目录表
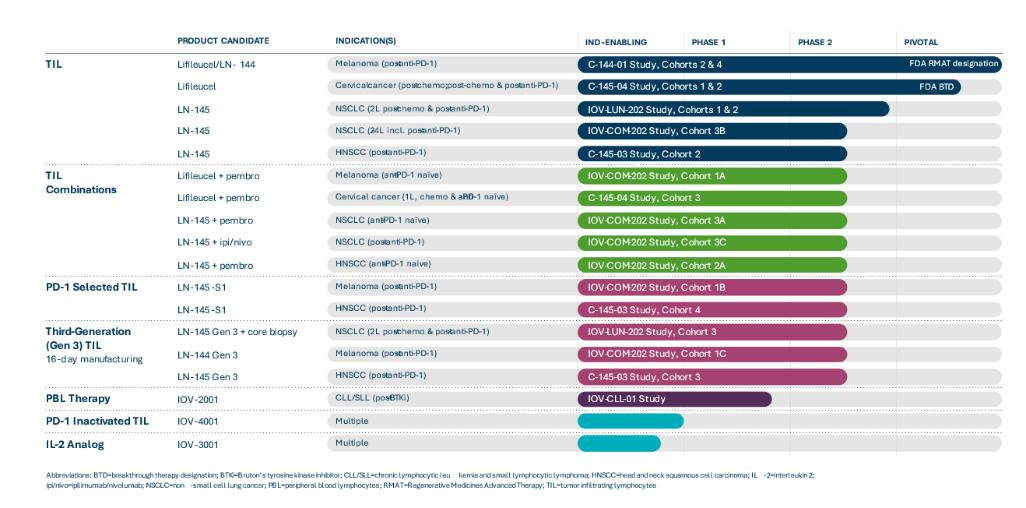
我们目前的候选产品如下图所示:

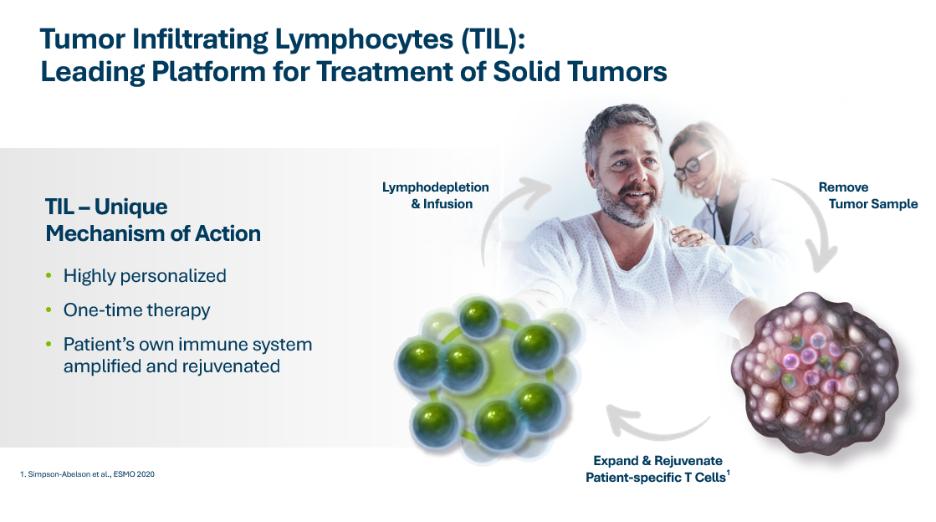
TIL单一疗法治疗转移性实体瘤
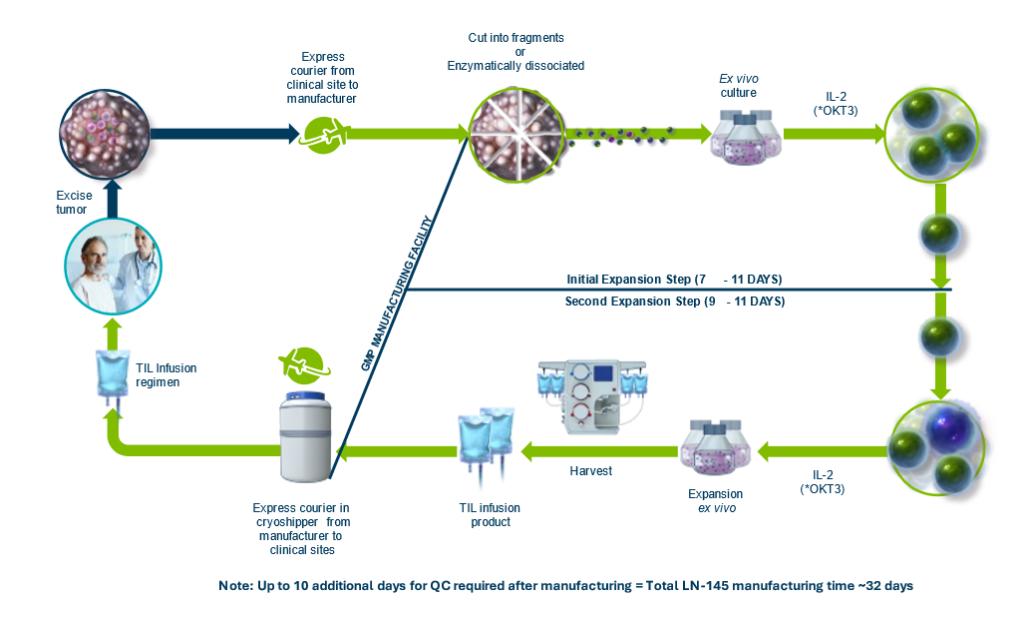
我们研究了TIL单一疗法在转移性黑色素瘤、宫颈癌、NSCLC和HNSCC中的应用。我们正在进行第二阶段临床试验,C-144-01,我们的主要TIL候选产品lifileucel,用于治疗转移性黑色素瘤。这项多中心关键试验纳入了经过至少一种系统治疗后病情恶化的黑色素瘤患者,包括PD-1抑制剂,如果BRAF突变,还包括BRAF抑制剂,或BRAF和MEK抑制剂的组合。C-144-01临床试验的Cohort 4是一个单臂队列,旨在支持向FDA提交生物制品许可证申请或BLA,以获得lifileucel。C-144-01试验的第二组和第四组使用我们的第二代制造工艺。我们于2018年完成并结束了C-144-01试验的患者入选队列2。C-144-01临床试验的队列2的结果最初在2019年6月的美国临床肿瘤学会年会(ASCO)上报告,最近一次在2021年6月在ASCO更新。截至2021年4月的数据摘录,在队列2中的66名转移性黑色素瘤患者中,根据研究人员的评估,Lifileucel治疗导致的客观有效率(ORR)为36%,其中3人完全缓解,21人部分缓解。