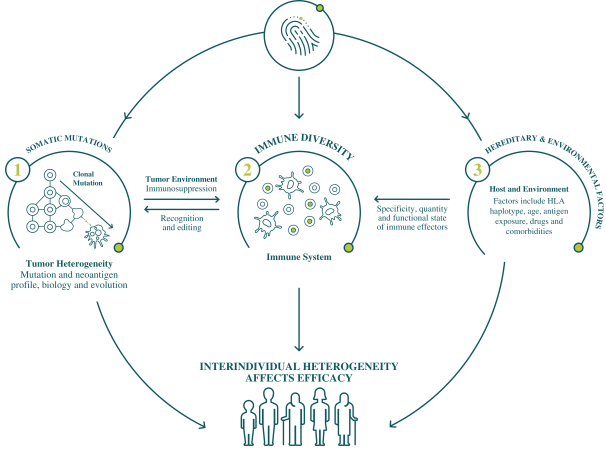
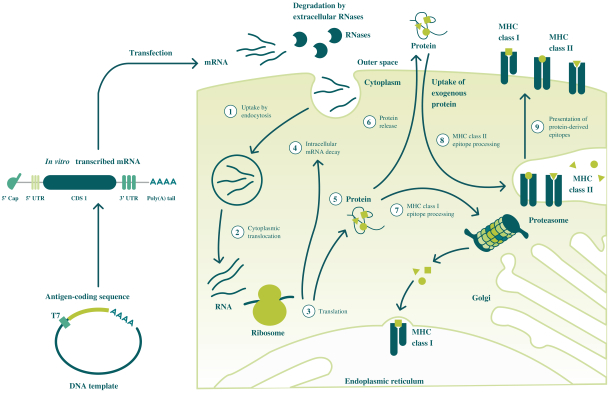
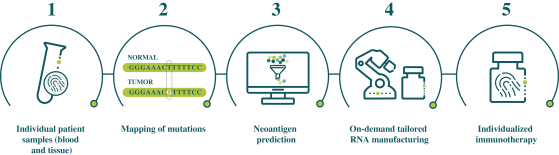
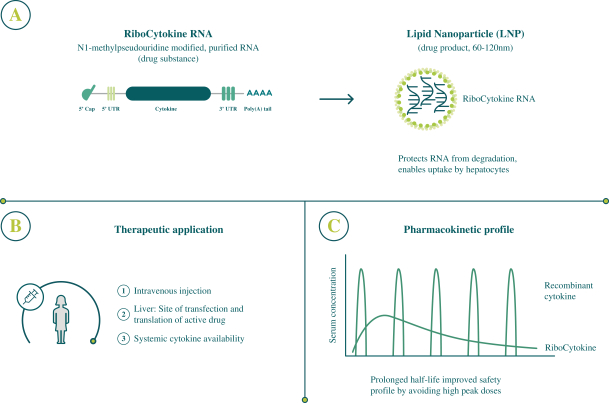
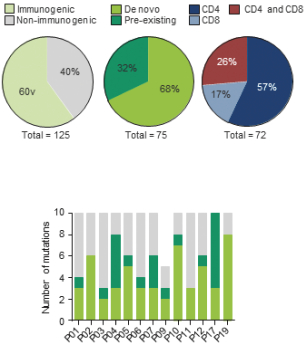
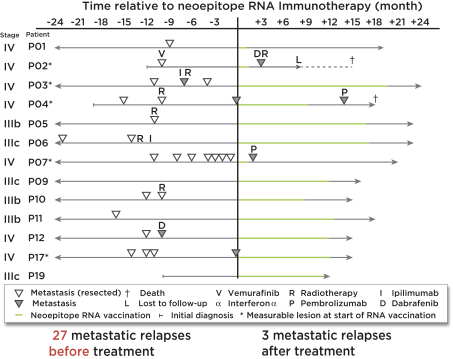
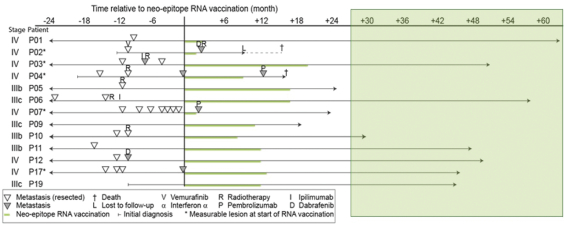
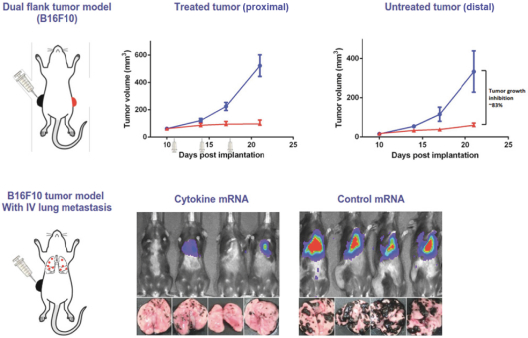
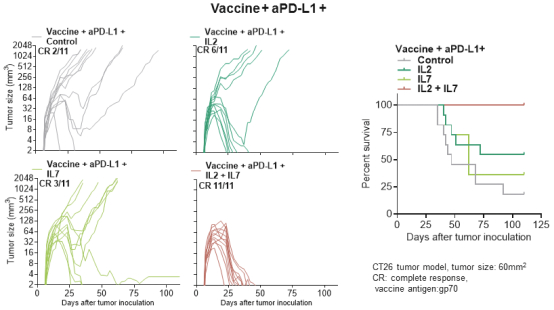
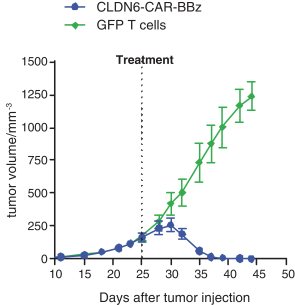
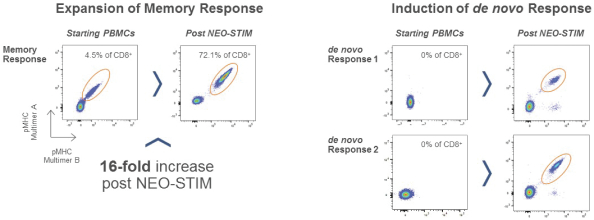
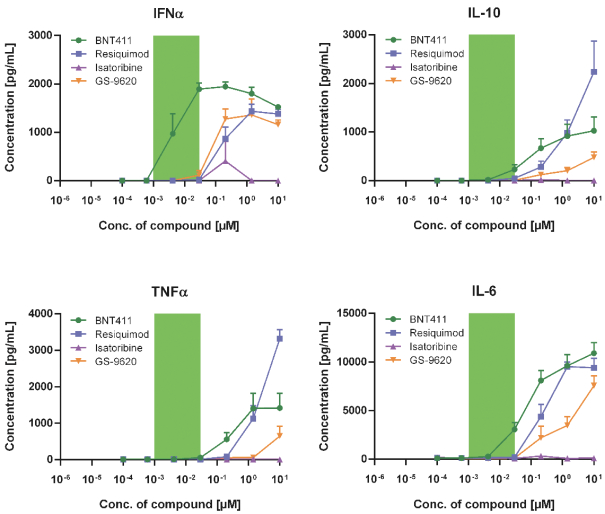
配股最多7,505,596股普通股,包括以美国存托股份为代表的普通股

我们向普通股东提供认购新普通股的权利,并通过纽约梅隆银行、我们的托管人和ADS 权利代理向美国存托股份(ADS)持有人提供根据供股或供股认购新ADS的不可转让权利。若干普通 股东已不可撤销地同意在本次发售中不转让或行使其购买总计5,616,407股普通股的权利,代表我们普通股的5,500,000股新美国存托凭证以承销 公开发售或包销发售的方式发售,价格与本次供股中发售的新普通股和新美国存托凭证相同。因此,我们预计在供股中发行最多1,889,189股新普通股,包括以美国存托凭证为代表的 股普通股。每一股新的ADS代表一股新的普通股,每股没有面值。这些美国存托凭证在纳斯达克全球精选市场上市,代码是BNTX。2020年7月22日,纳斯达克全球精选市场上美国存托凭证的最新报告售价为104.12美元/ADS。我们的普通股没有在任何国家的证券市场或交易所上市。
