--
美國
證券交易委員會
華盛頓特區20549
形式
根據1934年證券交易法第13或15(d)條提交的年度報告 |
截至本財政年度止
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission File Number:
(Exact name of registrant as specified in its charter)
|
|
|
|
||
(State or other jurisdiction of incorporation or organization) |
|
(I.R.S. Employer Identification No.) |
(Address of principal executive offices) (Zip Code)
(
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
|
|
|
|
|
Title of each class |
|
Trading Symbol |
|
Name of each exchange on which registered |
|
|
The |
Securities registered pursuant to Section 12(g) of the Act:
None
(Title of Class)
Indicate by a check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate by a check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
|
|
|
|
|
|
Large accelerated filer |
|
☐ |
|
Accelerated filer |
|
☐ |
|
|
|
|
|||
|
☒ |
|
Smaller reporting company |
|
||
|
|
|
|
|||
|
|
|
|
Emerging growth company |
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant's executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No
The aggregate market value of the voting and non-voting common equity held by non-affiliates, based on the closing price per share of Registrant's Common Stock on the Nasdaq Capital Market was approximately $
As of September 13, 2024, there were
MEI PHARMA, INC.
TABLE OF CONTENTS
|
|
|
|
|
|
|
|
|
|
|
Page |
|
|
|
|
|
|
|
|
|
Item 1: |
|
|
|
5 |
|
|
Item 1A: |
|
|
|
20 |
|
|
Item 1B: |
|
|
|
30 |
|
|
Item 1C: |
|
|
|
30 |
|
|
Item 2: |
|
|
|
31 |
|
|
Item 3: |
|
|
|
31 |
|
|
Item 4: |
|
|
|
32 |
|
|
|
|
|
||||
|
|
|
|
|
|
|
Item 5: |
|
|
|
32 |
|
|
Item 6: |
|
|
|
33 |
|
|
Item 7: |
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
|
|
33 |
|
Item 7a: |
|
|
|
38 |
|
|
Item 8: |
|
|
|
39 |
|
|
Item 9: |
|
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
|
|
63 |
|
Item 9A: |
|
|
|
63 |
|
|
Item 9B: |
|
|
|
63 |
|
|
Item 9C: |
|
Disclosure Regarding Foreign Jurisdictions that Prevent Inspections |
|
|
64 |
|
|
|
|
||||
|
|
|
|
|
|
|
Item 10: |
|
|
|
65 |
|
|
Item 11: |
|
|
|
71 |
|
|
Item 12: |
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
|
|
85 |
|
Item 13: |
|
Certain Relationships and Related Transactions, and Director Independence |
|
|
86 |
|
Item 14: |
|
|
|
86 |
|
|
|
|
|
||||
|
|
|
|
|
|
|
Item 15: |
|
|
|
87 |
|
|
Item 16: |
|
|
|
89 |
|
|
Forward-Looking Statements
This Annual Report on Form 10-K (Annual Report) includes forward-looking statements, which involve a number of risks and uncertainties. These forward-looking statements can generally be identified as such because the context of the statement will include words such as “may,” “will,” “intend,” “plan,” “believe,” “anticipate,” “expect,” “estimate,” “predict,” “potential,” “continue,” “likely,” or “opportunity,” the negative of these words or other similar words. Similarly, statements that describe our future plans, strategies, intentions, expectations, objectives, goals or prospects and other statements that are not historical facts are also forward-looking statements. Discussions containing these forward-looking statements may be found, among other places, in “Business” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in this Annual Report. For such statements, we claim the protection of the Private Securities Litigation Reform Act of 1995. Readers of this Annual Report are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the time this Annual Report was filed with the Securities and Exchange Commission, or SEC. These forward-looking statements are based largely on our expectations and projections about future events and future trends affecting our business, and are subject to risks and uncertainties that could cause actual results to differ materially from those anticipated in the forward-looking statements. These risks and uncertainties include, without limitation, those discussed in “Risk Factors” and in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” of this Annual Report. Other sections of this report and our other filings with the SEC may include additional factors which could adversely impact our business and financial performance. Moreover, we operate in a very competitive and rapidly changing environment. There is substantial uncertainty regarding the impact of activist investors, rising inflation and the increase in interest rates as a result, a potential economic downturn, industry, global economic conditions and government policy. New risk factors emerge from time to time and it is not possible for us to predict all risk factors, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements. In addition, past financial or operating performance is not necessarily a reliable indicator of future performance, and you should not use our historical performance to anticipate results or future period trends. We can give no assurances that any of the events anticipated by the forward-looking statements will occur or, if any of them do, what impact they will have on our results of operations and financial condition. Except as required by law, we undertake no obligation to update publicly or revise our forward-looking statements to reflect events or circumstances that arise after the filing of this Annual Report or documents incorporated by reference herein that include forward-looking statements.
Unless the context requires otherwise, references in this Annual Report to “MEI Pharma,” “MEI,” “we,” “us” and “our” refer to MEI Pharma, Inc.
MEI Pharma, Inc. and our corporate logo are registered service marks of MEI Pharma. Any other brand names or trademarks appearing in this Annual Report are the property of their respective holders.
4
PART I
Item 1. Business
Overview
MEI Pharma, Inc. (Nasdaq: MEIP) is a pharmaceutical company that has been developing novel and differentiated cancer therapies. We built our pipeline by acquiring promising cancer agents and creating value in programs through clinical development, strategic partnerships, and out-licensing or commercialization, as appropriate. Our approach to oncology drug development has been to evaluate our drug candidates in combinations with standard-of-care therapies to overcome known resistance mechanisms and address clear medical needs to provide improved patient benefit. Our drug candidate pipeline includes voruciclib, an oral cyclin-dependent kinase 9 (CDK9) inhibitor, and ME-344, an intravenous small molecule mitochondrial inhibitor targeting the oxidative phosphorylation pathway.
Strategic Alternatives
On July 22, 2024, we announced that our Board of Directors (Board) had determined unanimously to begin the evaluation of our strategic alternatives, including potential transactions as well as an orderly wind down of operations, if appropriate, to maximize the value of our assets for our stockholders. We commenced a reduction-in-force beginning August 1, 2024, which will continue in stages as our operational and strategic direction evolves. We have discontinued the clinical development of voruciclib, while certain nonclinical activities related to our drug candidate assets will continue to be conducted by us. As part of the review of strategic alternatives, we may consider options such as out-licensing opportunities for existing programs and merger and acquisition opportunities. Consistent with our intention to preserve cash, David M. Urso, our President and Chief Executive Officer, and Richard Ghalie, M.D., our Chief Medical Officer, have stepped down effective August 1, 2024. Mr. Urso also left the Board at that date. We have entered into consulting agreements with both Mr. Urso and Dr. Ghalie under which they will remain available to assist us in our strategic efforts. Charles V. Baltic III, the Chairperson of the Board, also stepped down from the Board contemporaneously with the announcement on July 22, 2024. Our Board has appointed Justin J. File, our current Chief Financial Officer, to assume the position of Acting Chief Executive Officer and has appointed Frederick W. Driscoll as Chairperson of the Board.
Cooperation Agreement
On October 31, 2023, we announced our entry into a Cooperation Agreement (Cooperation Agreement) with Anson Funds Management LP and Cable Car Capital LLC (Anson and Cable Car, respectively), which, among other non-financial related items, provided for a capital return to stockholders in the form of a dividend in the amount of $1.75 per share of common stock, as further discussed below. Additionally, the Cooperation Agreement contemplated a potential second return of capital not to exceed $9.33 million (Potential Second Return of Capital) if authorized by our Board should our ongoing ME-344 Phase 1b trial fail to meet certain defined endpoints or our Board determines not to proceed with a second cohort.
As part of the Cooperation Agreement, Anson and Cable Car withdrew their consent solicitation and agreed to abide by customary standstill provisions. Additionally, we reimbursed Anson's and Cable Car’s fees and expenses related to their engagement with us as of the date of the Cooperation Agreement in an amount of $1.1 million, which is recorded within general and administrative expenses in the consolidated statements of operations for the fiscal year ended June 30, 2024.
In April 2024, the Board unanimously determined not to proceed with the Potential Second Return of Capital under the Cooperation Agreement in order to conserve resources and align strategic investment, and thereby extend our operational runway.
Cash Dividend
On November 6, 2023, pursuant to the Cooperation Agreement, the Board declared a special cash dividend of $1.75 per share of common stock to stockholders of record at the close of business on November 17, 2023 (Capital Return). The total dividend of $11.7 million was paid on December 6, 2023, and was recorded as a reduction of additional paid-in capital in the consolidated statements of stockholders' equity, as we have an accumulated deficit, rather than retained earnings.
Other Events
Subject to approval by our stockholders, we, Infinity Pharmaceuticals, Inc. (Infinity), and Meadow Merger Sub, Inc., our wholly owned subsidiary (Merger Sub) entered into an agreement and plan of merger (Merger Agreement) in February 2023. At a special meeting of our stockholders held on July 23, 2023, the transaction did not obtain the necessary approval from our stockholders and, accordingly, on July 23, 2023, we sent Infinity a notice terminating the Merger Agreement.
5
In December 2022, we announced plans to realign our clinical development efforts after jointly deciding with our development partner, Kyowa Kirin Co., Ltd. (KKC), to discontinue development of our lead drug candidate, zandelisib, outside of Japan. In connection with the realignment, we focused ongoing development efforts on our two clinical assets in Phase 1 and Phase 1b clinical programs, voruciclib and ME-344, respectively. Additionally, we initiated a staggered workforce reduction, affecting 28 employees in December 2022 and an additional 26 employees through June 2023.
Drug Candidate Development Programs
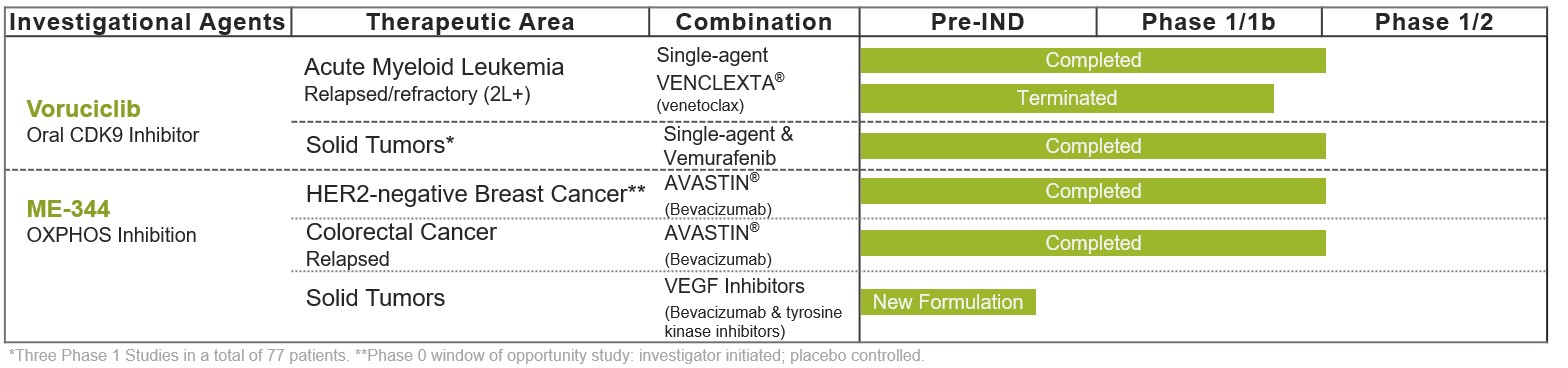
Our drug candidate pipeline includes voruciclib, an oral CDK9 inhibitor, and ME-344, an intravenous small molecule inhibitor mitochondrial oxidative phosphorylation (OXHPHOS). Each program has completed several clinical studies.
Voruciclib: Potent Orally Administered CDK9 Inhibitor in Phase 1 Studies
All ongoing clinical trial efforts for voruciclib have been ceased as of July 22, 2024. Voruciclib is a selective orally administered CDK9 inhibitor. Voruciclib recently completed a Phase 1 trial evaluating dose and schedule in patients with acute myeloid leukemia (AML) in combination with the B-cell lymphoma 2 (BCL-2) inhibitor venetoclax (marketed as Venclexta®). Voruciclib is also being evaluated in pre-clinical studies to explore potential activity in various solid tumor cancers including in combination with therapies that target the RAS signaling pathway, such as KRAS inhibitors.
Voruciclib Scientific Overview: Cell Cycle Signaling
CDK9 has important functions in cell cycle regulation, including the modulation of two therapeutic targets in cancer:
Directly inhibiting MCL1 and MYC has historically been difficult, but CDK9 is a promising approach to indirectly target these oncogenes.
Voruciclib: Inhibition of MCL1
CDK9 is a known transcriptional regulator of MCL1. Over expression of MCL1 is frequently observed in many tumor types and is closely associated with tumorigenesis, poor prognosis and drug resistance. In AML, MCL1 is upregulated in about half of patients with relapsed and refractory (R/R) disease and is associated with poor prognosis in these patients. Also important, high levels of MCL1 expression are associated with resistance to venetoclax.
In pre-clinical studies, voruciclib shows dose-dependent suppression of MCL1; in December 2017, a study of voruciclib published in the journal Nature Scientific Reports reported that the combination of voruciclib plus the BCL-2 inhibitor venetoclax was capable of inhibiting two master regulators of cell survival, MCL-1 and BCL-2, and achieved synergistic antitumor effect in an aggressive subset of DLBCL cells.
In a peer reviewed manuscript published in 2020, it was reported that the inhibition of CDK9 by voruciclib synergistically enhances cell death induced by the BCL-2 inhibitor venetoclax in preclinical models of AML. The data demonstrated that voruciclib synergizes with venetoclax to induce programmed cell death, or apoptosis, in both AML cell lines and primary patient samples. It was
6
also demonstrated that voruciclib downregulates MCL1, which is relevant for the synergy between voruciclib and venetoclax, and further that voruciclib downregulates MYC, which also contributes to the synergies with venetoclax.
Subsequently, and consistent with the reported pre-clinical studies, data from an ongoing Phase 1 study evaluating voruciclib as a single agent and in combination with venetoclax in patients with relapsed or refractory (R/R) AML have also demonstrated the anticipated decreases in Mcl-1 protein.
The research suggests that voruciclib is potentially an attractive therapeutic agent for treating cancers in combination with venetoclax or other BCL-2 inhibitors, to address potential resistance associated with MCL1, and is supportive of our ongoing clinical evaluation of voruciclib in AML.
Voruciclib: Inhibition of MYC
Many cancers are associated with over expression of MYC, a transcription factor regulating cell proliferation and growth. CDK9 is a known regulator of MYC transcription and a modulator of MYC protein phosphorylation. Data reported at the American Association for Cancer Research (AACR) Annual Meeting 2021 in preclinical models demonstrated that voruciclib:
The research presented suggests that voruciclib could be an attractive therapeutic agent for both hematological cancers, as well as solid tumors, dependent on the activity of MYC.
Clinical Programs
In a recent Phase 1 clinical trial, we evaluated the dose and schedule of voruciclib in combination with venetoclax, a BCL-2 inhibitor, in patients with R/R AML. The trial started with the evaluation of dose and schedule of voruciclib as a monotherapy in patients with relapsed and refractory B-cell malignancies and AML after failure of prior standard therapies to determine the safety, preliminary efficacy and maximum tolerated dose. The primary objectives of the study were to determine the safety and biologic effective dose of voruciclib monotherapy or voruciclib in combination with venetoclax. Secondary objectives of the study included assessing the preliminary efficacy, pharmacokinetics, pharmacodynamics, and biomarkers of voruciclib monotherapy or voruciclib in combination with venetoclax.
As we reported in a poster presented at the American Society of Hematology (ASH) Annual Meeting in December 2023, the voruciclib monotherapy dose escalation/expansion stage of the study enrolled a total of 40 patients and is complete. The majority of patients (n=21) had AML and the remaining patients (n=19) had B-cell malignancies. Of the 40 patients enrolled, the first 16 were dosed daily continuously at 50 and 100 mg and the following 24 patients were dosed on an intermittent schedule (14 consecutive days on therapy in a 28-day cycle) at 100, 150 and 200 mg. All patients were heavily pre-treated with a median of three prior therapies (range 1-9), and five patients had prior hematopoietic stem cell transplant. Voruciclib at doses up to 200 mg administered on 14 consecutive days in a 28-day cycle (Cohort 2) was well tolerated with no dose limiting toxicities (DLT) reported. The most common adverse events (≥20% of patients) were diarrhea, nausea, anemia and fatigue. The large majority of adverse events were Grade 1-2; of note, the only Grade 3-4 adverse events in Cohort 2 were diarrhea (n=1) and anemia (n=5). Pharmacokinetics were dose proportional and a mean half-life of approximately 24 hours supports once daily dosing.
On the intermittent dosing schedule selected for further development, no DLTs were observed, there were no Grade 3 or higher drug related toxicities, and dose escalation was stopped at 200 mg before reaching the maximum tolerated dose because plasma concentrations reached levels considered sufficient for target inhibition. In the 21 patients enrolled with AML, one patient at 100 mg achieved a morphologic leukemia-free state and nine patients had disease stabilization, which lasted at least three months in two patients. In the 19 patients enrolled with B-cell malignancies, four patients had stable disease with a decrease in tumor size. Initial results from correlative studies assessing myeloid leukemia cell differentiation protein (Mcl-1) and RNA Pol II phosphorylation on Ser2 (RNA Pol II p-S2) demonstrated reduction in expression consistent with the anticipated on-target pharmacodynamic effect of voruciclib on Mcl-1 and RNA Pol II p-S2.
The next stage of the study evaluated seven voruciclib dose levels from 50 mg every other day to 300 mg daily for 14 consecutive days in a 28-day cycle in combination with standard dose venetoclax in patients with R/R AML. A total of 41 patients with R/R AML, median age 67 years (range 34-89), enrolled in this dose escalation stage of the study evaluating voruciclib in combination with venetoclax. These patients were generally heavily pretreated; the median number of prior therapies was 2 (range 1-7), and 18 (44%) patients had ≥3 prior lines. Almost all patients (39/41) were treated with venetoclax in an earlier line of therapy.
7
Additionally, 30 (73%) patients were noted as being in an adverse 2017 ELN Risk Category due to adverse cytogenetics and molecular mutations.
Of the 32 patients administered voruciclib at doses ≥ 100 mg in combination with venetoclax 10 (31%) achieved disease control. Three patients achieved a response, including two patients that achieved a complete response with incomplete hematologic recovery (CRi) and one patient that achieved a morphologic leukemia-free state (MLFS), in each case having received venetoclax in an earlier line of treatment. Responses lasted 6 months in one patient, 9 months and ongoing in the second patient, and the third patient was referred to stem cell transplant. Further, an additional 7 patients had stable disease which lasted more than 90 days and 13 had stable disease < 3 months.
In the 28 patients administered voruciclib in combination with venetoclax and with blood samples available for analysis, initial results from correlative biomarker assay studies demonstrated anticipated decreases of Mcl-1, including a greater decrease in Mcl-1 in responding patients. This supports our hypothesis that voruciclib, as an inhibitor or CDK9, regulates Mcl-1 and therefore may address the upregulation of MCL1 associated with venetoclax. Additional evidence of anti-leukemic activity was also demonstrated including decreases in bone marrow blast counts post voruciclib/venetoclax administration versus pre drug administration in ~50% (11/21) of evaluable patients.
Voruciclib at doses up to 300 mg administered on 14 consecutive days in a 28-day cycle in combination with standard dose venetoclax was well tolerated with no dose limiting toxicities observed. The maximum tolerated dose of voruciclib administered on this schedule with venetoclax has not been established. There were no discontinuations due to drug-related adverse events and no evidence of overlapping toxicity has been observed to date. The most common (≥5% of patients) grade 3 adverse events were myelosuppression associated with AML. Only 1 patient was observed as having a non-hematologic grade 3 drug-related adverse event (diarrhea).
Before ending the study, three patients were administered 150 mg voruciclib over 21 consecutive days in a 28-day cycle in combination with venetoclax to increase dose intensity and potentially optimize patient response based upon the rebound of peripheral blast counts in 44% (8/18) of the patients between Day 14 and Day 28 when voruciclib was stopped while continuing venetoclax.
