404条の要求を遵守するために設定された適用期間を満たすために、タイムリーに救済できない可能性のある重大な弱点を見つけることができます。我々の財務報告の内部統制に重大な弱点があることが発見された場合、または404条の要求を直ちに遵守できない場合、または財務報告の内部統制が有効であると断言したり、独立した公認会計士事務所が財務報告の内部統制の有効性に意見を述べることができない場合、投資家は私たちの財務報告の正確性および完全性に自信を失う可能性がある。したがって、私たちの普通株の市場価格は実質的な悪影響を受けるかもしれない。
私たちの開示統制と手続きはすべてのミスや詐欺を阻止したり検出できないかもしれない。
私たちは“取引所法案”の定期報告書の要求事項を守らなければならない。私たちは、取引所法案に基づいて提出または提出された報告書で開示されなければならない情報が蓄積され、管理層に伝達され、米国証券取引委員会規則および表で指定された期間内に記録、処理、まとめ、報告されなければならないことを保証するために、合理的な保証を提供するために、開示制御および手続きを継続している。いかなる開示規制と手続きも、構想や運営がどのように詳細であっても、絶対的な保証ではなく合理的な保証しか提供できず、規制制度の目標が達成されることを確保するしかないと信じている。
これらの固有の限界は,意思決定過程における判断が誤りである可能性があり,簡単な誤りや誤りによって故障が発生する可能性があるという現実を含む.さらに、ある人の個人的な行動、2人以上の結託、または無許可超越制御は、制御を回避することができる。したがって,我々の制御システム固有の制限により,誤りや詐欺による誤り陳述が発生し,発見されない可能性がある.
私たちは予測可能な未来に私たちの普通株に現金配当金を支払わないと予想されるので、資本付加価値(あれば)があなたの唯一の収益源になるかもしれません。
私たちは普通株式に対するどんな現金配当金も発表したり支払ったりしたことがない。私たちは現在、すべての利用可能な資金と将来の収益を維持し、業務の発展、運営、拡張のために、予測可能な未来に現金配当金を発表または支払うことはないと予想している。したがって、予測可能な未来に、私たちの普通株の資本増加は、あなたが私たちの普通株に投資する唯一の収益源かもしれません。
121
項目1 B。取消解析Dスタッフがコメントした。
適用されません。
プロジェクト2.専門家ペルテスです。
私たちの主なオフィスはマサチューセッツ州ボストンフランクリン通り225番地にあります。そこで17,544平方フィートのオフィススペースを借りました。私たちは転貸契約に基づいてこの空間を借り、レンタル期間は2022年1月1日から2026年12月31日までです。私たちの施設は私たちの現在の需要を満たすのに十分で、必要な時に適切な追加空間を提供すると信じている。
項目3.法律法律手続き。
私たちは実質的な法的手続きの制約を受けないだろう。
4つ目:地雷の安全TYが披露する。
適用されません。
122
第II部
項目5.登録者普通株式市場、関連株持株者は重要で発行者が株式証券を購入する。
普通株取引に関するいくつかの情報は
私たちの普通株は2020年10月30日からナスダック全世界精選市場に上場し、取引コードはAVIRである。
私たち普通株保有者
2023年2月25日現在、我々の普通株には21人の登録株主がおり、その株が仲介人によって代名人や街頭名義で保有されていることは反映されていない。
配当をする
私たちは普通株式に対するどんな現金配当金も発表したり支払ったりしたことがない。私たちは現在、すべての利用可能な資金と任意の将来の収益を維持し、私たちの業務の発展と拡張に資金を提供するつもりですので、予測可能な未来に現金配当金は支払われません。いかなる未来の配当金の決定は私たちの取締役会が適宜決定し、私たちの財務状況、経営結果、資本要求、業務の見通し、契約の手配、任意の未来の債務協定における配当金の支払いに対するいかなる制限、取締役会が関連すると考える可能性のある他の要素に依存し、任意の未来の融資ツールに含まれる制限を受けるだろう。
収益の使用
2020年11月3日,普通株の初公開(IPO)を完了し,これにより,1株24.00ドルで14,375,000株の普通株を一般公開·売却した。
初公開で発行·売却されたすべての株式は、米国証券取引委員会が2020年10月29日に発効を発表したS-1表登録声明(第333-249404号文書)(以下、“登録声明”)に基づいて、証券法に基づいて登録されている。
引受割引と手数料および発売費用を差し引いた後、約3億176億ドルの純収益を得た。
私たちのIPOの純収益は主に通貨市場口座と有価証券に投資されている。我々は、証券法第424(B)(4)条に基づいて2020年10月29日に米国証券取引委員会に提出された目論見書に記載されているように、初めて公募して得られた金の期待用途に大きな変化はない。
株式表現グラフ
業績グラフは,2020年10月30日(我々普通株の株式初公開取引)から2022年12月31日までの,我々の普通株と標準プール500指数とナスダックバイオテクノロジー指数の表現を比較した。この比較は,2020年10月30日終値後,我々の普通株と上記各指数に100ドルが投資され,配当金(あれば)が再投資されると仮定している。この図に含まれる株価表現は,必ずしも将来の株価表現を予測するつもりもないことを示しているとは限らない.
123
以下の業績グラフ及び関連情報は提供されており、“募集材料”とみなされてはならないし、取引法第18条に基づいて米国証券取引委員会に“届出”されているものとみなされてはならないし、引用によってそのような届出文書に明示的に組み込まれない限り、そのような情報を取引法または証券法に基づいて提出された任意の届出文書に組み込むものとみなされてはならない。

第六項です。保留されている
124
項目7.経営陣の以下の問題の議論と分析財務状況と経営実績。
以下、我々の財務状況および経営結果の検討および分析は、当社の財務諸表および本年度報告書の他の部分がForm 10-K形式で含まれるこれらの報告書に関する注釈と共に読まなければならない。歴史財務情報を除いて、以下の議論と分析には、リスク、不確定性、および仮説に関連する前向きな陳述が含まれている。多くの要素、第1の部分1 A項“リスク要因”および本年度報告10-K表の他の部分で議論された要因を含むため、選択されたイベントに対する実際の結果および時間は、これらの前向き陳述において予想されるものと大きく異なる可能性がある。“前向きな陳述に関する特別な説明”を参照されたい
概要
著者らは臨床段階の生物製薬会社であり、深刻なウイルス感染患者の生活を改善するために、抗ウイルス治療薬の発見、開発と商業化に専念している。我々は新冠肺炎の治療に用いられる主要候補製品であるフェニフブビルを開発しており,SARS−CoV−2とその変種に感染することによる疾患である。フェニルニホブビルとルザスビルの併用によるC型肝炎ウイルスの治療も開発されている。
新冠肺炎は全世界の健康危機を引き起こし、数百万人の人が死亡し、多くの生存者の医療問題が頭から離れない。新冠肺炎の予防と治療において多くの迅速な進展を得たが、現在ワクチンと治療選択の制限のため、アメリカと全世界の大量のハイリスク群に対する医療需要はまだ満足されていない。われわれの新冠肺炎戦略の中核は,ベネホブビルを単一療法の開発とし,新冠肺炎療法の一部とすることが可能であり,ベネホブビルと別の抗ウイルス薬を組み合わせ,これらの現在のワクチンや治療法が依然として適用されていないハイリスク患者に集中することである。我々の目標は,依然としてSARS−CoV−2感染により入院·死亡しやすい個人に安全で有効かつ便利な治療選択を提供することである。
ワクチンや治療法があっても,新冠肺炎は米国で3番目の死亡原因であり,心臓病や癌に次ぐ。アメリカ疾病コントロール·予防センターの報告によると、2023年2月15日まで、アメリカでは毎日400人を超える人が新冠肺炎或いは関連合併症で死亡した。この人たちの75%以上の人の年齢が65歳以上だ。また,最近,2023年2月15日現在,60歳以上が米国の現在の新冠肺炎に関連する入院患者の70%を占めることが報告されている。
米国政府は最近、新冠肺炎に関連する公衆衛生緊急事態の発表を終了する計画を発表したが、新冠肺炎は今後しばらく深刻な地方的脅威になると予想される。新冠肺炎がまだ地方的脅威になる可能性がある原因は:(1)症状出現前のウイルス伝播;(2)全世界のワクチンの発売にばらつきがある;(3)ワクチンの持続的な躊躇;(4)自然感染とワクチン接種による免疫持続時間は限られている;(5)あるSARS-CoV-2変種に対するワクチンの効力は限られている;(6)薬物と薬物の相互作用、安全性と耐性などの既存の抗ウイルス薬物の局限性を内服する;(7)ワクチンの伝播に対する不確定な影響;(8)ウイルスは進化し続け、内因性免疫とワクチン誘導の免疫を逃避する;(9)マスク着用や社交距離のようなウイルス伝播の緩和行動を減少させる.
SARS-CoV-2変種の絶えずの出現はより大きな伝播性を持ち、更に深刻な疾病を招く可能性があり、更に普通の人群中の他のウイルス緩和行為の減少、及び公衆衛生緊急休暇の終了に関連する結果が予想され、現在の治療方法はこれらの患者にとって限られており、特にウイルスと関連疾病の影響を受けやすい。これらの要因を考慮して,我々はこれらの脆弱な患者のニーズを満たすために潜在的な治療法としてbemnifosbuvirを開発している。
新冠肺炎が引き続き深刻な全世界風土病になることに伴い、私たちはアメリカが引き続き最も重要な商業市場を占めることに伴い、新冠肺炎治療市場は今後数年間も数十億ドルの機会になると信じている。アメリカでは、新冠肺炎商業市場はすぐに単一の政府支払者からより伝統的な支払者ルート、例えば連邦医療保険、連邦医療補助、個人商業保険に移行することが予想される。私たちは予定しています
125
これらの第三者支払者の補償を決定する際には,コスト/価値分析を行うことが考えられ,これはある程度入院の経済的負担によって推進され,特にハイリスク群である。
ベニホブビル
著者らのチームは数十年の革新的な抗ウィルス治療で得られた専門知識と経験を利用して、Bemnifosbuvirを設計し、1種の研究中の、独自の、有効かつ選択的なヌクレオチドポリメラーゼ阻害剤は、単一療法のすべての薬物として開発することができ、他の抗ウィルス薬物と共同開発することもできる。Bemnifosbuvir(AT-527)は著者らの内部発見計画に由来し、この計画は独特なヌクレオチドステントを新型二プロドラッグと結合し、目的はウイルス複製の中心酵素を抑制することである。このような二重プロドラッグ部分を利用した方法では、ベンゼンフォブビルの活性代謝物を最大限に形成し、単鎖RNAウイルスの複製を防止しながら宿主細胞への毒性を回避することを目的とした経口抗ウイルス製品を創出することができると信じている。また,非臨床研究において,bemnifosbuvirはRdRp鎖の中止とSARS−CoV−2ウイルスの抑制とその変種のNiranを含む独自の作用機序を有することが証明された。このようなユニークな二重作用機序によりこれらの高度に保守的な部位を標的にすることにより,bemnifosbuvirは高い耐性障害を生じる可能性がある。さらにここでは体外培養我々が行った研究では,ベニフォブビルは新冠肺炎揮発性有機化合物中でその抗ウイルス活性を保持し,試験されたすべてのオミク戎亜型を含む。
新冠肺炎の臨床研究
2022年11月、著者らは世界的、多中心、無作為、二重盲検、プラセボ対照の3期臨床試験であるSunISE-3を開始した。日の出3日には,少なくとも1,500名の軽中度新冠肺炎を有するハイリスク非入院患者に対して,フェニホブビル(550 mg,bid,5日間)の評価を行っている。この試験は米国,ヨーロッパ,日本,世界の他地域の臨床試験地点で行われる。患者群は疾患進展リスクが最も高い群から構成され、80歳の患者80歳、1つ以上の主要なリスク因子を有する65歳患者および18歳の免疫機能低下患者を含み、これらはすべて新冠肺炎接種状態と関係がない。
“日の出3号”の設計目的は,ベネホブビルを単一療法として評価(予備分析)することであるが,抗ウイルス薬をベネホブビルとともに治療を受ける比較的小さい患者のサブセットにおける併用療法の効果も探索する(二次分析)。この実験には、1)“支持性看護集団”(承認された抗ウイルス治療を受ける資格のない患者、または現地で抗ウイルス薬を有さない患者)の2つの群が含まれ、単一療法としてベンゼンフォブビルに投与された患者(予備分析)および2)“併用抗ウイルス群”が評価され、SOCが他の互換性のある抗ウイルス薬と共に新冠肺炎に対する治療を含む場合には、併用療法(二次分析)が評価される。患者は、Bemnifosbuvir 550 mg Bidプラスローカル利用可能SOCまたはプラセボBIDプラスローカル利用可能SOCを1:1のランダム割合で5日間受け入れる。
日の出-3研究の主な終点は、支持性看護集団中の少なくとも1300人の患者のすべての原因が入院または死亡して29日目までであり、この集団においてプラセボと比較して臨床的意義のある入院/死亡の減少を検出する能力があることである。試験参加患者の中で最も疾患進展リスクの高い患者を増加させることにより,入院率/死亡率は~4−6%を目標とした。60%の患者が研究ARMに登録し,支持性看護群に組み入れた後,DSMBによる中期分析を行った。各支持性看護患者群と連合抗ウイルス群中の副次的な終点は新冠肺炎合併症、受診、症状リバウンド/再発とウイルス負荷リバウンドを含む。
日の出3号の臨床試験を行うとともに,候補蛋白加水分解酵素阻害剤製品の発見に努めており,フェニニホブビルと併用し,免疫反応が生じず併用治療が必要な特定の新冠肺炎患者集団の治療に用いることができる。私たちはすでに行いました体外培養ベニホブビルとネマレビルを含むプロテアーゼ阻害剤類の抗ウイルス薬を併用した場合,付加的な抗ウイルス作用があることが明らかになった。私たちは、共同治療を受けた患者のサブセットから行われる日の出−3号臨床試験から得られるデータは、Bemnifosbuvirといくつかの他の現在許可されている抗ウイルス治療との組み合わせを評価する第1の臨床データであると信じている。
126
総合療法
多種の異なる作用機序を有する直接作用抗ウィルス薬物を利用して連合治療を行うことは既定の策略であり、HIV、B型肝炎ウイルスとC型肝炎を含む多くの生命を脅かすウイルス疾患の治療において歴史上の成功を得た。核種(T)類似体は多くの成功した併用療法の柱である。有利なことに,薬物の組み合わせはウイルス複製周期における複数の点に対して同時に抗ウイルス活性を増加させる効果があり,単一の薬物を用いることで時間の経過とともに形成される可能性のある薬剤耐性にも対抗することができる。
C型肝炎ウイルスの臨床研究
慢性C型肝炎ウイルス感染の治療にはbemnifosbuvirとRuzasvirの併用が進められており,Ruzasvirは研究中のNS 5 A阻害剤である。全世界で約5800万人が慢性C型肝炎ウイルスを患っており,その中で米国は約240万人である。世界保健機関の報告によると、全世界の毎年の発病率は150万例であり、死亡者数は39.9万人である。C型肝炎の発病率の上昇に伴い、治療を受けた新しい患者の数量を相殺し、今後数年のアメリカのC型肝炎の流行率は変わらないと予想される。
著者らはbemnifosbuvirとruzasvirの結合は現在の看護標準を改善する可能性があると信じ、肝硬変を合併或いは合併しないC型肝炎ウイルス感染患者に差別化された短い治療コース、汎遺伝子型プロテアーゼ保留方案を提供する。
