目录
HPV阳性者3例,HPV阴性者2例,HPV状态不明者2例。观察到的治疗紧急不良事件(TEAE)与潜在的晚期疾病和已知的培溴利珠单抗、淋巴枯竭和IL-2方案的不良事件概况是一致的。最常见的TEAE是寒战、贫血、低血压、恶心、发热和血小板减少,发生在超过50%的可评估受试者中。
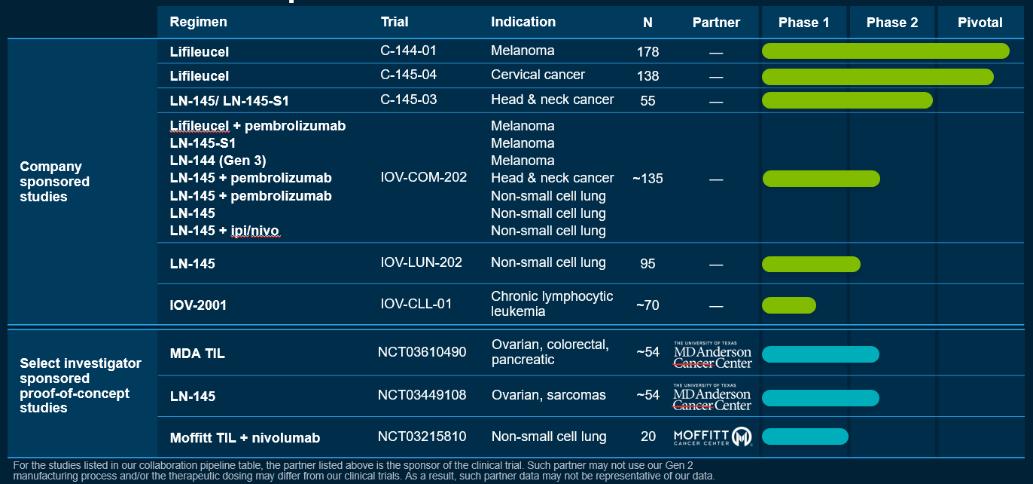
2019年11月,我们宣布,我们的PBL疗法IOV-2001的研究新药申请(IND)获得了FDA的授权,我们赞助的使用这种疗法的临床试验IOV-CLL-01获准继续进行。IOV-2001是一种非转基因的多克隆T细胞产品,从患者的血液中提取50毫升,用9天的工艺制造而成。IOV-CLL-01是一项1/2期临床试验,评估IOV-2001在复发或难治性CLL或SLL患者中的安全性和有效性。IOV-CLL-01试验预计将招募大约70名患者。
作为我们与MD Anderson癌症中心(MDACC)合作计划的一部分,2018年启动了两项第二阶段试验。这两项试验都是由MDACC赞助的。第一项试验NCT03449108旨在研究我们使用我们的制造工艺生产的LN-145,用于治疗软组织肉瘤、骨肉瘤、耐铂卵巢癌和甲状腺癌患者。与MDACC合作的第二个试验NCT03610490也在进行中。这项试验正在治疗对铂耐药的卵巢癌、胰腺癌和结直肠癌患者。这项试验使用MDACC使用urelumab制造的TIL,urelumab是一种4-1BB激动型抗体,作为制造过程的一部分。使用此制造过程获得的数据可能不能代表我们使用第二代制造过程的数据。
我们还与蒙特利尔大学医院中心(CHUM)、耶鲁大学(Yale University)和莫菲特(Moffitt)合作,对其他适应症的TIL疗法进行研究人员赞助的临床试验。CHUM和Moffitt赞助的临床试验使用或将使用由不同制造工艺制造的TIL,这可能不能代表我们使用第二代制造工艺的数据。
下图汇总了我们目前的候选产品和选定的研究人员赞助的概念验证研究:
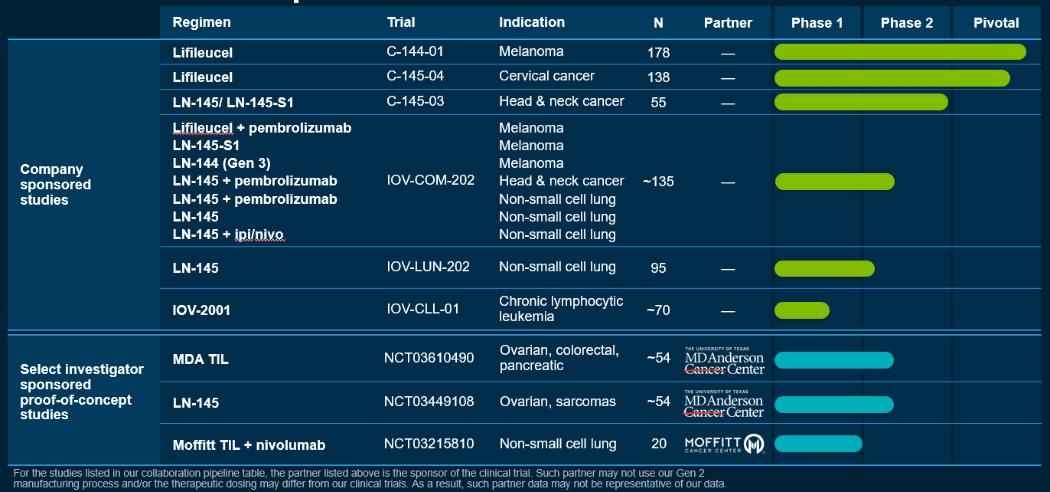
我们开发了名为Gen 3的第三代制造流程。Gen 3的流程比Gen 2更短。我们在NSCLC的IOV-COM-202试验的1C组和NSCLC的IOV-LUN-202试验的3组中使用了Gen 3制造流程,并曾在HNSCC的C-145-03试验中使用过它。
我们目前拥有20多项已授权或允许的美国和国际专利,用于治疗与我们的第二代制造工艺相关的广泛癌症的组合物和治疗方法,包括美国专利第10,130,659号,10,166,257号,
6