我们将这些指导原则应用于一系列广泛的制药平台,每个平台的设计都有以下目标:独特的作用模式、高精度靶向、高效力和治疗或预防潜力。我们希望每个平台都能产生一系列候选产品,以供进一步开发。凭借这种平台和候选产品的技术不可知的组合,我们相信我们是当今肿瘤学和传染病个性化、以患者为中心的治疗方法领域的先驱,并致力于在未来的其他疾病领域。
我们对抗人类疾病的颠覆性技术工具包
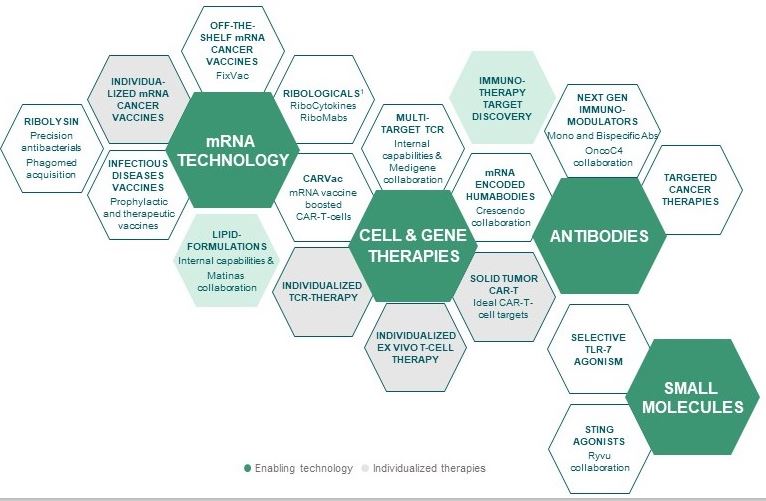
1mRNA编码的肿瘤靶向抗体和细胞因子
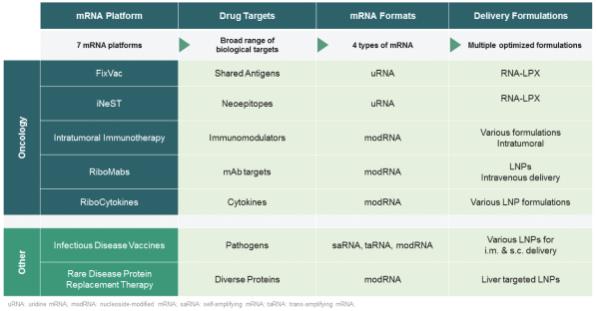
我们目前正在临床试验中测试的候选免疫治疗产品涵盖四种不同的药物类别:
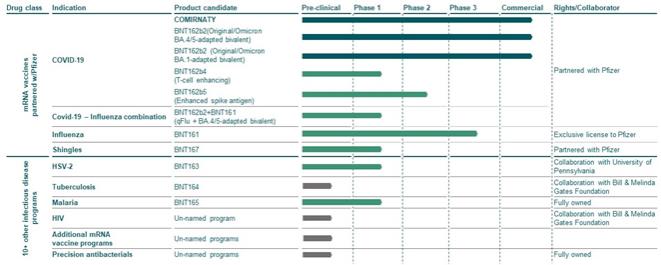
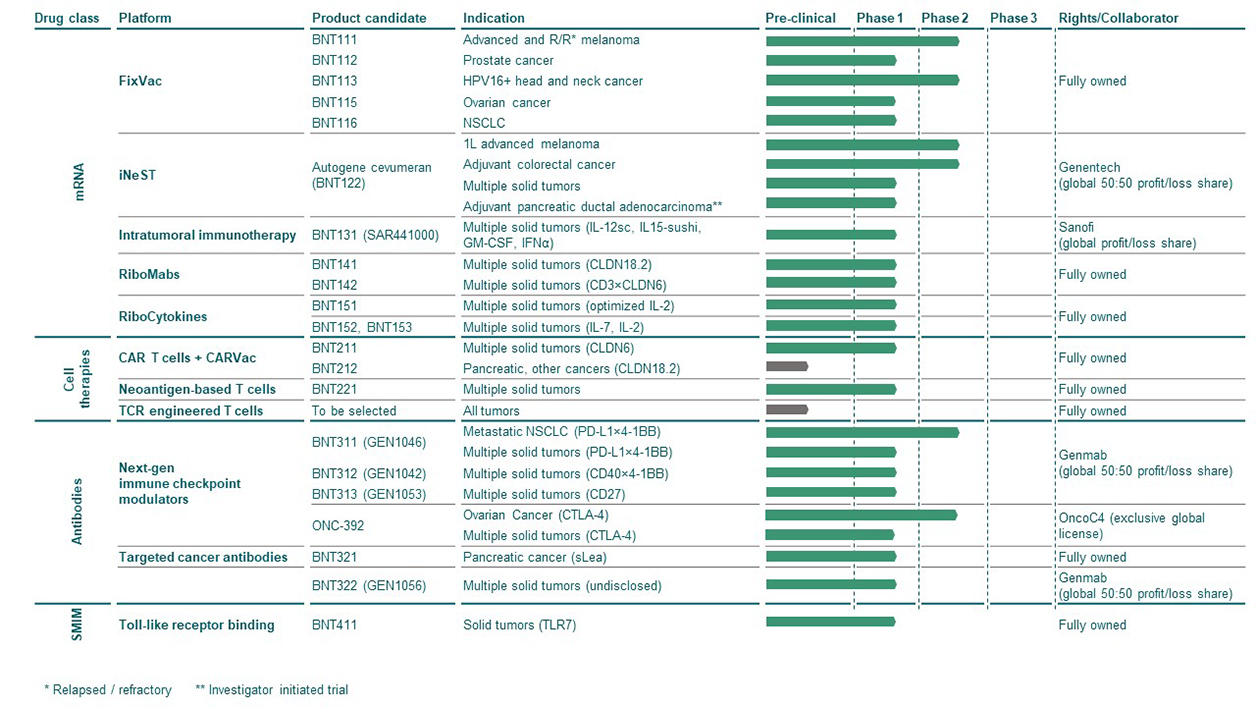
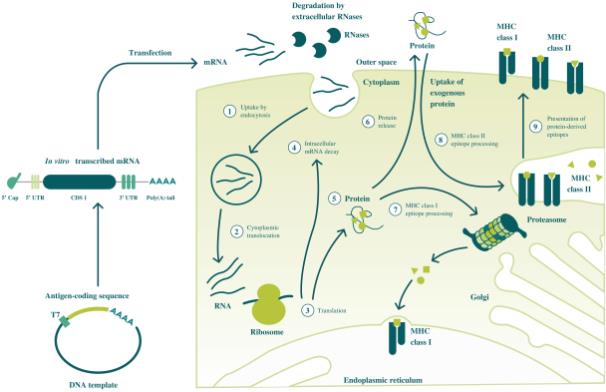
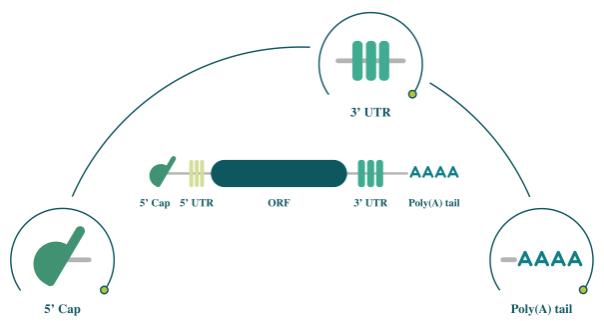