2023年第2四半期に,bemnifosbuvirとruzasvirを併用して単純なC型肝炎ウイルス感染患者を治療する第2段階臨床試験を開始する予定であり,肝硬変なしでも代償性肝硬変でも。本研究では,汎遺伝子組合せ治療8週間後の安全性と有効性を評価することを目的とし,550 mg/dのベネホブビルと180 mgのruzasvirからなる。約60名の患者を含む全遺伝子型の約280名のC型肝炎ウイルスに感染した未治療患者が,この第2段階の臨床試験に参加する予定である。研究の主な終点は安全性と治療後12週間の持続ウイルス学的応答(SVR)である。他のウイルス学的終点はウイルス学的失敗、治療後24週間のSVRと薬剤耐性を含む。
デング熱と呼吸器合胞体ウイルス
2023年2月,AT−752を第2段階臨床試験に進めた後,デング熱の治療および予防にAT−752をさらに開発しないことにした。この行動をとったのは,患者登録予定期間が長く,感染直後に抗ウイルス薬の使用に成功した挑戦を含む期待される臨床操作課題であり,現在の診断テストでは不可能であり,大量のコストを含む個々のデング熱治療や予防のための抗ウイルス薬のさらなる開発に関する推定資源負担があるからである。
われわれは最近,呼吸器合胞体ウイルス(RSV)を治療する候補製品を決定するために,われわれの発見作業をさらに行わないことにした。この行動は私たちの管理チームのより集中を促進し、私たちの他の資源を私たちが計画しているより先進的な治療適応に配置するためです。
財務資源
私たちは私たちが現在の計画を推進するための十分な資本を持っていると信じている。2022年12月31日現在、私たちは6.467億ドルの現金、現金等価物、有価証券を持っている。私たちの現在の計画によると、これらの財務資源は、現在と計画中の臨床プロジェクトを重要な転換点に推進し、重要な転換点を通過し、2026年まで私たちの活動に資金を提供することができると予想される。
羅氏許可協定
2020年10月,我々はF.Hoffmann−La Roche LtdとGenentech,Inc.と羅氏許可協定を締結し,化合物,製品,Companion Diagnosticsの世界開発,製造,商業化に触れた。羅氏許可協定の条項と条件に基づき、羅氏と我々は世界規模で新冠肺炎のためのベノホビルを共同開発し、このような開発活動に関連するコストを折半した。
127
羅氏許可協定に基づいて羅氏に付与された権利の一部の対価格として、羅氏は2020年11月に3億5千万ドルを前払いした。また、2021年6月に発展マイルストーンを実現した後、私たちは羅氏から5000万ドルを追加した。
羅氏が2021年11月に出した終了通知を受けた後、羅氏許可協定は2022年2月10日に終了した。私たちが羅氏と費用を分担する義務もその時に終わった。羅氏許可協定の終了に伴い、私たちは羅氏がすべての使用分野で研究、開発、製造、商業化化合物、製品、および診断に伴う世界的な独占的権利を再獲得した。
財務運営の概要
2022年12月31日現在、私たちは6.467億ドルの現金、現金等価物、有価証券を持っている。2022年12月31日までの1年間、経営活動のための現金純額は1.21億ドルだった。
私たちは、臨床前と臨床開発を通じて私たちの候補製品を推進し、規制部門の承認を求め、承認されれば、商業化を開始する準備と、より多くの候補製品を獲得、発見、検証、開発すること、私たちの知的財産権の組み合わせを獲得、維持、保護、実行すること、およびより多くの人員を募集することで、私たちが経営活動で使用する純現金は大幅に増加すると予想している。また、私たちは上場企業として運営を続けているので、追加コストが発生する可能性があります。私たちは私たちの利用可能な現金と現金等価物が2026年まで私たちの計画中の業務に資金を提供するのに十分だと信じている。
私たちはどんな製品も販売を許可されておらず、設立以来何の製品収入も発生していない。予測可能な未来に、私たちは製品販売から何の収入も得られないと予想される。私たちが製品収入を作る能力は、私たちの1つ以上の候補製品の成功的な開発、規制承認、最終商業化にかかっているだろう。私たちが製品販売から相当な収入を得ることができる前に、もしあれば、私たちは私募または公共株式または債務融資、第三者との協力または他の手配、または他の融資源を通じて私たちの運営に資金を提供する予定だ。受け入れ可能な条件で、私たちは十分な資金を得ることができないかもしれないし、全く持っていないかもしれない。もし私たちが必要な時に資金を調達したり、このような合意に到達できなければ、私たちは候補製品の開発と商業化を大幅に延期、削減、または停止しなければならないかもしれない。
我々はCROを含む第三者サービスプロバイダを引き続き使用して、私たちの臨床前と臨床開発を行い、CMOは私たちの候補製品開発過程で使用される材料を製造し、供給する予定である。また、私たちはCMOに依存して、私たちが開発に成功する可能性のある任意の候補製品の商業供給を製造したい。
私たちが引き続き私たちの計画を進めるにつれて、私たちは今後数年以内に著しく高い費用が発生すると予想する。私たちは持続的な活動に関連する費用が大幅に増加すると予想しています
128
経営成果の構成部分
収入.収入
私たちは何の製品も販売を許可されていません。今まで、私たちは製品販売から何の収入も得ていません。
我々の収入は、2020年10月に発効し、2022年2月10日に終了する羅氏許可協定からの完全な協力収入である。もし私たちが候補製品の開発に成功して商業化を実現すれば、私たちは将来的に製品販売や第三者と締結された協力またはライセンス契約支払いの組み合わせによって追加収入を生成するかもしれない。
運営費
研究と開発費
私たちのほとんどの研究と開発費用は私たちの候補製品開発に関する費用です。これらの費用には、CROおよびCMOを含む第三者に支払われる代表者が特定の研究開発活動を行う費用および相談費用、および我々の研究開発者の給料およびボーナス、従業員福祉コスト、株式ベースの給与支出、および賃貸料、設備、減価償却、情報技術コスト、研究開発者が占めるべき公共事業費を含む分配された管理費用が含まれる。私たちの内部と外部の研究開発費は実際に発生した方式で支出されている。前払い金額または発生したコストを超えた場合、サービスを提供するか、または貨物を渡す際に支出する前払い費用を記録します。
われわれの研究開発コストの大部分は外部コストであり,治療領域に応じて追跡している。私たちは複数の計画に配置されているので、私たちの内部研究開発費を治療分野で追跡していない。
我々の総合財務諸表付記3で述べたように、羅氏許可協定が2022年2月に終了した間、私たちと羅氏は50/50の割合でいくつかの製造と臨床開発コストを分担した。羅氏は私たちがこのような費用に占める割合について私たちに出した勘定書を研究開発費に記録しています。2021年12月31日までの1年間で、これらのコストは私たちの研究開発費の大きな部分を占め、私たちの総支出の大きな部分を占めています。2022年12月31日までの1年間に、羅氏から得られた信用により、我々が記録した研究開発費は690万ドル純減少した。これらの積分はロ氏許可プロトコル終了後の変更と調整の結果である[作者:羅氏]羅氏は我々と羅氏にフェニフブビルの開発に関する費用を分担している間に報告した見積費用金額を報告した。
次の表は指示に従って私たちの外部研究開発費と内部研究開発費をまとめました
129
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
新冠肺炎の外部コスト |
|
$ |
19,546 |
|
|
$ |
93,508 |
|
|
$ |
23,043 |
|
デング熱外コスト |
|
|
11,150 |
|
|
|
9,396 |
|
|
|
2,167 |
|
C型肝炎ウイルスの外部コスト |
|
|
5,817 |
|
|
|
27,514 |
|
|
|
1,831 |
|
RSV外部コスト |
|
|
2,135 |
|
|
|
1,887 |
|
|
|
1,127 |
|
内部研究開発コスト |
|
|
43,288 |
|
|
|
34,900 |
|
|
|
9,855 |
|
研究開発費総額 |
|
$ |
81,936 |
|
|
$ |
167,205 |
|
|
$ |
38,023 |
|
私たちはほとんどの資源を私たちの候補製品の開発、特にベニホブビルに集中しています。私たちは、少なくとも今後数年以内に、私たちの候補製品のためにより多くの臨床試験を開始し、私たちの臨床計画を完成させ、規制部門の私たちの候補製品の承認を求め、これらの候補製品の商業化に準備することを求めているので、私たちの研究と開発費用は大幅に増加すると予想される。私たちの臨床計画を完成したり、私たちの商業製造と供給過程の時間或いはコストを検証することは困難であり、多くの要素によって遅延が発生する可能性があり、私たちがコントロールできない要素を含む。例えば、FDAや他の規制機関が現在予想されている臨床試験を行うことを要求した場合、臨床開発を達成するために多くの追加の財政資源と時間が必要になるかもしれない。しかも、私たちは私たちの候補製品がいつ、あるいは規制部門の承認を受けるかどうかを確実に予測することができない。
一般と行政費用
一般及び行政支出は主に賃金及び人件費を含み、賃金及び花紅、福祉及び株式給与支出、コンサルティング、会計及び税務サービスの専門費用、法律費用及び知的財産権及び会社事務に関連する支出、分配された間接費用(家賃、設備、減価償却、情報科学技術コスト及び一般及び行政人員が占めるべき光熱費を含む)、及びその他の研究開発支出に分類されない一般運営支出を含む。
人員コストの増加、インフラの拡大、コンプライアンス、コンサルティング、法律および会計サービスコストの増加、および増加していく研究·開発活動の支援に関連するコスト、我々の候補製品のより高度な臨床開発、および私たちが成功する可能性のある任意の候補製品の潜在的商業化に備えた活動の開始により、私たちの一般的かつ行政的費用は増加し続けると予想される。上場企業として増加している経営コストは、追加の一般的かつ行政費用にもつながると予想される。
利子収入とその他の純額
利息収入とその他の純額は主に私たちの現金、現金等価物、有価証券が稼いだ利息収入からなります。
所得税
所得税は主に連邦と州の当期所得税で構成される。
130
経営成果
2022年と2021年12月31日終了年度比較
以下の表に示した期間の業務成果をまとめる
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
|
|
(単位:千) |
|
|||||||||
協力収入 |
|
$ |
— |
|
|
$ |
351,367 |
|
|
$ |
(351,367 |
) |
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
81,936 |
|
|
|
167,205 |
|
|
|
(85,269 |
) |
一般と行政 |
|
|
48,714 |
|
|
|
45,785 |
|
|
|
2,929 |
|
総運営費 |
|
|
130,650 |
|
|
|
212,990 |
|
|
|
(82,340 |
) |
営業収入(赤字) |
|
|
(130,650 |
) |
|
|
138,377 |
|
|
|
(269,027 |
) |
利子収入とその他の純額 |
|
|
11,151 |
|
|
|
213 |
|
|
|
10,938 |
|
所得税前収入 |
|
|
(119,499 |
) |
|
|
138,590 |
|
|
|
(258,089 |
) |
所得税の割引 |
|
|
3,590 |
|
|
|
(17,400 |
) |
|
|
20,990 |
|
純収益(赤字) |
|
$ |
(115,909 |
) |
|
$ |
121,190 |
|
|
$ |
(237,099 |
) |
収入.収入
2021年12月31日までの年間協力収入は、2020年10月に署名され、2022年2月に終了した羅氏許可協定から来ている。本年度報告10−K表の他の部分の連結財務諸表付記3で述べたように、2021年11月に羅氏の合意終了通知を受けた後、2021年の残りの繰延収入をすべて確認した。2022年12月31日までの1年間、私たちは協力収入を持っていない。
研究と開発費
研究·開発費は2021年12月31日までの年度の1億672億ドルから2022年12月31日までの年度の8190万ドルに減少し、8530万ドル減少した。研究·開発費が減少した要因は,外部費用が9370万ドル減少したことである。この減少は,主に我々が2022年2月にフェノブビルの開発による羅氏のコスト分担義務を停止したためである。この費用分担義務は羅氏許可協定の終了とともに同時に終了します。羅氏許可協定に関連して、2022年12月31日までの年間融資先は690万ドルを記録したが、2021年12月31日までの年間支出は7660万ドルであった。また、2021年12月31日までの1年間に、メルク許可協定の一括払いに関連した2500万ドルの費用を記録した。内部コスト増加840万ドルが部分的に相殺されたのは、主に給与とボーナス、福祉、株式ベースの報酬支出320万ドル、研究·製品開発従業員や相談費、その他の研究·開発費100万ドルを含む関係者関連の支出が増加したためだ。
一般と行政費用
一般·行政費は290万ドル増加し、2021年12月31日現在の4,580万ドルから2022年12月31日現在の4,870万ドルに増加した。一般および行政費用増加の主な原因は、賃金、福祉、株式ベースの報酬支出330万ドルを含む賃金と人事関連費用の420万ドル増加を反映した本組織の拡大であり、専門費用は120万ドル減少し、その他の一般的および行政費用は60万ドル増加した。
利子収入とその他の純額
2022年12月31日までの年間で、利息収入やその他の純額は、2021年12月31日までの年間より1090万ドル増加しており、これは主に収益の高い有価証券とより高い金利に投資しているためである。
131
所得税
2022年12月31日現在の事業年度では、所得税の純収益は360万ドルであるのに対し、2021年12月31日現在の事業年度は1740万ドルに充てられている。2022年12月31日までの年間記録された純収益は,主に我々の2021年の所得税の原始支出と申告された所得税申告書に反映された実金額との推定が変化した結果である。2021年12月31日までの年度に,主に羅氏許可協定から受け取った金額で稼いだ税前収入に関する所得税支出を記録した。
2021年12月31日までと2020年12月31日までの年度比較
以下の表に示した期間の業務成果をまとめる
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2021 |
|
|
2020 |
|
|
変わる |
|
|||
|
|
(単位:千) |
|
|||||||||
協力収入 |
|
$ |
351,367 |
|
|
$ |
48,633 |
|
|
$ |
302,734 |
|
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
167,205 |
|
|
|
38,023 |
|
|
|
129,182 |
|
一般と行政 |
|
|
45,785 |
|
|
|
21,640 |
|
|
|
24,145 |
|
総運営費 |
|
|
212,990 |
|
|
|
59,663 |
|
|
|
153,327 |
|
運営損失 |
|
|
138,377 |
|
|
|
(11,030 |
) |
|
|
149,407 |
|
利子収入とその他の純額 |
|
|
213 |
|
|
|
83 |
|
|
|
130 |
|
所得税前収入 |
|
|
138,590 |
|
|
|
(10,947 |
) |
|
|
149,537 |
|
所得税費用 |
|
|
(17,400 |
) |
|
|
— |
|
|
|
(17,400 |
) |
純収益(赤字) |
|
$ |
121,190 |
|
|
$ |
(10,947 |
