目录表
已实现,以促进PAX-101用于某些神经学适应症的加速开发计划,包括ASD、ME/CFS和LCS。根据我们在2021年3月与FDA举行的IND前会议,以及部分基于我们从卫生部、马拉维共和国和Lwala医院(乌干达索洛蒂)获得的关于使用苏拉明治疗东非HAT患者的独家许可数据的分析,我们相信我们已经制定了一项强大的发展战略,我们计划在寻求批准PAX-101用于治疗东非HAT时采用该战略。根据我们之前与FDA的互动,包括我们与FDA的Pre-IND和Type B会议,我们进一步认为,如果东非HAT获得批准,我们可能会获得FDA的优先审查凭证(PRV),我们可能会将其货币化,为我们未来的临床计划提供资金。我们预计将需要对PAX-101进行进一步的临床研究,以治疗ASD、FXS、FXTAS和ME/CFS,并需要类似的临床开发才能使PAX-102达到商业阶段。2020年11月,FDA批准PAX-101为治疗东非HAT的孤儿药物。然而,不能保证我们将获得FDA对用于治疗东非HAT的PAX-101的批准,即使PAX-101获得FDA的批准,也不能保证我们将获得PRV。有关PRV过程以及我们如何从中受益的更多信息,请参阅本年度报告中Form 10-K的部分。“第1项。商业-政府监管 - 优先审查代金券计划。”
开发管道
下表总结了我们目前的候选产品和适应症渠道。
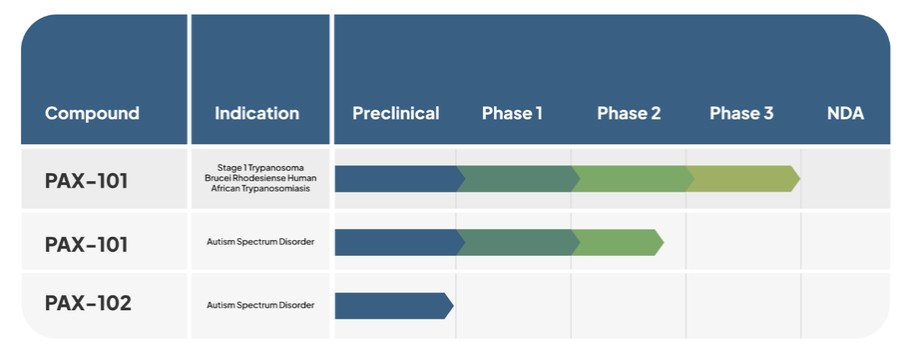
PAX-101(静脉注射苏拉明)
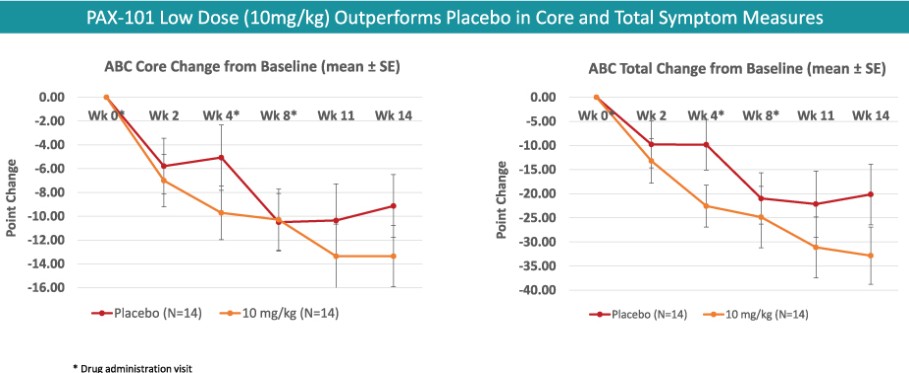
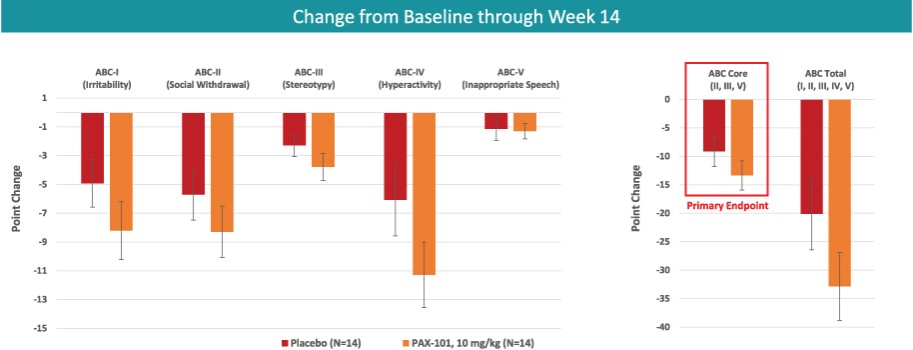
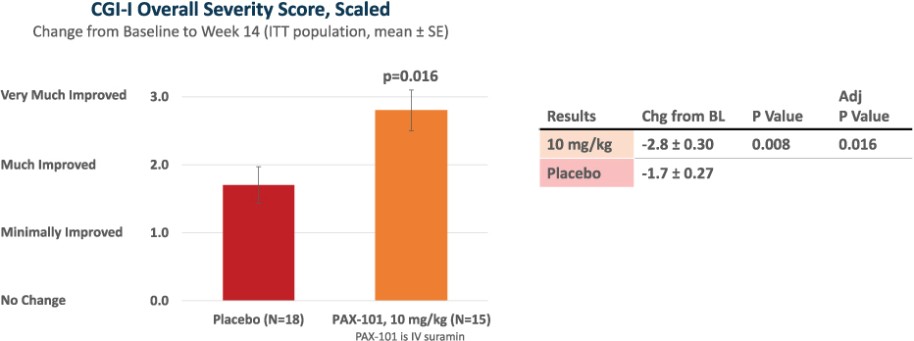
我们正在开发的主要候选产品是PAX-101,这是一种苏拉明的静脉制剂,我们正在开发用于多种适应症的PAX-101,包括东非HAT、ASD和ME/CFS。
我们正在开发PAX-101的最先进的适应症是用于“第一阶段”的治疗。布氏锥虫(东非)HAT,HAT临床病程的阶段,在外周循环中发现寄生虫,但尚未渗透到中枢神经系统。我们拥有在全球范围内独家授权使用苏拉明治疗1期东非HAT的患者级别数据的权利,我们打算利用这些数据来证明PAX-101的安全性和有效性。我们已经就PAX-101的实施和开发与FDA举行了三次正式会议。根据这些对话,并满足FDA的要求,证明了实质性的有效性,我们进行了新的前瞻性临床试验。2023年7月,我们完成了对2000年至2020年期间接受苏拉明治疗的东非HAT患者的回顾数据的分析和展示,我们获得了独家许可。除了这些回顾性数据,我们还将完成临床前和临床安全性研究,以支持提交PAX-101‘S东非HAT适应症的NDA。我们预计这项工作将在未来10个月内完成,打算在2024年下半年提交保密协议。此外,我们正在完成药物物质和药物产品专有供应链的开发,这将成为我们提交保密协议的基础。如果不建立这一供应链,我们将无法提交东非HAT标志的保密协议。有关我们建立供应链的预期时间表的其他信息,请参阅“制造”。2020年11月,FDA批准PAX-101为治疗东非HAT的孤儿药物。预计PAX-101如果被FDA批准用于东非HAT适应症,将有资格获得新的化学实体独家经营权(向该公司提供关于任何含有苏拉明的产品在美国的独家营销权,最长可达七年),此外还有孤儿药物独家经营权,
4