Voruciclib was also previously evaluated in more than 70 patients with solid tumors in multiple Phase 1 studies. The totality of the clinical data, along with data from pre-clinical studies, suggests voruciclib’s ability to inhibit its molecular target at a projected dose as low as 150 mg daily. In one clinical study, voruciclib was evaluated in combination with vemurafenib (marketed as Zelboraf®) in nine patients with BRAF mutated advanced/ inoperable malignant melanoma. All three BRAF/MEK naive patients achieved a response: two partial responses and one complete response. In this study voruciclib was dosed at 150 mg daily plus vemurafenib 720 mg or 960 mg twice daily in 28-day cycles. The most common adverse events were fatigue, constipation, diarrhea, arthralgia and headache. One instance of grade 3 fatigue was dose limiting and no serious adverse events related to voruciclib were reported. Other clinical studies evaluated voruciclib at doses up to 850 mg in patients with solid tumors, demonstrating additional evidence of potential biologic activity and an adverse event profile generally consistent with other drugs in its class.
ME-344: Mitochondrial Inhibitor with Combinatorial Potential
Nonclinical activities related to ME-344 are to continue following the announcement of the exploration of strategic alternatives on July 22, 2024. MEI-344 is a novel drug candidate that inhibits mitochondrial OXPHOS, a fundamental metabolic pathway involved in the production of adenosine triphosphate (ATP) in the mitochondria. ATP provides energy to drive many metabolic cell processes, including division, proliferation, and growth. By disrupting the production of ATP, ME-344 has been shown to induce cancer cell death in nonclinical models and was associated with antitumor activity in clinical studies. ME-344 has also demonstrated clinical activity in multiple clinical studies in combinations, including with bevacizumab (Avastin®).
Recently, we were advancing ME-344 via development of a new formulation with the goal of increasing biological activity, improving patient convenience of administration and increasing commercial opportunity. We believe a new formulation represents the optimal approach to pursue the potential of the program after observing encouraging data in two clinical studies evaluating the prior ME-344 formulation in combination with bevacizumab (Avastin®).
ME-344 Scientific Overview: Cancer Metabolism
Energy supplied in the form of ATP fuels tumor metabolism supporting cell division and growth. Accordingly, tumor cells often display a high metabolic rate to support tumor cell survival and proliferation. This heightened metabolism requires a continual supply of energy in the form of ATP.
Anti-angiogenics, such as the vascular endothelial growth factor (VEGF) inhibitor bevacizumab, have the potential to normalize vasculature and decrease reliance on glycolysis for ATP. The resulting reduction in glycolysis may trigger an increased dependence on mitochondrial ATP production for energy to support continued tumor proliferation. In such cases of tumor plasticity, the combination of ME-344 and bevacizumab may induce metabolic synthetic lethality, providing a novel therapeutic strategy. Specifically, leveraging the ability of antiangiogenics like bevacizumab to reduce glycolysis and force tumor cells to switch to mitochondrial respiration via OXPHOS, which is inhibited by ME-344, may reduce access to ATP needed for cell division and growth in tumors.
8
We obtained initial clinical data on this approach in a completed investigator-initiated, multi-center, randomized, controlled, window of opportunity clinical trial evaluating ME-344 in combination with bevacizumab that enrolled a total of 42 patients with human epidermal growth factor receptor 2 (HER2) negative breast cancer. Further clinical support for the combination of ME-344 in combination with bevacizumab was reported in April 2024 from a Phase 1b study of patients with relapsed metastatic colorectal cancer (mCRC) after failure of standard therapies. This study demonstrated clinical activity, including an effect on progression free survival in a cohort of 23 patients.
An earlier Phase 1 clinical study evaluating ME-344 as a single-agent in patients with refractory solid tumors also demonstrated anti-tumor activity, further validating the potential of mitochondrial inhibition as a promising therapeutic modality.
Clinical Program
ME-344 has been evaluated pre-clinically and clinically as a single agent and in combination with anti-angiogenics such as bevacizumab. When evaluated as a single agent, ME-344 demonstrated evidence of activity against refractory solid tumors in a Phase 1b trial, and in pre-clinical studies tumor cells treated with ME-344 resulted in a rapid loss of ATP and cancer cell death. In addition to single agent activity, ME-344 has also demonstrated significant potential in combination with anti-angiogenic therapeutics.
Pre-clinical studies have shown that one outcome of anti-angiogenics is a reduced rate of glycolysis in tumors as a mechanism to slow tumor growth. However, when faced with reduced glycolysis and reduced ATP production, tumor metabolism was able to shift to mitochondrial metabolism for energy production to support continued tumor proliferation. In such cases of tumor plasticity in the presence of treatment with anti-angiogenics, contemporaneously targeting the mitochondria as an alternative metabolic source of ATP with ME-344 may open an important development opportunity.
Support for this combinatorial use of ME-344 was first published in the June 2016 edition of Cell Reports; pre-clinical data from a collaboration with the Spanish National Cancer Research Centre in Madrid demonstrated mitochondria-specific effects of ME-344 in cancer cells, including substantially enhanced anti-tumor activity when combined with agents that inhibit the activity of VEGF. These data demonstrating the potential anti-cancer effects of combining ME-344 with a VEGF inhibitor due to an inhibition of both mitochondrial and glycolytic metabolism provided a basis for commencement of an investigator-initiated trial of ME-344 in combination with bevacizumab in HER2 negative breast cancer patients.
Results published in the November 2019 issue of Clinical Cancer Research from a multi-center, investigator-initiated, randomized, controlled, clinical trial that evaluated the combination of ME-344 and bevacizumab in 42 women with early HER2-negative breast cancer provided evidence for the combinatorial use of ME-344 with anti-angiogenic therapeutics.
The primary objective of the trial was to show proof of ME-344 biologic activity as measured by reductions in the nuclear protein Ki67 (expression of which is strongly associated with tumor cell proliferation and growth) from days 0 to 28 compared to the control group who received bevacizumab alone. Secondary objectives included determining whether ME-344 biologic activity correlates with vascular normalization. The data demonstrated significant biologic activity in the ME-344 treatment group:
Treatment was generally well tolerated; three grade 3 adverse events of high blood pressure were reported, two in the ME-344 arm and one in the bevacizumab monotherapy arm.
Building on the clinical study evaluating patients with breast cancer, a Phase 1b study evaluating ME-344 in combination with bevacizumab in patients with relapsed metastatic colorectal cancer (mCRC) after failure of standard therapies was initiated. The study was designed to evaluate ME-344 plus bevacizumab in up to two cohorts of approximately 20 patients each. The option to enroll the second cohort was conditioned upon Cohort 1 reaching a predetermined non-progression threshold of at least 20% at four months. Patients in the study were treated until disease progression or intolerability. The primary endpoint of the study was 16-week progression free survival (PFS), and secondary endpoints included overall PFS, duration of response, overall survival and safety.
ME-344 was administered once weekly on Days 1, 8 and 15 combined with bevacizumab on Days 1 and 15 of each 28-day cycle. Cohort 1 enrolled a total of 23 patients with relapsed mCRC. Patients were generally heavily pretreated; the median number of prior lines of therapy was 4 (range 1-8), 18 (78%) patients had ≥3 prior lines, and all patients had previously received bevacizumab and standard chemotherapy. As reported in April 2024, the combination was generally well tolerated with no overlapping toxicities observed. Two patients (9%) discontinued therapy due to an adverse event: fatigue considered related to study drugs and sepsis considered unrelated. The most common (≥10% of patients) drug-related adverse events (all grades/grade ≥3) were fatigue in 8 (35%) / 3 (13%) patients and abdominal pain in 3 (13%) / 2 (9%) patients.
9
It was further reported that in the first cohort, 5 of 20 (25%) evaluable patients completed 16 weeks of therapy without evidence of disease progression, exceeding the 20% predetermined threshold as set forth in the Clinical Study Protocol to proceed to Cohort 2. The median PFS was 1.9 months, the 4-month PFS rate was 31.2%, and the median overall survival was 6.7 months with 15 patients censored at the time of analysis. Nine (45%) of the 20 evaluable patients had stable disease. Although Cohort 1 exceeded the predetermined PFS threshold, we decided not to initiate enrollment in a second cohort.
Results from an earlier, first-in-human, single-agent Phase 1 clinical trial of ME-344 in patients with refractory solid tumors were published in the April 1, 2015 edition of Cancer. The results indicated that eight of 21 evaluable patients (38%) treated with ME-344 achieved stable disease or better, including five who experienced progression-free survival that was at least twice the duration of their last prior treatment before entry into the trial. In addition, one of these patients, a heavily pre-treated patient with small cell lung cancer, achieved a confirmed partial response and remained on study for two years. ME-344 was generally well tolerated at doses equal to or less than 10 mg/kg delivered on a weekly schedule for extended durations. Treatment-related adverse events included nausea, dizziness and fatigue. Dose-limiting toxicities were observed at both the 15 mg/kg and 20 mg/kg dose levels, consisting primarily of grade 3 peripheral neuropathy.
We were continuing to pursue ME-344 via development of a new formulation to advance our novel approach to inducing synthetic lethality in tumors in combination with VEGF inhibitors such as bevacizumab (Avastin®). We have already initiated research and development activity of the new formulation, with the goal of increasing biological activity, improving patient convenience of administration and increasing commercial opportunity.
Zandelisib: PI3Kδ Inhibitor Overview
Zandelisib is an oral, once-daily, selective PI3Kδ inhibitor that we were jointly developing with KKC under a global license, development and commercialization agreement entered into in April 2020.
In March 2022, we and KKC reported the outcome of an end of Phase 2 meeting with the FDA wherein the agency discouraged a filing based on data from a single-arm Phase 2 TIDAL trial. At this meeting, the FDA stated that data generated from single arm studies such as the Phase 2 TIDAL trial are insufficient to adequately assess the risk/benefit of PI3Kδ inhibitors evaluating indolent non-Hodgkin lymphoma. At that time, the FDA emphasized that we continue efforts with the ongoing randomized Phase 3 COASTAL trial evaluating patients with relapsed or refractory follicular or marginal zone lymphomas. Subsequently, at an April 2022 meeting of the FDA Oncology Drugs Advisory Committee, the committee voted that future approvals of PI3Kδ inhibitors for hematologic malignancies should be supported by randomized data.
In November 2022, we and KKC met with the FDA in a follow-up meeting to the March 2022 end of Phase 2 meeting. At this meeting, the FDA provided further guidance regarding the design and statistical analysis for the Phase 3 COASTAL trial. Following the November meeting, the companies jointly concluded that a clinical trial consistent with the recent FDA guidance, including modification of the ongoing COASTAL trial, would likely not be feasible to complete within a time period that would support further investment or with sufficient certainty of the regulatory requirements for approval to justify continued global development efforts. As a result, we and KKC jointly decided to discontinue global development of zandelisib for indolent forms of non-Hodgkin lymphoma outside of Japan. The discontinuation of zandelisib development outside of Japan was a business decision based on the most recent regulatory guidance from the FDA and is not related to the zandelisib clinical data generated to date. After making the joint decision to terminate development outside of Japan, we and KKC began closing all ongoing zandelisib clinical studies outside of Japan, including the Phase 3 COASTAL trial, the Phase 2 TIDAL trial, and the Phase 2 CORAL trial.
Subsequently, in May 2023, KKC decided to discontinue development of zandelisib in Japan. The discontinuation of zandelisib in Japan was a business decision by KKC based on the most recent regulatory guidance from the Pharmaceuticals and Medical Devices Agency in Japan and was not related to the zandelisib clinical data generated to date.
On July 14, 2023, we entered into a Termination Agreement (the Termination Agreement) with KKC to terminate all agreements between the parties and cease further zandelisib clinical development globally. Activities associated with the compassionate use supply and wind down of the KKC Commercialization Agreement were completed in fiscal year 2024.
KKC License, Development and Commercialization Agreement
In April 2020, we entered into the KKC Commercialization Agreement under which we granted to KKC a co-exclusive, sublicensable, payment-bearing license under certain patents and know-how controlled by us to develop and commercialize zandelisib and any pharmaceutical product containing zandelisib for all human indications in the U.S. (the U.S. License), and an exclusive (subject to certain retained rights to perform obligations under the KKC Commercialization Agreement), sublicensable, payment- bearing, license under certain patents and know-how controlled by us to develop and commercialize zandelisib and any pharmaceutical product containing zandelisib for all human indications in countries outside of the U.S. (the Ex-U.S. and the Ex-U.S. License). Also under the KKC Commercialization Agreement, we were granted a co-exclusive, sublicensable, license under certain patents and know-how controlled by KKC to develop and commercialize zandelisib for all human indications in the U.S., and a co-exclusive, sublicensable, royalty-free, fully paid license under certain patents and know-how controlled by KKC to perform our obligations in the Ex-U.S. and were paid an initial non-refundable payment of $100.0 million. Additionally, in Japan, the KKC
10
Commercialization Agreement included potential regulatory and commercialization milestone payments plus royalties on net sales of zandelisib in Japan, which are tiered beginning in the teens. Prior to the execution of the Termination Agreement on July 14, 2023, KKC was responsible for the development and commercialization of zandelisib in the Ex-U.S. and, subject to certain exceptions, solely responsible for all costs related thereto. We also provided to KKC certain drug supplies necessary for the development and commercialization of zandelisib in the Ex-U.S., with the understanding that KKC would have assumed responsibility for manufacturing for the Ex-U.S. as soon as practicable.
As noted above, on July 14, 2023, we entered into a Termination Agreement with KKC to mutually terminate the KKC Commercialization Agreement and all other related agreements between the parties. Pursuant to the Termination Agreement:
As of June 30, 2023, we had $64.9 million of aggregate deferred revenue associated with the KKC Commercialization Agreement, of which $64.5 million was allocated to the U.S. License and $0.3 million was allocated to the Development Services performance obligations which were recognized based on the proportional performance of these development activities through wind-down of the associated trials. As further discussed in Note 7. License Agreements, in connection with the execution of the Termination Agreement during the three months ended September 30, 2023, we recognized the $64.5 million of noncash long-term deferred revenue associated with the U.S. License as well as the remaining $0.3 million noncash deferred revenue associated with the completion of the underlying proportional performance activities. As of September 30, 2023, all deferred revenue associated with the KKC Commercialization Agreement had been recognized.
Competition
The marketplace for our drug candidates is highly competitive. A number of other companies have products or drug candidates in various stages of pre-clinical or clinical development that are intended for the same therapeutic indications for which our drug candidates are being developed. Some of these potential competing drug candidates are further advanced in development than our drug candidates and may be commercialized sooner. Even if we are successful in developing products that receive regulatory approval, such products may not compete successfully with products produced by our competitors or with products that may subsequently receive regulatory approval.
Our competitors include pharmaceutical companies and biotechnology companies, as well as universities and public and private research institutions. In addition, companies active in different but related fields represent substantial competition for us. Many of our competitors developing oncology drugs have significantly greater capital resources, larger research and development staffs and facilities, and greater experience in drug development, regulation, manufacturing, marketing and commercialization than we do. They compete with us in recruiting sites and eligible patients to participate in clinical studies and in attracting development and/or commercialization partners. They also license technologies that are competitive with our technologies. As a result, our competitors may be able to more easily develop technologies and products that would render our technologies or our drug candidates obsolete or non-competitive.
Intellectual Property
We own, by assignment or exclusive license, worldwide rights to each of our current drug candidates. Our intellectual property portfolio includes approximately 38 issued U.S. patents, 201 issued foreign patents, 10 pending U.S. patent applications, and 83 pending foreign applications.
We have acquired exclusive worldwide rights to develop, manufacture and commercialize voruciclib from Presage Biosciences, Inc. (Presage). The U.S. Patent and Trademark Office (USPTO) has allowed or issued 19 U.S. patents covering the composition of matter, pharmaceutical compositions, and methods of use to treat cancer which are projected to expire between 2026 and 2037, not including any patent term extension. There are approximately 90 allowed or issued foreign patents, 3 pending U.S. provisional patent applications, and approximately 60 pending foreign patent applications for voruciclib, related compounds, and related methods of use.
We have acquired, by assignment, patents and patent applications from Novogen, our former majority shareholder, relating to a family of isoflavonoid compounds, including ME-344. The USPTO has issued 12 patents covering ME-344 as composition of matter,
11
pharmaceutical compositions, and methods of use to treat cancer. There are approximately 61 foreign patents granted or allowed. The issued U.S. patents with composition of matter claims covering ME-344 are expected to expire between 2025 and 2031, not including patent term extension. There are 5 pending U.S. patent applications, 1 pending Patient Cooperation Treaty (PCT) and 10 pending foreign patent applications directed to ME-344 and related compounds or methods of use thereof.
We have acquired, by assignment, worldwide rights to zandelisib and other related compounds from Pathway Therapeutics, Inc. The USPTO has issued seven patents covering zandelisib as composition of matter, pharmaceutical compositions, and methods of use to treat cancer. The issued U.S. patents with composition of matter claims covering zandelisib are projected to expire between 2031 and 2032, not including any patent term extension. There are approximately 50 foreign patents granted or allowed. There are 2 pending U.S. patent applications, 1 pending PTC application and approximately 11 pending foreign patent applications directed to zandelisib and related compounds or methods of use thereof.
Our success depends in large part on our ability to protect our proprietary technologies, compounds and information, and to operate without infringing the proprietary rights of third parties. We rely on a combination of patent, trade secret, copyright, and trademark laws, as well as confidentiality, licensing and other agreements, to establish and protect our proprietary rights. We seek patent protection for our key inventions, including drug candidates we identify, routes for chemical synthesis and pharmaceutical formulations. There is no assurance that any of our pending patent applications will issue, or that any of our patents will be enforceable or will cover a drug or other commercially significant product or method. In addition, we regularly review our patent portfolio to identify patents and patent applications that we deem to have relatively low value to our ongoing business operations for potential abandonment. There is also no assurance that we will correctly identify which of our patents and patent applications should be maintained and which should be abandoned. The term of most of our other current patents commenced, and most of our future patents, if any, will commence, on the date of issuance and terminate 20 years from the earliest effective filing date of the non-provisional patent application. Because any marketing and regulatory approval for a drug often occurs several years after the related patent application is filed, the resulting market exclusivity afforded by any patent on our drug candidates and technologies will likely be substantially less than 20 years.
As most patent applications in the U.S. are maintained as confidential until published by the USPTO at 18 months from filing for all cases filed after November 29, 2000, or at issue, for cases filed prior to November 29, 2000, we cannot be certain that we or Presage were the first to make the inventions covered by the patents and applications referred to above. Additionally, publication of discoveries in the scientific or patent literature often lags behind the actual discoveries. Moreover, pursuant to the terms of the Uruguay Round Agreements Act, patents filed on or after June 8, 1995 have a term of twenty years from the date of such filing except for provisional applications, irrespective of the period of time it may take for such patent to ultimately issue. This may shorten the period of patent protection afforded to therapeutic uses of zandelisib, voruciclib or ME-344 as patent applications in the biopharmaceutical sector often take considerable time to issue. However, in some countries the patent term may be extended.
In order to protect the confidentiality of our technology, including trade secrets and know-how and other proprietary technical and business information, we require all of our consultants, advisors and collaborators to enter into agreements that prohibit the use or disclosure of information that is deemed confidential. These agreements also oblige our consultants, advisors and collaborators to assign to us, or negotiate a license to developments, discoveries and inventions made by such persons in connection with their work relating to our products. We cannot be sure that confidentiality will be maintained by those from whom we have acquired technology or disclosure prevented by these agreements. We also cannot be sure that our proprietary information or intellectual property will be protected by these agreements or that others will not independently develop substantially equivalent proprietary information or intellectual property.