) |
|
$ |
132,137 |
|
収入.収入
2021年12月31日と2020年12月31日までの年度の連携収入は,2020年10月に署名された羅氏許可協定から来ている。ロ氏許可契約の会計処理については、羅氏終了通知を受けた後のすべての残り繰延収入の確認を含め、本年度報告に他の場所のForm 10−Kに含まれる連結財務諸表付記3を参照されたい。
研究と開発費
研究開発費は1億292億ドル増加し、2020年12月31日までの年度の3800万ドルから2021年12月31日までの年度の1億672億ドルに増加した。研究開発費の増加は、主に新冠肺炎、C型肝炎およびデング熱候補治療製品の推進に関連するCROおよびCMOサービスによる外部費用の1.041億ドルの増加であり、その中には、私たちが分担すべき羅氏費用7,660万ドルとメルク社のライセンス契約前払いに関連する2,500万ドルの費用が含まれている。また、内部支出が2,500万ドル増加したのは、主に給与·ボーナス、福祉、株式ベースの報酬支出1,460万ドル、我々の研究·製品開発従業員·相談費、その他の研究·開発費240万ドルを含む人員関連支出の増加によるものである。研究·開発費は730万ドル減少し、これは羅氏がASC 808規格で発生したいくつかの費用シェアの精算であり、これらの費用は、本年度報告Form 10−Kの他の部分を含む当社が監査した連結財務諸表付記3で検討されている。
一般と行政費用
一般·行政費は2410万ドル増加し、2020年12月31日までの年度の2160万ドルから2021年12月31日までの年度の4580万ドルに増加した。一般費用と行政費用増加の主な原因は本組織の拡大であり,賃金と人員関連費用の2 060万ドルの増加を反映しており,賃金,福祉,株を含む
132
報酬支出は1760万ドル、専門費用は260万ドル増加し、その他の一般的で行政費用は90万ドル増加した。
利子収入とその他の純額
2021年12月31日までの年間で、利息収入やその他の純額が2020年12月31日までの年間より10万ドル増加したのは、主に現金等価物残高の増加によるものだ。
所得税
2021年12月31日と2020年12月31日までの年間所得税はそれぞれ1740万ドルと200万ドル。2021年12月31日までと2020年12月31日までの実質税率はそれぞれ12.5%と0%である。所得税支出の増加は主に私たちが以前羅氏と協力して2021年に確認した収入によるものだ。
流動性と資本資源
流動資金源
2022年12月31日現在、私たちは6.467億ドルの現金、現金等価物、有価証券を持っている。私たちは、私たちの利用可能な現金、現金等価物、および有価証券が、2026年まで私たちの計画中の業務に資金を提供するのに十分であると信じている。
私は2021年にJeffries,LLC(“Jeffries”)と公開市場販売プロトコル(“販売プロトコル”)を締結し、この合意によると、吾らは時々販売代理や依頼者を務めるJeffriesに普通株式を発売または販売することができ、総発行価格は最高2億ドルに達する。私たちはJeffriesに株式販売総収益の3.0%の手数料を支払い、法的費用と支出を返済し、Jeffriesに常習的な賠償と出資権を提供することに同意した。2022年12月31日現在、販売契約に基づいていかなる株式も発行されていない。
将来の資金需要
今まで、私たちはまだどんな製品収入も発生していない。私たちは規制部門から任意の候補製品の承認を得て商業化されない限り、いかなる製品収入も生じないと予想され、これがいつ、または発生するかどうかを知らない。私たちは予測可能な未来により多くの支出が発生することを予想し、私たちが引き続き私たちの候補製品を開発し、規制部門の承認を求め、任意の承認された製品を商業化し始めるにつれて、私たちの支出は増加すると予想される。私たちは新製品の候補製品開発に関連するすべてのリスクに支配されています。私たちは予測できない費用、困難、複雑な状況、遅延、その他私たちの業務に悪影響を及ぼす可能性のある未知の要素に遭遇する可能性があります。また、我々が上場企業として運営を継続し、候補製品のより高度な臨床開発を支援するための組織を拡大し、候補製品の潜在的な商業化に備えた活動の開始に伴い、追加の一般的かつ行政的コストが生じることが予想される。
私たちは引き続き私たちの候補製品を開発し、予測可能な未来に運営に資金を提供するために追加の資金を必要とするだろう。私たちは、公共またはプライベート·エクイティまたは債務融資、第三者との協力計画、または他の融資源による資金調達を求めることができる。私たちは多くの追加資本を集める必要があるかもしれません。必要な資金は多くの要素に依存します
133
我々の任意の候補製品の開発に関連するこれらまたは他の変数の結果の変化は、私たちの1つまたは複数の候補製品の開発に関連するコストおよび時間を著しく変化させることができる。また、私たちの運営計画は将来的に変化する可能性があり、運営需要やそのような運営計画に関連する資本要求を満たすための追加の資本が必要となります。もし私たちが株式証券を発行することでより多くの資金を調達すれば、私たちの株主は希釈されるかもしれない。私たちが将来参加する任意の債務融資は、私たちに留置権または追加債務の発生、配当金の支払い、私たちの普通株の買い戻し、特定の投資または特定の合併、合併、または資産売却取引に従事する能力の制限を含む、私たちの業務を制限する追加契約を加えるかもしれない。私たちが調達した任意の債務融資または追加株式には、私たちまたは私たちの株主に不利な条項が含まれているかもしれない。
受け入れ可能な条件で、私たちは十分な資金を得ることができないかもしれないし、全く持っていないかもしれない。私たちは必要な時に資金を集めることができず、私たちの財務状況と私たちの業務戦略を実施する能力にマイナス影響を与えるかもしれません。もし私たちが必要な時により多くの資金を集めることができない場合、私たちは私たちの開発計画および臨床試験の一部または全部を延期、減少または終了することを要求されるかもしれないし、あるいは他の人が特定の地域または私たちが自分で開発し商業化したい適応で私たちの候補製品を使用することができるかもしれない権利を販売することを要求されるかもしれない。もし私たちが私たちの資金を補充するために協力や他の手配を要求された場合、私たちは私たちの候補製品を開発して商業化する能力を制限するいくつかの権利を放棄しなければならないかもしれないし、私たちまたは私たちの株主に不利な他の条項があるかもしれません。これは私たちの業務や財務状況に大きな影響を与えるかもしれません。
市場変動、インフレ、金利変動及び新冠肺炎疫病と地政学事件に関連する懸念は、内乱或いは政治動乱(例えばウクライナとロシア間の持続的な衝突)を含み、資金源の獲得可能性及び任意の資金を得ることができる条項に重大な影響を与える可能性がある。
我々の巨額資本要件に関連する追加リスクについては、第1部、第1 A項、“リスク要因”を参照されたい。
現金流量集計表
以下の表に、以下の各期間の現金、現金等価物、および限定的な現金の主なソースおよび用途を示す
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
経営活動提供の現金純額 |
|
$ |
(120,982 |
) |
|
$ |
(87,005 |
) |
|
$ |
296,734 |
|
投資活動のための現金純額 |
|
|
(455,410 |
) |
|
|
(4 |
) |
|
|
(26 |
) |
融資活動が提供する現金純額 |
|
|
370 |
|
|
|
1,465 |
|
|
|
531,748 |
|
現金·現金等価物と現金純増(マイナス) |
|
$ |
(576,022 |
) |
|
$ |
(85,544 |
) |
|
$ |
828,456 |
|
経営活動のキャッシュフロー
2022年12月31日までの年度の経営活動に用いられる現金純額は1.21億ドル。経営活動で使用されている現金は主に純損失1.159億ドル,有価証券割増と割引により550万ドル増加し,前払い費用とその他の資産は7.7ドル増加した
134
売掛金と売掛金は3960万ドル減少し、4670万ドルの株式報酬部分で相殺された。
2021年12月31日までの年度の経営活動に用いられる現金純額は8700万ドル。業務活動で使用されている現金は主に純収入が1.212億ドル増加し,株式報酬が3960万ドル増加し,売掛金や売掛金が5420万ドル増加し,前払い費用やその他の資産が60万ドル増加し,これらは繰延収入の3.014億ドル減少によって相殺されている。
2020年12月31日までの年度、経営活動が提供する現金純額は2兆967億ドル。経営活動が提供する現金は、主に羅氏許可協定に関する繰延収入が3.014億ドル増加し、売掛金と売掛金が1190万ドル増加し、株式ベースの報酬が750万ドル増加したが、一部は我々の業務における候補製品開発のための資金によって相殺され、1090万ドルの純損失となった。この期間の現金の他の用途は、前払い費用および他の流動資産の730万ドルの増加、および未開請求書の売掛金の580万ドルの増加を含む。繰延収入の純増加301.4ドルは前金3.5億ドルが確認された収入4,860万ドルで相殺された結果である。
投資活動によるキャッシュフロー
2022年12月31日までの年度の投資活動用現金純額は4.554億ドルで、190万ドルの固定資産の購入と5.454億ドルの有価証券の購入を含むが、一部は9190万ドルの有価証券の販売と満期日によって相殺されている。
2021年12月31日と2020年12月31日までの毎年、投資活動のための純現金は、財産や設備の購入を含む10万ドル未満。
融資活動によるキャッシュフロー
2022年12月31日までの1年間、融資活動が提供する現金純額は40万ドルで、株式オプションを行使する収益と、我々の従業員の株式購入計画に基づいて普通株を発行する収益を含む。
融資活動が提供する現金純額は、2021年12月31日までの1年間で140万ドルで、株式オプションを行使して普通株を発行する収益を含む。
2020年12月31日までの年度、融資活動が提供する現金純額は5.318億ドルで、主にDシリーズ転換可能優先株を売却する1.066億ドルの純収益、D-1シリーズの転換可能な優先株を売却する1.075億ドルの純収益、私たちの初公募株の3.176億ドルの純収益を含む。
契約義務と約束
私たちはマサチューセッツ州ボストンでキャンセルできない経営性転貸方式で私たちのオフィススペースを貸しています。分譲期間は2022年1月1日から2026年12月31日まで満了する。
次の表は、2022年12月31日までの契約義務をまとめています
|
|
期限どおりの支払い |
|
|||||||||||||||||
|
|
少ないです |
|
|
1から3まで |
|
|
3から3まで |
|
|
超過 |
|
|
合計する |
|
|||||
|
|
(単位:千) |
|
|||||||||||||||||
経営リース義務 |
|
$ |
805 |
|
|
$ |
1,659 |
|
|
$ |
855 |
|
|
$ |
— |
|
|
$ |
3,319 |
|
我々は正常業務過程でCROと臨床前と臨床研究と試験契約を締結し、CMOと臨床試験材料を製造·供給する契約を締結し、他の第三者と運営目的のための他のサービスと製品の契約を締結した。これらの契約には最低購入約束は含まれておらず、通常、通知後の一定期間後に終了することが規定されているので、これらの合意の下での撤回不可能な義務は実質的ではないと考えられる。キャンセル時に支払うべき金額には、提供されたサービスの支払いとキャンセルの日までに発生した費用のみが含まれます。
135
2021年12月、私たちはメルク社とRuzasvirの開発、製造、商業化について許可合意に達した。Ruzasvirは我々が開発しているNS 5 A阻害剤であり,ベネホブビルと併用してC型肝炎ウイルスの治療に用いられている。
メルク許可協定の条項によれば、私たちは、研究、開発、製造、製造、使用、輸入、輸出、販売、販売、または他の方法で商業化されたRuzasvirまたはRuzasvirを含む製品を提供するために、いくつかのメルク特許および技術の下で独占的(内部研究を行ういくつかの留保権利の制約を受ける)、再許可および世界的許可を取得し、人間のすべての治療または予防用途のために使用する。
私たちがメルク許可協定によって得た権利を考慮して、私たちはメルクに2500万ドルの払い戻しできない前払いを支払った。私たちはメルクがある開発と規制のマイルストーンを実現した時に合計1.35億ドルのマイルストーン支払いを支払い、いくつかの販売ベースのマイルストーンを実現したときにメルクに合計3億ドルの支払いを支払う義務がある。また、製品の年間純売上高に応じてメルクに等級版税を支払い、高い1桁から10代程度の割合で範囲を拡大する。我々の特許使用料支払い義務は、(I)製品(または製品に含まれる化合物)を要求するメルク特許の有効な権利主張が最後に満了した日および(Ii)当該製品が国/地域で初めて商業販売されてから数年後まで続くであろう。便宜上、事前に書面で通知された場合、メルク許可プロトコルを終了することができます。最初の潜在的なマイルストーンは第3段階の臨床試験の開始時に支払われるだろう。次の表にはメルク許可協定に基づいて潜在的なマイルストーンまたは特許使用料の支払いが義務付けられている可能性があります。
上の表には、私たちがあるコンサルタントと締結した合意に基づいて、私たちが支払うべき潜在的なマイルストーンと成功費用も含まれていません。私たちはあるコンサルタントと、ある製品の売上のパーセントで計算された成功費の支払いを要求し、累計最高支払額は500万ドルであるという合意に達した。この成功的な支払いは未来の事件の発生にかかっており、このような支払いの時間と可能性は不可能であり、見積もることもできない。
重要な会計政策と試算
我々の財務諸表は米国公認会計原則(“GAAP”)に基づいて作成されている。これらの財務諸表を作成する際には、報告された資産および負債の報告金額、財務報告日、または有資産および負債の開示、および報告期間内に発生する報告費用に影響を与える推定および仮定を行う必要がある。我々の見積もりは,我々の歴史的経験と,当時の状況では合理的な様々な他の要因に基づいており,これらの要因の結果は資産や負債の帳簿価値を判断する基礎を構成しているが,これらの資産や負債の帳簿価値は他の源からは明らかではないように見える。異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。重大な改訂が予想される場合、その影響は推定変動の日から連結財務諸表に反映されることになる。
私たちの重要な会計政策は財務諸表を作成する際に最も重大な判断と見積もりが必要な政策です。経営陣は、私たちの最も重要な会計政策は、収入確認、計算すべき研究開発費、株式ベースの報酬に関する政策であることを確認した。
収入確認
2022年12月31日現在、我々のこれまでのすべての収入は、羅氏許可協定による協力収入である。
会計基準がASCトピック808をコードする範囲内にあるかどうかを評価するために、私たちの協力計画を分析します協力手配(“ASC 808‘)は、このような手配が双方によって展開される共同経営活動に関連するかどうかを決定するために、これらの締約国は、活動の積極的な参加者であり、そのような活動の商業成功に依存する重大なリスクおよびリターンに直面している。このスケジュールの一部または全部がASC 808の範囲内であり、顧客との取引を代表しないと結論した場合、共同で行われた活動に関連する分担コストは、発生した期間に関連する費用の構成要素であることを確認する。もし私たちがこの計画の一部または全ての側面が
136
顧客との取引は、ASC 606の範囲内でスケジュールの態様を考慮している取引先と契約した収入 (“ASC 606”).