向美国存托凭证持有人提供
美国存托凭证持有者将 在下午5:00收到每一个已登记ADS的ADS版权。(纽约市时间)2020年7月24日。31 ADS权利持有人将有权以每ADS 93美元的价格认购和购买一个新的ADS。 要认购新的美国存托凭证,ADS权利持有人必须向纽约梅隆银行支付该认购价。部分美国存托凭证将不会发行。ADS转播权将于上午12:01到期。(纽约时间)2020年8月14日。请参阅向美国存托凭证持有人提供的配股说明。
向普通股持有人要约
普通股持有者将在晚上11点59分之后的一分钟内,以每股登记在册的普通股换取一股普通股权利。(德国美因茨 时间)2020年7月29日。31股普通股将使此类权利的持有人有权认购和购买一股新普通股,认购价为每股新普通股80.32欧元,相当于每股新ADS的 美元价格,根据2020年7月22日的有效汇率换算。或者,普通股持有者可以改为支付每股新普通股93美元,这是每股新ADS的美元价格。不会发行 股零碎普通股。认购新普通股的权利将在晚上11点59分后一分钟到期。(德国美因茨时间)2020年8月14日。请参阅向 普通股持有人提供配股的说明。
全球服务
如本招股说明书所述,供股和包销发售是最多7,505,596股普通股(包括由美国存托凭证代表的普通股 )的单一全球发售的一部分。
投资我们的普通股和代表我们普通股的美国存托凭证涉及高度风险。请参阅本招股说明书第22页开始的风险因素。
我们是一家新兴的成长型公司,也是一家根据美国联邦证券法定义的外国私人发行人,因此,我们有资格 降低上市公司的披露要求。有关更多信息,请参阅招股说明书摘要和作为新兴成长型公司和外国私人发行商的影响。
美国证券交易委员会(Securities And Exchange Commission)和任何州证券委员会都没有批准或不批准这些证券,也没有确定本 招股说明书是否真实或完整。任何相反的陈述都是刑事犯罪。
配股中的普通股和美国存托凭证预计将于2020年8月27日左右交付。
经销商经理
| 摩根大通 |
美国银行证券 |
贝伦伯格 |
日期为2020年7月23日的招股说明书