The pharmaceutical industry is highly competitive, and patents may have been applied for by, and issued to, other parties relating to products competitive with zandelisib, voruciclib or ME-344. Use of these compounds and any other drug candidates may give rise to claims that they infringe the patents or proprietary rights of other parties, existing now and in the future. An adverse claim could subject us to significant liabilities to such other parties and/or require disputed rights to be licensed from such other parties. We cannot be sure that any license required under any such patents or proprietary rights would be made available on terms acceptable to us, if at all. If we do not obtain such licenses, we may encounter delays in product market introductions, or may find that the development, manufacture or sale of products requiring such licenses may be precluded.
Research and Development
The objective of our research and development program is the generation of data sufficient to achieve regulatory approval of our drug candidates in one or more dosage forms in major markets such as the U.S., to meet medical needs and develop a clinical and commercial profile with attractive attributes, and/or to allow us to enter into a development and/or commercial relationship with another party. The data are generated by our pre-clinical studies and clinical trial programs.
12
The key aspects of our research and development program have been to provide more complete characterization of the following:
Government Regulation
U.S. Regulatory Requirements
The U.S. Food and Drug Administration (FDA), and comparable regulatory agencies in other countries, regulate and impose substantial requirements upon the research, development, nonclinical and clinical testing, labeling, manufacture, quality control, storage, approval, advertising, promotion, marketing, distribution, import, and export of pharmaceutical products, as well as significant reporting and record-keeping obligations. State governments may also impose obligations in these and other areas. These requirements are extensive and are frequently changing.
In the U.S., pharmaceutical products are regulated by the FDA under the Federal Food, Drug, and Cosmetic Act (FDCA) and other laws. The process required by the FDA before drugs may be marketed in the U.S. generally involves the following:
The testing and approval process requires substantial time, effort, and financial resources, and we cannot be certain that we will be able to ultimately submit marketing applications for any of our product candidates, that our development efforts will prove to be successful, that our studies will have positive outcomes, or that any approval will be granted on a timely basis, if at all.
The results of the nonclinical studies, together with initial specified manufacturing information, the proposed clinical trial protocol, and information about the participating investigators are submitted to the FDA as part of an investigational new drug (IND) application, which must become effective before we may begin human clinical trials in the U.S. Clinical trials must be conducted in accordance with federal regulations and Good Clinical Practice (GCP) requirements, and with investigational products that follow cGMP. GCPs include, among other requirements, the requirements related to monitoring, drug accountability, data integrity, and that all research subjects provide their informed consent in writing for their participation in any clinical trial. Recently, the FDA has issued a number of new guidance regarding the conduct of clinical studies. For instance, the FDA issued an updated guidance on good clinical practices, which is intended to modify the agency’s GCP guidelines, including with respect to clinical trial quality, the use of digital health technologies, computerized systems, and data governance. The FDA also issued guidance regarding the conduct of decentralized clinical trials, use of electronic records, systems and signatures in clinical trials, and the conduct of risk-based clinical trial monitoring. Following issuance of a final guidance, the FDA will further be requiring diversity action plans for certain clinical studies.
Additionally, an independent IRB must review and approve each study protocol and oversee conduct of the trial. An IND becomes effective 30-days after receipt by the FDA, unless the FDA, within the 30-day period, raises concerns or questions about the conduct of the trials as outlined in the IND and imposes a clinical hold. If the FDA imposes a clinical hold at any time before or
13
during clinical trials, the IND sponsor must resolve the FDA’s concerns before clinical trials can begin or continue. Nonclinical tests and studies can take several years to complete, and there is no guarantee that an IND that is submitted based on such tests and studies will become effective within any specific time period, if at all.
Sponsors must make certain reports and submissions to the FDA and global health authorities, as appropriate, and to clinical investigators who, in turn, make certain reports and submissions to the IRB or ethics committee, including annual reports, and reports of investigator financial interests, serious adverse events and other significant safety information, study amendments, and new study protocols. Information about certain clinical trials, including a description of the study and study results, must also be submitted within specific time frames to the National Institutes of Health (the NIH), for public dissemination on the clinicaltrials.gov website. Sponsors of investigational products for serious diseases must also have a publicly available policy on requests for expanded access.
Investigational drugs and active ingredients imported into the U.S. are also subject to regulation by the FDA. Further, the export of investigational products outside of the U.S. is subject to regulatory requirements of the receiving country as well as U.S. export requirements under the FDCA.
Human clinical trials are typically conducted in three sequential phases that may overlap.
Concurrent with clinical trials, companies usually complete additional nonclinical and toxicology studies and must also develop additional information about the chemistry, manufacturing and controls (CMC) of the product candidate.
Some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data monitoring committee. This group reviews data and advises the study sponsor regarding the continuing safety of the trial. This group may also review interim data to assess the continuing validity and scientific merit of the clinical trial. The data monitoring committee may advise the sponsor to halt the clinical trial, modify the clinical trial, or continue the clinical trial depending on safety results and the trial’s likelihood of success.
We cannot be certain that we will successfully complete clinical testing of our products within any specific time period, if at all. Furthermore, the FDA, the IRB or we may suspend or terminate clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable safety risk or noncompliance with applicable regulatory requirements.
Results of nonclinical and toxicology studies, and clinical trials, as well as detailed information about the manufacturing process, quality control methods, and product composition, among other things, are submitted to the FDA as part of an NDA seeking approval to market and commercially distribute the product on the basis of a determination that the product is safe and effective for its intended use. Once the FDA receives an application, it has 60 days to review the NDA to determine if it is substantially complete to permit a substantive review, before it accepts the application for filing. The FDA may request additional information rather than accept an application for filing. In this event, the application must be resubmitted with the additional information. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. Under the goals agreed to by the FDA under the Prescription Drug User Fee Act (PDUFA), the agency currently aims to review 90% of all applications for new molecular entities within ten months of the 60-day filing date for a standard review. The PDUFA date is only a goal, thus, the FDA does not always meet its PDUFA dates. The PDUFA date may also be extended if the FDA requests or the sponsor provides substantial additional information regarding the submission.
The FDA may refer certain applications to an advisory committee, which is a panel of experts that make a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
14
Before approving an NDA, the FDA will inspect the facilities at which the product is manufactured and may inspect the sponsor, clinical study vendors, and clinical sites at which the product candidate was studied and will not approve the product unless cGMP and GCP compliance are satisfactory. Inspections may be in-person or conducted remotely. If applicable regulatory criteria are not satisfied, the FDA may issue a complete response letter (CRL) to the sponsor requiring additional nonclinical or clinical studies or data or additional CMC information. If a CRL is issued, the applicant may either: resubmit the marketing application, addressing all of the deficiencies identified in the letter; withdraw the application; or request an opportunity for a hearing.
Once the FDA determines that the approval requirements are met, it will issue an approval letter that authorizes commercial marketing of the product with specific prescribing information for specific indications. As a condition of approval, the FDA also may require post-marketing commitments and requirements, including studies, and/or surveillance to monitor the product’s safety or efficacy. The FDA also may require a Medication Guide and also a risk evaluation and mitigation strategy (REMS), or other conditions for a product’s approval or following approval to ensure that the benefits of the product candidate outweigh the risks. Moreover, even if the FDA approves a product, it may limit the approved indications or populations for use of the product, require that contraindications, warnings, or precautions be included in the product labeling, including a black box warning, impose other conditions, such as post-approval studies, or may not approve label statements that are necessary for successful commercialization and marketing.
Even after an NDA is approved, the FDA may impose additional obligations or restrictions (such as labeling changes, or clinical post-marketing requirements), or even suspend or withdraw a product approval or require additional testing or label revisions on the basis of data that arise after the product reaches the market, or if compliance with regulatory standards is not maintained. We cannot be certain that any NDA we submit will be approved by the FDA for full or accelerated approval on a timely basis, if at all. Also, any such approval may limit the indicated uses for which the product may be marketed. Any refusal to approve, delay in approval, suspension or withdrawal of approval, or restrictions on indicated uses could have a material adverse impact on our business prospects.
Each NDA must be accompanied by a substantial user fee pursuant to the requirements of the PDUFA and its amendments. Fee waivers or reductions are available in certain circumstances. Following product approval, drug products are also subject to annual program fees. The FDA adjusts the PDUFA user fees on an annual basis. A written request can be submitted for a waiver for the application fee for the first human drug application that is filed by a small business, but there are no small business waivers for program fees. Product candidates that are designated as orphan products are not subject to application user fees unless the application includes an indication other than the orphan indication and may be exempt from program fees if certain criteria are met. We are not at the stage of development with our products where we are subject to these fees, but they are significant expenditures that may be incurred in the future and must be paid at the time of application submissions to the FDA.
Satisfaction of FDA requirements typically takes many years. The actual time required varies substantially, based upon the type, complexity, and novelty of the pharmaceutical product, among other things. Government regulation imposes costly and time-consuming requirements and restrictions throughout the product life cycle and may delay product marketing for a considerable period of time, limit product marketing, or prevent marketing altogether. Success in nonclinical or early-stage clinical trials does not ensure success in later stage clinical trials. Data obtained from nonclinical and clinical activities are not always conclusive and may be susceptible to varying interpretations that could delay, limit, or prevent marketing approval. Even if a product receives marketing approval, the approval is limited to specific clinical indications. Further, even after marketing approval is obtained, the discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market.
After product approval, there are continuing significant regulatory requirements imposed by the FDA, including record-keeping requirements, obligations to report adverse side effects in patients using the products, and restrictions on advertising and promotional activities. Quality control and manufacturing procedures must continue to conform to cGMPs, and the FDA periodically inspects facilities, via in person inspections and remote regulatory assessments, to assess cGMP compliance. Additionally, post-approval changes in ingredient composition, manufacturing processes or facilities, product labeling, or other areas may require submission of an NDA Supplement to the FDA for review and approval. New indications will require additional clinical studies and submission of an NDA Supplement. Commercially distributed products are also subject to a variety of additional requirements, including requirements regarding tacking, tracing, and supply chain integrity; and requirements related to drug shortages and drug shortage prevention.
Failure to comply with the FDA’s regulatory requirements may result in an enforcement action by the FDA, including clinical holds, refusal to approve marketing applications or supplements, Warning Letters, product recalls, suspension or revocation of product approval, seizure of product to prevent distribution, impositions of injunctions prohibiting product manufacture or distribution, and civil and criminal penalties, among other actions. Maintaining compliance is costly and time-consuming. We cannot be certain that we, or our present or future suppliers or third-party manufacturers, will be able to comply with all FDA regulatory requirements, and potential consequences of noncompliance could have a material adverse impact on our business prospects.
15
The FDA’s policies may change, and additional governmental regulations may be enacted that could delay, limit, or prevent regulatory approval of our products, that require that we implement additional compliance steps, or affect our ability to manufacture, market, or distribute our products after approval.
Our activities also may be subject to state laws and regulations that affect our ability to develop and sell our products. We are also subject to numerous federal, state, and local laws relating to such matters as safe working conditions, clinical, laboratory, and manufacturing practices, environmental protection, fire hazard control, and disposal of hazardous or potentially hazardous substances. We may incur significant costs to comply with such laws and regulations now or in the future, and the failure to comply may have a material adverse impact on our business prospects.
The FDCA includes provisions designed to facilitate the development and expedite the review of drugs intended for treatment of serious or life-threatening conditions that demonstrate the potential to address unmet medical needs for such conditions or present a significant improvement over existing therapy. These provisions set forth a procedure for designation of a drug as a fast track product. The fast track designation applies to the combination of the product and specific indication for which it is being studied. A product designated as fast track is ordinarily eligible for additional programs for expediting development and review, such as increased FDA interactions and rolling submission of the application.
Products that are intended to treat serious or life-threatening conditions and that provide a meaningful therapeutic benefit over existing treatments may also be eligible for accelerated approval. Drug approval under the accelerated approval regulations may be based on evidence of clinical effect on a surrogate endpoint that is reasonably likely to predict clinical benefit. A post-marketing clinical study will be required to be completed to verify clinical benefit, and other restrictions to assure safe use may be imposed. By the date of approval of an accelerated approval product, the FDA must specify the conditions for the required post approval studies, including enrollment targets, the study protocol, milestones, and target completion dates. The FDA may also require that the confirmatory Phase 4 studies be commenced prior to the FDA granting a product accelerated approval. Reports on the progress of the required Phase 4 confirmatory studies must be submitted to the FDA every 180 days after approval. Failure to conduct required post-approval studies, or confirm a clinical benefit, will allow the FDA to withdraw the drug or biologic from the market on a statutorily defined expedited basis. Failure to conduct the required Phase 4 confirmatory studies or to conduct such studies with due diligence, as well as failure to submit the required update reports can subject a sponsor to penalties. In recent years, the accelerated approval pathway has come under significant FDA and public scrutiny. Accordingly, the FDA may be more conservative in granting accelerated approval or, if granted, may be more apt to withdrawal approval if clinical benefit is not confirmed or the risk benefit assessment changes.
A third potential designation that may be available is breakthrough therapy designation. A breakthrough therapy is a product that is intended, alone or in combination with one or more other products, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. Products designated as breakthrough therapies are eligible for intensive FDA guidance, a commitment from the FDA to involve senior managers and experienced review staff in a proactive collaborative and cross-disciplinary review, rolling submission of the application, and the facilitation of cross-disciplinary review.
Finally, if a product is intended to treat a serious condition and, if approved, would provide significant improvements in the safety or effectiveness of the treatment, diagnosis, or prevention of the condition, the product may be eligible for priority review meaning that the FDA’s goal for the review of an NDA is shortened to six months (after a two month period during which the FDA decides whether the application is ready for filing) rather than the standard review of ten months from application acceptance. If we should seek additional designations for any of our programs, we cannot be assured that it will be granted by the FDA. There is also no guarantee that we will be able to maintain any designation that we have received or may receive.
Following the FDA’s approval of an NDA, sponsors are required to list with the FDA each patent with claims that cover the applicant’s drug or a method of using the drug. These patents are published in the FDA’s list of Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can be cited by potential competitors as a reference listed drug in support of a 505(b)(2) NDA or an Abbreviated New Drug Application, (ANDA). In an effort to clarify which patents must be listed in the Orange Book, in January 2021, Congress passed the Orange Book Transparency Act of 2020, which largely codified the FDA’s existing practices into the FDCA.
A 505(b)(2) NDA is an application that contains full reports of investigations of safety and efficacy but where at least some of the information required for approval comes from investigations that were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted. This regulatory pathway enables the applicant to rely, in part, on the FDA’s prior findings of safety and efficacy for an existing product, or published literature. An ANDA provides for marketing of a generic drug product that has the same active ingredients, dosage form, strength, route of administration, labeling, performance characteristics and intended use as a previously approved product. ANDA
16
applicants generally must only scientifically demonstrate that their product is bioequivalent to, or performs in the same manner as, the innovator drug and can often be substituted by pharmacists under prescriptions written for the reference listed drug.
Generally, the FDA may not approve an abbreviated new drug application (ANDA) or 505(b)(2) NDA unless the reference listed drug’s Orange Book listed patents have expired and/or if the applicant certifies that it is not seeking approval for a patented method of use. The FDA may approve these applications, however, if the 505(b)(2) NDA or ANDA sponsor certifies that the Orange Book listed patents for the reference listed drug are invalid or will not be infringed upon by the manufacture, use or sale of the drug product for which the application is submitted. This later certification is called a paragraph IV certification. If the ANDA or 505(b)(2) NDA applicant has made a paragraph IV certification, following notice to the NDA and patent holders, the NDA and patent holders may then initiate a patent infringement lawsuit. If a lawsuit is brought, the FDA may not make an approval effective until the earlier of 30 months from the patent or application owner’s receipt of the notice of the paragraph IV certification, the expiration of the patent, when the infringement case concerning each such patent is favorably decided in the applicant’s favor or settled, or such shorter or longer period as may be ordered by a court.
Congress and U.S. federal administrative agencies have taken certain measures to increase drug competition and thus decrease drug prices, including by facilitating 505(b)(2) NDAs and ANDAs, and by introducing additional products into the U.S. market. For example, the FDA finalized a rule and a guidance to facilitate drug importation. Congress also passed a bill requiring sponsors of NDA products to provide sufficient quantities of drug product on commercially reasonable market-based terms to entities developing generic and 505(b)(2) products. This bill also included provisions on shared and individual REMS for generic drug products.
Under the Drug Price Competition and Patent Term Restoration Act of 1984, a sponsor may obtain marketing exclusivity for a specified period of time following FDA approval of certain drug applications. For example, new drugs containing new chemical entities that have not been previously approved by the FDA may obtain five years of exclusivity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the therapeutic activity of the drug substance. During the exclusivity period, the FDA may not accept for review an ANDA or a 505(b)(2) NDA submitted by another company that contains the previously approved active moiety. However, an ANDA or 505(b)(2) NDA may be submitted after four years if it contains a paragraph IV certification. This exclusivity is not absolute. For instance, it will not delay the submission or approval of a full NDA; though, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and efficacy.
Following NDA approval, a patent owner may obtain an extension of a single unexpired patent that has not previously been extended for a period equal to one-half the period of time elapsed between the filing of an IND and the filing of the corresponding NDA plus the period of time between the filing of the NDA and FDA approval, with a five year maximum patent extension. The total patent life of the product with the extension cannot exceed fourteen years from the product’s approval date. The period of patent extension may also be reduced for any time that the applicant did not act with due diligence. We cannot be certain that we will be able to take advantage of either the patent term extension or marketing exclusivity provisions of these laws or that, if received, they will adequately protect any approved products from competition.
The Best Pharmaceuticals for Children Act (BPCA) was reauthorized and amended by the FDA Amendments Act of 2007 (FDAAA). The reauthorization of BPCA adds an additional six months of marketing exclusivity and patent protection to unexpired exclusivities and unexpired patents listed with the FDA for NDA applicants that conduct acceptable pediatric studies of new and currently marketed drug products for which pediatric information would be beneficial, as identified by the FDA in a Pediatric Written Request. The data do not need to show the product to be effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly address the agreement between the sponsor and the FDA in the Pediatric Written Request, the additional protection is granted.
The Pediatric Research Equity Act (PREA) also was reauthorized and amended by the FDAAA. The reauthorization of PREA requires that most applications for drugs include a pediatric assessment (unless waived or deferred) to ensure the drugs’ safety and effectiveness in children. Such pediatric assessment must contain data, gathered using appropriate formulations for each age group for which the assessment is required, that are adequate to assess the safety and effectiveness of the drug product for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the drug product is safe and effective. The pediatric assessments can only be deferred provided there is a timeline for the completion of such studies. The FDA may waive (partially or fully) the pediatric assessment requirement for several reasons, including if the applicant can demonstrate that reasonable attempts to produce a pediatric formulation necessary for that age group have failed. Orphan products are also exempt from the PREA requirements. The Food and Drug Administration Safety and Innovation Act signed into law on July 9, 2012, permanently renewed and strengthened BPCA and PREA.
17
Under the FDA Reauthorization Act of 2017, sponsors submitting original applications on or after August 18, 2020, for product candidates intended for the treatment of adult cancer which are directed at molecular targets that the FDA determines to be substantially relevant to the growth or progression of pediatric cancer must submit, prior to marketing application submission, an initial Pediatric Study Plan for FDA agreement, and with the application, reports from molecularly targeted pediatric cancer clinical investigations designed to yield clinically meaningful pediatric study data, using appropriate pediatric formulations, to inform potential pediatric labeling. While orphan products are not exempt from this requirement, the FDA may grant full or partial waivers, or deferrals, for submission of data.
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition, which generally is a disease or condition that affects fewer than 200,000 individuals in the U.S. Additionally, sponsors must present a plausible hypothesis for clinical superiority to obtain orphan drug designation if there is a product already approved by the FDA that is considered by the FDA to be the same as the already approved product and is intended for the same indication. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. Orphan drug designation does, however, entitle a party to financial incentives such as opportunities for grant funding towards clinical study costs, tax advantages, and certain user-fee waivers. The tax advantages, however, were limited in the 2017 Tax Cuts and Jobs Act. If a product which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, i.e., the FDA may not approve any other applications to market the same drug for the same indication for a period of seven years, except in limited circumstances. By example, if there is already a product approved by the FDA that is the same product for the same indication, the orphan designated product will only receive orphan drug exclusivity if the prior hypothesis of clinical superiority is demonstrated. Competitors may also be able to receive approval for different drugs for the indication for which the orphan product has exclusivity or the same drug for a different indication.