ASC 606の範囲内の手配によって確認されるべき適切な収入金額を決定するために、(I)顧客との契約を決定するステップと、(Ii)契約における履行義務を決定するステップと、(Iii)取引価格を決定するステップと、(Iv)契約の履行義務に取引価格を割り当てるステップと、(V)各履行義務を履行する際に収入を確認するステップと、を実行する。ASC 606は、以下の事項について重大な判断、推定、および結果の変更を要求するが、これらに限定されないが、(I)可変対価格の推定を含む取引価格の決定、(Ii)推定販売価格の決定を含む取引価格の割り当て、および(Iii)サービス関連コミットメント進展の測定基準としての適用比率実績を含む確認モード、および供給関連コミットメント適用時点確認を含む。
取引価格には、一般に、契約開始時に満了する前金と、私たちのサービスおよび材料を支払う形態での可変対価格と、指定されたイベントを実現する際に満了するマイルストーン支払いが含まれます。お客様がライセンス製品の純売上を確認した場合、私たちは等級版税を含む他の支払いを受ける権利があります。私たちの計画には任意の重要な融資部分が存在すると考えられ、実質的なビジネス目的が存在するため、顕著な融資メリットを提供するのではなく、支払い構造を支援するために、私たちのスケジュールに重要な融資部分が存在しないことが確認された。私たちは、約束された商品および/またはサービスを顧客に移転するために、私たちが獲得する権利があると予想された対価格金額に基づいて取引価格を測定します。期待値手法や最も可能な金額法を用いて可変対価金額を推定することは,どの方法が獲得する権利のある対価格額をより良く予測することが期待できるかに依存する.可変対価格額は取引価格に計上され,可変対価格に関する不確実性がその後解決されれば,確認された累積収入が大きく逆転しない可能性が高い。開発や規制のマイルストーン支払いを含む手配については、関連イベントが実現可能であると考えられているかどうかを評価し、可能な金額法を用いて取引価格に含まれる金額を推定する。規制の承認を受けた支払いに依存するような、私たちまたは許可者の制御範囲内のマイルストーン支払いではありません, トリガイベントが発生するまで実現可能であるとは考えられない.各報告期間の終了時に、各マイルストーンの実現確率と任意の関連制限を再評価し、必要に応じて全体の取引価格の推定を調整する。いずれの調整も累積追跡原則で入金されており、これは調整期間中の収入と純損失に影響する。販売ベースの特許使用料を含む手配については、あるレベルの製品販売を達成することに基づくマイルストーン支払いを含み、ライセンスは、支払いに関連する唯一または主要な項目とみなされ、(I)関連販売が発生した場合、または(Ii)支払いによって割り当てられた履行義務が履行された(または部分的に履行された)場合に、以下の遅い場合に収入を確認する。オプション商品および(または)サービスの対価格は、契約開始時の取引価格には含まれない。
私たちは通常相対的に独立した販売価格に基づいて契約履行義務ごとに取引価格を割り当てます。我々は、適用される市場条件と関連する特定の実体要因に基づいて、顧客と合意する際に考慮する要因と推定される研究開発コストを含み、判断すべき仮定を作成し、契約義務毎の独立販売価格を決定する。しかしながら、場合によっては、可変対価格の条項が義務履行状況に関連し、割り当てられた金額が、義務履行のために予想される金額と一致する場合があれば、可変対価格を1つまたは複数の履行義務に全て割り当てる。
我々は,義務履行時や義務履行時に約束した貨物やサービスを顧客に譲渡して義務を履行する際に義務履行ごとに割り当てられた取引価格の金額に基づいて収入を確認する.ある時点で履行された履行義務については,商品および/またはサービスの制御権をクライアントに移行する際に収入を確認する.時間の経過とともに履行される業績義務については、単一の進捗を測る方法を用いて、業績義務の完全履行への進捗を測定することで、収入を確認する
137
これは、関連する商品および/またはサービスの制御権を顧客に転送する性能を説明する。私たちは通常、入力方法を使用して、一定期間にわたって義務を完全に履行する進捗状況を測定する。我々の知的財産権許可を含む手配については、手配で決定された他の履行義務とは異なると判断され、許可が被許可者に譲渡され、被許可者が許可を使用して利益を得ることができる場合には、許可に割り当てられた金額の収入を確認する。他の承諾とバンドルされたライセンスについては,合併履行義務が時間の経過とともに履行されているか,ある時点で履行されているかを決定するために,合併履行義務の性質を判断するために利用し,時間の経過とともに収入を確認するために進展を測定する適切な方法を決定する。1つの手配に必要な努力の程度と費用を決定し、1つの手配に基づいて義務履行の期限を達成することを期待する際には、重大な管理判断が必要である。私たちは報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整する。いずれの調整も累積追跡原則で入金されており、これは調整期間中の収入と純損失に影響する。
2021年11月に羅氏が羅氏許可協定を終了する通知(2022年2月から発効)を受ける前に、羅氏が何らかの活動(総合履行義務と呼ぶ)の達成に向けた進展に基づき、予想業績期間中の協力収入を確認する。私たちの結論は、終了通知は会計目的で契約を修正することだ。私たちはまた、終了通知を受けた後、合併履行義務が完全に履行されたと結論した。そこで,2021年12月31日までの年度総合運営報告書と全面収入において,すべての残りの繰延収入を連携収入と確認した(詳細については,付記3,連携収入を参照)。
契約費用
顧客との契約の増分コストを資産として確認し、これらのコストが回収されることが期待できれば。我々は、最初に確認された資産の予想償却期間が1年以下であれば、取得契約の増額コストを発生時の費用として確認する実際の便宜策を選択した。羅氏許可協定に関連して,2021年12月31日までの年度運営報告書における一般と行政コストおよび本年度報告Form 10−Kの他の部分の全面損失に含まれる700万ドルの増分コストが発生した。
研究と開発に応じる
私たちはCMOとCROと様々な合意に到達した。我々の研究·開発費用は、提供されたサービスレベル、研究進展(活動の段階または完了を含む)、および契約コストに基づいて推定されるべきである。請求書が発行されていないが提供されている研究および開発された推定コストは、貸借対照表の計算すべき負債に計上される。実際にサービスを提供する時間や努力の程度が従来予想されていたものと異なる場合には,対応する項目を調整する.関連サービスを提供する前に、これらの手配に従って関連サービス提供前にパートナーおよびパートナーに支払われたお金は、前払い費用および他の流動資産として入金される。実際に発生した金額と実質的に異なることはないと予想されるが、提供されたサービスの実際の状態および時間に対する提供サービスの状態および時間の理解が異なる可能性があり、報告された金額が任意の特定の時期に高すぎたり、過小になったりする可能性がある。これまで,我々が計算した研究や開発費の先行推定には何の大きな調整もなかった.
株に基づく報酬
私たちは公正な価値に基づく方法を使用して、株式オプションと株式奨励を含む従業員および非従業員とのすべての株式ベースの報酬スケジュールを計算する。付与されたオプションの公正価値は、オプション報酬と引き換えにオプション譲渡者がサービスを提供することが要求されている期間内に直線的に確認され、この期間は必要なサービス期間と呼ばれ、通常は授権期間である。付与された株式オプションの公正価値を決定する際には,主観的仮説の入力を要求するブラック-スコアズモデルを用いる.これらの仮説には公平さを見積もることがあります
138
普通株の市場価値は、従業員が行使前に既得株式オプションの時間長(予想期間)、予想期間内の我々の普通株価格の推定変動率(予想変動率)、無リスク金利、および期待配当を保持すると推定される。ブラック·スコアーズオプション定価モデルを適用して、2022年、2021年、2020年12月31日までの年度にそれぞれ付与された株式オプションの推定公正価値を決定する際に使用される特定の仮定の情報については、本年度報告に含まれる他の部分に含まれる監査された総合財務諸表の付記10を参照されたい。私たちの初めての公募株が完成する前に、私たちの普通株の公正価値は重大な判断と推定の使用に関連すると推定される。
普通株式公正価値の試算
初公募前には、ブラック·スコアーズオプション定価モデルを用いて公正価値計算を行う際に、株式奨励の基礎となる普通株の公正価値を推定することが求められていた。私たちが初めて公募する前に、私たちの株式オプションの基礎となる普通株の公正価値は、私たちの取締役会が授与日ごとに経営陣の意見に基づいて、私たちの普通株の最新の第三者推定値を考慮して決定されました。私たちの普通株を購入するオプションは、付与日に我々が知っている情報に基づいて、付与日のこれらのオプションに関連する普通株1株当たりの公正価値を推定する行使価格を下回らないように付与される。
初公募前に我々の普通株が公開取引市場を有していない場合には、授権日ごとに、独立第三者推定会社の推定値に基づいて、我々が認可日に知っている情報、最近発生した任意の事件及び普通株当たりの公正価値の潜在的影響の審査を用いて、我々の普通株の公正価値を推定する。
我々普通株の第三者推定値は、米国公認会計士協会“執行援助--補償として発行された個人保有会社株式証券の推定値”(“勤務援助”)で概説された基準に基づいて決定される。
私たちの普通株式の推定公正価値を決定するための仮定は、多くの客観的かつ主観的な要素に基づいており、管理層の判断に合わせて、以下のようになっている
“実践支援マニュアル”は、各値推定日における普通株式の推定公正価値を決定するために、異なるカテゴリおよびシリーズの株式に企業価値を割り当てる様々な利用可能な方法を決定する。実践援助計画によると、以下の方法が考えられる
139
我々の初期発展段階と他の関連要因に基づいて、OPM方法およびOPMとPWERM方法の混合方法は、我々の普通株の推定公正価値を決定するために我々の企業価値を割り当てる最適な方法であることを決定した。私たちの普通株の推定公正価値を決定する際に、私たちの取締役会はまた、私たちの株主が公開市場で私たちの普通株を自由に取引できないという事実を考慮した。そのため、私たちは加重平均予想流動性時間に基づいて、割引を適用して、私たちの普通株の市場性が不足していることを反映した。私たちの普通株の各付与日の推定公正価値は、未来の流動性イベントの予想可能性とタイミングに部分的に基づく非市場的割引を反映している。
初公募株が完成した後、私たちの普通株の公正価値は私たちの普通株の毎日の終値市場オファーに基づいています。
我々はまた、ASC主題718に基づいて、共有ベースの支払いの任意の修正を考慮する報酬--株式報酬 ("ASC 718").
現金と現金等価物
すべての原始満期日または購入時の残り満期日が3カ月以下の高流動性投資は現金等価物であると考えられる。私たちの現金等価物は通貨市場基金と商業手形を含み、それらは高い流動性と強力な信用評価を持っている。このような投資の信用と市場の危険はわずかだ。現金と現金等価物はコスト別に記載され、市場価値に近づいている。
有価証券
私たちの投資戦略は保証に重点を置いている。私たちは私たちの投資政策で概要された信用品質基準に適合するツールに投資する。有価証券には満期日が三ヶ月を超える投資が含まれています。有価証券には、米国債、米国機関債券、社債、商業手形、資産支援証券が含まれる。私たちは私たちのすべての有価証券を販売可能な証券に分類します。したがって、このような投資は公正な価値で入金される。未実現損益は株主権益内に他の全面収益の構成要素を計上する。割引と割増の償却と増額利息収入を計上する。
公正価値計量
公正価値は、資産を売却する際に受信された価格または計量日に市場参加者間で負債を移転するために支払われる価格として定義される。ASC 820、公正価値計測公正な価値によって計量するツールのために三級評価階層構造を構築する。この階層構造は、計量日までの資産または負債推定値の投入の透明性に基づく。この3つのクラスは以下のように定義される
第1レベル-推定方法の投入は、アクティブ市場における同じ資産または負債の見積もり(調整されていない)である。
第2レベル--推定方法の投入は、市場における同様の資産および負債のオファーをアクティブにすることと、金融商品期間全体にわたって実質的に直接的または間接的に観察可能な資産または負債の投入を含む。
第3級-推定方法の投入は観察できず、公正価値計量に重要な意義がある。
信用リスクと表外リスク集中
私たちを集中的な信用リスクに直面させる可能性のある金融商品には、主に現金等価物と有価証券が含まれる。私たちの投資政策によると、信用格付け、満期日、業界グループ、投資タイプと発行者によってこのような証券の投資額を制限し、アメリカ政府が発行した証券を除く。私たちは私たちがこのような金融商品がもたらすどんな重大な信用リスクの影響を受けるとは思わない。外国為替契約、オプション契約、その他の外国ヘッジ手配のような表外リスクのある金融商品はありません。
140
賠償協定
私たちは正常な業務過程で標準的な賠償計画を達成した。これらの手配によると、私たちは無害を賠償し、維持し、補償された側が受けたまたは発生した損失を賠償することに同意し、任意の第三者がその技術について提出した任意の商業秘密、著作権、特許または他の知的財産権侵害請求に関連する損失を含む。このような賠償協定の期限は一般的に協定締結後のいつでも永久的に有効だ。このような計画によると、私たちが未来に支払わなければならない最高金額は確定できないかもしれない。私たちは訴訟を弁護したり、このような賠償協定に関連したクレームを解決する費用を負担したことがない。したがって、私たちはこのような合意の公正な価値がわずかだと思う。
また、取締役や上級管理者が私たちの要求に応じてこのような身分でサービスする場合、ある事件や事件が発生した場合、私たちの役員と上級管理者に賠償を行うことに同意します。賠償期間には役員サービス期間中に発生したすべての関連事件と事件が含まれる。これらの賠償協定によると、私たちが将来支払う必要がある可能性のある最大潜在金額は協定に規定されていません。しかし、私たちは将来支払うべき任意の金額の一部を回収できるように、私たちのリスクを減らすことができる役員と役人保険があります。保険適用範囲を超えたこれらの賠償協定の推定公正価値は最も低いと考えられる。
141
第七A項。数量化と高質VE市場リスクの開示について
金利感度
我々の金融商品や財務状況に固有の市場リスクとは、金利や為替レートの不利な変動による潜在的損失を指す。2022年12月31日現在、私たちは6.467億ドルの現金、現金等価物、および有価証券を持っており、その公正な価値は米国金利の全体的なレベルの変化の影響を受けるだろう。しかし、私たちの現金等価物の短期満期日と低リスクイメージのため、金利の即時10%の相対的な変化は、私たちの現金等価物の公正な価値または私たちの未来の利息収入に実質的な影響を与えないだろう。
インフレ、金利の変化、あるいは外貨為替レートの変動が、本稿で述べたいかなる時期の経営業績に大きな影響を与えるとは思いません。
プロジェクト8.財務諸表Sと補足データ。
本項目8の要求に基づいて提出された財務諸表は、本年度報告のテーブル10−Kの後に添付される。これらの財務諸表のインデックスは、本年度報告表格10-Kの第15項に記載されている。
項目9.Accouとの変更と分岐会計と財務情報開示の専門。
ない。
第9条。制御するプログラムがあります
経営陣の私たちの開示制御と手続きの評価
我々の開示制御およびプログラムを設計·評価する際に、管理層は、任意の制御およびプログラムは、設計および動作がどんなに良好であっても、予想される制御目標を達成するために合理的な保証を提供することしかできないことを認識している。さらに、開示制御およびプログラムの設計は、リソース制限が存在することを反映しなければならず、管理層は、そのコストに対する可能な制御およびプログラムの利益を評価する際に判断することを要求する。
我々の経営陣は、我々の最高経営責任者および最高財務責任者(それぞれ私たちの最高経営責任者およびCEO)の参加の下で、2022年12月31日までの開示制御プログラムの有効性を評価した。このような評価に基づき、我々の最高経営責任者及び最高財務責任者は、2022年12月31日まで、我々の開示統制及び手続が合理的な保証水準で有効であると結論した。
財務報告の内部統制の変化
2022年12月31日までの四半期内に、財務報告の内部統制(“取引法”第13 a-15(F)および15 d-15(F)規則の定義による)に大きな影響を与えなかったか、または合理的に財務報告の内部統制に大きな影響を与える可能性のある変化はなかった。
経営陣財務報告内部統制年次報告書
我々の経営陣は、“取引法”の下のルール13 a-15(F)で定義されている財務報告の十分な内部統制の確立と維持を担当している。
最高経営責任者と最高財務責任者の監督と参加の下、我々の経営陣は、テレデビル委員会後援組織委員会が発表した内部統制-総合枠組み(2013)に規定されている基準に基づいて、2022年12月31日までの財務報告書の内部統制に対する有効性を評価した。この評価に基づき、経営陣は、財務報告書に対する内部統制は2022年12月31日から有効であると結論した。
2022年12月31日現在、財務報告の内部統制の有効性は、独立公認会計士事務所ピマウェイ会計士事務所が監査しており、その報告は以下のとおりである。
142
独立公認会計士事務所報告
株主や取締役会に
ATEA製薬会社:
財務報告の内部統制については
トレデビル協賛組織委員会が発表した“内部統制−総合枠組み(2013)”で確立された基準に基づき,ATEA製薬会社とその子会社(当社)が2022年12月31日までの財務報告内部統制を監査した。トレデビル委員会後援組織委員会が発表した“内部統制−総合枠組み(2013)”で確立された基準によると,2022年12月31日現在,会社はすべての実質的な面で財務報告に対する有効な内部統制を維持していると考えられる。
我々はまた、米国上場会社会計監督委員会(PCAOB)の基準に従って、当社の2022年12月31日と2021年12月31日までの総合貸借対照表、2022年12月31日までの3年間の各年度の関連総合経営報告書と全面収益(赤字)、転換可能な優先株と株主権益(損失)、現金流量および関連付記(総称して総合財務諸表と呼ぶ)を監査し、2023年2月28日の報告でこのなどの総合財務諸表に対して留保のない意見を表明した。
意見の基礎
当社経営陣は、効果的な財務報告内部統制を維持し、付随する経営陣財務報告内部統制年次報告に含まれる財務報告内部統制の有効性の評価を担当する。私たちの責任は私たちの監査に基づいて、会社の財務報告書の内部統制に意見を述べることです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務報告の有効な内部統制がすべての重要な面で維持されているかどうかを決定するために、合理的な保証を得るために監査を計画し、実行することを要求する。我々の財務報告の内部統制の監査には、財務報告の内部統制を理解すること、重大な弱点があるリスクを評価すること、評価されたリスクテストに基づいて内部統制の設計と運営有効性を評価することが含まれる。私たちの監査はまた、私たちがこのような状況で必要だと思う他の手続きを実行することを含む。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
財務報告の内部統制の定義と限界
会社の財務報告に対する内部統制は、公認された会計原則に基づいて、財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供することを目的としたプログラムである。会社の財務報告に対する内部統制には、(1)合理的で詳細かつ正確かつ公平に会社の資産を反映した取引および処分に関する記録の保存、(2)公認された会計原則に従って財務諸表を作成するために必要な取引が記録されている合理的な保証を提供し、会社の収入および支出は会社の経営陣と取締役の許可のみに基づいて行われる。(三)財務諸表に重大な影響を及ぼす可能性のある不正買収、使用、または処分会社の資産を防止またはタイムリーに発見する合理的な保証を提供する。
143
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
/s/ピマウェイ法律事務所
ボストン、マサチューセッツ州
2023年2月28日
プロジェクト9 B。他にも情報です。
ない。
プロジェクト9 Cです。展示する検査を阻止した外国司法管区について
適用されません。
144
第三部
プロジェクト10.役員·役員職ICERSと会社管理。
道徳的規則
我々の取締役会は、我々の最高経営責任者、最高財務官、首席会計官、財務総監、または同様の機能を実行する者を含む、すべての上級管理者、取締役および従業員に適用される書面による商業行為および道徳基準を採択している。私たちのウェブサイトにビジネス行動と道徳基準の最新コピーを掲示しましたサイトはWwww.ateapharma.com“コーポレート·ガバナンス”の下での“投資家”部分。我々は、Form 8-K第5.05項のビジネス行為および道徳基準条項の修正または免除に関する開示要件、および開示役員および役員免除に関するナスダックの要求を満たす予定であり、このような情報を私たちのサイトに公開する方法であり、アドレスおよび位置は上記のように指定されている。我々のサイト上の情報は,本Form 10-K年次報告に引用的に組み込まれていない.
