目录表
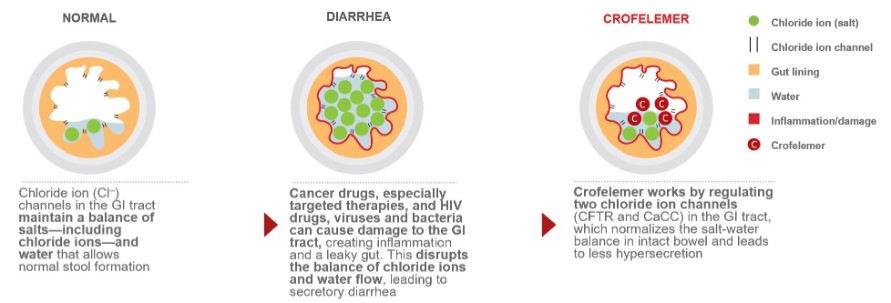
下图说明了克罗非莱姆的作用机制,它可以使肠道的氯离子分泌和液体含量正常化,以改善大便的稠度。
业务战略
我们的目标是成为一家领先的制药公司,拥有一流的可持续衍生产品,满足全球重大的未得到满足的胃肠道医疗需求。为达致这个目标,我们计划:
通过利用我们重要的胃肠产品知识、经验和知识产权组合来扩展Mytesi
Mytesi(克罗非勒姆125 mg缓释片)是一种新型的、一流的抗分泌性止泻药,它对肠道内的电解质和液体平衡具有正常化作用,这种作用机制可能有利于多种胃肠道疾病。我们的Mytesi(CroFelemer)产品获得FDA批准,用于抗逆转录病毒治疗的成人艾滋病毒/艾滋病患者的非感染性腹泻的症状缓解。捷豹通过Napo和Napo Treateutics拥有Mytesi的全球未受约束的权利。Mytesi正在开发多种可能的后续适应症,包括与使用或不使用标准化疗的靶向治疗相关的腹泻预防。CroFelemer缓释片也在评估IBS-D和特发性/功能性腹泻。
正在开发用于口服液的克罗非勒粉,以支持患有SBS和/或CDD(如MVID)的婴儿和/或儿童的孤儿或罕见疾病适应症。
此外,一种NP-300正在开发中,用于缓解症状和治疗中到重度腹泻,包括细菌、病毒和寄生虫感染引起的细菌、病毒和寄生虫感染霍乱弧菌,引起霍乱的细菌。
我们的管理团队集体在处方药开发方面拥有丰富的经验。这种经验涵盖了产品开发的方方面面,包括发现、临床前和临床开发、GMP制造、监管事务和商业化。这个团队的主要成员成功地开发了Mytesi。
在Mytesi销售和营销工作中保持商业能力
NAPO的直销组织由Mytesi现场销售代表组成,这些销售代表战略性地部署在不同的地区,以覆盖最具潜力的美国地理区域。在随之而来的营销、促销活动、患者赋权计划(包括整合的社交数字活动)和下文所述的医学教育倡议的支持下,我们预计接受Mytesi治疗的患者数量将出现比例反应。
12
