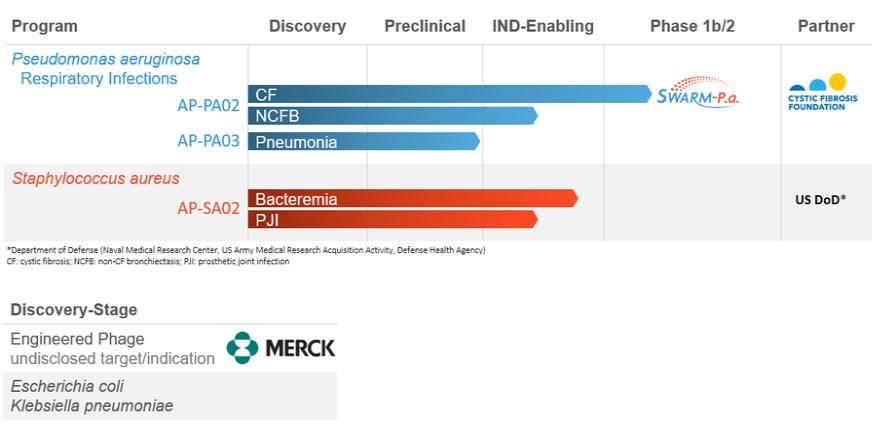
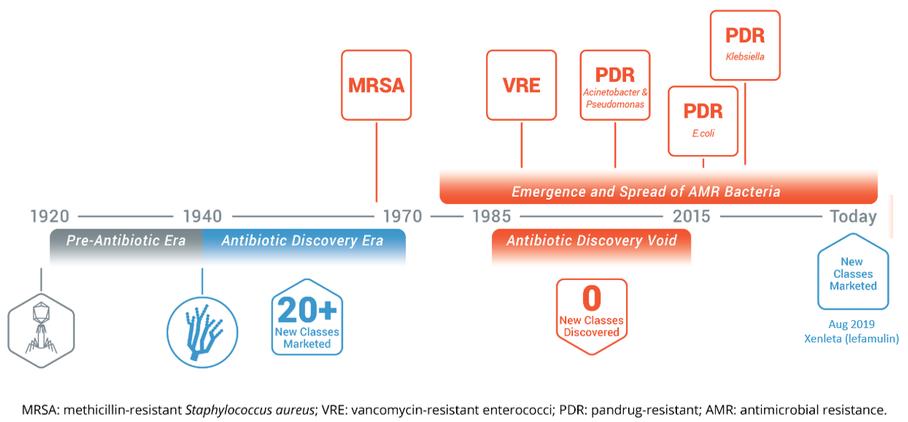
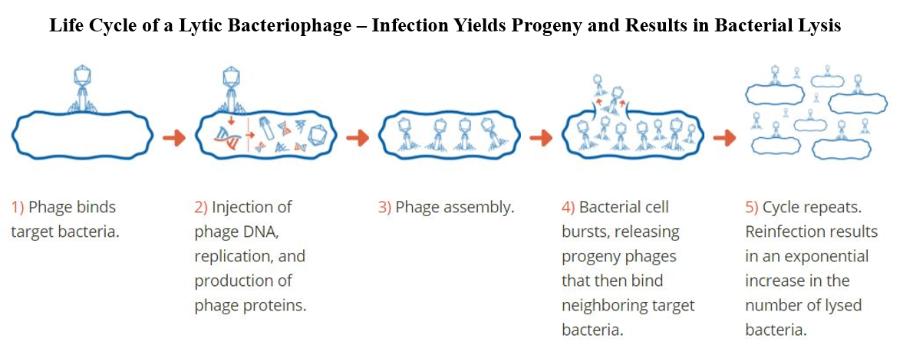
(4)由(A)约13,889股普通股及(B)约14,501股普通股组成,根据股票期权的行使,巴斯蒂亚尼先生有权在2021年3月1日起60天内向我们收购普通股。
(5)包括(A)根据股票期权的行使,科斯塔斯博士有权在2021年3月1日起60天内从我们手中收购的13,692股普通股,(B)8,710,800股普通股和可为Innoviva,Inc.持有的8,710,800股普通股行使的认股权证,以及(C)1,867,912股普通股和可为1,867,912股持有的普通股行使的认股权证Innoviva,Inc.和Innoviva Strategic Opportunities,LLC是因担任Innoviva,Inc.董事的身份而与其有关联的实体。Kostas博士可能被认为对Innoviva,Inc.和Innoviva Strategic Opportunities,LLC实益拥有的股票拥有共同投票权和处置权,但放弃这种实益所有权,除非他们在其中有金钱利益(如果有的话)。
(6)由13,692股普通股组成,根据股票期权的行使,帕蒂博士有权在2021年3月1日起60天内从我们手中收购普通股。
(7)由13,692股普通股组成,根据股票期权的行使,彼得森博士有权在2021年3月1日起60天内从我们手中收购普通股。
(8)包括(A)施莱辛格博士根据股票期权的行使有权在2021年3月1日起60天内从我们手中收购的13,692股普通股,(B)8,710,800股普通股和可为Innoviva,Inc.持有的8,710,800股普通股行使的认股权证,以及(C)1,867,912股普通股和可为持有的1,867,912股普通股行使的认股权证。Innoviva,Inc.和Innoviva Strategic Opportunities,LLC是与施莱辛格博士有关联的实体,因为她是Innoviva,Inc.和Innoviva Strategic Opportunities,LLC的董事。施莱辛格博士可能被认为对Innoviva,Inc.和Innoviva Strategic Opportunities,LLC实益拥有的股票拥有共同投票权和处置权,但放弃这种实益所有权,除非他们在其中有金钱利益(如果有的话)。
(9)包括(A)49,058股普通股,(B)155,028股限制性普通股,以及(C)根据股票期权的行使,帕特里克先生有权在2021年3月1日起60天内从我们手中收购的130,737股普通股。
(10)由(A)约203股普通股、(B)约31,851股限制性普通股及(C)Varnum博士有权于2021年3月1日起60天内向吾等收购的116,510股普通股组成。
(11)由(A)约26股普通股及(B)约46,952股普通股组成,根据股票期权的行使,马丁先生有权于2021年3月1日起60天内向吾等收购该等普通股。
(12)包括(A)约5,612股普通股,(B)约20,532股限制性普通股及(C)约26,069股普通股,根据购股权的行使,莫礼时先生有权于2021年3月1日起60个交易日内向吾等收购。
(13)代表截至2021年3月1日我们现任董事和高管作为一个集团持有的我们普通股的实益所有权,包括2021年3月1日起60天内可行使的任何期权和认股权证。
股权薪酬计划信息
2009年3月,我们的董事会和股东通过了我们的2009股票激励计划,或者说2009计划。根据2009年计划,没有剩余的普通股可供未来奖励。
2012年10月,我们的董事会批准并通过了我们的2012年股票激励计划,即2012年计划。根据2012年计划,没有剩余的普通股可供未来奖励。