行政員および役員
本項目10に要求される執行要項及び取締役に関する情報は、本年度報告10−Kテーブル第I部、項目I、業務、“我々の執行者及び取締役に関する情報”というタイトルに掲載されている。本第10項の規定で開示しなければならない残りの資料は、2023年の株主総会のために作成された最終委託書に含まれ、タイトルは“会社管治”、“延滞第16条(A)条報告書”(例えば、適用される)及び“取締役会委員会”であり、ここに組み込まれて参考となる。
プロジェクト11.行政官イー補償します。
第11項に要求される情報は、2023年年次総会に関する我々が米国証券取引委員会に提出した最終委託書に含まれ、参照により本明細書に組み込まれる。
プロジェクト12.特定の受益者の保証所有権従業員と経営陣と関連した株主問題。
第12項に要求される情報は、2023年年次総会に関する我々が米国証券取引委員会に提出した最終委託書に含まれ、参照により本明細書に組み込まれる。
第13条に要求される情報は、2023年年次総会に関する我々が米国証券取引委員会に提出した最終委託書に含まれ、引用により本明細書に組み込まれる。
第14項.元金口座弁護士代とサービス料です。
第14条要求された情報は、2023年年次総会に関する我々が米国証券取引委員会に提出した最終委託書に含まれ、引用により本明細書に組み込まれる。
独立公認会計士事務所はピマウェイ会計士事務所ボストンマサチューセッツ州監査役事務所ID:
145
第4部
プロジェクト15.展示品、資金エーアールアイチェックリストです。
一財務諸表
以下の文書は、本文書に添付されているF−1~F−26ページに含まれ、本年度報告の10−Kテーブルの一部として提出される。
独立公認会計士事務所報告 |
|
F-2 |
|
|
|
合併貸借対照表 |
|
F-3 |
|
|
|
合併経営表と全面損益表(赤字) |
|
F-4 |
|
|
|
転換可能優先株と株主権益連結報告書(損失) |
|
F-5 |
|
|
|
統合現金フロー表 |
|
F-6 |
|
|
|
連結財務諸表付記 |
|
F-7 |
(2)財務諸表添付表
スケジュールは必要ではないか,適用されないか,あるいは情報が他の方法で本明細書に含まれるため省略される.
(3)展示品
|
|
|
|
引用で編入する |
|
提出済み/ |
||||||
展示品 番号をつける |
|
展示品説明 |
|
表 |
|
書類番号. |
|
展示品 |
|
保存する 日取り |
|
家具を完備する ここから声明する |
|
|
|
|
|
|
|
|
|
|
|
|
|
3.1 |
|
再記載した会社登録証明書。 |
|
8-K |
|
001-39661 |
|
3.1 |
|
11/5/2020 |
|
|
3.2 |
|
添付例を改訂及び再編成する。 |
|
8-K |
|
001-39661 |
|
3.2 |
|
11/5/2020 |
|
|
4.1 |
|
普通株式の証明書証明書サンプル |
|
S-1 |
|
333-249404 |
|
4.2 |
|
10/9/2020 |
|
|
4.2 |
|
株本説明 |
|
10-K |
|
001-39661 |
|
4.2 |
|
3/30/2021 |
|
|
4.3 |
|
改正された第4回改正と再署名された株主協定 |
|
S-1/A |
|
333-249404 |
|
4.1 |
|
10/23/2020 |
|
|
10.1# |
|
2020年インセンティブ奨励計画とそのプロトコルフォーマット |
|
S-1/A |
|
333-249404 |
|
10.2 |
|
10/26/2020 |
|
|
10.1-1# |
|
2020年度インセンティブ·インセンティブ計画における業績に基づく限定的な株式単位奨励協定(CEO)の形態 |
|
10-K |
|
001-39661 |
|
10.1.1 |
|
2/28/2022 |
|
|
10.1-2# |
|
2020年度インセンティブ·インセンティブ計画の下での業績に基づく限定的な株式単位奨励協定(非CEO執行者)形式 |
|
10-K |
|
001-39661 |
|
10.1.2 |
|
2/28/2022 |
|
|
10.2# |
|
2020年従業員株購入計画 |
|
S-1/A |
|
333-249404 |
|
10.3 |
|
10/26/2020 |
|
|
10.3# |
|
非従業員役員報酬計画 |
|
|
|
|
|
|
|
|
|
* |
10.4# |
|
役員及び上級者の弁済協議形式 |
|
S-1/A |
|
333-249404 |
|
10.5 |
|
10/26/2020 |
|
|
10.5^ |
|
登録者とMSD International GmbHとの間のライセンス契約は,期日は2021年12月23日である |
|
10-K |
|
001-39661 |
|
10.5 |
|
2/28/2022 |
|
|
10.6# |
|
会社がJean-Pierre Sommadossi博士と締結した雇用契約は、2020年10月25日となっている |
|
S-1/A |
|
333-249404 |
|
10.9 |
|
10/26/2020 |
|
|
146
|
|
|
|
引用で編入する |
|
提出済み/ |
||||||
展示品 番号をつける |
|
展示品説明 |
|
表 |
|
書類番号. |
|
展示品 |
|
保存する 日取り |
|
家具を完備する ここから声明する |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.7# |
|
会社とAndrea Corcoranの雇用協定は2020年10月25日 |
|
S-1/A |
|
333-249404 |
|
10.10 |
|
10/26/2020 |
|
|
10.8# |
|
会社がジャネット·ハモンド博士と締結した雇用契約は、2020年11月3日となっている |
|
10-K |
|
001-39661 |
|
10.8 |
|
3/30/2021 |
|
|
10.9# |
|
メリーランド州Arantxa Horgaとの雇用協定は、2020年11月3日となっている |
|
10-K |
|
001-39661 |
|
10.9 |
|
3/30/2021 |
|
|
10.10# |
|
会社とJohn Vavrickaの間の雇用協定は2020年11月3日です |
|
10-K |
|
001-39661 |
|
10.10 |
|
3/30/2021 |
|
|
10.11# |
|
会社とウェイン·フォスターの雇用協定は2020年11月3日 |
|
10-K |
|
001-39661 |
|
10.11 |
|
3/30/2021 |
|
|
10.12# |
|
改訂された2013年株式インセンティブ計画とその合意のフォーマット |
|
S-1 |
|
333-249404 |
|
10.1 |
|
10/9/2020 |
|
|
10.13 |
|
当社とDataRobot,Inc.との転貸プロトコルは,2021年7月19日である. |
|
8-K |
|
001-39661 |
|
10.1 |
|
7/23/2021 |
|
|
10.13-1 |
|
当社とDataRobot,Inc.が2022年4月11日に締結した転貸協定修正案。 |
|
10-Q |
|
001-39661 |
|
10.1 |
|
8/8/2022 |
|
|
10.14# |
|
相談協定は、2021年5月18日に、会社と上流健康·健康有限責任会社が署名した。 |
|
8-K |
|
001-39661 |
|
10.1 |
|
5/20/2021 |
|
|
10.15 |
|
当社はBruce Polsky,M.D.と2022年6月10日に締結した諮問協定である |
|
10-Q |
|
001-39661 |
|
10.2 |
|
8/8/2022 |
|
|
21.1 |
|
登録者の子会社リスト |
|
|
|
|
|
|
|
|
|
* |
23.1 |
|
独立公認会計士事務所の同意 |
|
|
|
|
|
|
|
|
|
* |
31.1 |
|
ルール13 a−14(A)/15 d−14(A)に従って首席実行幹事証明書が発行される。 |
|
|
|
|
|
|
|
|
|
* |
31.2 |
|
財務チーフ幹事は、細則13 a~14(A)/15 d−14(A)に従って認証される。 |
|
|
|
|
|
|
|
|
|
* |
32.1 |
|
“米国法典”第18編第1350条に基づく最高経営責任者の証明。 |
|
|
|
|
|
|
|
|
|
** |
32.2 |
|
“米国法典”第18編第1350条に基づく首席財務官の証明。 |
|
|
|
|
|
|
|
|
|
** |
101.INS |
|
連結されたXBRLインスタンス文書であるインスタンス文書は、そのXBRLタグがイントラネットXBRL文書に埋め込まれているので、対話データファイルには表示されない。 |
|
|
|
|
|
|
|
|
|
* |
101.書院 |
|
イントラネットXBRL分類拡張アーキテクチャ文書 |
|
|
|
|
|
|
|
|
|
* |
101.カール |
|
インラインXBRL分類拡張計算リンクライブラリ文書 |
|
|
|
|
|
|
|
|
|
* |
101.def |
|
インラインXBRL分類拡張Linkbase文書を定義する |
|
|
|
|
|
|
|
|
|
* |
101.介護会 |
|
XBRL分類拡張ラベルLinkbase文書を連結する |
|
|
|
|
|
|
|
|
|
* |
147
|
|
|
|
引用で編入する |
|
提出済み/ |
||||||
展示品 番号をつける |
|
展示品説明 |
|
表 |
|
書類番号. |
|
展示品 |
|
保存する 日取り |
|
家具を完備する ここから声明する |
|
|
|
|
|
|
|
|
|
|
|
|
|
101.Pre |
|
インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント |
|
|
|
|
|
|
|
|
|
* |
104 |
|
表紙相互データファイル(添付ファイル101に含まれるイントラネットXBRLのフォーマット) |
|
|
|
|
|
|
|
|
|
* |
*アーカイブをお送りします。
**関数で提供されます。
S-K条例第601(B)(10)(Iv)項によれば、本展示品の一部の内容は省略されている。
# 契約または補償計画を管理すること。
項目16.表10-Kの概要
ない。
148
登録する解決策
1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者が代表して本報告書に署名することを正式に手配した.
|
|
ATEA製薬会社 |
|
|
|
|
|
日付:2023年2月28日 |
|
差出人: |
ジャン·ピエール·ソマドシ |
|
|
|
ジャン·Pierre Sommadossi博士 |
|
|
|
社長と最高経営責任者 |
本報告書は、改正された1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
ジャン·ピエール·ソマドシ |
|
社長、CEO兼取締役会長(最高経営責任者) |
|
2023年2月28日 |
ジャン·Pierre Sommadossi博士 |
|
|
|
|
|
|
|
|
|
/s/Andrea Corcoran |
|
首席財務官兼常務副総裁、法律兼秘書(首席財務官) |
|
2023年2月28日 |
アンドレア·コクラン |
|
|
|
|
|
|
|
|
|
/s/ウェイン·フォスター |
|
常務副総裁、首席会計官(首席会計官) |
|
2023年2月28日 |
ウェイン·フォスター |
|
|
|
|
|
|
|
|
|
/s/フランクリン·バージャー |
|
重役(筆頭取締役) |
|
2023年2月28日 |
フランクリン·バージャー |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ジェローム·アダムス |
|
役員.取締役 |
|
2023年2月28日 |
ジェローム·アダムス医学博士M.P.H |
|
|
|
|
|
|
|
|
|
/s/Barbara Duncan |
|
役員.取締役 |
|
2023年2月28日 |
バーバラ·ダンカン |
|
|
|
|
|
|
|
|
|
/s/ブルーノ·ルシディ |
|
役員.取締役 |
|
2023年2月28日 |
ブルーノ·ルシディ |
|
|
|
|
|
|
|
|
|
/s/Polly A.Murphy |
|
役員.取締役 |
|
2023年2月28日 |
ボリー·A·マーフィーD.V.M博士 |
|
|
|
|
/s/Bruce Polsky |
|
役員.取締役 |
|
2023年2月28日 |
ブルース·ボルスキー医学博士 |
|
|
|
|
149
連結財務諸表索引
独立公認会計士事務所報告 |
F-2 |
|
|
合併貸借対照表 |
F-3 |
|
|
合併経営表と全面損益表(赤字) |
F-4 |
|
|
転換可能優先株と株主権益連結報告書(損失) |
F-5 |
|
|
統合現金フロー表 |
F-6 |
|
|
連結財務諸表付記 |
F-7 |
F-1
独立公認会計士事務所報告
株主や取締役会に
ATEA製薬会社:
連結財務諸表に対するいくつかの見方
ATEA製薬会社とその子会社(当社)が2022年12月31日までと2021年12月31日までの連結貸借対照表,2022年12月31日までの3年間の各年度の関連総合経営報告書と全面収益(赤字),転換可能優先株と株主権益(損失)とキャッシュフロー,および関連付記(総称して合併財務諸表と呼ぶ)を監査した。総合財務諸表は、すべての重要な面で、会社の2022年12月31日と2021年12月31日までの財務状況、および2022年12月31日までの3年間の毎年の経営結果と現金流量を公平に反映しており、米国公認会計原則に適合していると考えられる。
また、米国上場企業会計監督委員会(PCAOB)の基準に基づき、テレデビル委員会後援組織委員会が発表した“内部統制-総合枠組み(2013)”で確立された基準に基づいて、2022年12月31日までの財務報告内部統制を監査し、2023年2月28日の報告書は社内統制の有効性について保留のない意見を発表した。
意見の基礎
これらの連結財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいてこのような連結財務諸表に意見を発表することだ。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、連結財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。我々の監査には、連結財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであっても詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれる。これらの手続きは、連結財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、合併財務諸表の全体列報の評価も含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
重要な監査事項とは、連結財務諸表を当期監査する際に生じた、伝達された、または監査委員会に伝達された事項である:(1)総合財務諸表に対して重大な意義を有する勘定または開示に関連する;(2)私たちが特に挑戦的で、主観的または複雑な判断に関連する。私たちは重要な監査事項が存在しないと確信する。
/s/
2014年以来、当社の監査役を務めてきました。
2023年2月28日
F-2
ATEA製薬会社
合併B割当書
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産 |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
有価証券 |
|
|
|
|
|
— |
|
|
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
制限現金 |
|
|
|
|
|
|
||
その他の資産 |
|
|
|
|
|
— |
|
|
経営的リース使用権資産純額 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債 |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用とその他の流動負債を計算しなければならない |
|
|
|
|
|
|
||
賃貸負債の当期部分を経営する |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
リース負債を経営する |
|
|
|
|
|
— |
|
|
所得税に対処する |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
株主権益: |
|
|
|
|
|
|
||
優先株、$ |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
その他の総合損失を累計する |
|
|
( |
) |
