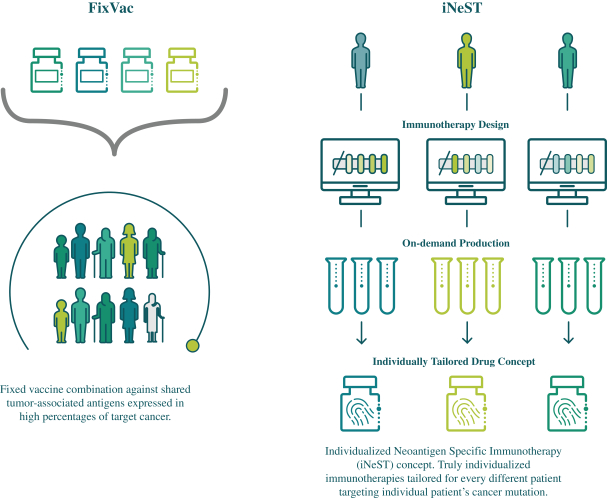
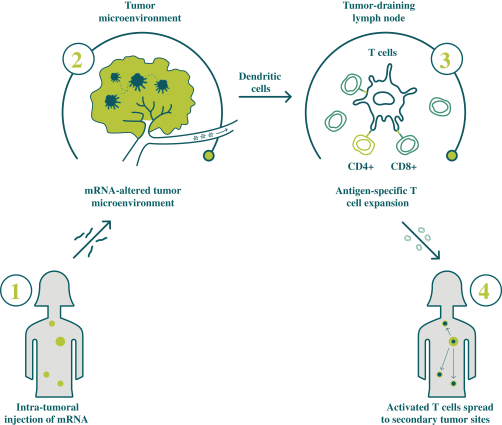
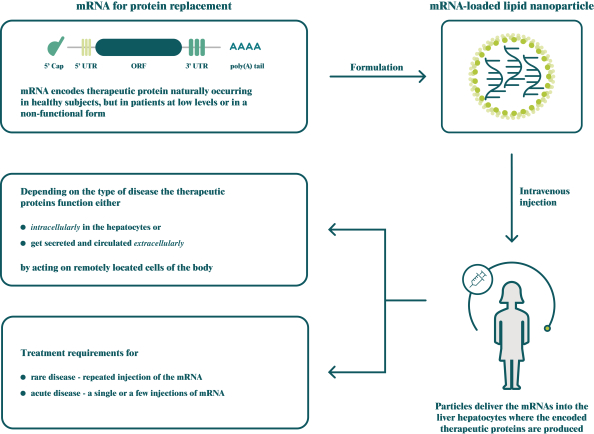
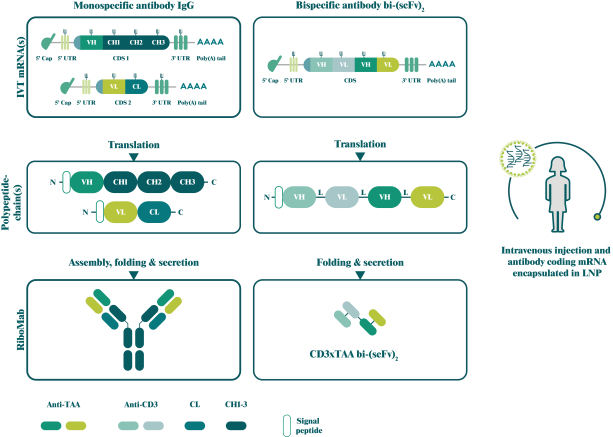
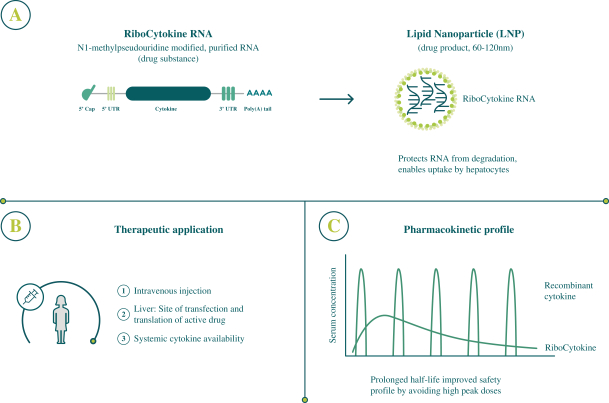
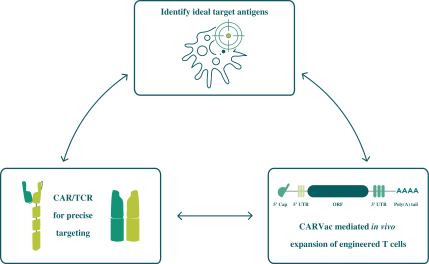
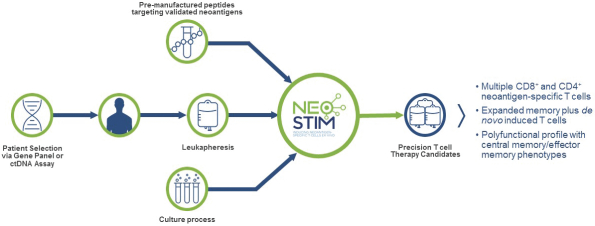
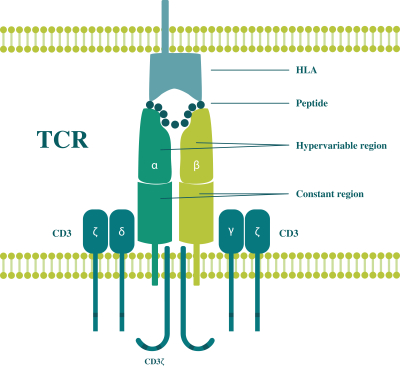
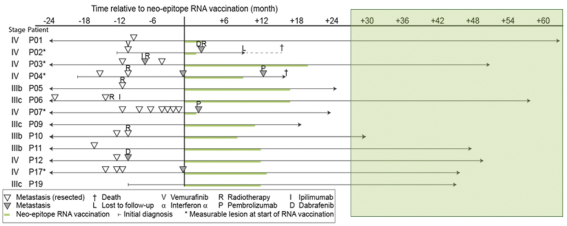
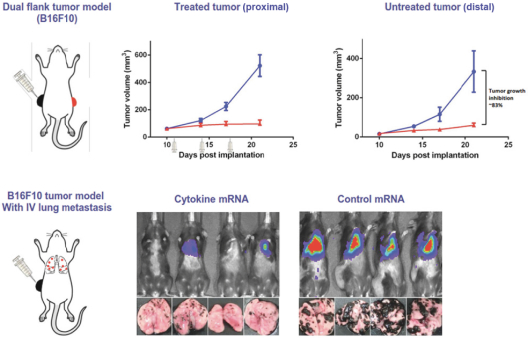
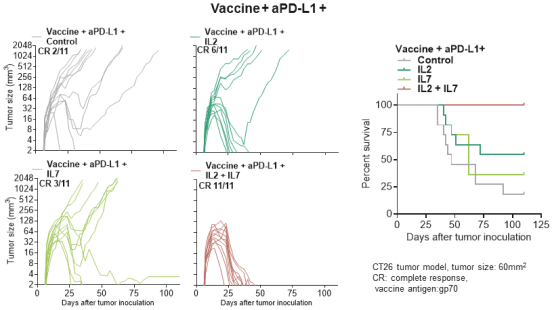
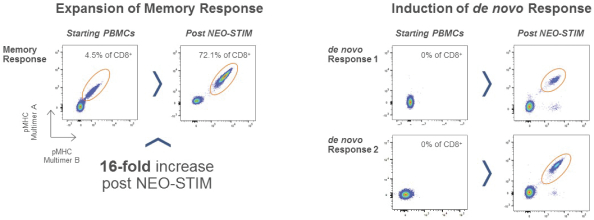
此初步招股说明书中包含的信息不完整,可能会 更改。在提交给美国证券交易委员会的注册声明生效之前,我们不能出售这些证券。
| 初步招股说明书 | 完成日期为2020年7月22日 |
500万股美国存托股份

相当于500万股普通股
我们将发行500万股美国存托股票(ADS),每股ADS相当于一股普通股,即包销发行。如果承销商行使购买以下所述额外美国存托凭证的选择权,本招股说明书中确定的出售 股东将提供750,000份美国存托凭证。我们不会收到出售股东 出售ADS的任何收益。公开发行价为每ADS 1美元。代表我们普通股的美国存托凭证(ADS)在纳斯达克全球精选市场挂牌上市,代码是#BNTX。2020年7月21日,美国存托凭证在纳斯达克全球精选市场的最新报告售价为每ADS 91.6美元。
本次发行是 全球发行的一部分,包括配股发行和本次承销发行,总计涵盖多达6,681,850股普通股(包括由美国存托凭证代表的普通股),如本招股说明书中进一步描述的那样。本招股说明书所提供的美国存托凭证(ADS)是基于我们普通股的某些持有人(占我们已发行普通股(包括由 ADS代表的普通股)的74.83%)根据德国法律订立的不可撤销的、具有约束力的协议,不转让或行使权利认购我们打算在本招股说明书向公众公布与招股说明书相关的要约价格 之后进行的供股中授予的新普通股。此次发售与配股发售一起构成了全球发售。
