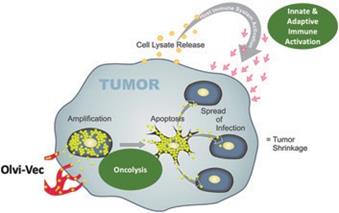
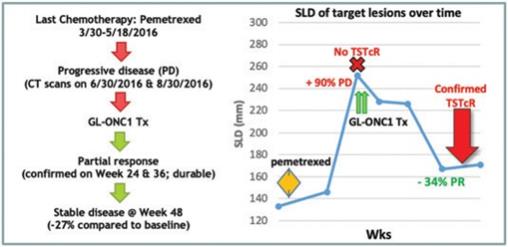
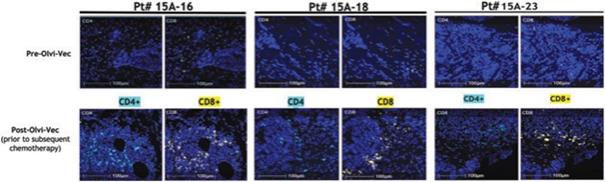
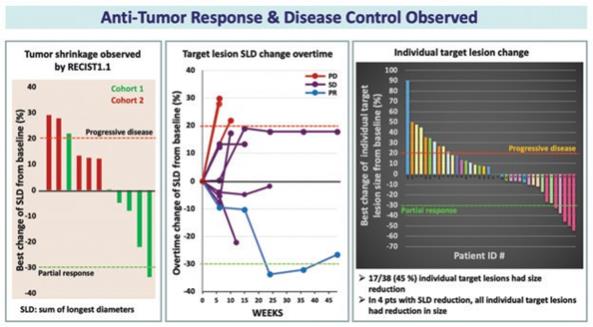
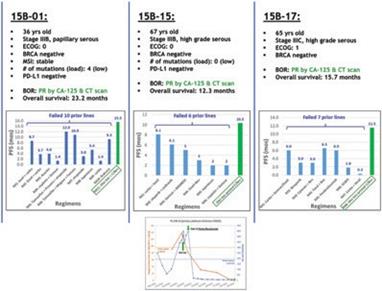
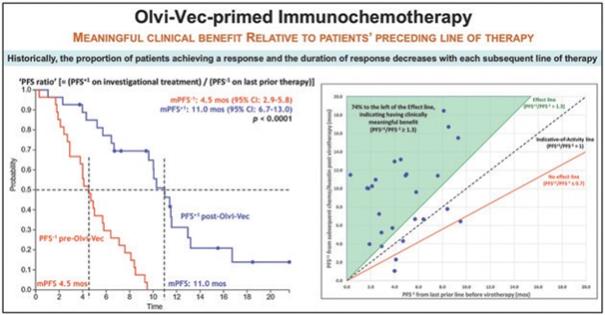
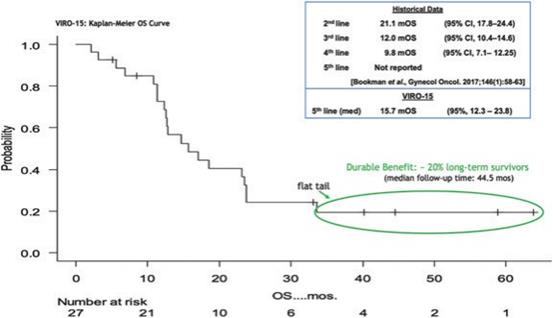
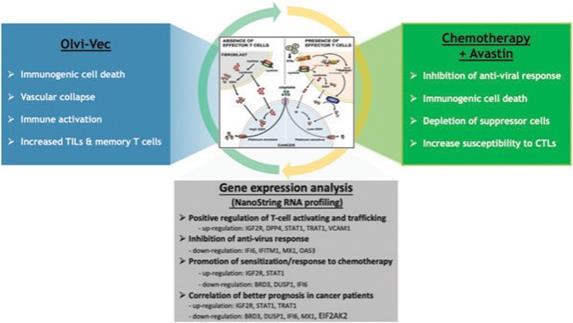
下表总结了我们的临床开发流程:
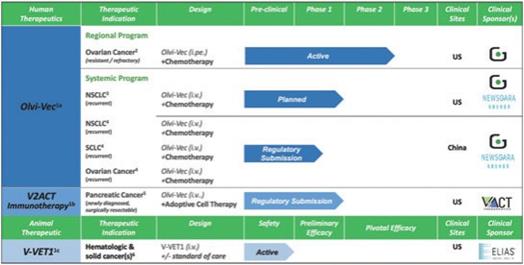
| 1 | 商业权利 |
1a Genelux: 全球(不包括大中国);Newsoara(大中国)
1bV2ACT免疫疗法:全球(不包括大中国)
1cELIAS:全球
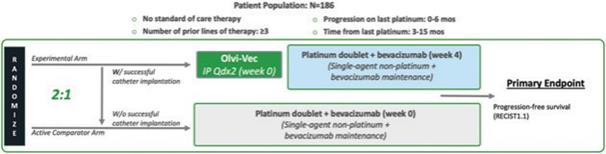
| 2 | 我们在我们的3期临床试验中招募了第一名患者。 |
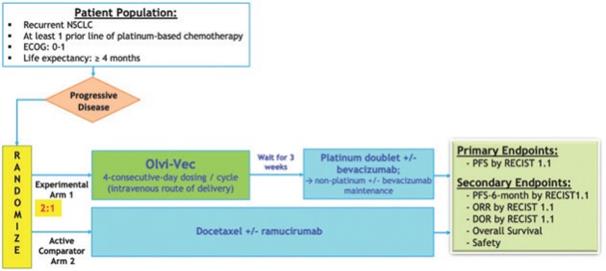