|
|
— |
|
利益剰余金(累積損失) |
|
|
( |
) |
|
|
|
|
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
F-3
ATEA製薬会社
業務所合併報告書減価償却および総合収益(赤字)
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
協力収入 |
|
$ |
— |
|
|
$ |
|
|
$ |
|
||
運営費 |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
|
|
|
|
|
|
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
総運営費 |
|
|
|
|
|
|
|
|
|
|||
営業収入(赤字) |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
利子収入とその他の純額 |
|
|
|
|
|
|
|
|
|
|||
所得税前収入 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
所得税の割引 |
|
|
|
|
|
( |
) |
|
|
— |
|
|
純収益(赤字) |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
その他全面収益(赤字) |
|
|
|
|
|
|
|
|
|
|||
売却可能投資の未実現損失 |
|
|
( |
) |
|
|
— |
|
|
|
— |
|
総合収益(赤字) |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
1株当たり普通株の純収益に帰することができる |
|
|
|
|
|
|
|
|
|
|||
基本的な情報 |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
薄めにする |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
加重-使用された平均普通株式数 |
|
|
|
|
|
|
|
|
|
|||
基本的な情報 |
|
|
|
|
|
|
|
|
|
|||
薄めにする |
|
|
|
|
|
|
|
|
|
|||
付記はこれらの連結財務諸表の構成要素である。
F-4
ATEA製薬会社
変換可能印刷機統合報告書株と株主権益を繰延する
(単位は千で、シェアは含まれていない)
|
|
オープンカー |
|
|
|
普通株 |
|
|
その他の内容 |
|
|
積算 |
|
|
利益を残す |
|
|
合計する |
|
||||||||||||||
|
|
株 |
|
|
金額 |
|
|
|
株 |
|
|
金額 |
|
|
資本 |
|
|
保監所 |
|
|
(累積赤字) |
|
|
権益 |
|
||||||||
残高-2020年1月1日 |
|
|
|
|
$ |
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
( |
) |
|||||
Dシリーズ転換車を発行する |
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
D-1シリーズオープンカーを発行します |
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
行使のために普通株を発行する |
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
株式ベースの報酬は |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
初公開で、発行後の純額を差し引く |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
優先株を |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
残高-2020年12月31日 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||||
行使のために普通株を発行する |
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
純収入 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
残高-2021年12月31日 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
|
|||||
行使のために普通株を発行する |
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
ESPPにより普通株式を発行する |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
その他総合損失 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
純損失 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
残高-2022年12月31日 |
|
|
— |
|
|
$ |
— |
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
付記はこれらの連結財務諸表の構成要素である。
F-5
ATEA製薬会社
合併状態キャッシュフロープロジェクト
(単位:千)
|
|
現在までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
経営活動のキャッシュフロー |
|
|
|
|
|
|
|
|
|
|||
純収益(赤字) |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
純収益(赤字)を純現金に調整する |
|
|
|
|
|
|
|
|
|
|||
株に基づく報酬費用 |
|
|
|
|
|
|
|
|
|
|||
減価償却および償却費用 |
|
|
|
|
|
|
|
|
|
|||
有価証券の割増と割引の増加 |
|
|
( |
) |
|
|
— |
|
|
|
— |
|
経営性資産と負債の変動 |
|
|
|
|
|
|
|
|
|
|||
未開売掛金 |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
前払い費用と他の流動資産 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その他の資産 |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
売掛金 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
費用とその他の負債を計算すべきである |
|
|
( |
) |
|
|
|
|
|
|
||
収入を繰り越す |
|
|
— |
|
|
|
( |
) |
|
|
|
|
リース負債を経営する |
|
|
|
|
|
— |
|
|
|
— |
|
|
経営活動提供の現金純額 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
投資活動によるキャッシュフロー |
|
|
|
|
|
|
|
|
|
|||
物件と設備の追加料金 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
有価証券を購入する |
|
|
( |
) |
|
|
— |
|
|
|
— |
|
有価証券の売却と満期日 |
|
|
|
|
|
— |
|
|
|
— |
|
|
投資活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
融資活動によるキャッシュフロー |
|
|
|
|
|
|
|
|
|
|||
転換優先株を発行して得た金の純額 |
|
|
— |
|
|
|
— |
|
|
|
|
|
株式オプションを行使するために普通株で得た金を発行する |
|
|
|
|
|
|
|
|
|
|||
ESPPにより普通株式を発行して得た金 |
|
|
|
|
|
— |
|
|
|
— |
|
|
普通株の純利益を初公開する |
|
|
— |
|
|
|
— |
|
|
|
|
|
融資活動が提供する現金純額 |
|
|
|
|
|
|
|
|
|
|||
現金、現金等価物および制限現金純増加(マイナス) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
現金、現金等価物、制限された現金 |
|
|
|
|
|
|
|
|
|
|||
期末現金、現金等価物、および制限現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
期末現金、現金等価物、および制限現金 |
|
|
|
|
|
|
|
|
|
|||
現金と現金等価物 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
制限現金 |
|
|
|
|
|
|
|
|
|
|||
現金総額、現金等価物、および限定現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|||
非現金融資活動を補充開示する |
|
|
|
|
|
|
|
|
|
|||
初公募終了時に優先株を普通株に変換します |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
経営性賃貸負債と引き換えに使用権資産 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
付記はこれらの連結財務諸表の構成要素である。
F-6
ATEA製薬会社
連結財務諸表付記
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
1.ビジネスの性質
ATEA製薬会社とその子会社ATEA製薬証券会社は合併に基づいて“会社”と呼ばれている。
同社は臨床段階の生物製薬会社であり,抗ウイルス療法の発見,開発,商業化に専念し,生命を脅かすウイルス感染患者の生活を改善している。
2020年10月、同社はF.Hoffmann-La Roche LtdとGenentech,Inc.(総称して“羅氏”と呼ぶ)と許可協定(“羅氏許可協定”)を締結し、羅氏に米国以外の地域で同社のある化合物を開発·商業化する独占許可を付与し、その主要な候補製品Bemnifosbuvirを含む。
対価格の一部として、羅氏は同社に#ドルを前払いすることに同意した
2021年11月12日、羅氏は羅氏許可協定を終了する通知を会社に提供した。羅氏許可協定の条項によると、終了は2022年2月に発効する。終了後,会社は羅氏許可協定により羅氏に付与された権利と許可を会社に返還し,会社は世界的な臨床開発と将来的にBemnifosbuvirの商業化を継続するすべての権利を持っている。
2020年11月3日、会社は普通株式の初公開(IPO)を完了した。初公募について当社は発行した
同社は臨床段階の生物製薬会社によく見られるリスクと不確定要素の影響を受けている。これらのリスクには、臨床前および臨床研究の潜在的な失敗、一般的な研究および開発活動に関連する不確実性、他社からの技術革新の競争、重要な人員への依存、政府規定の遵守、会社が開発する可能性のある任意の候補製品の市場承認を得る必要があり、患者、支払者、および保健提供者の広範な受け入れを得て、市場の承認を得た任意の製品を商業化することに成功し、企業の独自技術および製品に対する十分な知的財産権保護を確保し、維持する必要があるが、これらに限定されない。また、同社の現在の多くの臨床前研究、臨床開発と製造活動は第三者サービスプロバイダに依存している。現在開発中の候補製品は大量の追加資本と、広範な臨床前と臨床テスト及び監督許可を含む追加的な研究と開発努力を必要とし、その後商業化することができる。同社がその候補品の販売から収入を得ることができても、承認されれば利益を上げることができない可能性がある。同社が利益を達成できなかったり、継続的に利益を上げられなかったりすると、計画通りの運営を継続できず、運営の減少を余儀なくされる可能性がある。
会社は、追加株式証券融資、債務融資、またはそれと達成可能な任意の新しい協力関係または他の手配に関連する資金を売却することによって、1つまたは複数の融資方法によって追加資本を求めることができる。当社が当社が受け入れ可能な条項に従ってタイムリーまたは根本的にそのような追加資金を得ることができる保証はありません。どんな融資条項も、会社の既存株主の持株や権利に悪影響を及ぼす可能性がある。 当社はその現金と現金等価物及び有価証券を信じています
F-7
当社は2021年11月にJeffries LLC(“Jeffries”)と公開市場販売協定(“販売協定”)を締結し、この合意により、当社はその普通株式を随時発売·販売することができ、総発行価格は最高$に達する
同社はまた、予想されるいくつかの試験に関連する実際および潜在的な遅延、および同社の業務運営および追加資本の調達がその運営に資金を提供する能力の潜在的な負の影響を含む、新冠肺炎の世界的大流行に関連するリスクの影響を受けている。国内や政治的な動揺(ウクライナとロシアの間で続く戦争など)を含む地政学的事件は、世界の商業·金融市場を深刻に混乱させている。また、最近または将来の市場変動、インフレ激化、より高い金利は、継続すれば、私たちの融資コストを増加させ、将来の潜在的な流動性源の獲得を制限する可能性がある。
2.主な会計政策の概要
予算の列報と使用根拠
公認会計基準に従って財務諸表を作成することは、合併財務諸表およびこれらの付記報告書の金額に影響を与えるために、管理層に推定および仮定を要求する。当社は過去の経験、既知の傾向及びその他の市場の特定或いはその他の関連要素及びその当時の状況に属すると考えられる合理的な仮定に基づいて推定します。経営陣は、研究開発費を計算すべき推定数、有価証券の推定値、株式奨励の推定値、経営リース使用権資産および賃貸負債の推定値、所得税など、その推定数を評価し続けている。推定された変化は、それらが知られている期間に記録される。
合併原則
連結財務諸表にはATEA製薬会社とその完全子会社ATEA製薬証券会社の勘定が含まれる。すべての会社間金額は合併中に販売されました。
再分類する
前年の財務諸表のいくつかの項目は、現在の列報方式に適合するように再分類された。
収入確認
同社のすべての収入は、2021年12月31日現在、羅氏許可協定による協力収入である。
2021年11月に羅氏から通知を受けるまで、羅氏は2022年2月に羅氏許可協定を終了する前に、会社はある活動の進展状況の測定に基づいて、予想業績期間の協力収入を確認し、この指標を総合業績義務と呼ぶ。同社の結論は、終了通知は会計目的で契約を修正したものである。当社はさらに、終了通知を受けた後、合併履行義務が完全に履行されたと判断した。そこで,会社は2021年12月31日までの年度総合運営報告書と全面収入において,すべての残りの繰延収入を連携収入と確認した(詳細は付記3,連携収入を参照)。
当社は、会計基準がASCテーマ808を編集した範囲内にあるかどうかを評価するために、その連携スケジュールを分析した協力手配(“ASC 808‘)は、このような手配が双方によって展開される共同経営活動に関連するかどうかを決定するために、これらの締約国は、活動の積極的な参加者であり、そのような活動の商業成功に依存する重大なリスクおよびリターンに直面している。もし会社が結論を出したら
F-8
ASC 808の範囲内に配置され、顧客との取引を代表するものではなく、会社は、ASC 730による共同活動による分担コストの割り当てを確認する研究と開発それは.そのため、会社は、発生時に費用を支出し、いずれの精算済み費用も含めて、受け取った精算を研究開発費の減少と確認する。会社がスケジュールのいくつかまたは全部が顧客との取引を表していると判断した場合、同社はASC 606の範囲内でスケジュールの態様を説明している取引先と契約した収入 (“ASC 606”).
会社がASC 606の範囲内で確認すべき適切な収入額を決定するために、(I)顧客との契約を決定するステップと、(Ii)契約における履行義務を決定するステップと、(Iii)取引価格を決定するステップと、(Iv)契約に取引価格を割り当てる履行義務と、(V)各履行義務が履行されたときに収入を確認するステップと、を実行する。ASC 606は、以下の事項について重大な判断、推定、および結果の変更を要求するが、これらに限定されないが、(I)可変対価格の推定を含む取引価格の決定、(Ii)推定販売価格の決定を含む取引価格の割り当て、および(Iii)サービス関連コミットメント進展の測定基準としての適用比率実績を含む確認モード、および供給関連コミットメント適用時点確認を含む。
