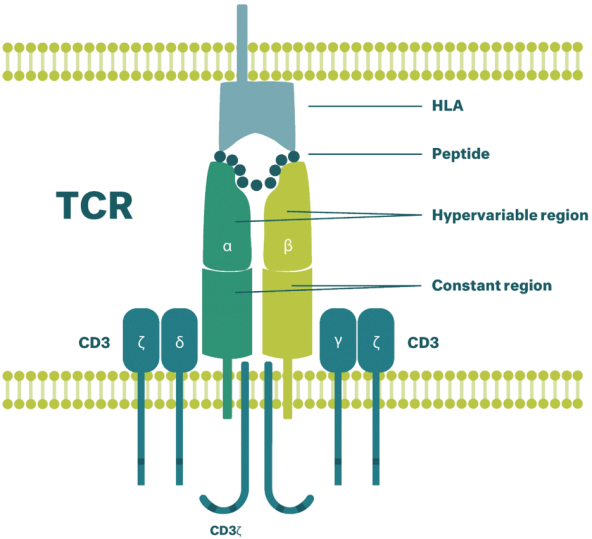
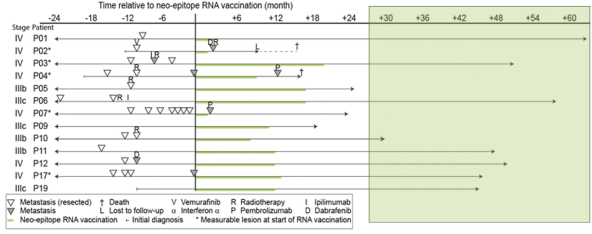
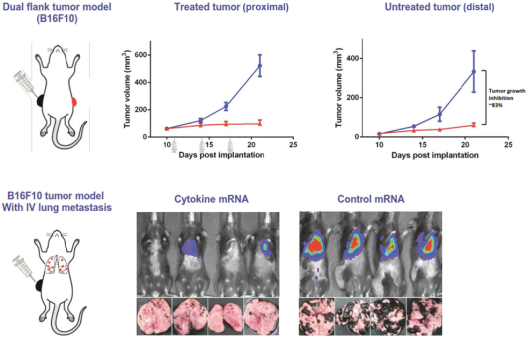
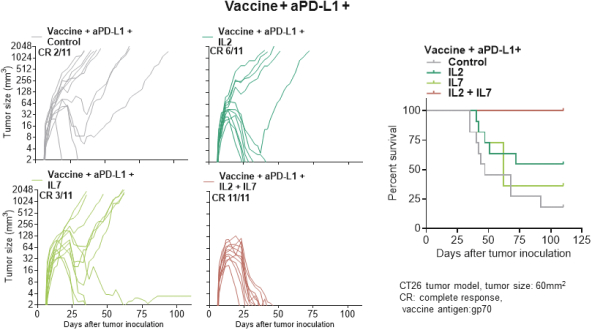
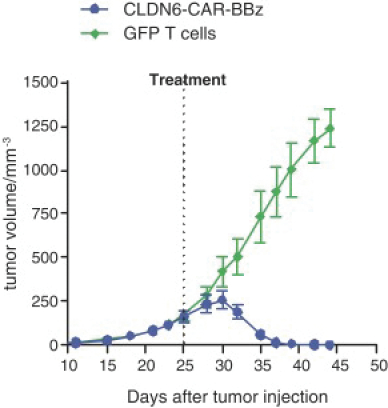
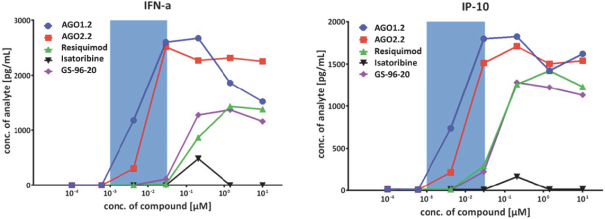
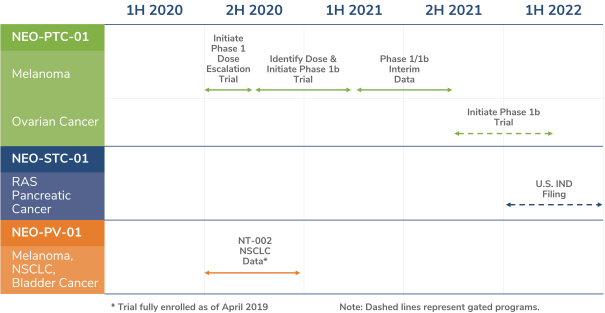
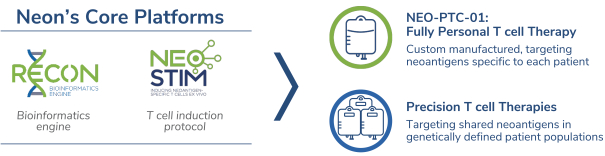
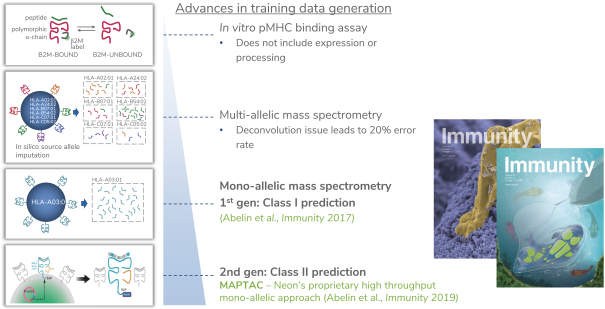
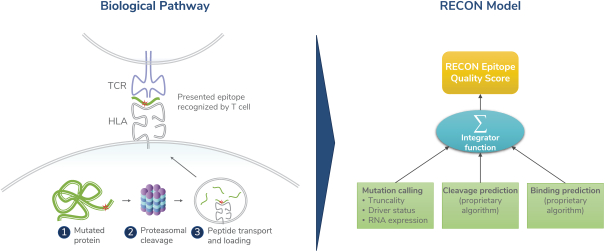
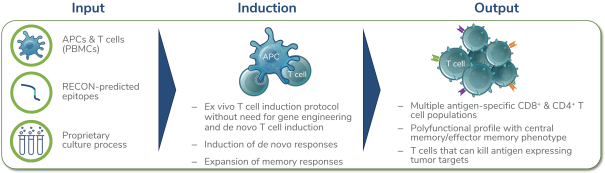
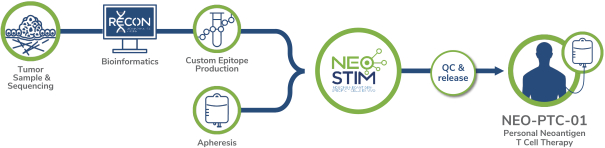