| 3 | 根据我们之前完成的对实体肿瘤患者静脉注射Olvi-Vec的第一阶段临床试验的结果,我们计划启动Olvi-Vec治疗复发NSCLC的第二阶段临床试验。 |
| 4 | Newsoara已向中国医疗产品协会提交了IND和方案。 |
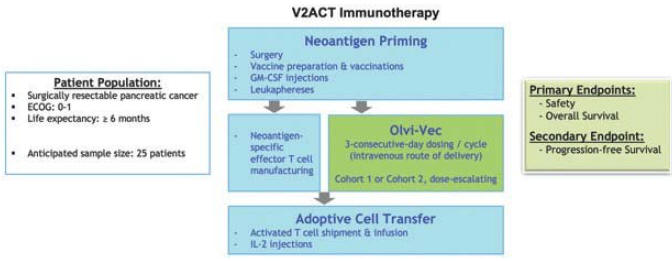
| 5 | V2ACT具有此候选产品的活动IND。1b/2a期临床试验尚未计划启动。 |
| 6 | 伊莱亚斯正在开发一种疗效试验。 |
我们是由洛马林达大学的一个学术团队于2001年创立的,该团队由Aladar A.Szalay博士领导,Aladar A.Szalay博士是监测基因调控以及使用发光蛋白质或蛋白质融合进行全细胞和活体成像方面的国际公认的领导者。我们组建了一支经验丰富的业务领导团队,在肿瘤治疗方面拥有丰富的经验,包括推动候选产品从临床前研究到临床开发和商业化。Thomas D.Zindrick,J.D.,首席执行官兼董事长总裁此前曾在阿米泰克治疗解决方案公司担任总裁兼首席执行官和董事 职位,并在安进公司担任过各种高管管理职位,包括协理副总裁总裁、总法律顾问和首席合规官,并在陶氏化学公司担任责任日益增加的法律职位。董事首席独立董事詹姆斯·L·泰里曾在雅培担任过多个高管职务,其中包括环球医药执行副总裁总裁,曾在Sugen,Inc.担任总裁一职,还曾在百时美施贵宝公司和辉瑞公司担任过管理职务。
3