Notably, the exact scope of any period of orphan drug exclusivity may change. Specifically, 2021 judicial decision, Catalyst Pharms., Inc. v. Becerra, challenged and reversed an FDA decision on the scope of orphan product exclusivity for the drug, Firdapse. Under this decision, orphan drug exclusivity for Firdapse blocked approval of another company’s application for the same drug for the entire disease or condition for which orphan drug designation was granted, not just the disease or condition for which approval was received. In a January 2023 Federal Register notice, however, the FDA stated that it intends to continue to apply its regulations tying the scope of orphan-drug exclusivity to the uses or indications for which a drug is approved. The exact scope of orphan drug exclusivity will likely be an evolving area.
Pharmaceutical Coverage, Pricing and Reimbursement & Healthcare Reform
In addition, future sales of our products, if approved for marketing, will depend, in part, on the availability and extent of coverage and reimbursement by third-party payors, such as government health programs, including Medicare and Medicaid, commercial insurance, and managed healthcare organizations. These third-party payors are increasingly challenging the price and limiting the coverage and reimbursement amounts for medical products and services. There may be significant delays in obtaining coverage and reimbursement for approved products, and coverage may be more limited than the purposes for which the product is approved by the FDA or regulatory authority in other countries. It is time-consuming and expensive to seek reimbursement from third- party payors. Moreover, eligibility for reimbursement does not imply that any product will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. In the United States, third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies, but they also have their own methods and approval process apart from Medicare coverage and reimbursement determinations.
In addition, the containment of healthcare costs has become a priority for federal and state governments, and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on coverage and reimbursement, and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Decreases in third-party reimbursement for our product candidates or a decision by a third-party payor to not cover our product candidates could reduce physician usage of the product candidate and have a material adverse effect on our sales, results of operations and financial condition. Moreover, there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. Individual states in the United States have also increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and drug price transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. On August 16, 2022, President Biden signed into the law the Inflation Reduction Act of 2022, or the IRA. Among other things, the IRA
18
has multiple provisions that may impact the prices of drug products, such as negotiated ceiling prices and penalties for price increases that exceed the rate of inflation, that are both sold into the Medicare program and throughout the United States.
Foreign Regulatory Requirements
Outside the U.S., our ability to market our products will also be contingent upon receiving marketing authorizations from the appropriate regulatory authorities and compliance with applicable post-approval regulatory requirements. Although the specific requirements and restrictions vary from country to country, as a general matter, foreign regulatory systems include risks similar to those associated with the FDA’s regulations, described above.
Under European Union (European Union) regulatory systems, marketing authorizations may be submitted either under a centralized or a decentralized procedure (DCP). Under the centralized procedure, a single application to the European Medicines Agency (EMA) leads to an approval granted by the European Commission which permits the marketing of the product throughout the EU. The centralized procedure is mandatory for certain classes of medicinal products such as new substances for the treatment of oncology. In addition, all medicinal products developed by certain biotechnological means, and those developed for cancer and other specified diseases and disorders, must be authorized via the centralized procedure. The centralized procedure will apply to any of our products that are developed by means of a biotechnology process or are intended for treatment of cancer. The DCP is used for products that are not eligible or not required to be authorized by the centralized procedure. The centralized procedure is optional for certain other products. Since the exit of the UK from the European Union, the UK has been excluded from the centralized procedure. It will be necessary for applicants to make a separate application to the UK Medicines and Healthcare products Regulatory Agency (MHRA) for a UK marketing authorization. There is currently no procedure for mutual EU/UK recognition of new medicinal products although there is an expedited review procedure (the EC Decision Reliance Procedure) for approval in the UK of EU approved products which is currently to run to December 31, 2023. Thereafter a new international recognition framework will be in place, which will have regard to decisions already made by the EMA. This means applications with a positive opinion from the Committee for Medicaid Products for Human Use (CHMP) received after December 31, 2023 will be eligible.
As with FDA approval, we may not be able to secure regulatory approvals in the EU in a timely manner, if at all. Additionally, as in the U.S., post-approval regulatory requirements, such as those regarding product manufacture, marketing, or distribution, would apply to any product that is approved in the EU, and failure to comply with such obligations could have a material adverse effect on our ability to successfully commercialize any product.
The conduct of clinical trials in the EU is governed by the European Clinical Trials Regulation (CTR), which was implemented in June 2022. This CTR governs how regulatory bodies in member states control clinical trials. No clinical trial may be started without a clinical trial authorization granted by the national competent authority and favorable ethics approval. Under the Regulation, clinical trial sponsors were able to use the Clinical Trials Information System (CTIS) since January 31, 2022, but are not obliged to use it immediately, in line with a three-year transition period. National regulators in the EU Member States and European Economic Area (EEA) countries could use the CTIS since January 31, 2022. With the exit of the UK from the EU, the UK did not implement the CTR and the UK provisions implementing the previous law as set out in the previous Clinical Trial Directive (which fundamentally covered the same area as the CTR but was far less detailed and predated the CTIS) will continue to apply until amended by the UK.
Accordingly, there is a marked degree of change and uncertainty both in the regulation of clinical trials and in respect of marketing authorizations which we face for our products in the EU.
Manufacturing
We do not have the facilities or capabilities to commercially manufacture any of our drug candidates. We are and expect to continue to be dependent on contract manufacturers for supplying our existing and future candidates for clinical trials and commercial scale manufacturing of our candidates in accordance with regulatory requirements, including cGMP. Contract manufacturers may utilize their own technology, technology developed by us, or technology acquired or licensed from third parties. FDA approval of the manufacturing procedures and the site will be required prior to commercial distribution.
Human Capital Management
As of June 30, 2024, we had 28 employees, 3 of whom hold a Ph.D. or M.D. degree, all of which reside in the United States. Of the 28 employees, 12 were engaged in research and development activities and 16 were engaged in business development, finance, information systems, facilities, human resources or administrative support. Other personnel resources are used from time to time as consultants or third-party service organizations on an as-needed basis. All members of our senior management team have prior experience with pharmaceutical, biotechnology or medical product companies. We believe that we have been successful in attracting skilled and experienced personnel, but there can be no assurance that we will be able to attract and retain the individuals needed.
19
Our people are a critical component in our continued success. We strive to create a workplace of choice to attract, retain and develop top talent to achieve our strategic goals. We strive to maximize the potential of our human capital resources by creating a respectful, rewarding, and inclusive work environment that enables our employees to further our mission. We adhere to a philosophy that includes, among other things, commitments to create ongoing job opportunities, pay fair wages, and protect worker health and safety.
We invest in our workforce by offering competitive salaries and benefits. We endeavor to foster a strong sense of ownership by offering stock options under our equity incentive plan. We also offer comprehensive and locally relevant benefits for all eligible employees.
We focus on our culture through a combination of regular training for employees at all levels, policies and practices in support of these goals, and a variety of internal and community-based events and actions that reinforce the power of our values and the unique characteristics of each of our employees.
None of our employees are represented by a labor union or covered by collective bargaining agreements. We have never experienced a work stoppage and believe our relationship with our employees is good. Management considers our relations with employees to generally be positive.
Available Information
Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed with or furnished to the SEC pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, are available free of charge through our website at www.meipharma.com as soon as reasonably practicable after they are electronically filed with, or furnished to, the SEC. Further, the SEC maintains an internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC and can be found on our EDGAR page at http://www.sec.gov.
Item 1A. Risk Factors
Investment in our securities involves a high degree of risk. You should consider carefully the risks described below, together with other information in this Annual Report and other public filings, before making investment decisions regarding our securities. If any of the following events actually occur, our business, operating results, prospects or financial condition could be materially and adversely affected. This could cause the trading price of our common stock to decline and you may lose all or part of your investment. Moreover, the risks described below are not the only ones that we face. Additional risks not presently known to us or that we currently deem immaterial may also affect our business, operating results, prospects or financial condition.
Risks Related to Our Review of Strategic Alternatives
We may not be successful in identifying and implementing any potential strategic alternatives in a timely manner, or at all, and any strategic transactions that we may consummate in the future could have negative consequences.
In July 2024, we announced that we are undertaking a comprehensive exploration of strategic alternatives focused on maximizing stockholder value. We expect to devote substantial time and resources to exploring strategic alternatives that our Board believes will maximize stockholder value. Despite management devoting significant efforts to identify and evaluate potential strategic alternatives, there can be no assurance that this strategic review process will result in us pursuing any transaction or that we will be able to successfully consummate any particular strategic transaction on attractive terms, on a timely basis, or at all. For example, certain types of strategic transactions may require third-party consents, such as stockholder approval, which could be difficult or costly to obtain. We have not set a timetable for completion of this strategic review process, and our Board has not approved a definitive course of action. Additionally, there can be no assurance that any particular course of action, business arrangement or transaction, or series of transactions, will be pursued, successfully consummated or lead to increased stockholder value or that we will make any cash distributions to our stockholders.
The process of continuing to evaluate our strategic alternatives may be costly, time-consuming and complex, and we may incur significant legal, accounting and advisory fees and other expenses, some of which may be incurred regardless of whether we successfully enter into a transaction. We may also incur additional unanticipated expenses in connection with this process. Any such expenses will decrease the remaining cash available for use in our business. Our ability to pursue or consummate strategic transactions also depends upon our ability to retain certain of our employees, the loss of whose services may adversely impact the ability to
20
identify, negotiate and consummate such transaction. If we are unable to successfully retain certain of our key remaining personnel, we are at risk of a disruption to our exploration and consummation of one or more strategic transactions.
In addition, potential counterparties in a strategic transaction involving us may place minimal or no value on our assets and our public listing. Further, should we resume the development of future drug candidates, such as one or more of the programs in our pipeline for which we halted further development, the development and any potential commercialization of our future drug candidates will require substantial additional cash to fund the costs associated with conducting the necessary preclinical and clinical testing and obtaining regulatory approval. Consequently, any potential counterparty in a strategic transaction involving us may choose not to spend additional resources to resume or continue development of our future drug candidates and may attribute little or no value, in such a transaction, to our future drug candidates.
In addition, any strategic transactions that we may pursue could have a variety of negative consequences, and we may enter into a transaction that yields unexpected results that adversely affect our business and decreases the remaining cash available for use in our business. Any potential transaction would be dependent on a number of factors that may be beyond our control, including, among other things, market conditions, industry trends, the interest of third parties in a potential transaction with us, obtaining stockholder approval and the availability of financing to third parties in a potential transaction with us on reasonable terms. There can be no assurance that any particular course of action, business arrangement or transaction, or series of transactions, will be pursued, successfully consummated, lead to increased stockholder value, or achieve the anticipated results.
If we are not successful in setting forth a new strategic path for us, or if our plans are not executed in a timely fashion, this may cause reputational harm with our stockholders and the value of our securities may be adversely impacted. In addition, speculation regarding any developments related to the review of strategic alternatives and perceived uncertainties related to the future of us could cause our stock price to fluctuate significantly.
Even if we successfully consummate any strategic transaction, or series of transactions, from our strategic assessment, we may fail to realize all or any of the anticipated benefits of any such transaction, such benefits may take longer to realize than expected, we may encounter integration difficulties or we may be exposed to other operational and financial risks.
Our ability to realize the anticipated benefits of any potential strategic transaction will depend on a number of factors, including our ability to integrate with any future business partner, our ability to obtain value for portions of our business, if divested, and our ability to generate future stockholder value. The process may be disruptive to our business, and the expected benefits may not be achieved within the anticipated time frame, or at all. The failure to overcome the challenges involved and to realize the anticipated benefits of any potential transaction could adversely affect our business and financial condition. The negotiation and consummation of any potential strategic transaction will require significant time on the part of our management, and the diversion of management’s attention may disrupt our business.
The negotiation and consummation of any such transaction may also require more time or greater cash resources than we anticipate and expose us to other operational and financial risks, including, but not limited to, increased near-term and long-term expenditures, exposure to unknown liabilities, higher than expected acquisition or integration costs, incurrence of substantial debt or dilutive issuances of equity securities to fund future operations, including financings in connection with a strategic transaction, write-downs of assets or goodwill or incurrence of non-recurring, impairment or other charges, increased amortization expenses, difficulty and cost in combining the operations and personnel of any acquired or acquiring business with our operations and personnel, impairment of relationships with key suppliers or customers of any acquired or acquiring business due to changes in management and ownership, inability to retain our key employees or any acquired or acquiring business and possibility of future litigation. Any of the foregoing risks could have a material adverse effect on our business, financial condition and prospects.
If a strategic transaction is not consummated, our Board may decide to pursue a dissolution and liquidation. In such an event, the amount of cash available for distribution to our stockholders will depend significantly on the timing of such liquidation as well as the amount of cash that may need to be reserved for commitments and contingent liabilities.
If we do not successfully consummate a strategic transaction, our Board may decide to pursue a dissolution and liquidation. In such an event, the amount of cash available for distribution to our stockholders will depend heavily on the timing of such decision and, with the passage of time, the amount of cash available for distribution will be reduced as we continue to fund our operations. In addition, if our Board were to approve and recommend, and our stockholders were to approve, a dissolution and liquidation, we would be required under Delaware corporate law to pay our outstanding obligations, as well as to make reasonable provision for contingent and unknown obligations, prior to making any distributions in liquidation to our stockholders. As a result of this requirement, a portion of our assets may need to be reserved pending the resolution of such obligations and the timing of any such resolution is uncertain. In addition, we may be subject to litigation or other claims related to a dissolution and liquidation. If a dissolution and liquidation were pursued, our Board, in consultation with our advisors, would need to evaluate these matters and make a determination
21
about a reasonable amount to reserve. Accordingly, holders of our common stock could lose all or a significant portion of their investment in the event of a liquidation, dissolution or winding up.
The value to stockholders in the event of a strategic transaction or dissolution may depend on the extent to which we will be able to successfully satisfy our existing contractual obligations to third parties and regulatory commitments on favorable terms, which may include the outcome of our negotiations to reduce or terminate such commitments.
We are currently subject to certain contractual and regulatory obligations and commitments. In connection with our comprehensive exploration of strategic alternatives, we may seek to negotiate with third parties in order to reduce or eliminate such obligations and commitments. Our ability to successfully negotiate such obligations or commitments on favorable terms, or at all, or our ability to satisfy any such obligations may impact our ability to pursue a strategic transaction on terms favorable to us, the resulting value to stockholders in a strategic transaction or the cash available for distribution to our stockholders in the event of our dissolution. We may also incur substantial costs in connection with or as a result of such negotiations or termination of any of our commitments. There can be no assurance that we will be successful in negotiating to reduce or eliminate any of our existing contractual or regulatory obligations and commitments, or that we will be able to satisfy any such obligations on a timetable that will allow us to maximize potential value to our stockholders.
We may become involved in litigation, including securities class action litigation, that could divert our management’s attention and harm our business, and insurance coverage may not be sufficient to cover all costs and damages.
In the past, litigation, including securities class action litigation, has often followed certain significant business transactions, such as the sale of a company or announcement of any other strategic transaction, or the announcement of negative events. These events may also result in investigations by the SEC. We may be exposed to such litigation even if no wrongdoing occurred. Litigation is usually expensive and diverts management’s attention and resources, which could adversely affect our business and cash resources and our ability to consummate a potential strategic transaction or the ultimate value our stockholders receive in any such transaction.
Our workforce reduction may not achieve our intended outcome and may result in significant adverse consequences.
In July 2024, in connection with our evaluation of strategic alternatives, our Board approved a reduction-in-force to continue in stages as our operational and strategic direction evolves. This reduction-in-force may result in unintended consequences and costs, such as the loss of institutional knowledge and expertise, attrition beyond the intended number of employees, decreased morale among our remaining employees, and the risk that we may not achieve the anticipated benefits of the reduction-in-force. In addition, while positions have been eliminated, certain functions necessary to our operations remain, and we may be unsuccessful in distributing the duties and obligations of departed employees among our remaining employees. The reduction-in-force could also make it difficult for us to pursue, or prevent us from pursuing, new opportunities and initiatives due to insufficient personnel, or require us to incur additional and unanticipated costs to hire new personnel to pursue such opportunities or initiatives, including any potential strategic alternatives. If we are unable to realize the anticipated benefits from the reduction-in-force, or if we experience significant adverse consequences from the reduction-in-force, our business, financial condition, and results of operations may be materially adversely affected.
Risks Related to Our Financial Condition and Capital Requirements
We have incurred significant losses from our inception, and we anticipate that we may incur losses in the foreseeable future.
We are a clinical-stage pharmaceutical company. Until recently, we had focused our efforts primarily on developing voruciclib, a selective orally administered CDK9 inhibitor, and ME-344, an intravenous small molecule mitochondrial inhibitor targeting the oxidative phosphorylation pathway, with the goal of achieving regulatory approval. In connection with our decision to undertake a comprehensive exploration of strategic alternatives, we have discontinued our clinical programs involving voruciclib and ME-344.
Since inception, we have incurred significant operating losses. During the fiscal year ended June 30, 2024, we had net income of $17.8 million, while during the fiscal year ended June 30, 2023, we incurred a net loss of $31.8 million. As of June 30, 2024, we have an accumulated deficit of $388.2 million. In connection with the termination of all ongoing clinical programs noted above, our research and development expenses have decreased. We expect to continue to incur costs and expenditures in connection with the process of evaluating our strategic alternatives.
Should we resume development activities in the future, we expect that research and development costs would increase significantly and we would continue to incur significant expenses and operating and net losses, as we develop and seek regulatory approval for such drug candidates.
Our financial results may fluctuate significantly from year to year, depending on whether we resume development of our drug candidates or any future drug candidates, the timing of any clinical trials, the receipt of payments under any future agreements we may
22
enter into, and our expenditures on other research and development (R&D) activities as well as any payments owed under the License Agreement with Presage and any future similar agreements.
Should we resume the development activities in the future, we expect we would to continue to incur significant losses for the foreseeable future as we:
Because of the numerous risks and uncertainties associated with pharmaceutical drug development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. If we are required by the FDA or foreign regulatory authorities, to perform studies in addition to those currently expected, or if there are any delays in completing our clinical trials or the development of our drug candidate, our expenses could increase.
If we decide to resume development of our drug candidate or any future drug candidate, we will need additional funding and may be unable to raise capital when needed, which would force us to delay, reduce or eliminate our drug development programs.
Conducting clinical trials, pursuing regulatory approvals, establishing outsourced manufacturing relationships, and successfully manufacturing and commercializing drugs and drug candidates is expensive. If we resume development of our drug candidates or any future drug candidate, we will need to raise additional capital to continue such development.
In July 2024, we discontinued the clinical program in our pipeline in connection with our undertaking a comprehensive exploration of strategic alternatives focused on maximizing stockholder value. In connection with our streamlined operating plan, we commenced a reduction-in-force.
After taking into account the discontinuation of our clinical development programs, reduction-in-force and comprehensive exploration of strategic alternatives, we expect that our current unrestricted cash and cash equivalents and short-term investments will be sufficient to fund our currently anticipated operating plan for at least the next 12 months. In connection with the termination of all ongoing clinical programs, our research and development expenses have decreased. We expect to continue to incur costs and expenditures in connection with the process of evaluating our strategic alternatives. Should we resume development activities in the future, we expect that research and development costs would increase significantly. It is possible that the assumptions upon which we have based this estimate may prove to be wrong, and we could use our capital resources sooner than we presently expect.
Our future funding requirements will depend on many factors, including, but not limited to:
Future capital requirements will also depend on the extent to which we acquire or invest in additional complementary businesses, products and technologies. Until we can generate a sufficient amount of product revenue, if ever, we may seek to finance future cash needs through public or private equity offerings, debt financings, milestone and royalty payments from corporate collaboration and licensing arrangements, as well as through interest income earned on cash and investment balances. We cannot be certain that additional funding will be available on acceptable terms, or at all, and our ability to raise additional capital may be adversely impacted by potential worsening global economic conditions, including high rates of inflation and interest rates, the continuing disruptions to
23
and volatility in the credit and financial markets in the United States and worldwide, including resulting from the ongoing conflicts between Russia and the Ukraine, conflicts in the Middle East, and increasing tensions between China and Taiwan.