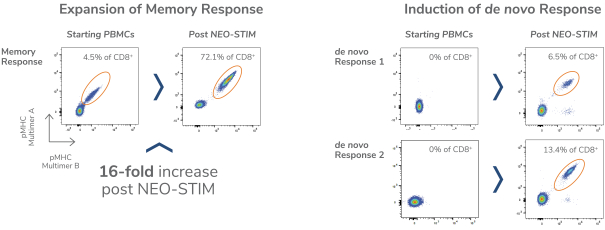
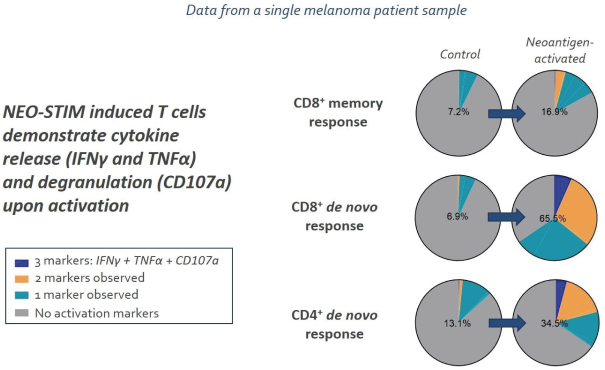
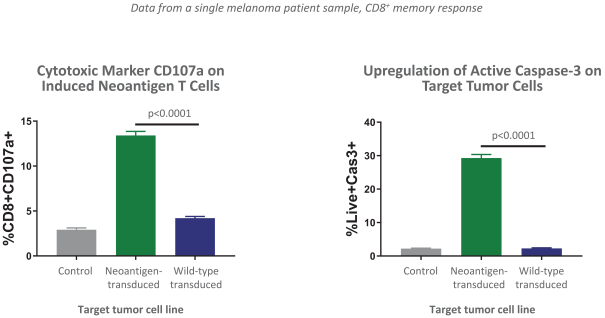
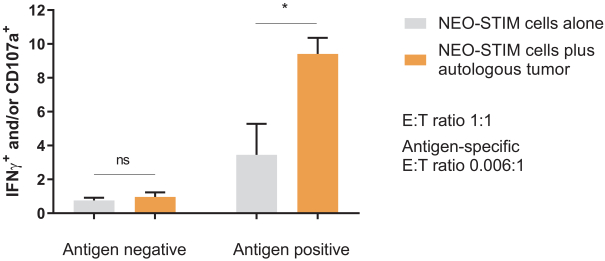
除非(除其他事项外)霓虹灯股东以有权就该事项投票的霓虹灯普通股过半数流通股持有人的赞成票批准合并提议。
霓虹灯董事会(或霓虹灯董事会)一致认为(1)根据协议所载条款及条件,合并 协议及拟进行的交易(包括合并)符合霓虹灯股东的最佳利益;(2)根据特拉华州公司法的规定,批准及宣布合并协议及拟进行的交易;及(3)建议霓虹灯股东在霓虹灯特别大会上批准及采纳合并协议。Neon 董事会一致建议Neon股东投票支持合并提案,投票支持休会提案。
你的投票很重要。无论您是否希望在线参加Neon特别会议,我们都敦促您通过以下方式尽快投票:(1)访问您的代理卡上指定的互联网网站;(2)拨打您的代理卡上指定的免费电话;或(3)签署并退回所提供的邮资已付信封中随附的代理卡,以便您的Neon普通股可以在Neon特别会议上代表并投票。如果您持有的霓虹灯普通股是以银行、经纪人或其他受托机构的名义持有的,请按照记录持有人提供的投票指导卡上的说明进行操作。