取引価格には、一般に、契約開始時に満了した前金と、会社サービスおよび材料の形態で支払いされる可変対価格と、指定されたイベントを実現する際に支払われるべきマイルストーン支払いとが含まれる。顧客がライセンス製品の純売上を確認した場合、同社は等級別印税を含む他の支払いを受ける権利がある。当社はその手配に任意の重大な融資構成部分が存在すると考え、その手配に重大な融資構成部分が存在しないことを確定した。重大な融資メリットを提供する以外に、支払い構造を支援する実質的な業務目的が存在するからである。同社は、約束された貨物および/またはサービスを顧客に譲渡するために、その予想される対価格金額に基づいて取引価格を測定する。当社は予想値法または最可能金額法を用いて可変対価金額を推定し、どの方法が当社が獲得する権利のある対価格金額を予測できるかによります。可変対価格額は取引価格に計上され,可変対価格に関する不確実性がその後解決されれば,確認された累積収入が大きく逆転しない可能性が高い。開発または規制マイルストーン支払いを含む手配について、会社は、関連イベントが実現可能であると考えられているかどうかを評価し、最も可能な金額法を用いて取引価格に含まれる金額を推定する。監督管理部門の承認を受けた支払いに依存するような、会社または顧客統制範囲内でないマイルストーン支払い, トリガイベントが発生するまで実現可能であるとは考えられない.各報告期間の終了時に、当社は、各マイルストーンの実現確率および任意の関連制限を再評価し、必要に応じて全体の取引価格の推定を調整する。いずれの調整も累積追跡原則で入金されており、これは調整期間中の収入と純損失に影響する。販売ベースの特許使用料を含む手配については、製品販売に基づいて一定のレベルに達するマイルストーン支払いを含み、ライセンスは、支払いに関連する唯一または主要な項目とみなされ、会社は、以下のより遅い場合に収入を確認するであろう:(I)関連販売が発生した場合、または(Ii)一部または全部の支払いによって割り当てられた履行義務が履行された(または部分的に履行されている)。オプション商品および(または)サービスの対価格は、契約開始時の取引価格には含まれない。
F-9
当社は一般に相対的に独立した販売価格に基づいて取引価格を契約履行義務ごとに割り当てています。適用される市場条件や関連する特定の実体要因を考慮して、顧客と合意する際に考慮する要因や見積もりの研究·開発コストを含めて、会社は契約履行義務ごとの独立販売価格を決定するために判断すべき仮定を策定している。しかしながら、場合によっては、可変対価格の条項がそれぞれの義務を履行する場合に関連し、割り当てられた金額が、義務を履行するために意図されて受信されると予想される金額と一致する場合があれば、会社は、可変対価格を1つまたは複数の履行義務に全て割り当てる。
当社が収入を確認した根拠は、約束した貨物やサービスを顧客に譲渡することで義務を履行した場合に、義務を履行する取引価格ごとに割り当てられた金額である。ある時点で履行された履行義務に対して,会社は商品および/またはサービスの制御権を顧客に移転する際に収入を確認する.長期的に履行された履行義務について、会社は、関連商品および/またはサービスの制御権を顧客に移転する実績を記述した単一の進捗を測定する方法を用いて、履行義務の完全履行の進展を測定することによって収入を確認する。会社は通常、入力法を用いて一定期間にわたって義務を完全に履行する進捗状況を測定する。その知的財産権許可を含む手配については、手配中に決定された他の履行義務とは異なると判断され、許可が被許可者に譲渡され、被許可者が許可を使用して利益を得ることができる場合には、会社は、許可に割り当てられた金額の収入を確認する。他の承諾とバンドルされたライセンスについて、会社は、合併履行義務が時間の経過とともに履行されるか、ある時点で履行されるかを決定するために、判断を利用して、合併履行義務の性質を評価し、時間が経過するにつれて、収入を確認するための進展を測定する適切な方法を決定する。一つの手配に必要な努力の程度及び当社が一つの手配の下でその履行責任を達成することを期待する期間を決定する際には、重大な経営陣の判断が必要である。企業が必要であれば各報告期間内に進捗の評価基準を評価する, 業績と関連収入確認の測定基準を調整する。いずれの調整も累積追跡原則で入金されており、これは調整期間中の収入と純損失に影響する。
契約費用
現金と現金等価物
当社はすべての原始満期日または購入時の残り満期日が三ヶ月以下の高流動性投資を現金等価物と見なしています。同社の現金等価物には通貨市場基金と商業手形が含まれており、それらは高い流動性と高い信用評価を持っている。このような投資の信用と市場の危険はわずかだ。現金と現金等価物はコスト別に記載され、市場価値に近づいている。
有価証券
♪the the the会社の投資戦略は保本に重点を置いています。当社は当社の投資政策で概説した信用品質基準に適合するツールに投資しています。有価証券には満期日が三ヶ月を超える投資が含まれています。現金等価物に分類されず、満期日が12ヶ月未満の投資は、総合貸借対照表において流動資産として分類される。当社は12ヶ月を超える投資期限が12ヶ月を超える投資を意図的に保有している投資を分類しています
F-10
当面ではない合併した貸借対照表にあります。有価証券には、米国債、米国機関債券、社債、商業手形、資産支援証券が含まれる。その会社はそのすべての有価証券を販売可能なものに分類した。したがって、このような投資は公正な価値で入金される。未実現損益は株主権益内に他の全面収益の構成要素を計上する。利息、配当金及び償却並びに割引及び保険料の増加は利息収入及びその他の純額に計上される。当社は有価証券の減値状況を定期的に審査し、市場価値の低下が非一時的とされた場合にはこれらの投資を公正価値に調整している。有価証券の公正価値低下が一時的でないと判定された場合は,利子収入とその他の純額を計上する。
信用リスクと重要なサプライヤーの集中度
会社を集中的な信用リスクに直面させる可能性のある金融商品には、主に現金等価物と有価証券が含まれる。その投資政策によると、同社は信用格付け、満期日、業界グループ、投資タイプ、発行者によってこのような証券に投資する金額を制限しており、米国政府が発行した証券は除く。当社はこのような金融商品がそれに対していかなる重大な信用リスク集中を構成しているとは考えていない。当社には外国為替契約、オプション契約、その他の海外ヘッジ手配など、表外リスクのある金融商品はありません。
同社は第三者メーカーに依存してその計画中の研究·開発活動に製品を提供している。特に,同社は少数のメーカーがこれらの計画に関連する活性医薬成分や処方薬の供給要求に依存していることに依存し続けると予想される。これらのプロジェクトは活性薬物成分や製剤薬物供給の深刻な中断の悪影響を受ける可能性がある。
公正価値計量
公正価値は、資産を売却する際に受信された価格または計量日に市場参加者間で負債を移転するために支払われる価格として定義される。ASC 820、公正価値計測公正な価値によって計量するツールのために三級評価階層構造を構築する。この階層構造は、計量日までの資産または負債推定値の投入の透明性に基づく。この3つのクラスは以下のように定義される
第1レベル-推定方法の投入は、アクティブ市場における同じ資産または負債の見積もり(調整されていない)である。
第2レベル--推定方法の投入は、市場における同様の資産および負債のオファーをアクティブにすることと、金融商品期間全体にわたって実質的に直接的または間接的に観察可能な資産または負債の投入を含む。
第3級-推定方法への投入は観察できず、資産と負債の公正価値計量に重要な意義がある。口座の帳簿金額。短期満期のため、対応、計算、前払い費用及びその他の流動資産はその公正価値に近い。
財産と設備
財産と設備はコストから減価償却累計と償却を差し引く。財産·設備は資産の推定耐用年数内に直線減価償却を使用する
資産 |
|
使用寿命を見込む |
実験室装置 |
|
|
オフィス家具及び固定装置 |
|
|
コンピュータハードウェア |
|
|
賃借権改善 |
|
対応する資産寿命を改善または延長していないメンテナンスおよびメンテナンスは、発生時に運用費用に計上される。資産を処分する際には、関連コスト及び減価償却が勘定から差し引かれ、それによって生じる収益又は損失はいずれも連結経営報告書に含まれる。
F-11
その他の資産
他の資産には#ドルの売主保証金が含まれている
長期資産減価準備
事件や環境変化により資産の帳簿価値が回収できない可能性がある場合、当社は長期資産を審査します。回収能力は,資産の帳簿価値と資産予想による推定未割引将来のキャッシュフローを比較することで測定した。もし推定された未割引未来の純現金流量が帳簿価値より少ない場合、資産減値は、収益の中で確認された減値損失は資産帳簿価値がその公正価値を超える金額で計量すべきであり、公正価値は資産予想による推定に基づいて未来の現金流量を割引して計量する
賃貸借証書
当社はASCテーマ842に基づいてレンタルを会計処理しているリース会計それは.手配開始時に、当社はその手配が賃貸借契約であるか否か又は賃貸借契約を含むか否かを決定する。年間年間を超えるリースは、総合貸借対照表で使用権(ROU)資産および流動および非流動賃貸負債であることが確認されています(場合によっては)。レンタル負債およびそれに対応するROU資産は、予想される残りのレンタル期間内の将来のレンタル支払いの現在値に基づいて入金される。運営リースのリースコストは直線法でレンタル期間内に運営費用として確認されている。レンタル契約に隠されている金利は一般的に確定しにくい。したがって、当社はその逓増借入金利を使用しており、この金利は、当社が類似経済環境下で同じ通貨、類似期限で担保方式で借金した固定金利を反映している。
研究開発コスト
研究·開発コストは発生時に費用を計上する。研究開発支出は主にアウトソーシング研究開発活動に関連するコストを含み、臨床前と臨床開発、契約研究機関と学術機関による製造と研究、従業員の報酬(株式給与とコンサルティング費用を含む)及び関連費用、専門費用及び施設と間接費用を含む。施設と間接費用には、主に賃貸料の分配、光熱費、研究開発者によるオフィス関連の費用が含まれる。前払い金額又は発生したコストを超えた場合には、当社は、サービスの提供又は貨物の交付時に支出する前払い費用を記録する。
同社はすでに第三者と様々な研究開発契約を締結している。これらのプロトコルは通常キャンセル可能であり、関連支払いは発生時に研究および開発費用として記録される。同社は想定されている進行中の研究コストの計上項目を記録している。計算すべき負債の十分性を評価する際に、会社は、イベント完了段階、受信された請求書、および契約コストを含む研究進展を分析する。いずれの報告期間終了時の当計残高を決定する際には,重大な判断と見積もりを行う。実際の結果は会社の見積もりとは違うかもしれません。
特許費用
会社の特許を取得·維持するコストは,発生時に費用を計上し,会社の総合経営報告書では一般·行政費用に分類される。
株に基づく報酬
株式補償費用の総合業務報告書と総合収益(損失)表における分類方式は,受賞者の賃金コストやサービス支払分類の方式と同様である。従業員および非従業員に付与される株式報酬は、ブラック·スコアーズオプション定価モデル(“ブラック·スコルス”)に基づいて報酬の推定公正価値に基づいて測定される。サービス条件のある報酬に対する株式報酬費用は、サービス期間中に直線法で確認される。株式ベースの報酬:
F-12
業績条件を満たす可能性がある場合には、業績条件のある奨励を認める。株式ベースの報酬は最終的に付与されると予想されるボーナスに基づいているため、没収により減少する。当社は発生した没収行為を計算します。
ブラック·スコアーズ法は株式報酬公正価値を決定する主観的仮定を使用することを要求する。これらの仮説には
普通株主公正価値
初公募前には、会社普通株が公開市場を持っていなかったため、取締役会は授与日ごとに会社普通株関連株奨励の公正価値を推定した。同社は、独立第三者評価会社の推定値に基づいて、当社の授出日に既知の情報と、最近発生した任意の事件及び普通株当たりの公正価値の潜在的影響の審査を利用して、授出日ごとに普通株式の公正価値を推定した。同社の普通株の第三者推定値は、米国公認会計士協会“勤務援助”、補償として発行された私持株会社の株式証券推定値または“勤務援助”で概説された基準に基づいて決定された。会社の普通株式の公正価値を決定するための仮定は、多くの客観的かつ主観的な要素に基づいており、管理層の判断に合わせて、以下のようになっている
“実践支援マニュアル”は、各値推定日における普通株式の推定公正価値を決定するために、異なるカテゴリおよびシリーズの株式に企業価値を割り当てる様々な利用可能な方法を決定する。“実践援助”によると、同社は以下のような方法を考えている
当社の初期発展段階及びその他の関連要素に基づいて、当社はOPM方法及びOPMとPWERM方法の混合方法がその企業価値を分配してその普通株の公正価値を決定する最適な方法であることを決定した。同社の普通株の推定公正価値を決定する際には、その取締役会は、その株主が公開市場でその普通株を自由に取引できないことも考慮している。そのため、同社は加重平均予想流動資金時間に基づいて割引を行い、その普通株の販売可能性の欠如を反映している。公正価値を推定しています
F-13
各社の付与日ごとの普通株は、将来の流動性事件の予想可能性とタイミングに部分的に基づく非市場的割引を反映している。
当社の初公開株式が完了した後、その普通株の公正価値は、その普通株の1日終値市場見積に基づいている。
無リスク金利·無リスク金利は、付与時に有効な米国財務省ゼロ金利債券に基づいており、期間は、株式ベースの報酬の予想期限に対応する。
所期期限·予想期間は、株式ベースの報酬が突出すると予想される期間を表す。同社には具体的な履歴がないことから、オプション付与の期待期限は簡略化方法を用いて決定されている。簡略化方法は、期間を、株式に基づく報酬の帰属時間および契約期間の平均値とする。
予想変動率·同社は2020年10月29日まで個人持株であるため、その普通株は取引履歴を有さず、予想変動率は、株式ベースの報酬の予想期間と等しい期間における上場バイオテクノロジー企業の平均変動率に基づいて推定される。比較可能な会社は、類似した規模、ライフサイクル、または専門分野の段階に基づいて選択される。同社は2022年12月31日までの財政年度中に、その予想変動率の計算に自身の株価の過去の変動率を盛り込むようになった。
期待配当収益率−会社は、その普通株に配当金を支払ったこともなく、その普通株に配当金を支払う計画もない。そこで同社が使用している予想配当収益率は
会社はまた、ASCテーマ718に基づいて、株式ベースの支払いの修正を説明した報酬--株式報酬 (ASC 718).
会社の2020年従業員株購入計画(“ESPP”)下の会社普通株の買い取り価格は
所得税
当社は貸借対照法を用いて所得税を計算します。繰延税金資産および負債は、既存の資産および負債の帳簿金額とそのそれぞれの税ベースとの一時的な違いによる将来の税金項目の影響を推定することができることを確認することができる。繰延税項資産は制定された税率計量を採用し、これらの臨時差額の回収または決済が予想される年度の課税所得額に適用される予定だ。繰延税金支出または利益は繰延税金資産と負債変化の結果である。既存の証拠によると、当社は繰延税金資産が現金にならない可能性が高いと考えていれば、繰延税金資産を減少させるために推定値を設定する。繰延税金資産を回収する能力を評価する際に、当社はその経営業績、持続的な税務計画及び将来の課税収入の予測を含むすべての利用可能なプラス及び負の証拠を考慮する。
準備金は不確実な税金優遇を達成するために準備されている。税務機関が基本税収状況がより持続可能であると審査した場合にのみ、このメリットを確認する。不確定税収状況に関連した利息と罰金は所得税引当金で確認された。
F-14
総合収益(赤字)
包括収益(赤字)には、純収益(赤字)および株主権益(赤字)の他の変化が含まれており、これらの変化は、株式所有者との取引や経済事件ではなく、取引や経済事件によるものである。2022年12月31日現在、総合赤字には売却投資可能な未実現損失が含まれている。2021年12月31日および2020年12月31日までに、純収益(赤字)を除いて、当社には全面収益や赤字項目はありません。
普通株主の1株当たり純収益(赤字)
普通株株主が1株当たり基本純収入(損失)を占めるべき計算方法は、普通株株主が純収益(損失)を当期発行普通株の加重平均株式数で割るべきである。普通株株主の1株当たり純収入の算出方法は、普通株株主は、当期発行済み普通株の加重平均で割った純収入を占めるべきであり、その中には、発行済み株式オプションの希薄化の影響を仮定した潜在希釈性普通株を含む。
会社が初めて公募する前に、普通株株主は1株当たりの基本純損失と希釈後の1株当たり純損失を2段階法で確定すべきであり、これは証券参加に必要である。当社は優先株を参加証券に転換できると考えているが、普通株が配当を出すと、転換可能な優先株保有者は普通株株主と一致する基準で配当を受け取る権利があるからだ。両法では、普通株株主が占めるべき純損失は転換可能優先株に割り当てられず、転換可能優先株の保有者は損失を分担する契約義務がないためである。
2段階法の下で、普通株株主が1株当たり基本純損失を占めるべき計算方法は普通株株主が純損失を占めるべきであり、普通株加重平均株式数を割るべきである。2022年12月31日までと2020年12月31日までの純損失により、普通株株主が1株当たりの純損失を占めるのは基本的に赤字と同じであり、すべての潜在的な希薄化証券の影響が逆になっているからである.