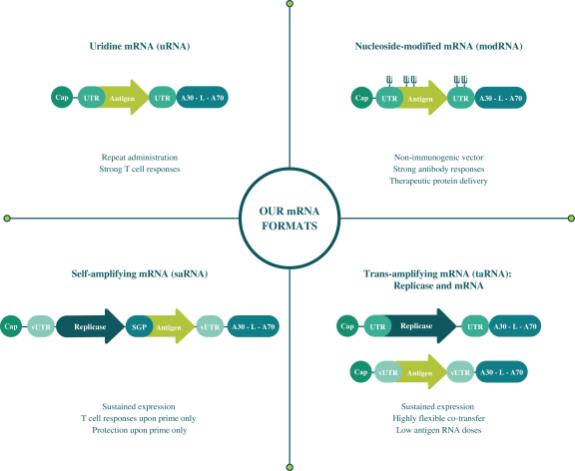
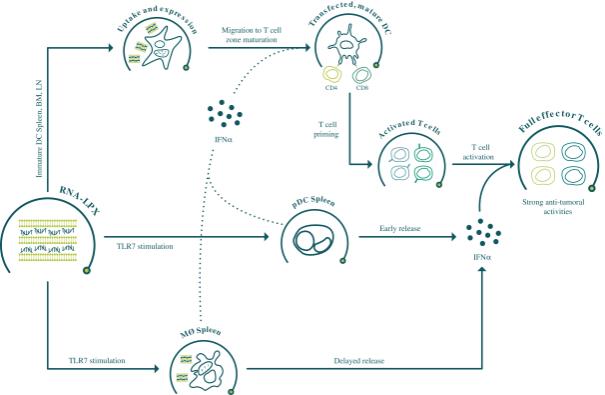
| • | mRNA疫苗和治疗。我们利用mRNA向细胞传递核酸信息,在细胞中这些信息用于表达蛋白质以达到药理作用。在传染病方面,我们正在开发基于mRNA的预防性疫苗, COVID-19,带状疱疹,疟疾,肺结核, HSV—2型,和其他传染病。在肿瘤学方面,我们正在开发一系列基于mRNA的疗法来治疗癌症,包括 固定真空与Genentech合作的iNeST,以及mRNA编码细胞因子和抗体,或抗菌剂。 |
| • | 细胞疗法我们正在开发一系列针对实体瘤的细胞疗法,包括 CAR-T细胞疗法,基于新抗原的 T细胞治疗和TCR治疗,其中患者的T细胞被修饰或引发以靶向癌症特异性抗原。我们还结合了mRNA 固定真空平台与我们的第一个 CAR-T候选产品,以增强持久性 CAR-T细胞体内. |
| • | 抗体的除了我们的基于信使核糖核酸的抗菌药外,我们还在利用我们的内部该公司正在与Genmab合作开发下一代抗体,旨在调节患者对癌症的免疫反应。 |