Risks Related to Any Future Development and Commercialization of Our Drug and Potential Drug Candidates
Should we resume development of our drug candidate or future drug candidates, if we are unable to successfully complete clinical development, obtain regulatory approvals and commercialize our drug candidate or future drug candidates, or experience significant delays in doing so, our business will be materially harmed.
In July 2024, we discontinued the clinical programs in our pipeline in connection with our undertaking a comprehensive exploration of strategic alternatives. Should we resume development activities in the future, we cannot be certain that any such drug candidates will be successful in clinical trials or receive regulatory approval. Regulatory authorities may interpret our data differently than we do. We are not permitted to market or promote any of our drug candidates before we receive regulatory approval from the FDA or comparable foreign regulatory authorities; should we resume development activities in the future we may never receive such regulatory approval for voruciclib or any future drug candidates.
Should we resume development of our drug candidate or any future drug candidates, the success of such drug candidates will depend on many factors, including but not limited to:
● successful enrollment in, and completion of, clinical trials, as well as completion of preclinical studies;
● favorable efficacy and acceptable safety data from our clinical trials and other studies;
● receipt of additional regulatory approvals;
● managing our reliance on sole-source third parties such as our third-party vendors, suppliers, and manufacturers;
● the performance by CROs or other third parties and consultants we may retain of their duties to us in a manner that complies with our protocols and applicable laws and that protects the integrity of the resulting data;
● obtaining and maintaining patent, trade secret and other intellectual property protection and regulatory exclusivity;
● ensuring we do not infringe, misappropriate or otherwise violate the valid patent, trade secret or other intellectual property rights of third parties;
● successfully launching, either alone or with a commercial partner, any drug candidate for which regulatory approval is received;
● obtaining and maintaining favorable reimbursement from third-party payers and governments for drugs and drug candidates;
● competition with other drugs;
● post-marketing commitments, if any, to regulatory agencies following regulatory approval of any drug candidate;
● continued acceptable safety profile following regulatory approval; and
● manufacturing or obtaining sufficient supplies of our drugs and any drug candidate that may be necessary for use in clinical trials for evaluation of any drug candidate and commercialization of any approved drug.
If we do not achieve and maintain one or more of these factors in a timely manner or at all, we could experience significant delays in our ability to, or be unable to obtain regulatory approvals for, and/or to successfully commercialize any drugs or drug candidates, which would materially harm our business and we may not be able to generate sufficient revenues and cash flows to continue our operations.
Risks Related to Our Intellectual Property
The value of our intellectual property is dependent, in part, on obtaining and maintaining patent protection and preserving trade secrets, which cannot be guaranteed.
Patent protection and trade secret protection are important to our business and our future will depend, in part on our ability to maintain trade secret protection, obtain patents and operate without infringing the proprietary rights of others both in the U.S. and abroad. Litigation or other legal proceedings may be necessary to defend against claims of infringement, to enforce our patents or to protect our trade secrets. Such litigation could result in substantial costs and diversion of our management’s attention.
The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and involve complex legal and factual questions. We acquired patents and patent applications related to voruciclib from Presage in 2017, and acquired both issued patents and pending patent applications related to ME-344 from Novogen in relation to its Isoflavone-based compounds, which we previously licensed from Novogen, in 2011. Additionally, Novogen had previously applied for patents in a number of countries with
24
respect to the use of their isoflavone compounds, including ME-344. Finally, in September 2013, we acquired patents and patent applications related to zandelisib from Pathway Therapeutics, Inc.
The patent applications may not proceed to grant or may be amended to reduce the scope of protection of any patent granted. The applications and patents may also be opposed or challenged by third parties. Should we resume development of our drug candidate or any future drug candidates, our commercial success will depend, in part, on our ability to obtain and maintain effective patent protection for our compounds and their use in treating, preventing, or curing cancer, and to successfully defend patent rights in those technologies against third-party challenges. As patent applications in the U.S. are maintained in secrecy until published or issued and as publication of discoveries in the scientific or patent literature often lag behind the actual discoveries, we cannot be certain that we or Presage were the first to make the inventions covered by the pending patent applications or issued patents referred to above or that we or they were the first to file patent applications for such inventions. Additionally, the breadth of claims allowed in biotechnology and pharmaceutical patents or their enforceability cannot be predicted. We cannot be sure that, should any patents issue, we will be provided with adequate protection against potentially competitive products. Furthermore, we cannot be sure that should patents issue, they will be of commercial value to us, or that private parties, including competitors, will not successfully challenge our patents or circumvent our patent position in the U.S. or abroad.
General Business Risks
Our employees, independent contractors, consultants, commercial partners, principal investigators, or CROs may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could have a material adverse effect on our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees, independent contractors, consultants, commercial partners, manufacturers, investigators, or CROs could include intentional, reckless, negligent, or unintentional failures to comply with FDA regulations, comply with applicable fraud and abuse laws, provide accurate information to the FDA, properly calculate pricing information required by federal programs, comply with federal procurement rules or contract terms, report financial information or data accurately or disclose unauthorized activities to us. This misconduct could also involve the improper use or misrepresentation of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter this type of misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. Moreover, it is possible for a whistleblower to pursue a False Claims Act (FCA), case against us even if the government considers the claim unmeritorious and declines to intervene, which could require us to incur costs defending against such a claim. Further, due to the risk that a judgment in an FCA case could result in exclusion from federal health programs or debarment from government contracts, whistleblower cases often result in large settlements. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, financial condition, and results of operations, including the imposition of significant fines or other sanctions.
Our business and operations would suffer in the event of system failures.
Our internal computer systems and those of our CROs and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war, and telecommunication and electrical failures. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our drug candidate development and, if such drug candidates are approved commercialization programs. For example, the loss of clinical trial data from completed, ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of or damage to our data or applications, or inappropriate disclosure of personal, confidential or proprietary information, we could incur liability and regulatory enforcement actions, and the further development of any of our drug candidates could be delayed.
Our efforts will be seriously jeopardized if we are unable to retain and attract key employees.
Our success depends on the continued contributions of our principal management, development and scientific personnel. We face competition for such personnel, and we believe that risks and uncertainties related to our business, including the timing and risk associated with research and development, our available and anticipated cash resources, and the volatility of our stock price, may impact our ability to hire and retain key and other personnel. The loss of services of our Acting Chief Executive Officer, Chief Financial Officer or other key employees could adversely impact our operations and ability to generate or raise additional capital.
25
Negative U.S. and global economic conditions may pose challenges to our business strategy, which relies on funding from the financial markets or collaborators.
Negative conditions in the U.S. or global economy, including financial markets, may adversely affect our business and the business of current and prospective vendors, licensees and collaborators, and others with whom we do or may conduct business. The duration and severity of these conditions is uncertain. If negative economic conditions occur, we may be unable to secure funding on terms satisfactory to us to sustain our operations or to find suitable collaborators to advance our internal programs, even if we achieve positive results from our drug development programs.
Laws, rules and regulations relating to public companies may be costly and impact our ability to attract and retain directors and executive officers.
Laws and regulations affecting public companies, including rules adopted by the SEC and by Nasdaq, may result in increased costs to us. These laws, rules and regulations could make it more difficult or costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our Board, on our board committees or as executive officers. We cannot estimate accurately the amount or timing of additional costs we may incur to respond to these laws, rules and regulations.
Security breaches and privacy issues could compromise our information and expose us to liability, which would cause our business and reputation to suffer.
In the ordinary course of our business, we collect and store sensitive data, including intellectual property, our proprietary business information and that of our suppliers, as well as personally identifiable information of clinical trial participants and employees. Similarly, our third-party providers possess certain of our sensitive protected health data. The secure maintenance of this information is critical to our operations and business strategy. Despite our reasonable security measures, our information technology and infrastructure may be vulnerable to cyber-attacks or breached due to employee error, malfeasance or other disruptions. Cyber-attacks and other security incidents are increasing in their frequency, levels of persistence, sophistication and intensity, and are being conducted by sophisticated and organized groups and individuals with a wide range of motives and expertise. Although we develop and maintain systems and controls designed to prevent these events from occurring, and we have a process to identify and mitigate threats, the development and maintenance of these systems, controls and processes is costly and requires ongoing monitoring and updating as technologies change and efforts to overcome security measures become more sophisticated, and such systems, controls and processes may not be successful in preventing a breach or other incident. Any such security incident could compromise our networks and the information stored there could be accessed, publicly disclosed, encrypted, lost or stolen. We could be required to expend significant amounts of money and other resources to repair or replace information systems or networks. In addition, our liability insurance may not be sufficient in type or amount to cover us against claims related to security breaches, cyber-attacks and other related security incidents.
The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing amount of focus on privacy and data protection issues with the potential to affect our business, including compliance with the Health Insurance Portability and Accountability Act of 1996 and state laws requiring security breach notification. The collection and use of personal health data of individuals in the European Union is also governed by strict data protection laws. In addition to existing laws, since May 25, 2018, the General Data Protection Regulation (GDPR) has imposed obligations with respect to European Union data and substantial fines for breaches of the data protection rules. The GDPR increased our responsibility and potential liability in relation to personal data that we process, and we were required to implement additional mechanisms to comply with the GDPR and related European Union data protection rules. Enforcement uncertainty and the costs associated with ensuring GDPR compliance may be onerous and adversely affect our business, operating results, prospects and financial condition.
We continue to evaluate the legal issues that arise concerning transfer of personal data of residents of the European Economic Area (EEA) member states or the U.K. to the U.S. or other jurisdictions that are not deemed adequate by the European Commission. Among other steps, we are implementing the new standard contractual clauses issued on June 4, 2021 by the European Commission. It remains uncertain how these standard contractual clauses will be implemented by the data exporters and data importers and whether they will ultimately be deemed sufficient by European courts. MEI Pharma observes the developments and will agree to the appropriate data transfer mechanism. In addition to standard contractual clauses, we may rely on individual contents of the patients where appropriate and necessary to safeguard the data flow from the EU to the U.S. Present solutions to legitimize transfers of personal data from the EEA may be challenged or deemed insufficient. We may, in addition to other impacts, experience additional costs associated with increased compliance burdens, and we and our customers face the potential for regulators in the EEA or U.K. to apply different standards to the transfer of personal data from the EEA/U.K. to the U.S., and to block, or require ad hoc verification of measures taken with respect to, certain data flows from the EEA or U.K. to the U.S. We also may be required to engage in new contract negotiations with third parties that aid in processing data on our behalf. We may experience reluctance or refusal by current or
26
prospective European clinical trial sites and CROs to use our products, and we may find it necessary or desirable to make further changes to our processing of personal data of EEA or U.K. data subjects.
Additionally, California has the California Consumer Privacy Act (CCPA), which creates individual privacy rights for consumers (as that word is broadly defined in the law) and places increased privacy and security obligations on entities handling personal data of consumers or households. The CCPA may significantly impact our business activities and require substantial compliance costs that adversely affect business, operating results, prospects and financial condition. Amendments to the CCPA mandated by the California Privacy Rights Act (CPRA) will impose additional privacy requirements, effective on January 1, 2023. Similarly comprehensive state consumer privacy laws in other states, such as Virginia, Utah, Connecticut and Colorado will also become effective in 2023. These new state privacy measures may reflect the start of a movement in other state legislatures to enact more comprehensive privacy laws, which would create a more complex privacy regulatory landscape for our business in the U.S. In addition, there is privacy legislation and rule making efforts at the federal level which may increase our privacy obligations in the U.S.
Thus, any access, disclosure or other loss of information, including our data being breached at our partners or third-party providers, along with violations of privacy laws that exist and are increasing around the world, could result in legal claims or proceedings and liability under laws that protect the privacy of personal information, disrupt our operations and damage our reputation, which could adversely affect our business.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could harm our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. From time to time and in the future, our operations may involve the use of hazardous and flammable materials, including chemicals and biological materials, and may also produce hazardous waste. Even if we contract with third parties for the disposal of these materials and waste, we cannot completely eliminate the risk of contamination or injury resulting from these materials. In the event of contamination or injury resulting from the use or disposal of our hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
We maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, but this insurance may not provide adequate coverage against potential liabilities. However, we do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. Current or future environmental laws and regulations may impair our research, development or production efforts. In addition, failure to comply with these laws and regulations may result in substantial fines, penalties or other sanctions.
We or the third parties upon whom we depend may be adversely affected by natural disasters and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
Events outside of our control, including natural disasters and public health emergencies, could severely disrupt our operations and have a material adverse effect on our business, operating results, prospects or financial condition. If a natural disaster, or public health emergency such as COVID-19, power outage or other event occurred that prevented us from conducting our clinical trials, including by damaging our critical infrastructure, such as third-party facilities, or that otherwise disrupted operations and travel, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. The disaster recovery and business continuity plans we have in place may prove inadequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which could have a material adverse effect on our business, operating results, prospects or financial condition.
Limitations on the deductibility of net operating losses could adversely affect our business and financial condition.
We have a history of net operating losses. In December 2017, the U.S. government enacted comprehensive tax legislation commonly referred to as the Tax Cuts and Jobs Act (the Tax Act). The Tax Act limits the deduction of net operating losses to 80% of current year taxable income. The limitations on the net operating loss deduction, as well other changes in tax policy, may subject us to additional taxation, adversely affecting our results of operations and financial condition.
Risks Related to Securities Markets and Investment in our Stock
We are currently operating in a period of capital markets disruption and economic uncertainty.
The U.S. capital markets are currently experiencing extreme volatility and disruption following the global outbreak of COVID-19, high inflation and the government response thereto, potential economic downturn, publicized failures in the regional banking
27
sector, the war in Ukraine, the upcoming U.S. presidential election, and other global events. Disruptions in the capital markets in the past have resulted in illiquidity in parts of the capital markets. Future market disruptions and/or illiquidity would be expected to have an adverse effect on our business, financial condition, results of operations and cash flows. Unfavorable economic conditions also would be expected to increase our funding costs, limit our access to the capital markets or result in a decision by lenders not to extend credit to us should that become required for us to fund ongoing operations. These events have limited and could continue to limit our capital investment considerations, limit our ability to fund further clinical development, limit our ability to identify and implement any potential strategic alternatives and have a material negative impact on our operating results.
If we fail to comply with the continued listing standards of the Nasdaq Capital Market, we may be delisted and the price of our common stock, our ability to access the capital markets and our financial condition could be negatively impacted.
Our common stock is currently listed on Nasdaq under the symbol MEIP. To maintain the listing of our common stock on the Nasdaq Capital Market, we are required to meet certain listing requirements, including, among others, maintaining a minimum closing bid price of $1.00 per share. As we continue to explore strategic alternatives, we intend to actively monitor the bid price of our common stock and its compliance with the listing requirement. If we fail to comply with the continued listing standards and the Nasdaq Capital Market delists our securities from trading on its exchange, we and our stockholders could face significant negative consequences including: reducing the liquidity and market price of our common stock; reducing the number of investors willing to hold or acquire our common stock, which could negatively impact our ability to raise equity financing; decreasing the amount of news and analyst coverage of us; and limiting our ability to issue additional securities or obtain additional financing in the future. In addition, delisting from Nasdaq may negatively impact our reputation and, consequently, our business.
Our business could be negatively impacted as a result of any future activism campaigns by Anson Advisors Inc. and Cable Car Capital LLC and other activist investors.
Anson Advisors Inc. holds approximately 16.4% of our outstanding common stock and Cable Car Capital LLC holds approximately 9.2% of our outstanding common stock, and each have sought to exert influence on our business operations and Board, and we expect that each will or may continue to do so.
In July 2023, Anson Advisors Inc. and Cable Car Capital LLC initiated a consent solicitation to seek the consent of our stockholders holding at least a majority of our outstanding shares of common stock to, among other things, remove and replace all members of our Board. On September 18, 2023, we filed our definitive consent revocation statement urging stockholders to revoke their consents and reject the Consent Solicitation.
The Consent Solicitation and our response to it resulted in significant distraction for management and additional capital outlays by us. Continued pursuit or further activities by Anson Advisors Inc. and Cable Car Capital LLC, or by other activist shareholders, could result in yet additional distractions and costs and could lead to a materially adverse impact on our business or operating results.
The trading price of the shares of our common stock has been and may continue to be highly volatile and could decline in value and we may incur significant costs from class action litigation.
The trading price of our common stock could be highly volatile in response to various factors, many of which are beyond our control, including, but not limited to, the following:
28
Equity markets in general, and the market for biotechnology and life sciences companies in particular, have experienced substantial price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of companies traded in those markets. In addition, changes in economic conditions in the U.S., the Europe or globally, particularly in the context of current global events, could impact upon our ability to grow profitably. Adverse economic changes are outside our control and may result in material adverse impacts on our business or our results of operations. These broad market and industry factors may materially affect the market price of shares of our common stock, regardless of our development and operating performance. In the past, following periods of volatility in the market price of a company’s securities, securities class-action litigation has often been instituted against that company. Such litigation, if instituted against us, could cause us to incur substantial costs and divert management’s attention and resources.
Future sales of our common stock, including common stock issued upon exercise of outstanding warrants or options, may depress the market price of our common stock and cause stockholders to experience dilution.
The market price of our common stock could decline as a result of sales of substantial amounts of our common stock in the public market, including upon exercise of outstanding warrants or stock options, and any subsequent sales of such shares. As of June 30, 2024, we had outstanding warrants exercisable to purchase 102,513 shares of common stock at an exercise price of $6.80 per share, which expire in October 2027. We also have outstanding options to purchase 1,357,213 shares of common stock . We may seek additional capital through one or more additional equity transactions in the future; however, such transactions will be subject to market conditions and there can be no assurance any such transactions will be completed. If we sell shares in the future, the prices at which we sell these future shares will vary, and these variations may be significant. Stockholders will experience significant dilution if we sell these future shares at prices significantly below the price at which such previous stockholders invested.
Other than as described below or in connection with a strategic transaction we do not intend to pay, and we have not paid, any cash dividends on our shares of common stock. Our stockholders will not be able to receive a return on their shares unless the value of our common stock appreciates and they sell their shares.
Other than the Capital Return, we have never paid or declared any cash dividends on our common stock, and we intend to retain any future earnings to finance the development and expansion of our business. We do not anticipate paying any cash dividends on our common stock in the foreseeable future other than in connection with a strategic transaction. Therefore, our stockholders will not be able to receive a return on their investment unless a strategic transaction that requires a dividend is successful or the value of our common stock appreciates and they sell their shares.
We will have broad discretion over the use of the net proceeds from any exercise of outstanding warrants and options.
We will have broad discretion to use the net proceeds to us upon any exercise of outstanding warrants and options, and investors in our stock will be relying on the judgment of our Board and management regarding the application of these proceeds. Although we expect to use a substantial portion of the net proceeds from any exercise of the warrants and options for general corporate purposes and progression of our clinical trial programs, we have not allocated these net proceeds for specific purposes.
We are authorized to issue blank check preferred stock, which could adversely affect the holders of our common stock.
Our amended and restated certificate of incorporation allows us to issue blank check preferred stock with rights potentially senior to those of our common stock without any further vote or action by the holders of our common stock. The issuance of a class of preferred stock could decrease the amount of earnings and assets available for distribution to the holders of our common stock or could adversely affect the rights and powers, including voting rights, of such holders. In certain circumstances, such issuance could have the effect of decreasing the market price of our shares or making a change in control of the company more difficult.
Anti-takeover provisions contained in our amended and restated certificate of incorporation and sixth amended and restated bylaws, as well as provisions of Delaware law, could impair a takeover attempt.
Our amended and restated certificate of incorporation and sixth amended and restated bylaws contain provisions that may discourage unsolicited takeover proposals that stockholders may consider to be in their best interests. We are also subject to anti-takeover provisions under Delaware law, which could delay or prevent a change of control. Together, these provisions may make more difficult the removal of management and may discourage transactions that otherwise could involve payment of a premium over prevailing market prices for our securities. These provisions include:
29
Our sixth amended and restated bylaws require, to the fullest extent permitted by law, that derivative actions brought in our name, actions against our directors, officers, other employees or stockholders for breach of fiduciary duty and other similar actions may be brought only in the Court of Chancery in the State of Delaware and, if brought outside of Delaware, the stockholder bringing the suit will be deemed to have consented to service of process on such stockholder’s counsel, which may have the effect of discouraging lawsuits against our directors, officers, other employees or stockholders.