疾病控制率(DCR)为80.3%。在中位研究随访33.1个月后,队列2中的中位应答持续时间(DOR)尚未达到。2021年6月在ASCO会议上公布的队列2的结果也表明,在抗PD-1治疗的初始进展时早期使用脂细胞干预可能会获得最大益处。队列2中的患者接受了大量的预治疗,平均以前接受了3.3次治疗。我们之前曾报道过,在接受过抗CTLA-4和BRAF靶向治疗的转移性黑色素瘤患者中,各个年龄段的患者都有持久的反应, 无论是BRAF突变状态,还是PD-L1高、低状态患者。不良事件概况与潜在的晚期疾病以及淋巴枯竭和IL-2方案的概况基本一致。此外,详细的Cohort 2数据发布在临床肿瘤学杂志 in May 2021.
C-144-01试验的Pivotal Cohort 4被招募来评估独立审查委员会(IRC)读出的ORR,作为主要终点,这是基于我们对2018年第三季度与FDA举行的第二阶段结束会议(EOP2)中与FDA讨论的解释。2018年10月,根据在EOP2会议期间提供给FDA的数据,我们宣布lifileucel已获得FDA的再生药物高级治疗(RMAT)称号。C-144-01试验的队列4于2019年3月开始招募,患者剂量于2020年1月完成。共有87名患者服用了为第四队列发布的第二代产品。2020年5月,我们公布了第四队列中68名患者的初步结果,根据研究人员的确定,这些患者进行了两次放射学评估。
5