| • | 小分子免疫调节剂。我们的目标是使用小分子通过诱导特定和离散的免疫调节模式来增强其他药物类别的活性。第一个方案是一种小分子Toll样受体7,或TLR7,用于治疗实体肿瘤的免疫调节剂。 |
在这四个药物类别中,我们拥有强大的临床流水线,包括肿瘤学领域的20种候选产品和传染病领域的6种候选产品。我们计划在2023-24年加快我们肿瘤学商业能力的建设,目标是在美国、欧盟和其他选定地区做好商业准备,以支持从2026年起推出第一批肿瘤学。从长远来看,我们将看到我们的技术在自身免疫性疾病、炎症性疾病、心血管疾病、神经退行性疾病和再生医学领域的应用。
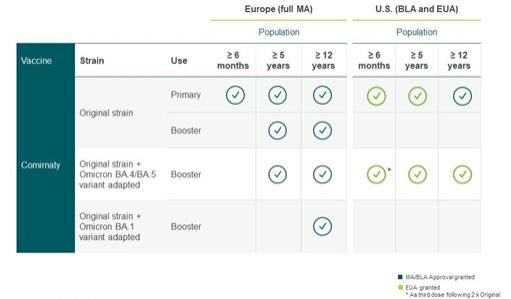
三、上市产品:喜剧性,我们的新冠肺炎疫苗计划(BNT162)
我们的第一个商业产品,好笑,是有史以来第一个获得批准的基于信使核糖核酸的产品,据我们所知,它代表了从病毒样本到批准的最快开发的预防性疫苗。截至2022年12月,我们的原版新冠肺炎疫苗产品已在全球100多个国家和地区获得授权或批准紧急或临时使用,或获得营销授权,我们的努力已导致超过40亿剂疫苗运往全球。
84