Our sixth amended and restated bylaws provide that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware will, to the fullest extent permitted by law, be the sole and exclusive forum for any stockholder to bring (i) any derivative action or proceeding brought on our behalf, (ii) any action asserting a claim of breach of a fiduciary duty owed by any director, officer or other employee of ours to us or our stockholders, (iii) any action asserting a claim pursuant to any provision of the Delaware General Corporation Law, or (iv) any action asserting a claim governed by the internal affairs doctrine, and, if brought outside of Delaware, the stockholder bringing the suit will be deemed to have consented to service of process on such stockholder’s counsel, provided, however, that, in each case, if the Court of Chancery does not have jurisdiction, the forum for such action shall be another state court located within the State of Delaware or, if no state court located within the State of Delaware has jurisdiction, the federal district court for the District of Delaware, in all cases subject to the court having personal jurisdiction over the indispensable parties named as defendants therein.
Any person or entity purchasing or otherwise acquiring or holding any interest in our shares of capital stock shall be deemed to have notice of and consented to such provisions.
Notwithstanding the foregoing, the forum selection provision of our sixth amended and restated bylaws will not apply to suits brought to enforce any liability or duty created by the federal securities laws or any other claim for which the federal district courts of the U.S. of America shall be the sole and exclusive forum.
This choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or any of our directors, officers, other employees or stockholders, which may discourage lawsuits with respect to such claims. Alternatively, if a court were to find the choice of forum provision contained in our sixth amended and restated bylaws to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could harm our business, operating results and financial condition.
Our executive officers and directors may sell shares of their stock, and these sales could adversely affect our stock price.
Sales of our stock by our executive officers and directors, or the perception that such sales may occur, could cause the market price of our common stock to decline or could make it more difficult for us to raise funds through the sale of equity in the future, either as part, or outside, of trading plans under Rule 10b5-1 under the Securities Exchange Act of 1934, as amended (the Exchange Act).
Item 1B. Unresolved Staff Comments
None.
Item 1C. Cybersecurity
We recognize the importance cybersecurity has to the success of our business, as well as recognize the need to continually assess cybersecurity risks and evolve our responses in the face of a rapidly and ever-changing environment. Accordingly, we aim to protect our business operations, records and information against known and evolving cybersecurity threats.
Risk Management and Strategy
We have established policies and processes for assessing, identifying, and managing material risk from cybersecurity threats, and have integrated these processes into our overall risk management systems and processes. We routinely assess material risks from cybersecurity threats, including any potential unauthorized occurrence on or conducted through our information systems that may result in adverse effects on the confidentiality, integrity, or availability of our information systems or any information residing within these systems.
30
We conduct periodic risk assessments to identify cybersecurity threats, as well as assessments in the event of a material change in our business practices that may affect information systems that are vulnerable to such cybersecurity threats. These risk assessments include identification of reasonably foreseeable internal and external risks, the likelihood and potential damage that could result from such risks, and the sufficiency of existing policies, procedures, systems, and safeguards in place to manage such risks.
Following these risk assessments, we re-design, implement, and maintain reasonable safeguards to minimize identified risks, reasonably address any identified gaps in existing safeguards, and regularly monitor the effectiveness of our safeguards. Primary responsibility for assessing, monitoring and managing our cybersecurity risks rests with the Vice President of Information Technology who reports to our Acting Chief Executive Officer, Chief Financial Officer to manage the risk assessment and mitigation process.
As part of our overall risk management system, we monitor and test our safeguards and train our employees on these safeguards, in collaboration with our Information Technology department. Personnel at all levels and departments are made aware of our cybersecurity policies through trainings and internal communications.
If required, we engage consultants, or other third parties in connection with our risk assessment processes. These service providers, where appropriate, assist us in the assessment, testing or other aspects of our security controls to help identify material cybersecurity risks to our critical systems, information, products, services, and our broader enterprise IT environment, as well as assist us in designing and implementing our cybersecurity policies and procedures.
We have not encountered cybersecurity challenges that have materially impaired our operations or financial standing. For additional information regarding risks from cybersecurity threats, please refer to Item 1A, “Risk Factors,” in this annual report on Form 10-K.
Cybersecurity Governance
Our Board considers cybersecurity risk as part of its risk oversight function and has delegated oversight of cybersecurity and other information technology risks to the Audit Committee. The Audit Committee oversees management’s implementation of our cybersecurity risk management program and is responsible for monitoring and assessing strategic risk exposure, while our management team is responsible for the day-to-day operations over the material risks we face. Our management team, including our Acting Chief Executive Officer, Chief Financial Officer and Vice President of Information Technology, provide periodic briefings to the Audit Committee regarding our cybersecurity risks and activities, including any recent cybersecurity incidents and related responses, if applicable.
The Audit Committee receives annual reports from management on our cybersecurity risks. In addition, management updates the Audit Committee, as necessary, regarding any material cybersecurity incidents, as well as any incidents with lesser impact potential.
The Audit Committee reports to the full Board regarding its activities, including those related to cybersecurity. The full Board also receives briefings from management on our cyber risk management program, in the discretion of the Board and management. Board members may receive presentations on cybersecurity topics from external experts as part of the Board’s continuing education on topics that impact public companies.
Our Acting Chief Executive Officer, CFO and Vice President of Information Technology, are responsible for assessing and managing our material risks from cybersecurity threats and have decades of experience in overseeing operations, including information technology functions, in the public company environment. The team has primary responsibility for our overall cybersecurity risk management program and supervises our retained external cybersecurity consultants as needed.
Our management team supervises cybersecurity risk management efforts to prevent, detect, mitigate, and remediate cybersecurity risks and incidents through various means, which may include briefings from external consultants engaged by us; threat intelligence and other information obtained from governmental, public or private sources; and alerts and reports produced by security tools deployed in the IT environment. The cybersecurity risk management program also includes tools and activities to prevent, detect, and analyze current and emerging cybersecurity threats, and plans and strategies to address threats and incidents.
Item 2. Properties
We occupy approximately 45,100 square feet of office space in San Diego, California under a lease that expires September 30, 2024.
Item 3. Legal Proceedings
We are not currently party to a material legal proceeding that we believe will have a material adverse effect on our business or financial conditions.
31
Item 4. Mine Safety Disclosures
Not applicable.
PART II
Item 5. Market for the Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Our common stock is listed on the Nasdaq Capital Market under the symbol MEIP.
Holders
As of September 13, 2024, there were 6,662,857 shares of our common stock outstanding and 321 holders of record of our common stock. This number was derived from our stockholder records and does not include beneficial owners of our common stock whose shares are held in the name of various dealers, clearing agencies, banks, brokers and other fiduciaries.
For a discussion of outstanding warrants and other securities exercisable for or convertible into shares of our common stock, see Note 10 - Stockholders' Equity and Note 11 - Share-based Compensation under Item 8 Consolidated Financial Statements and Supplementary Data in this Annual Report.
Dividends
On November 6, 2023, pursuant to the Cooperation Agreement, the Board declared a special cash dividend of $1.75 per share of common stock to stockholders of record at the close of business on November 17, 2023. The total dividend of $11.7 million was paid on December 6, 2023, and was recorded as a reduction of additional paid-in capital in the consolidated statements of stockholders' equity, as we have an accumulated deficit, rather than retained earnings. We do not anticipate paying additional cash dividends in the foreseeable future other than in connection with a strategic transaction which requires it, and currently intend to retain all available funds and future earnings, if any, to support operations. Any future determination related to our dividend policy will be made at the discretion of our board of directors.
Securities authorized for issuance under equity compensation plans
The table below shows, as of June 30, 2024, information for equity compensation plans previously approved by stockholders and for compensation plans not previously approved by stockholders.
Plan Category |
|
Number of securities |
|
|
Weighted-average |
|
|
Number of securities |
|
|||
Equity compensation plans approved by security |
|
|
1,270,950 |
|
|
$ |
32.50 |
|
|
|
465,633 |
|
Equity compensation plans not approved by security |
|
|
86,263 |
|
|
|
18.25 |
|
|
|
130,737 |
|
Total |
|
|
1,357,213 |
|
|
$ |
31.60 |
|
|
|
596,370 |
|
32
Item 6. [Reserved]
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
The following discussion and analysis should be read in conjunction with Item 8. Consolidated Financial Statements and Supplementary Data included below in this Annual Report. Operating results are not necessarily indicative of results that may occur in future periods.
This discussion and analysis contains forward-looking statements that involve a number of risks, uncertainties and assumptions. Actual results may differ materially from those anticipated in the forward-looking statements as a result of many factors including, but not limited to, those set forth under Cautionary Statement About Forward-Looking Statements and Risk Factors in Item 1A. Risk Factors included above in this Annual Report. All forward-looking statements included in this Annual Report are based on the information available to us as of the time we file this Annual Report, and except as required by law, we undertake no obligation to update publicly or revise any forward-looking statements.
Overview
MEI Pharma, Inc. (Nasdaq: MEIP) is a pharmaceutical company that has been developing novel and differentiated cancer therapies. We built our pipeline by acquiring promising cancer agents and creating value in programs through clinical development, strategic partnerships, and out-licensing or commercialization, as appropriate. Our approach to oncology drug development has been to evaluate our drug candidates in combinations with standard-of-care therapies to overcome known resistance mechanisms and address clear medical needs to provide improved patient benefit.
Clinical Development Programs
Our drug candidate pipeline includes voruciclib, an oral cyclin-dependent kinase 9 (CDK9) inhibitor and ME-344, an intravenous small molecule mitochondrial inhibitor targeting the oxidative phosphorylation pathway.
For a more complete discussion of our business, see the section of this Annual Report Item 1. Business above.
Recent Developments
Notification of Strategic Alternatives Evaluation
On July 22, 2024, we announced that our Board had determined unanimously to begin the evaluation of our strategic alternatives, including potential transactions as well as an orderly wind down of operations, if appropriate, to maximize the value of our assets for our stockholders. We commenced a reduction-in-force beginning August 1, 2024, which will continue in stages as our operational and strategic direction evolves. We have discontinued the clinical development of voruciclib, while certain nonclinical activities related to MEI’s drug candidate assets will continue to be conducted by us. As part of the review of strategic alternatives, we may consider options such as out-licensing opportunities for existing programs and merger and acquisition opportunities. Consistent with our intention to preserve cash, David M. Urso, our President and Chief Executive Officer, and Richard Ghalie, MD, our Chief Medical Officer, have stepped down effective August 1, 2024. Mr. Urso also left the Board at that date. We have entered into consulting agreements with both Mr. Urso and Dr. Ghalie under which they will remain available to assist us in our strategic efforts. Charles V. Baltic III, the Chairperson of the Board, also stepped down from the Board contemporaneously with the announcement on July 22, 2024. Our Board has appointed Justin J. File, our current Chief Financial Officer, to assume the position of Acting Chief Executive Officer and has appointed Frederick W. Driscoll as Chairperson of the Board.
Cooperation Agreement
On October 31, 2023, we announced our entry into a Cooperation Agreement (Cooperation Agreement) with Anson Funds Management LP and Cable Car Capital LLC (Anson and Cable Car, respectively), which, among other non-financial related items provided for a capital return to stockholders in the form of a dividend in the amount of $1.75 per share of common stock, as further discussed below. Additionally, the Cooperation Agreement contemplated a potential second return of capital not to exceed $9.33 million (Potential Second Return of Capital) if authorized by Board should our ongoing ME-344 Phase 1b trial fail to meet certain defined endpoints or our Board determines not to proceed with a second cohort.
In April 2024, the Board unanimously determined not to proceed with the Potential Second Return of Capital under the Cooperation Agreement in order to conserve resources and align strategic investment, and thereby extend our operational runway.
As part of the Cooperation Agreement, Anson and Cable Car withdrew their consent solicitation and agreed to abide by customary standstill provisions. Additionally, we reimbursed Anson's and Cable Car’s fees and expenses related to their engagement
33
with us as of the date of the Cooperation Agreement in an amount of $1.1 million, which is recorded within general and administrative expenses in the consolidated statements of operations for the fiscal year ended June 30, 2024.
Cash Dividend
On November 6, 2023, pursuant to the Cooperation Agreement, the Board declared a special cash dividend of $1.75 per share of common stock to stockholders of record at the close of business on November 17, 2023. The total dividend of $11.7 million was paid on December 6, 2023, and was recorded as a reduction of additional paid-in capital in the consolidated statements of stockholders' equity, as we have an accumulated deficit, rather than retained earnings.
Equity Transactions
Shelf Registration Statement
We have a shelf registration statement (February 2024 Shelf Registration Statement) that permits us to sell, from time to time, up to $100.0 million of common stock, preferred stock, warrants rights and units subject to the "Baby Shelf Limitation" described below. The February 2024 Shelf Registration Statement was filed February 20, 2024 and declared effective February 28, 2024.
At-The-Market Equity Offering
On February 20, 2024, we entered into a capital on demand sales agreement with JonesTrading Institutional Services LLC, pursuant to which we can offer and sell shares having an aggregate offering price of up to $25.0 million (ATM Program). In no event will we sell securities registered on this registration statement in a public primary offering with a value exceeding more than one-third of our public float in any 12-month period so long as our public float remains below $75 million (Baby Shelf Limitation). As of January 2, 2024, the date used under applicable rules of the Securities and Exchange Commission to determine our public float at the commencement of the offering, one-third of our public float was equal to approximately $9.9 million. As of June 30, 2024, no shares have been issued and sold under our ATM Program.
Rights Agreement
On October 1, 2023, our Board approved and adopted a rights agreement (Rights Agreement) by and between us and Computershare, Inc., as Rights Agent (as defined in the Rights Agreement). Pursuant to the Rights Agreement, the Board declared a dividend of one preferred share purchase right (each, a Right) for each outstanding share of our common stock, par value $0.00000002 (each, a Common Share and collectively, the Common Shares). The Rights are distributable to stockholders of record as of the close of business on October 12, 2023. One Right also will be issued together with each Common Share issued by us after October 12, 2023, but before the Distribution Date (as defined in the Rights Agreement) (or the earlier of the redemption or expiration of the Rights) and, in certain circumstances, after the Distribution Date.
Warrants
In May 2023, outstanding warrants to purchase 802,949 shares of our common stock expired. The warrants were fully vested and exercisable at a price of $50.80 per share. Pursuant to the terms of the warrants, we could have been required to settle the warrants in cash in the event of an acquisition of the company and, as a result, the warrants were required, prior to their expiration, to be measured at fair value and reported as a liability in the consolidated balance sheets. As of June 30, 2022, the warrants were valued at $1.6 million. Prior to their expiration, the warrants had been revalued to $0, as of December 31, 2022. All corresponding changes in fair value were recorded as a component of other income (expense) in our consolidated statements of operations. The warrants expired in May 2023.
As of June 30, 2024, we have outstanding warrants to purchase 102,513 shares of our common stock, all issued to Torreya Partners LLC in fiscal year 2023. The warrants are fully vested, exercisable at a price of $6.80 per share and expire in October 2027. No warrants were exercised during the years June 30, 2024 and 2023.
Critical Accounting Estimates
Critical accounting policies are those most important to the portrayal of our financial condition and results of operations and require management’s difficult, subjective, or complex judgment, often as a result of the need to make estimates about the effect of matters that are inherently uncertain and may change in subsequent periods. Certain accounting estimates are particularly sensitive because of their significance to financial statements and because of the possibility future events affecting the estimate may differ significantly from management’s current judgments. We believe the following critical accounting policies involve the most significant estimates and judgments used in the preparation of our consolidated financial statements.
Except as provided below, there have been no material changes from the critical accounting estimates identified below nor our significant accounting policies set forth in Note 2. Summary of Significant Accounting Policies.
34
Impairment of Long-Lived Assets (Property and Equipment, and Intangible Assets)
In accordance with the authoritative guidance for impairment or disposal of long-lived assets Accounting Standards Codification (ASC) Topic 360, Property, Plant and Equipment (ASC 360), we assess potential impairments to our long-lived assets, including property, equipment and right-of-use assets, when there is evidence that events or changes in circumstances indicate that the carrying value may not be recoverable. We recognize an impairment loss when the undiscounted cash flows expected to be generated by an asset (or group of assets) are less than the asset’s carrying value. Any required impairment loss would be measured as the amount by which the asset’s carrying value exceeds its fair value and would be recorded as a reduction in the carrying value of the related asset and charged to results of operations. Assumptions and estimates used in evaluating our long-lived assets future values and remaining useful lives are complex and often subjective. They can be affected by a variety of factors, including external factors such as industry and economic trends, and internal factors such as changes in our business strategy, internal forecasts and clinical trial results. For example, if we experience a sustained decline in our market capitalization determined to be indicative of a reduction in fair value of our enterprise, we may be required to record future impairment charges for our acquired technology intangible assets with finite lives.
Impairment charges could materially decrease our future net income and result in lower asset values on our balance sheet. Key assumptions include, but are not limited to, future cash flows, operating margins, capital expenditures, terminal growth rates and discount rates. We also consider our market capitalization as a part of our analysis. During the fiscal year ended June 30, 2024, we recorded long-lived asset impairment charges of $10.9 million. During the fiscal year ended June 30, 2023, we had no similar charge. For additional details regarding our intangible assets and related impairments see Note 3—Balance Sheet Details and Note 9—Leases, to our consolidated financial statements and related notes included elsewhere in this Annual Report.
Revenue
We apply the five-step revenue recognition model within the scope of ASC Topic 606, Revenue from Contracts with Customers (ASC 606). Under this model, we: (i) identify the contract, (ii) identify the performance obligations in the contract, (iii) determine the transaction price, (iv) allocate the transaction price to the performance obligations in the contract, and (v) recognize revenue when, or as, a company satisfies a performance obligation. A performance obligation is a promise in a contract to transfer a distinct good or service and is the unit of accounting in ASC 606. A contract’s transaction price is allocated among each distinct performance obligation based on relative standalone selling price and recognized as revenue when, or as, the applicable performance obligation is satisfied.
The terms of our arrangements include upfront and license fees, research and development services, milestone and other contingent payments for the achievement of defined objectives and certain preclinical, clinical, regulatory and sales-based events, as well as royalties on sales of commercialized products. Agreements with certain upfront payments may require deferral of revenue recognition to a future period until we perform the obligations under these agreements. We use the most likely amount method to estimate variable consideration for event-based milestones and other contingent payments. Given the high degree of uncertainty around the occurrence of such events, the event-based milestones and other contingent payments have been fully constrained until any uncertainty associated with these payments is resolved. Revenue from sales-based milestones and royalty payments is recognized at the later of when or as the sales occur or when the related performance obligation has been satisfied or partially satisfied. We continue to re-evaluate the transaction price in each reporting period as contingencies are resolved and other changes in circumstances occur.
Revenue recognition is subject to uncertainty due to the variable consideration estimates required to be made. These estimates include the level of effort required to satisfy our obligations under our research and development services arrangements. These amounts are estimated at the inception of the services arrangement and are re-evaluated at each reporting period. To accomplish this, we rely on management’s experience, relevant internal data reports and regulatory approvals. The recorded variable consideration is directly sensitive to the estimated inputs made by management used in the calculation. Changes in estimates are accounted for prospectively.
In response to the discontinuance of zandelisib development with KKC during the fiscal year ended June 30, 2023, we updated our estimated costs to complete each of the performance obligations, which resulted in a higher progress towards completion based on the ratio of costs incurred to date to the total estimated costs and a corresponding decrease in our deferred revenue. Additionally, we recognized revenue related to non-refundable payments for performance obligations that have not commenced and will no longer be initiated. During fiscal year 2024, in regard to the KKC Commercialization Agreement, all deferred revenue had been recognized and all wind-down activities were completed.