現在行われている研究と開発資産
公認会計原則の下での業務合併資格を満たさない取引で買収されている研究開発資産、及び将来他の用途のない資産は、資産買収の間に支出される。
細分化市場
運営分部は実体の構成要素として定義され、単独の財務情報を得ることができ、首席運営意思決定者(“CODM”)が個別支部にどのように資源を割り当てるかを決定し、業績を評価する際に定期的に審査する。会社のCODMはその最高経営責任者であり、会社全体に基づいて資源を管理·分配している。その結果1つは運営部門と
最近発表された会計公告
新しい会計声明は、時々、財務会計基準委員会(“FASB”)または会社が指定された発効日から採用された他の基準作成機関によって発表される。以下にさらに開示されない限り、当社は、最近発表された基準を採用することが、その総合財務諸表および開示に重大な影響を及ぼす可能性があるとは考えない。
最近採用された会計公告
2022年12月31日までの年間で、会社はいかなる会計声明も採択していない。
F-15
3.協調収益
背景
2020年10月、会社はF.Hoffmann-LaRoche Ltd.とGenentech,Inc.(総称して“羅氏”と呼ぶ)と許可協定(“羅氏許可協定”)を締結し、この協定に基づいて、会社は羅氏にBemnifosbuvirの米国国外でのいくつかの開発と商業化権利に関する独占許可(あるC型肝炎ウイルス用途を除く)を授与した。
2021年11月、羅氏は2022年2月に発効した羅氏許可協定の終了通知を当社に出した。終了後,会社は羅氏許可協定に基づいて羅氏の権利と許可を会社に返還し,会社にBemnifosbuvirの臨床開発と将来の商業化を継続する権利を持たせた。世界開発計画活動と締約国との関連費用分担は発効終了日まで続いている。
会社の結論は、終了通知は会計目的で契約を修正したものである。当社はさらに、終了通知を受けた後、当社のすべての履行義務が完全に履行されたと判断しました。そこで,会社は2021年12月31日までの年度の総合経営報告書と全面収益(赤字)のうち,すべての余剰繰延収入を連携収入として確認した。
終了通知を受ける前に、会社は羅氏許可協定によって確認されたすべての収入を、添付の総合経営報告書と全面収益(損失)における協力収入に分類する。同社が記録した収入は#ドルです
全世界開発計画を達成する活動はASC 808項目の下に列挙されている。発生した費用と羅氏から取得または支払われた費用はASC 730に従って入金されなければならない研究と開発それは.そのため、会社は発生した費用を羅氏に支払ういかなる補償も含めて計上し、羅氏から受け取った補償が発効日を終了する前に研究開発費を削減したことを確認した。
羅氏が返済すべき費用(研究開発費の減少に反映)は2021年12月31日と2020年12月31日までの年度で#ドルとなっている
2022年12月31日まで、当社は貸手#ドルを記録しました
F-16
4.公正価値計測
以下の表は、会社が公正価値によって日常的に計量する金融資産に関する情報を提供し、このような公正価値を決定するための公正価値レベルを示す
|
|
|
|
|
|
|
|
|
|
|
2022年12月31日まで |
|
||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
現金等価物 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
有価証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ財務省債務 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
アメリカ政府機関証券 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
商業手形 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
社債 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
資産支援証券 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|
2021年12月31日まで |
|
||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
現金等価物 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
現金等価物合計 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
同社の公正価値は、公開取引の共通基金であり、2022年12月31日および2021年12月31日までの総合貸借対照表に現金等価物として提示される公開価値レベルにおいて1級に分類される資産には、通貨市場基金に投資される通貨市場口座が含まれる。
2022年12月31日と2021年12月31日までの年間では,1級,2級または3級種別の間に移行はなかった。
5.有価証券
|
|
|
|
|
|
|
|
|
|
|
2022年12月31日まで |
|
||||
|
|
原価を償却する |
|
|
未実現収益 |
|
|
未実現損失 |
|
|
公正価値 |
|
||||
有価証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ財務省債務 |
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
|
||
アメリカ政府機関証券 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
商業手形 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
社債 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
資産支援証券 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
合計する |
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
|
||
2022年12月31日現在、会社は
F-17
2022年12月31日までに
同社が受け取った収益は#ドルだった
6.財産と設備、純額
財産と設備、純額は、以下を含む
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
実験室装置 |
|
$ |
|
|
$ |
|
||
オフィス家具及び固定装置 |
|
|
|
|
|
|
||
コンピュータハードウェア |
|
|
|
|
|
|
||
賃借権改善 |
|
|
|
|
|
|
||
原価で計算した財産と設備総額 |
|
|
|
|
|
|
||
減算:減価償却累計と償却 |
|
|
( |
) |
|
|
( |
) |
財産と設備、純額 |
|
$ |
|
|
$ |
|
||
減価償却と償却費用は#ドルです
7.課税費用およびその他の流動負債
計算すべき費用および他の流動負債には以下の項目が含まれる
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
研究と開発、製造と臨床支出を含む |
|
$ |
|
|
$ |
|
||
許可料 |
|
|
|
|
|
|
||
所得税 |
|
|
|
|
|
|
||
給与明細は給与明細と関係がある |
|
|
|
|
|
|
||
専門費用とその他の費用 |
|
|
|
|
|
|
||
費用とその他の流動負債総額を計算しなければならない |
|
$ |
|
|
$ |
|
||
8.支払いの引受や事項
経営賃貸契約
2021年7月、当社は取り消すことのできない経営リース協定を締結し、この合意に基づき、当社は
225レンタル開始について、会社は#ドルの使用権資産と経営リース負債を記録した
F-18
以下の資産と負債は、会社が2022年及び2021年12月31日までの総合貸借対照表に記録している。
|
|
|
|
12月31日まで |
|
|||
|
2022 |
|
|
|
2021 |
|
||
使用権資産 |
|
$ |
|
|
$ |
|
||
流動賃貸負債 |
|
|
|
|
|
|||
非流動賃貸負債 |
|
|
|
|
— |
|
||
2022年と2021年12月31日終了年度総合業務報告書に一般と行政および研究·開発費用の間に割り当てられるリース構成部分は以下のとおりである
|
|
|
|
|
十二月三十一日までの年度 |
|
||
|
|
2022 |
|
|
2021 |
|
||
リースコストを経営する |
|
$ |
|
|
$ |
|
||
可変リースコスト |
|
|
|
|
|
|
||
総賃貸コスト |
|
$ |
|
|
$ |
|
||
2022年12月31日と2021年12月31日までの年間の可変レンタルコストには、マサチューセッツ州ボストンの主要オフィス施設に関連する公共エリアメンテナンスと会社賃貸に関連する他の運営費用が含まれています。当社の賃貸は暗黙的な金利を提供していないため、当社はその逓増借入金利を利用して賃貸支払いを割引しており、これは、当社が類似経済環境下で同じ通貨で類似期限内に担保をもとに賃貸支払いの固定金利を借り入れていることを反映している。
|
|
|
|
|
|
12月31日まで |
|
|
||
|
2022 |
|
|
|
|
2021 |
|
|
||
残存期間(年単位) |
|
|
|
|
|
|
|
|||
割引率 |
|
|
% |
|
|
|
% |
|||
225借約は現在、同社が2022年12月31日までの唯一の経営賃貸契約であり、将来の最低支払いは以下の通り
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
賃貸支払総額 |
|
|
|
|
隠れた利息に相当する金額を差し引く |
|
|
( |
) |
リース総負債 |
|
$ |
|
|
賃貸負債の当期部分を経営する |
|
|
|
|
賃貸負債の非流動部分を経営する |
|
$ |
|
すべての経営賃貸契約で確認されたレンタル料は#ドルです
当社は225レンタル期間内に信用状を持っていなければなりません。同社の銀行預金は#ドルです
F-19
許可協定
背景
2021年12月、同社はメルク社(“メルク”)の付属会社MSD International GmbHとRuzasvir(“複方”)の開発、製造、商業化についてライセンス契約(“Merckライセンス契約”)を締結した。Ruzasvirは同社がbemnifosbuvirと共同開発しているNS 5 A阻害剤であり,C型肝炎ウイルスの治療に用いられている。
メルク許可協定の条項によれば、当社は、メルクのいくつかの特許および技術に基づいて、メルクから独占的(内部研究を行ういくつかの保持された権利の制約を受ける)、再許可および世界的許可を取得し、研究、開発、製造、製造、使用、輸入、輸出、販売、カプセル販売、またはその化合物を含む製品(各製品)を人間のすべての治療または予防用途のために販売または他の方法で商業化する(“分野”)。
会社がメルク許可協定によって得た権利の対価格として、会社はメルクに払い戻し不可能な前金#ドルを支払った
同社は研究と開発費を確認している
臨時相談料
同社はあるコンサルティング会社と、ある製品の売上のパーセントで計算された成功費の支払いを要求し、累計最高支払額は#ドルである契約を締結した
賠償する
当社はいくつかのタイプの契約を締結しており、これらの契約は当社に第三者のクレームについて各方面に賠償を要求する可能性があります。この等の契約は主に(I)当社の定款に関連し、当該等の細則に基づいて、当社は取締役及び高級管理者及びその他の高級管理者及び従業員がその関係により生じる責任について賠償しなければならない。(Ii)当社は取締役及びいくつかの上級職員及びコンサルタントがその関係により生じた責任について彼らに賠償する契約を行わなければならないこと、及び(Iii)当該等の契約に基づいて、当社は当社の製品、技術、知的財産権又はサービス面の役割又は不作為により彼らに提出した請求を含むいくつかの請求についてサプライヤー、サービスサプライヤー又は特許所持者に賠償しなければならない可能性がある。
正常な業務過程で、会社は時々これらの契約に基づいて賠償要求を受ける可能性がある。上記の1つまたは複数の事項が当社にクレームを提起し、結果的に判決または和解を含む場合、当社の将来の業務、経営業績または財務状況に重大な悪影響を及ぼす可能性がある。その会社は以前の賠償請求の歴史がなく、各特定のクレームに関連する独特の事実と状況が決定的になるので、これらの契約に基づいて支払われる最高潜在金額を決定することは不可能である。
F-20
9.優先株
2022年12月31日までに会社は
2020年5月、当社は
会社の初公募については、転換可能な優先株のすべての株式が転換された
10.普通株式
2022年12月31日現在、会社の法定資本は
2022年12月31日現在、会社は以下の普通株式準備株式を所有している
未平倉オプション |
|
|
|
|
発行された限定株式単位 |
|
|
|
|
業績に基づく優れた限定株式単位 |
|
|
|
|
2020年奨励奨励計画に基づいて将来のために保留株式を付与する |
|
|
|
|
ESPPにより保有された株式 |
|
|
|
|
|
|
|
|
11.株ベースの報酬
2020年10月、株主は会社の2020年インセンティブ奨励計画(“2020計画”)を承認した。2020年には最初に最大支給を規定する予定です
2020年計画は、改訂された当社の2013年株式インセンティブ計画(“2013計画”)に代わって継承されています。未完了のオプション奨励をキャンセルした後、最大でご購入いただけます
F-21
限定株単位
2022年12月31日までの年間で、当社は授与します
|
|
量 |
|
|
加重平均 |
|
||
2022年1月1日に返済されていません |
|
|
— |
|
|
$ |
— |
|
授与する |
|
|
|
|
$ |
|
||
発表する |
|
|
— |
|
|
$ |
— |
|
キャンセルします |
|
|
( |
) |
|
$ |
|
|
2022年12月31日現在の未帰属株式 |
|
|
|
|
$ |
|
||
業績に基づく限定株式単位
2022年12月31日までの年間で、当社は授与します
2022年12月31日までの年間で
|
|
量 |
|
|
加重平均 |
|
||
2022年1月1日に返済されていません |
|
|
— |
|
|
$ |
— |
|
授与する |
|
|
|
|
$ |
|
||
発表する |
|
|
— |
|
|
$ |
— |
|
キャンセルします |
|
|
( |
) |
|
$ |
|
|
2022年12月31日現在の未帰属株式 |
|
|
|
|
$ |
|
||
従業員株購入計画
2020年10月、株主は2020年11月の同社初公募終了時に発効する従業員持株計画を承認した。その会社は最初は全部保留した
2022年4月、会社はESPPに基づいて最初の引受期間を開始した。各発売期間は6ヶ月、購入日は発売期間の最終取引日となっている。2022年9月30日、初発期間が終わり、会社が発行します
F-22
株式オプション
以下に株式オプション活動のまとめを示す
|
|
量 |
|
|
重みをつける |
|
|
重みをつける |
|
|
骨材 |
|
||||
2022年1月1日に返済されていません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
授与する |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
キャンセルします |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
2022年12月31日に返済されていません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
すでに帰属しており,2022年12月31日に帰属する予定である |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年12月31日に帰属して行使可能です |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
オプションが付与された総内在価値は、会社普通株公正価値よりも低い価格を行使するオプションの行権価格と会社普通株推定公正価値との差額として計算される。
オプション付与のサービス期間は一般的にあるいは…
二零二二年十二月三十一日、二零二一年及び二零年十二月三十一日までに年度内に授与された各購入持分の加重平均授受日公許可価値は
|
|
この年度までに |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
無リスク金利 |
|
|
% |
|
|
% |
|
|
||||
所期期限 |
|
|
|
|
|
|
||||||
予想変動率 |
|
|
% |
|
|
% |
|
|
||||
期待配当収益率 |
|
|
% |
|
|
% |
|
|
% |
|||
株に基づく報酬費用
株式に基づく報酬費用は以下のように分類される
|
|
この年度までに |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
研究開発費 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
株式に基づく報酬総支出 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
F-23
株式ベースの報酬費用の構成は以下のとおりである
|
|
この年度までに |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
制限普通株 |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
制限株式単位 |
|
|
|
|
|
— |
|
|
|
— |
|
|
業績に基づく限定株式単位 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
株式オプション |
|
|
|
|
|
|
|
|
|
|||
ESPP |
|
|
|
|
|
— |
|
|
|
— |
|
|
株式に基づく報酬総支出 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
12.所得税
2022年12月31日までに,当社は所得税の利益を$と記録した
連邦法定所得税率と会社の有効所得税率の入金は以下のとおりである
|
|
この年度までに |
|||||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|
|||
連邦法定税率で計算される所期所得税割引 |
|
|
|
% |
|
|
% |
|
|
% |
|||
州税と地方税 |
|
|
|
|
|
|
|
|
( |
) |
|
||
戻り調整 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
研究開発単位 |
|
|
|
|
|
( |
) |
|
|
|
|
||
株に基づく報酬 |
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
海外で得られた無形収入 |
|
|
- |
|
|
|
( |
) |
|
|
— |
|
|
不確定税収状況 |
|
|
( |
) |
|
|
|
|
|
— |
|
|
|
他にも |
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
評価免除額を変更する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
合計する |
|
|
|
% |
|
|
% |
|
|
% |
|||
繰延所得税は、財務報告目的のための資産および負債の帳簿価値と所得税目的のための額との間の一時的な差異の純影響を反映する
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
繰延税金資産(負債) |
|
|
|
|
|
|
||
資本化研究と開発 |
|
$ |
|
|
$ |
— |
|
|
純営業損失が繰り越す |
|
|
|
|
|
— |
|
|
許可協定 |
|
|
|
|
|
|
||
株に基づく報酬 |
|
|
|
|
|
|
||
研究開発単位 |
|
|
|
|
|
— |
|
|
他にも |
|
|
|
|
|
|
||
前払い費用 |
|
|
( |
) |
|
|
( |
) |
繰延税金資産(負債) |
|
|
|
|
|
|
||
減算:推定免税額 |
|
|
( |
) |
|
|
( |
) |
繰延税項目純資産(負債) |
|
$ |
|
|
$ |
|
||
2022年1月1日に発効した2017年の減税と雇用法案の要求に基づき、我々の研究開発支出は資本化と償却を行い、納税資産の繰延を招いた。
F-24
2022年12月31日現在、同社の連邦純運営損失は
経営陣はすでに会社の繰延税金資産の現金化に影響するプラスと負の証拠について評価を行った。2023年以降の純営業損失に対する会社の予測によると、会社は繰延税金資産の収益を確認しない可能性が高いと考えている。そのため、同社は約#ドルの全額推定手当を記録した
米国国税法(IRC)第382及び383条の規定によると、当社の所有権が重大な変動(IRCで定義されているような)が発生した場合、経営損失純額、貸出項目の繰越及びその他の税務属性が制限される可能性がある。
同社はIRC第382節に基づいて2021年12月31日までの分析を行い、純営業損失や他の税務属性の使用に制限があるかどうかを決定した。この分析に基づき、当社は2021年に当社が純営業損失や信用繰越を利用する能力に影響を受けないことを決定した。同社は2022年度第382条の研究を完了している。 企業が追加の持分融資または主要株主の所有権権益を調達することが他に変化した場合、追加の税収属性は年間制限を受ける可能性がある。これは、将来の課税収入または納税義務を相殺するために毎年使用できる税金属性の数を制限することができるかもしれない。
その会社は連邦と各州の所得税申告書を提出した。米国国税局(“IRS”)と州税務機関が評価した訴訟時効は、会社設立以来のすべての納税年度内に開放されている。当社が税収属性の繰越を持っている場合、その属性を発生させた納税年度は、米国国税局や国家税務機関の審査を経て今後の期間中に使用できる程度に調整することができる。現在連邦や州税務監査は行われていない。
次の表は、2022年12月31日までの年度未確認税収割引総額の期初と期末入金状況をまとめています
年初残高 |
|
$ |
|
|
今年度の返品調整への支出に関する減少 |
|
|
( |
) |
応算利息に関する増加 |
|
|
|
|
期末残高 |
|
$ |
|
13.普通株主の1株当たり純収益(損失)
基本的な1株当たり収益と希釈後の1株当たり収益は以下のように計算される
|
|
現在までの年度 |
|
||||||||||||||
|
|
十二月三十一日 |
|
||||||||||||||
|
|
2022 |
|
|
|
2021 |
|
|
|
2020 |
|
||||||
純収益(赤字) |
|
$ |
|
( |
) |
|
|
$ |
|
|
|
|
$ |
|
( |
) |
|
加重平均は普通株式を発行し、基本株は |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
未償還株式オプションの希薄化効果 |
|
|
— |
|
|
|
|
|
|
|
|
|
— |
|
|||
加重平均は普通株式を発行し、希釈した後 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
1株当たりの純収益(損失)は,基本的に |
|
$ |
|
( |
) |
|
|
$ |
|
|
|
|
$ |
|
( |
) |
|
薄めて1株当たりの純収益 |
|
$ |
|
( |
) |
|
|
$ |
|
|
|
|
$ |
|
( |
) |
|
F-25
購入用株式オプション
購入用株式オプション
2020年12月31日までの年度、購入オプション
14.福祉計画
当社は2021年12月31日までの年間で、国内税法第401(K)節(“401(K)計画”)に基づいて固定供出計画を実施している。401(K)プログラムは、最低年齢およびサービス要件に適合するすべての従業員を実質的にカバーする。401(K)計画の条項によると、会社記録の一致入金は最高
15.関連するパーティ取引
当社は、2021年12月31日までの年度内に、その取締役1人が支配する1つのエンティティとコンサルティング契約を締結した。その協定は毎年の雇用費を#ドルと規定している
2022年5月、同社は取締役の一人とコンサルティング契約を締結した。会社が確認した費用は#ドルです
F-26