与安慰剂相比,蒙哥马利-阿斯伯格抑郁评定量表(MADRS)第6周总分的变化在统计学上有显著改善。在每项研究中,CAPLYTA 42 mg还显示出与双相情感障碍临床总体印象有关的关键次要终点在统计学上的显著改善。此外,CAPLYTA显示出良好的耐受性和安全性,与先前的精神分裂症临床研究结果一致。最常见的不良反应(发生率为5%或以上,至少是安慰剂的两倍)是嗜睡/镇静、头晕、恶心和口干。CAPLYTA和安慰剂在体重、空腹血糖、总胆固醇、甘油三酯和低密度脂蛋白胆固醇方面的平均变化与基线相似。
我们的发展计划
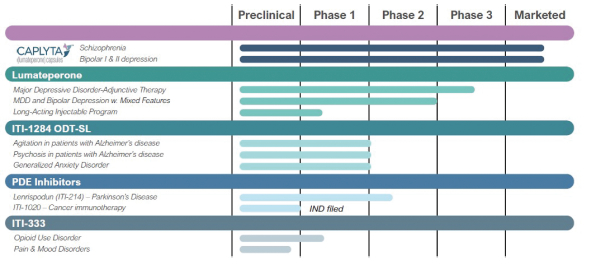
我们的产品线包括临床开发的几个候选产品和非临床测试的其他候选产品。我们相信,我们的候选产品提供了创新的治疗方法,并可能提供相对于当前疗法的优势。下表总结了我们的候选产品和计划:
我们的治疗渠道
研究试剂和/或批准产品的研究用途的安全性和有效性尚未确定。
Lumateperone开发计划
鲁马西培酮的疗效可能是通过中枢5-羟色胺拮抗剂的结合来实现的。5-羟色胺2A中枢多巴胺D2受体和突触后拮抗剂活性。在药效学方面,鲁马替培酮与5-羟色胺具有较高的结合亲和力5-羟色胺2A与多巴胺D2受体、5-羟色胺转运体、多巴胺D1受体、多巴胺D4受体以及肾上腺素能α1A和α1B受体具有中等的结合亲和力。它缺乏与其他受体的生物相关的相互作用,包括毒鼠碱和组胺能受体。因此,我们认为鲁马替培酮可能代表着一种跨越多种治疗适应症的潜在治疗方法。
鲁马替培酮治疗重度抑郁障碍和其他情绪障碍
作为一种强有力的5-羟色胺2A受体拮抗剂和5-羟色胺再摄取抑制剂,我们相信鲁马西培酮可以改善抑郁症的症状,副作用比目前市场上销售的抗精神病药物和
4