包括辉瑞公司在内的某些新的和现有的投资者已 表示有兴趣以公开发行价 $购买总计2亿美元的美国存托凭证(相当于本次发售的普通股),并按与本次发售中的其他购买者相同的条款和条件购买。然而,由于意向指示不是 具有约束力的协议或购买承诺,这些投资者可能决定购买少于他们表示有意购买或不购买本次发售中任何美国存托凭证的美国存托凭证。这些投资者也可能表示 有兴趣购买更多美国存托凭证。此外,承销商可以决定向这些投资者中的任何一个出售更少的美国存托凭证,而不是投资者表示有兴趣购买或不向这些投资者出售任何美国存托凭证。
投资代表我们普通股的美国存托凭证涉及高度风险。请参阅本招股说明书第23页 开始的风险因素。
我们是一家新兴的成长型公司,也是一家根据美国联邦证券法定义的外国私人发行人,因此有资格降低上市公司的披露要求。有关更多信息,请参阅招股说明书摘要和作为新兴成长型公司和外国私人发行商的影响。
美国证券交易委员会(Securities And Exchange Commission)和任何州证券委员会都没有批准或不批准这些证券,也没有确定 本招股说明书是否真实或完整。任何相反的陈述都是刑事犯罪。
| 每个ADS | 共计 | |||||||
| 公开发行价 |
$ | $ | ||||||
| 承保折扣和佣金 (1) |
$ | $ | ||||||
| 扣除费用前给我们的收益 |
$ | $ | ||||||
| (1) | 我们已同意赔偿承销商在承销发行中发生的某些费用。有关详细信息,请参阅 z承销。 |
承销商有权在本招股说明书发布之日起30天内 向出售股东额外购买750,000份美国存托凭证。
美国存托凭证预计将在2020年 左右交付。
| 摩根大通 | 美国银行证券 | 贝伦伯格 | ||
| 瑞银投资银行 | Canaccel Genuity | |||
| 德国商业银行 | 沃尔夫资本市场和咨询 | Bryan,Garnier&Co. |
招股说明书日期为 ,2020