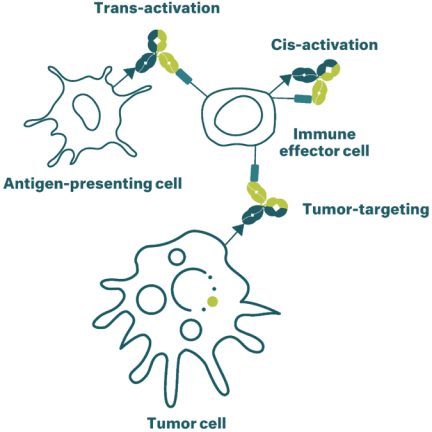
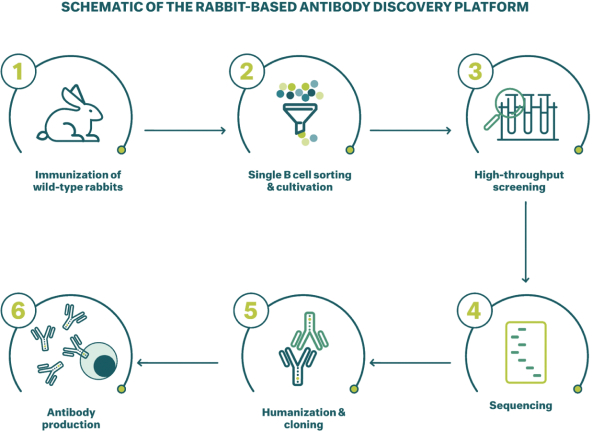
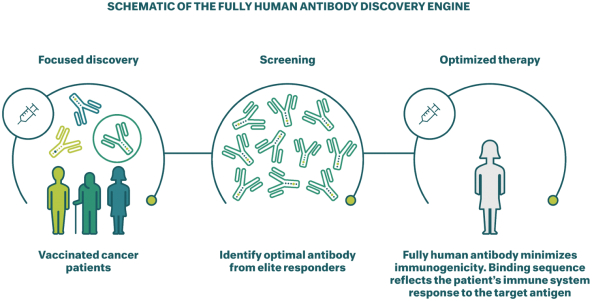
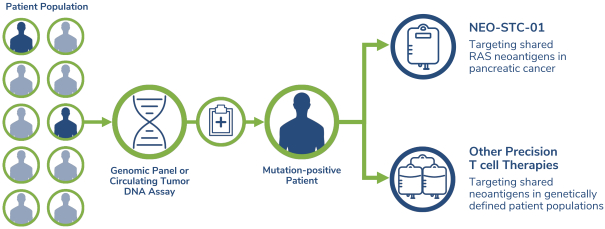
Neon和 BioNTech完成合并的义务取决于合并协议中规定的几个条件的满足或放弃。有关Neon、BioNTech和合并的更多信息包含在本委托书/招股说明书中。请仔细阅读本 完整的委托书/招股说明书,包括本委托书/招股说明书中其他位置的风险因素。
真诚地
休·奥多德
总裁兼首席执行官
SEC或任何州证券委员会均未批准或不批准本委托书/招股说明书中描述的合并,或 与合并相关的BioNTech美国存托凭证的发行,也未考虑本委托书/招股说明书的准确性或充分性。任何相反的陈述都是刑事犯罪。
本委托书/招股说明书注明日期[●],2020年,并将于 左右首次邮寄给Neon的股东 [●], 2020.