Research and Development Costs
Research and development costs are expensed as incurred and include costs paid to third-party contractors to perform research, conduct clinical trials and develop and manufacture drug materials. Clinical trial costs, including costs associated with third-party contractors, are a significant component of research and development expenses and we expense research and development costs based on work performed. In determining the amount to expense, management relies on estimates of total costs based on contract components completed, the enrollment of subjects, the completion of trials, and other events. Costs incurred related to the purchase or
35
licensing of in-process research and development for early-stage products or products that are not commercially viable and ready for use, or have no alternative future use, are charged to expense in the period incurred.
As part of the process of preparing the consolidated financial statements, we are required to estimate expenses resulting from obligations under contracts with vendors, clinical research organizations (CROs), consultants and under clinical site agreements relating to conducting clinical trials. The financial terms of these contracts vary and may result in payment flows that do not match the periods over which materials or services are provided under such contracts.
Our objective is to reflect the appropriate clinical trial expenses in our consolidated financial statements by recording those expenses in the period in which services are performed and efforts are expended. We account for these expenses according to the progress of the clinical trial as measured by patient progression and the timing of various aspects of the trial. Management determines accrual estimates through financial models and discussions with applicable personnel and outside service providers as to the progress of clinical trials.
During a clinical trial, we adjust the clinical expense recognition if actual results differ from our estimates. We make estimates of accrued expenses as of each balance sheet date based on the facts and circumstances known at that time. Our clinical trial accruals are partially dependent upon accurate reporting by CROs and other third-party vendors. Our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in changes in our estimates.
Results of Operations
Comparison of Fiscal Years Ended June 30, 2024 and 2023
The following table summarizes certain components of our results of operations (in thousands):
|
|
For the Fiscal Year Ended June 30, |
|
|
|
|
||||||||||
|
|
2024 |
|
|
2023 |
|
|
$ Change |
|
|
% Change |
|
||||
Revenues |
|
$ |
65,297 |
|
|
$ |
48,816 |
|
|
$ |
16,481 |
|
|
|
33.8 |
% |
Research and development |
|
|
16,561 |
|
|
|
52,450 |
|
|
|
(35,889 |
) |
|
|
(68.4 |
)% |
General and administrative |
|
|
23,295 |
|
|
|
33,130 |
|
|
|
(9,835 |
) |
|
|
(29.7 |
)% |
Impairment of long-lived assets |
|
|
10,899 |
|
|
|
— |
|
|
|
10,899 |
|
|
|
100.0 |
% |
Other income, net |
|
|
3,236 |
|
|
|
4,926 |
|
|
|
(1,690 |
) |
|
|
(34.3 |
)% |
Revenue: We recognized revenue of $65.3 million for the fiscal year ended June 30, 2024, compared to $48.8 million for the fiscal year ended June 30, 2023. The increase in revenue was due to the recognition of all remaining deferred revenue associated with the KKC Commercialization Agreement that was terminated in July 2023, offset by a decrease in revenue recognized related to cost sharing from the terminated KKC Commercialization Agreement.
Research and Development: The following table illustrates the components of our research and development expenses for the years presented (in thousands):
|
|
For the Fiscal Year Ended June 30, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
zandelisib |
|
$ |
435 |
|
|
$ |
25,900 |
|
voruciclib |
|
|
3,413 |
|
|
|
2,335 |
|
ME-344 |
|
|
4,724 |
|
|
|
1,137 |
|
Other |
|
|
7,989 |
|
|
|
23,078 |
|
Total research and development expenses |
|
$ |
16,561 |
|
|
$ |
52,450 |
|
Costs related to zandelisib decreased $25.5 million primarily as a result of the discontinuation of the program during fiscal year 2023 with lower costs in fiscal year 2024 associated with wind-down activities. Costs related to voruciclib increased $1.1 million due to higher clinical costs partially offset by lower manufacturing costs. Costs related to ME-344 increased $3.6 million due to higher clinical costs related to the Phase 1b study and manufacturing costs of ME-344 to support clinical and nonclinical studies. Other research and development costs decreased $15.1 million primarily due to a decrease of $14.1 million in personnel costs, including a $2.6 million decrease in one-time employee termination benefits, resulting from our reductions in workforce during fiscal year 2023 and a $0.4 million decrease in noncash stock-based compensation.
General and Administrative: General and administrative expenses decreased $9.8 million to $23.3 million for the fiscal year ended June 30, 2024, compared to $33.1 million for the fiscal year ended June 30, 2023. The net decrease was primarily due to $5.5 million less in personnel costs, which includes $1.7 million resulting from our reduction-in-force and other termination
36
related benefits, as well as $0.9 million less in noncash stock-based compensation, a $1.7 million decrease in external professional services, $1.5 million decrease in corporate overhead costs and a $0.2 million decrease in legal fees.
Impairment of Long-lived Assets: The impairment of our long-lived assets of $10.9 million consists of a $10.4 million loss recognized for our right-of-use (ROU) asset, as more fully described in Note 9. Leases, and a $0.5 million loss recognized related to the furniture and fixtures we agreed to sell to our landlord, as more fully described in Note 3 - Balance Sheet Details, recorded in accordance with Accounting Standards Codification 360 - Property. Plant and Equipment. During the fiscal year ended June 30, 2023, there were no similar transactions.
Other Income, Net: Other income, net, decreased by $1.7 million to $3.2 million for the fiscal year ended June 30, 2024, as compared to $4.9 million for the fiscal year ended June 30, 2023. We recorded a noncash gain of $1.6 million during the fiscal year ended June 30, 2023, due to a change in the fair value of our warrant liability. The warrants expired in May 2023.
New Accounting Pronouncements
See Note 2. Summary of Significant Accounting Policies, to the Consolidated Financial Statements included in Item 8. Consolidated Financial Statements and Supplementary Data of this Annual Report.
Liquidity and Capital Resources
We have accumulated losses of $388.2 million since inception and expect to incur operating losses and generate negative cash flows from operations for the foreseeable future. As of June 30, 2024, we had $38.3 million in cash, cash equivalents and short-term investments. On July 22, 2024, we announced that our Board had determined unanimously to begin the evaluation of our strategic alternatives, including potential transactions as well as an orderly wind down of operations, if appropriate, to maximize the value of our assets for our stockholders. In connection with the exploration of strategic alternatives, we commenced a reduction-in-force on August 1, 2024 and discontinued the clinical development of voruciclib. As a result of this announcement, we expect our research and development expenses to decrease significantly as we discontinued our clinical research and development activities. We will continue to incur research and development expenses in connection with clinical trial closing costs and the completion of certain ongoing nonclinical activities. We believe our cash balance, including our short-term investments, is sufficient to fund operations for at least the next 12 months.
To date, we have obtained cash and funded our operations primarily through equity financings and license agreements and to resume the development of our drug candidates we would require one or more capital transactions, whether through the sale of equity securities, debt financing, license agreements or entry into strategic partnerships at some point in the future. There can be no assurance that we will be able to continue to raise additional capital in the future.
Sources and Uses of Our Cash
Net cash used in operating activities for the fiscal year ended June 30, 2024, of $50.5 million consisted of our net income of $17.8 million and $84.3 million cash used by operating activities partially offset by $16.0 million for noncash items. Net cash used in in operating activities during the fiscal year ended June 30, 2023, of $52.5 million consisted of our net loss of $31.8 million and $24.9 million cash used in operating activities partially offset by $4.3 million of noncash items.
Net cash provided by investing activities for the fiscal year ended June 30, 2024, was $49.1 million compared to $53.7 million for the fiscal year ended June 30, 2023. The decrease in net cash provided by investing activities was due to fewer purchases of short-term investments offset by a lower amount of maturities in short-term investments being utilized to fund ongoing operations during the fiscal year ended June 30, 2024.
Net cash used in financing activities for the fiscal year ended June 30, 2024, was $11.9 million compared to $40,000 of cash used in financing activities for the fiscal year ended June 30, 2023. The increase from prior year was primarily due to the payment of $11.7 million in dividends agreed to under the Cooperation Agreement and the payment of approximately $0.2 million for issuance costs of our ATM. Cash used in financing activities during fiscal year 2023, was associated with the payment of tax withholdings related to vesting of restricted stock units.
Capital Resource Requirements
On June 18, 2024, we entered into a lease termination agreement (Agreement) with our landlord, for our offices at 11455 El Camino Real, Suite 200 and Suite 250, San Diego, California. Under the Agreement, the lease will be terminated as of September 30, 2024, rather than its scheduled expiration date of November 30, 2029. We paid the landlord a termination fee totaling approximately $11.1 million in addition to prepaying the remaining rent under the Agreement in the amount of approximately $0.2 million (collectively, the Termination Amounts). Prior to June 30, 2024, the Termination Amounts had been paid and we had no further financial obligations under the Agreement.
37
As of June 30, 2024, we have the following potential purchase obligations for which the timing and/or likelihood of occurrence is unknown; however, if such claims arise in the future, they could have a material effect on our financial position, results of operations, and cash flows.
Our future capital requirements will depend on many factors, including:
Item 7a. Quantitative and Qualitative Disclosures about Market Risk
As a smaller reporting company, the Company is not required to provide the information otherwise required by this Item.
38
Item 8. Consolidated Financial Statements and Supplementary Data
MEI Pharma, Inc.
Index to Consolidated Financial Statements
|
|
|
|
|
|
|
40 |
|
|
|
|
42 |
|
|
|
|
43 |
|
|
|
|
44 |
|
|
|
|
45 |
|
|
|
|
46 |
|
|
|
|
47 |
|
39
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the shareholders and the Board of Directors of MEI Pharma, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheet of MEI Pharma, Inc. and subsidiary (the "Company") as of June 30, 2024, the related consolidated statements of operations, stockholders' equity, and cash flows, for the year ended June 30, 2024, and the related notes (collectively referred to as the "financial statements"). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of June 30, 2024, and the results of its operations and its cash flows for the year ended June 30, 2024, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current-period audit of the financial statements that was communicated or required to be communicated to the audit committee and that (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Leases – Amendment of Lease - Refer to Note 1, Note 2, and Note 9 to the Financial Statements
Critical Audit Matter Description
As discussed in Note 1, Note 2, and Note 9 to the consolidated financial statements, on June 18, 2024, the Company agreed to terminate its existing lease agreement with AAT Torrey Plaza, LLC effective September 30, 2024 (“Agreement”), which was originally set to expire on November 30, 2029. In consideration for the early termination, the Company agreed to pay a termination fee of approximately $11.1 million and prepay the remaining monthly rent. The Company determined the Agreement should be accounted for as a modification of an existing lease
The principal consideration for our determination that performing procedures related to the lease modification constitute a critical audit matter is that there was a moderate degree of auditor judgment and subjectivity in evaluating the determination if the contract results in a lease modification or termination.
40
How the Critical Audit Matter Was Addressed in the Audit
Our audit procedures related to the accounting treatment and remeasurement of the lease liability and ROU asset included the following, among others:
/s/ Deloitte & Touche LLP
San Diego, California
September 19, 2024
We have served as the Company’s auditor since 2024.
41
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Stockholders and Board of Directors
MEI Pharma, Inc.
San Diego, California
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheet of MEI Pharma, Inc. (the Company) as of June 30, 2023, the related consolidated statements of operations, stockholders’ equity, and cash flows for the year then ended, and the related notes (collectively referred to as the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at June 30, 2023, and the results of its operations and its cash flows for the year then ended, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audit provides a reasonable basis for our opinion.
/s/ BDO USA, P.C.
We have served as the Company’s auditor from 2011 to 2023.
San Diego, California
September 26, 2023
42
MEI PHARMA, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except par value amounts)
|
|
June 30, |
|
|
June 30, |
|
||
|
|
2024 |
|
|
2023 |
|
||
|
|
|
|
|
|
|
||
ASSETS |
|
|
|
|
|
|
||
Current assets: |
|
|
|
|
|
|
||
Cash and cash equivalents |
|
$ |
|
|
$ |
|
||
Short-term investments |
|
|
|
|
|
|
||
Unbilled receivables |
|
|
|
|
|
|
||
Prepaid expenses and other current assets |
|
|
|
|
|
|
||
Total current assets |
|
|
|
|
|
|
||
Operating lease right-of-use asset |
|
|
|
|
|
|
||
Property and equipment, net |
|
|
|
|
|
|
||
Total assets |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
LIABILITIES AND STOCKHOLDERS’ EQUITY |
|
|
|
|
|
|
||
Current liabilities: |
|
|
|
|
|
|
||
Accounts payable |
|
$ |
|
|
$ |
|
||
Accrued liabilities |
|
|
|
|
|
|
||
Deferred revenue |
|
|
|
|
|
|
||
Operating lease liability |
|
|
|
|
|
|
||
Total current liabilities |
|
|
|
|
|
|
||
Deferred revenue, long-term |
|
|
|
|
|
|
||
Operating lease liability, long-term |
|
|
|
|
|
|
||
Total liabilities |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
Stockholders’ equity: |
|
|
|
|
|
|
||
Preferred stock, $ |
|
|
|
|
|
|
||
Common stock, $ |
|
|
|
|
|
|
||
Additional paid-in capital |
|
|
|
|
|
|
||
Accumulated deficit |
|
|
( |
) |
|
|
( |
) |
Total stockholders’ equity |
|
|
|
|
|
|
||
Total liabilities and stockholders’ equity |
|
$ |
|
|
$ |
|
||
See accompanying notes to consolidated financial statements.
43
MEI PHARMA, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share amounts)
|
|
For the Fiscal Year Ended June 30, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Revenues: |
|
|
|
|
|
|
||
Revenue from customers |
|
$ |
|
|
$ |
|
||
Revenue from collaboration agreements |
|
|
|
|
|
|
||
Total revenues |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Operating expenses: |
|
|
|
|
|
|
||
Research and development |
|
|
|
|
|
|
||
General and administrative |
|
|
|
|
|
|
||
Impairment of long-lived assets |
|
|
|
|
|
|
||
Total operating expenses |
|
|
|
|
|
|
||
Income (loss) from operations |
|
|
|
|
|
( |
) |
|
Other income (expense): |
|
|
|
|
|
|
||
Change in fair value of warrant liability |
|
|
|
|
|
|
||
Interest and dividend income |
|
|
|
|
|
|
||
Other expense, net |
|
|
( |
) |
|
|
( |
) |
Total other income, net |
|
|
|
|
|
|
||
Net income (loss) |
|
$ |
|
|
$ |
( |
) |
|
|
|
|
|
|
|
|
||
Net income (loss) per share - basic and diluted |
|
$ |
|
|
$ |
( |
) |
|
|
|
|
|
|
|
|
||
Weighted-average shares used in computing net income (loss) |
|
|
|
|
|
|
||
See accompanying notes to consolidated financial statements.
44
MEI PHARMA, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands)
|
|
Common |
|
|
Additional |
|
|
Accumulated |
|
|
Total |
|
||||
Balance at June 30, 2022 |
|
|
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
Net loss |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Issuance of common stock for vested restricted stock |
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
Issuance of warrants |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Share-based compensation expense |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Balance at June 30, 2023 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
Net income |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
Cash dividends declared ($ |
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
Share-based compensation expense |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Balance at June 30, 2024 |
|
|
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
See accompanying notes to consolidated financial statements.
45
MEI PHARMA, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
|
|
For the Fiscal Year Ended June 30, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Cash flows from operating activities: |
|
|
|
|
|
|
||
Net income (loss) |
|
$ |
|
|
$ |
( |
) |
|
Adjustments to reconcile net income (loss) to net cash used in operating activities: |
|
|
|
|
|
|
||
Change in fair value of warrant liability |
|
|
|
|
|
( |
) |
|
Share-based compensation |
|
|
|
|
|
|
||
Issuance of warrants |
|
|
|
|
|
|
||
Impairment of long-lived assets |
|
|
|
|
|
|
||
Noncash lease expense |
|
|
|
|
|
|
||
Depreciation expense |
|
|
|
|
|
|
||
Loss on disposal of property and equipment |
|
|
|
|
|
|
||
Changes in operating assets and liabilities: |
|
|
|
|
|
|
||
Unbilled receivables |
|
|
|
|
|
|
||
Prepaid expenses and other current assets |
|
|
|
|
|
( |
) |
|
Accounts payable |
|
|
( |
) |
|
|
( |
) |
Accrued liabilities |
|
|
( |
) |
|
|
|
|
Deferred revenue |
|
|
( |
) |
|
|
( |
) |
Operating lease liability |
|
|
( |
) |
|
|
( |
) |
Net cash used in operating activities |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
||
Cash flows from investing activities: |
|
|
|
|
|
|
||
Purchases of short-term investments |
|
|
( |
) |
|
|
( |
) |
Proceeds from maturity of short-term investments |
|
|
|
|
|
|
||
Purchases of property and equipment |
|
|
( |
) |
|
|
( |
) |
Net cash provided by investing activities |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Cash flows from financing activities: |
|
|
|
|
|
|
||
Payments of tax withholdings related to vesting of restricted stock units |
|
|
|
|
|
( |
) |
|
Payment of cash dividend |
|
|
( |
) |
|
|
|
|
Payment of financing costs |
|
|
( |
) |
|
|
|
|
Net cash used in financing activities |
|
|
( |
) |
|
|
( |
) |
Net (decrease) increase in cash and cash equivalents |
|
|
( |
) |
|
|
|
|
Cash and cash equivalents at beginning of the year |
|
|
|
|
|
|
||
Cash and cash equivalents at end of the year |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
Supplemental cash flow information: |
|
|
|
|
|
|
||
Operating lease right-of-use assets obtained in exchange for operating |
|
$ |
|
|
$ |
|
||
Re-measurement of right-of-use asset and related lease liability upon lease |
|
$ |
( |
) |
|
$ |
|
|
Re-measurement of right-of-use asset for direct costs associated with lease |
|
$ |
|
|
$ |
|
||
See accompanying notes to consolidated financial statements.
46
MEI PHARMA, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1. Description of Business and Basis of Presentation
Description of Business
MEI Pharma, Inc. (Nasdaq: MEIP) is a pharmaceutical company that has been developing novel and differentiated cancer therapies. We built our pipeline by acquiring promising cancer agents and creating value in programs through development, strategic partnerships, and out-licensing or commercialization, as appropriate. Our approach to oncology drug development has been to evaluate our drug candidates in combinations with standard-of-care therapies to overcome known resistance mechanisms and address clear medical needs to provide improved patient benefit. Our pipeline includes voruciclib, an oral cyclin-dependent kinase 9 (CDK9) inhibitor, and ME-344, an intravenous small molecule mitochondrial inhibitor targeting the oxidative phosphorylation pathway.
Basis of Presentation and Consolidation
We prepared the consolidated financial statements in accordance with accounting principles generally accepted in the United States (GAAP) and the rules and regulations of the Securities and Exchange Commission (SEC) related to annual reports on Form 10‑K. The accompanying consolidated financial statements include the accounts of MEI Pharma, Inc. and our wholly owned subsidiary, Meadow Merger Sub, Inc. We have eliminated all intercompany accounts and transactions in consolidation.
The Company has evaluated subsequent events through the date the consolidated financial statements were issued.
Current Events
Strategic Alternatives
On July 22, 2024, we announced that our Board of Directors (Board) had determined unanimously to begin the evaluation of our strategic alternatives, including potential transactions as well as an orderly wind down of operations, if appropriate, to maximize the value of our assets for our stockholders. We commenced a reduction-in-force beginning August 1, 2024, which will continue in stages as our operational and strategic direction evolves. We have discontinued the clinical development of voruciclib, while certain nonclinical activities related to MEI’s drug candidate assets will continue to be conducted by us. As part of the review of strategic alternatives, we may consider options such as out-licensing opportunities for existing programs and merger and acquisition opportunities.
Consistent with our intention to preserve cash, David M. Urso, our President and Chief Executive Officer, and Richard Ghalie, M.D., our Chief Medical Officer, stepped down effective August 1, 2024. Mr. Urso also left the Board at that date. We have entered into consulting agreements with both Mr. Urso and Dr. Ghalie under which they will remain available to assist us in our strategic efforts. Charles V. Baltic III, the Chairperson of the Board, also stepped down from the Board contemporaneously with the announcement on July 22, 2024. Our Board has appointed Justin J. File, our current Chief Financial Officer, to assume the position of Acting Chief Executive Officer and has appointed Frederick W. Driscoll as Chairperson of the Board.
