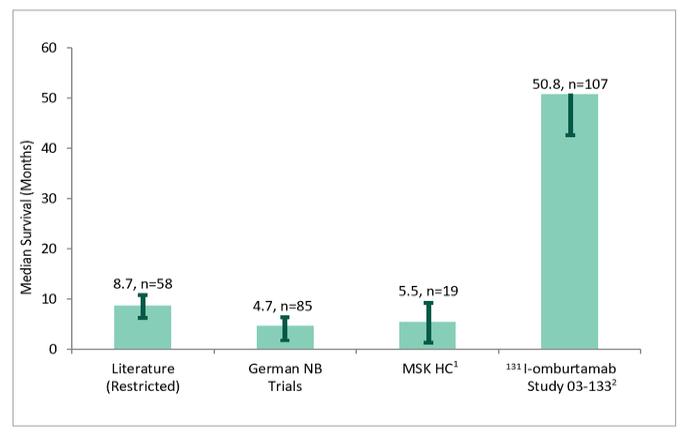
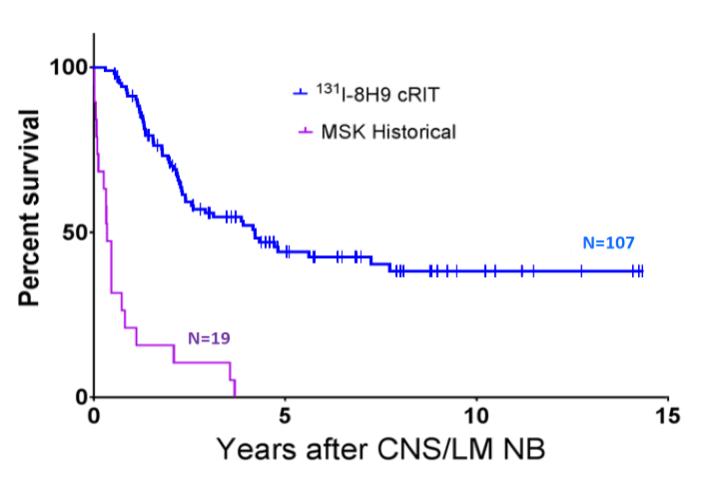
附注7-股东权益
授权股票
截至2020年12月31日和2019年12月31日,本公司共授权105,500,000股票,100,000,000其中为普通股,票面价值$0.0001每只股票,以及5,500,000其中优先股面值为$0.0001每股。
普通股
普通股每股有权一投票吧。普通股股东有权获得董事会宣布的股息,但须受优先股优先股息权的限制。无其中一些已经发行了。公司已经发布了40,688,447截至2020年12月31日的普通股股票以及39,728,416截至2019年12月31日的普通股。
优先股
优先股可不时以一个或多个系列发行,并附有本公司董事会批准的指定、优先及相对参与、可选择或其他特殊权利及资格、限制或限制。不是优先股已于2020年12月31日或2019年12月31日发行。
与非雇员签订的股票赠与协议
于2015年8月,我们与本公司非雇员订立若干股票授出协议。我们同意总共发行2,800,000共享至二参与从MSK获得许可的技术开发的非雇员研究人员,考虑到他们之前的服务。这些股票是根据归属时间表发行的。总计560,0002015年发行了股票,总共发行了448,0002016年和2017年分别发行的股票。2018年,共有544,000股票发行给了二研究人员,借此一的二补助金已全部发放。2019年,400,000股票发行给了一医生的名字,剩下的400,000股票于2020年8月发行,但须符合某些条件。根据这项奖励,未来不会发行任何股票。总奖励在2015年按其估计公允价值支出,因为未来不需要任何服务来继续授予和接受股票。
2020年4月,关于SADA许可协议,我们签订了若干股票授予协议,根据这些协议,我们同意总共发行213,996共享至二参与开发从MSK和麻省理工学院获得许可的SADA技术的非雇员研究人员,考虑到他们之前的服务。全213,996股票于2020年4月发行,托管于40发行时立即归属的股份的%,剩余的60应按比例在下一年按比例归属的股份的百分比三年在协议的周年纪念日。只要SADA许可协议在股份归属前终止,股份将被没收。没有现金结算功能,研究人员不需要未来的服务来授予和接收股票。虽然股票随着时间的推移而归属,但股票没有业绩条件。在2020年4月,我们记录了一笔研发费用,总额为$7,376,000与股份有关,代表股份于授出日的公允价值。
在2020年7月,根据股票授予协议,我们还借出了二研究人员总共花费$2,610,000与与股票赠与一起到期的个人纳税有关。每笔贷款都有一份三年担保本票。贷款的未偿还本金,连同按年利率计算的所有应计利息1年利率,在贷款到期日到期并支付。这些贷款由质押和担保协议(Pledge And Security Agreement)担保,根据这些协议,研究人员已将这些股票质押,作为偿还贷款的担保,利率为市场利率。贷款按摊销成本入账,由于短期性质和市场利率变动很小,这一成本接近公允价值。