Cooperation Agreement
On October 31, 2023, we announced our entry into a Cooperation Agreement (Cooperation Agreement) with Anson Funds Management LP and Cable Car Capital LLC (Anson and Cable Car, respectively), which, among other non-financial related items provided for a capital return to stockholders in the form of a dividend in the amount of $
As part of the Cooperation Agreement, Anson and Cable Car withdrew their consent solicitation and agreed to abide by customary standstill provisions. Additionally, we reimbursed Anson's and Cable Car’s fees and expenses related to their engagement with us as of the date of the Cooperation Agreement in an amount of $
In April 2024, the Board unanimously determined not to proceed with the Potential Second Return of Capital under the Cooperation Agreement in order to conserve resources and align strategic investment, and thereby extend our operational runway.
Cash Dividend
On November 6, 2023, pursuant to the Cooperation Agreement, the Board declared a special cash dividend of $
47
Infinity Merger
In February 2023, we, Infinity Pharmaceuticals, Inc. (Infinity), and Meadow Merger Sub, Inc., our wholly owned subsidiary (Merger Sub) entered into an agreement and plan of merger (Merger Agreement). The Merger Agreement provided that Merger Sub will merge with and into Infinity, with Infinity being the surviving entity as a wholly owned subsidiary of us (transaction referred to as the Merger) and was subject to approvals by our and Infinity’s stockholders, respectively. On July 23, 2023, we convened our Special Meeting of Stockholders at which time our stockholders did not approve the proposed transaction and subsequently, we delivered a letter to Infinity which terminated the Merger Agreement pursuant to Section 7.2(c) of the Merger Agreement, effective July 23, 2023.
KKC Termination Agreement
In November 2022, we and Kyowa Kirin Co., Ltd. (KKC) met with the U.S. Food and Drug Administration (FDA) in a follow-up meeting to the March 2022 end of Phase 2 meeting related to zandelisib. At this meeting, the FDA provided further guidance regarding the design and statistical analysis for the COASTAL trial. Following the November meeting, the companies jointly concluded that a clinical trial consistent with the recent FDA guidance, including modification of the ongoing COASTAL trial, would likely not be feasible to complete within a time period that would support further investment or with sufficient certainty of the regulatory requirements for approval to justify continued global development efforts. As a result, we and KKC jointly decided to discontinue global development of zandelisib for indolent forms of non-Hodgkin lymphoma outside of Japan.
The discontinuation of zandelisib development outside of Japan was a business decision based on the regulatory guidance from the FDA and was not related to the zandelisib clinical data generated to date. Although KKC continued certain ongoing Japanese clinical trials at that time, in May 2023 KKC decided to discontinue development of zandelisib in Japan after meeting with the Pharmaceuticals and Medical Devices Agency (PMDA), concluding that conducting a randomized study consistent with agency guidance to support a marketing application would likely not be feasible to complete within a time period that would support further investment.
Due to KKC’s decision to discontinue development of zandelisib in Japan, as well as the prior joint decision to discontinue zandelisib development outside of Japan, on July 14, 2023, the parties entered into a Termination Agreement to mutually terminate the global License, Development and Commercialization Agreement executed in April 2020.
Liquidity
We have accumulated losses of $
To date, we have obtained cash and funded our operations primarily through equity financings and license agreements and to continue the development of our drug candidates, we would require one or more capital transactions, whether through the sale of equity securities, debt financing, license agreements or entry into strategic partnerships at some point in the future. There can be no assurance that we will be able to continue to raise additional capital in the future.
Note 2. Summary of Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and disclosures made in the accompanying notes to the consolidated financial statements. We use estimates that affect the reported amounts (including assets, liabilities, revenues and expenses) and related disclosures. Actual results could materially differ from those estimates.
48
Cash and Cash Equivalents
Cash and cash equivalents consist of cash and highly liquid investments with original maturities of three months or less when purchased. Cash is maintained at financial institutions and, at times, balances may exceed federally insured limits. We have not experienced any losses related to these balances.
We attempt to minimize credit risk associated with our cash and cash equivalents by periodically evaluating the credit quality of our primary financial institutions. Our investment portfolio is maintained in accordance with our investment policy, which is designed to preserve capital, safeguard funds and limit exposure to risk. While we maintain cash deposits in FDIC insured financial institutions in excess of federally insured limits, we do not believe we are exposed to significant credit risk due to the financial position of the depository institutions in which those deposits are held. We have not experienced any losses on such accounts.
Short-term Investments
Short-term investments are marketable securities with maturities greater than three months but less than one year from date of purchase. As of June 30, 2024 and 2023, our short-term investments consisted of $
Fair Value Measurements
Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. The fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value is as follows:
Property and Equipment
Property and equipment are stated at cost and depreciated over the estimated useful lives of the assets (generally to
Leases
We account for our leases under ASC Topic 842, Leases (Topic 842). Leases which are identified within the scope of Topic 842 and which have a term greater than one year are recognized on our consolidated balance sheets as right-of-use (ROU) assets and lease liabilities. Operating lease liabilities and their corresponding ROU assets are recorded based on the present value of lease payments over the expected remaining lease term. The lease term includes any renewal options and termination options that we are reasonably certain to exercise. Certain adjustments to the right-of-use asset may be required for items such as initial direct costs paid or incentives received. The present value of lease payments is determined by using the interest rate implicit in the lease, if that rate is readily determinable; otherwise, we use our incremental borrowing rate. The incremental borrowing rate is determined based on the rate of interest that we would pay to borrow on a collateralized basis an amount equal to the lease payments for a similar term and in a similar economic environment. The interest rate implicit in lease contracts to calculate the present value is typically not readily determinable. As such, significant management judgment is required to estimate the incremental borrowing rate.
Operating lease expense for operating leases is recognized on a straight-line basis over the lease term based on the total lease payments. We have elected the practical expedient to not separate lease and non-lease components for our real estate leases. Our non-lease components are primarily related to property maintenance, which varies based on future outcomes, and thus is recognized in operating lease expense when incurred.
49
On June 18, 2024, we entered into a lease termination agreement (Agreement) with our landlord pursuant to which the parties agreed to terminate, as of September 30, 2024, the lease for our existing office space. See Note 3. Balance Sheet Details and Note 9. Leases for the impact of the Agreement on our consolidated financial statements.
Revenue Recognition
Revenue from Customers
In accordance with ASC Topic 606, Revenue from Contracts with Customers (Topic 606), we recognize revenue when control of the promised goods or services is transferred to our customers, in an amount that reflects the consideration we expect to be entitled to in exchange for those goods or services. For enforceable contracts with our customers, we first identify the distinct performance obligations – or accounting units – within the contract. Performance obligations are commitments in a contract to transfer a distinct good or service to the customer.
Payments received under commercial arrangements, such as licensing technology rights, may include non-refundable fees at the inception of the arrangements, milestone payments for specific achievements designated in the agreements, and royalties on the sale of products. At the inception of arrangements that include milestone payments, we use judgment to evaluate whether the milestones are probable of being achieved, and we estimate the amount to include in the transaction price using the most likely method. If it is probable that a significant revenue reversal will not occur, the estimated amount is included in the transaction price. Milestone payments that are not within our or the licensee’s control, such as regulatory approvals, are not included in the transaction price until those approvals are received. At the end of each reporting period, we re-evaluate the probability of achievement of development milestones and any related constraint and, as necessary, we adjust our estimate of the overall transaction price.
We may enter into arrangements that consist of multiple performance obligations. Such arrangements may include any combination of our deliverables. To the extent a contract includes multiple promised deliverables, we apply judgment to determine whether promised deliverables are capable of being distinct and are distinct in the context of the contract. If these criteria are not met, the promised deliverables are accounted for as a combined performance obligation. For arrangements with multiple distinct performance obligations, we allocate variable consideration related to our 50-50 cost share for development services directly to the associated performance obligation and then allocate the remaining consideration among the performance obligations based on their relative stand-alone selling price.
Stand-alone selling price is the price at which we would sell a promised good or service separately to the customer. When not directly observable, we typically estimate the stand-alone selling price for each distinct performance obligation. Variable consideration that relates specifically to our efforts to satisfy specific performance obligations is allocated entirely to those performance obligations. Other components of the transaction price are allocated based on the relative stand-alone selling price, over which management has applied significant judgment. We develop assumptions that require judgment to determine the stand-alone selling price for license-related performance obligations, which may include forecasted revenues, development timelines, reimbursement rates for personnel costs, discount rates and probabilities of technical, regulatory and commercial success. We estimate stand-alone selling price for research and development performance obligations by forecasting the expected costs of satisfying a performance obligation plus an appropriate margin.
In the case of a license that is a distinct performance obligation, we recognize revenue allocated to the license from non-refundable, up-front fees at the point in time when the license is transferred to the licensee and the licensee can use and benefit from the license. For licenses that are bundled with other distinct or combined obligations, we use judgment to assess the nature of the performance obligation to determine whether the performance obligation is satisfied over time or at a point in time and, if over time, the appropriate method of measuring progress for purposes of recognizing revenue. If the performance obligation is satisfied over time, we evaluate the measure of progress each reporting period and, if necessary, adjust the measure of performance and related revenue recognition. From time to time, we perform additional services for KKC, at their request, the costs of which are fully reimbursed to us. The cost of these services is recognized in the consolidated statements of operations as research and development expense.
The selection of the method to measure progress towards completion requires judgment and is based on the nature of the products or services to be provided. Revenue is recorded proportionally as costs are incurred. We generally use the cost-to-cost measure of progress because it best depicts the transfer of control to the customer which occurs as we incur costs. Under the cost-to-cost measure of progress, the extent of progress towards completion is measured based on the ratio of costs incurred to date to the total estimated costs at completion of the performance obligation (an input method under Topic 606). We use judgment to estimate the total cost expected to complete the research and development performance obligations, which include subcontractors’ costs, labor, materials, other direct costs and an allocation of indirect costs. We evaluate these cost estimates and the progress each reporting period and, as necessary, we adjust the measure of progress and related revenue recognition.
50
For arrangements that include sales-based or usage-based royalties, we recognize revenue at the later of (i) when the related sales occur or (ii) when the performance obligation to which some or all of the royalty has been allocated has been satisfied or partially satisfied. To date, we have not recognized any sales-based or usage-based royalty revenue from license agreements.
In connection with our License, Development and Commercialization Agreement (the KKC Commercialization Agreement) with KKC, we performed development services related to our 50-50 cost sharing arrangement for which revenue was recognized over time. Additionally, we performed services for KKC at their request, the costs of which were fully reimbursed to us. We recorded the reimbursement for such pass through services as revenue at
We recognized revenue associated with the KKC Commercialization Agreement for the periods presented (in thousands):
|
|
For the Fiscal Year Ended June 30, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Timing of Revenue Recognition: |
|
|
|
|
|
|
||
Services performed over time |
|
$ |
|
|
$ |
|
||
Pass through services at a point in time |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
Contract Balances
Accounts receivable are included in prepaid expenses and other current assets, and contract liabilities are included in deferred revenue and deferred revenue, long-term in our consolidated balance sheets. Our contract liabilities accounted for under Topic 606 relate to the amount of initial upfront consideration allocated to the development services performance obligations.
As of June 30, 2024 and 2023, we had
|
|
June 30, 2024 |
|
|
June 30, 2023 |
|
|
June 30, 2022 |
|
|||
Unbilled receivables |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Contract liabilities included in deferred revenue and |
|
$ |
|
|
$ |
|
|
$ |
|
|||
A reconciliation of the beginning and ending amount of contract liabilities, which are primarily related to the combined performance obligation for the transfer of development services under the KKC Commercialization Agreement and are a separate performance obligation in our contracts pursuant to research plans under the agreements, was as follows for the periods presented:
|
|
June 30, 2024 |
|
|
June 30, 2023 |
|
||
Beginning balance |
|
$ |
|
|
$ |
|
||
Recognized as revenue: |
|
|
|
|
|
|
||
Revenue recognized upon satisfaction of performance obligations |
|
|
( |
) |
|
|
( |
) |
Revenue recognized from change in estimate for performance |
|
|
|
|
|
( |
) |
|
Revenue recognized for performance obligations that will no longer |
|
|
|
|
|
( |
) |
|
Ending balance |
|
$ |
|
|
$ |
|
||
The timing of revenue recognition, invoicing and cash collections results in billed accounts receivable and unbilled receivables (contract assets) and deferred revenue (contract liabilities). We invoice our customers in accordance with agreed-upon contractual terms, typically at periodic intervals or upon achievement of contractual milestones. Invoicing may occur subsequent to revenue recognition, resulting in unbilled receivables. We may receive advance payments from our customers before revenue is recognized, resulting in contract liabilities. The unbilled receivables and deferred revenue reported on the consolidated balance sheets relate to the KKC Commercialization Agreement.
Revenue from Collaborators
At contract inception, we assess whether the collaboration arrangements are within the scope of Topic 808, to determine whether such arrangements involve joint operating activities performed by parties that are both active participants in the activities and exposed to significant risks and rewards dependent on the commercial success of such activities. This assessment is performed based on the responsibilities of all parties in the arrangement. For collaboration arrangements within the scope of Topic 808 that contain multiple units of account, we first determine which units of account within the arrangement are within the scope of Topic 808 and which elements are within the scope of Topic 606. For units of account within collaboration arrangements that are accounted for pursuant to Topic 808, an appropriate recognition method is determined and applied consistently, by analogy to authoritative
51
accounting literature. For elements of collaboration arrangements that are accounted for pursuant to Topic 606, we recognize revenue as discussed above. Consideration received that does not meet the requirements to satisfy Topic 606 revenue recognition criteria is recorded as deferred revenue in the accompanying consolidated balance sheets, classified as either current or long-term deferred revenue based on our best estimate of when such amounts will be recognized.
Research and Development
Research and development costs are expensed as incurred and include costs paid to third-party contractors to perform research, conduct clinical trials and develop and manufacture drug materials. Clinical trial costs, including costs associated with third-party contractors, are a significant component of research and development expenses. We expense research and development costs based on work performed. In determining the amount to expense, management relies on estimates of total costs based on contract components completed, the enrollment of subjects, the completion of trials, and other events. Costs incurred related to the purchase or licensing of in-process research and development for early-stage products or products that are not commercially viable and ready for use, or have no alternative future use, are charged to expense in the period incurred.
As part of the process of preparing the consolidated financial statements, we are required to estimate expenses resulting from obligations under contracts with vendors, clinical research organizations (CROs), consultants and under clinical site agreements relating to conducting clinical trials. The financial terms of these contracts vary and may result in payment flows that do not match the periods over which materials or services are provided under such contracts.
Our objective is to reflect the appropriate clinical trial expenses in our consolidated financial statements by recording those expenses in the period in which services are performed and efforts are expended. We account for these expenses according to the progress of the clinical trial as measured by patient progression and the timing of various aspects of the trial. Management determines accrual estimates through financial models and discussions with applicable personnel and outside service providers as to the progress of clinical trials.
During a clinical trial, we adjust the clinical expense recognition if actual results differ from our estimates. We make estimates of accrued expenses as of each balance sheet date based on the facts and circumstances known at that time. Our clinical trial accruals are partially dependent upon accurate reporting by CROs and other third-party vendors. Our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in changes in our estimates.
Share-based Compensation
Share-based compensation expense for stock options and restricted stock units (RSUs) granted to employees and directors is recognized in the consolidated statements of operations based on estimated amounts. The cost of stock options is measured at the grant date, based on the estimated fair value of the stock option using the Black-Scholes valuation model, which incorporates various assumptions including expected volatility, risk-free interest rate, the expected term of the award and the dividend yield on the underlying stock. Expected volatility is calculated based on the historical volatility of our stock over the expected option life and other appropriate factors. We use the simplified method to calculate the expected term of share options and similar instruments as we do not have sufficient historical exercise data to provide a reasonable basis upon which to estimate the expected term. Risk-free interest rates are calculated based on continuously compounded risk-free rates for the appropriate term. The dividend yield is assumed to be zero as we do not intend to do so in the foreseeable future. For RSUs, we estimate the grant date fair value using our closing stock price on the date of grant. The estimated fair value of stock options and RSUs is amortized over the requisite service period, adjusted for actual forfeitures at the time they occur. The requisite service period is generally the time over which our share-based awards vest.
Interest and Dividend Income
Interest on cash and investment balances is recognized when earned. Dividend income is recognized when the right to receive the payment is established.
Income Taxes
Our income tax expense consists of current and deferred income tax expense or benefit. Current income tax expense or benefit is the amount of income taxes expected to be payable or refundable for the current year. A deferred income tax asset or liability is recognized for the future tax consequences attributable to tax credits and loss carryforwards and to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets are reduced by a valuation allowance when, in the opinion of management, it is more likely than not that some portion or all of the deferred tax assets will not be realized. As of June 30, 2024 and 2023, we have established a valuation allowance to fully reserve our net deferred tax assets. Tax rate changes are reflected in income during the period such changes are enacted. Changes in our ownership may limit the amount of net operating loss carryforwards that can be utilized in the future to offset taxable income.
52
The Financial Accounting Standards Board (FASB) Topic on income taxes prescribes a recognition threshold and measurement attribute criteria for the financial statement recognition and measurement of tax positions taken or expected to be taken in a tax return. For those benefits to be recognized, a tax position must be more-likely-than-not to be sustained upon examination by taxing authorities.
Net Income (Loss) Per Share
Basic and diluted net income (loss) per share is computed using the weighted-average number of shares of common stock outstanding during the period, less any shares subject to repurchase or forfeiture. There were
For purposes of the diluted net income (loss) per share calculation for the fiscal years ended June 30, 2024 and 2023, potentially dilutive securities are excluded from the calculation of diluted net income (loss) per share because their weighted-average exercise prices were above our weighted-average share price for the fiscal years ended June 30, 2024 and 2023, respectively; therefore, basic and diluted net income (loss) per share were the same for the fiscal years ended June 30, 2024 and 2023.
The following table presents potentially dilutive shares that have been excluded from the calculation of net income (loss) per share because of their anti-dilutive effect (in thousands):
|
|
For the Fiscal Year Ended June 30, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Stock options |
|
|
|
|
|
|
||
Warrants |
|
|
|
|
|
|
||
Total anti-dilutive shares |
|
|
|
|
|
|
||
Segment Information
We have
Recent Accounting Pronouncement
Recently Adopted
In June 2016, the FASB issued Accounting Standards Update No. 2016-13, Financial Instruments–Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments (ASU 2016-13), as amended. The amendments in ASU 2016-13 require, among other things, financial assets measured at amortized cost basis to be presented at the net amount expected to be collected as compared to previous U.S. GAAP which delayed recognition until it was probable a loss had been incurred. The amendments in ASU 2016-13 are effective for fiscal years beginning after December 15, 2022, including interim periods within those fiscal years, with early adoption permitted. The adoption of ASU 2016-13 did not have a material impact on our consolidated financial statements and related disclosures.
Recently Issued
From time to time, new accounting pronouncements are issued by the FASB or other standards setting bodies that are adopted as of the specified effective date. We believe the impact of recently issued standards, other than those noted below, and any issued but not yet effective standards will not have a material impact on our consolidated financial statements upon adoption.
In November 2023, the FASB issued ASU No. 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures, which requires a public entity to disclose significant segment expenses and other segment items on an annual and interim basis and provide in interim periods all disclosures about a reportable segment’s profit or loss and assets that are currently required annually. Additionally, it requires a public entity to disclose the title and position of the Chief Operating Decision Maker. This ASU does not change how a public entity identifies its operating segments, aggregates them, or applies the quantitative thresholds to determine its reportable segments. The new standard is effective for fiscal years beginning after December 15, 2023, and interim periods within fiscal years beginning after December 15, 2024, with early adoption permitted. A public entity should apply the amendments in this ASU retrospectively to all prior periods presented in the financial statements. We expect this ASU to only impact our disclosures with no impact to our results of operations, cash flows and financial condition.
53
In December 2023, the FASB issued ASU No. 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures, which focuses on the rate reconciliation and income taxes paid. ASU No. 2023-09 requires a public business entity (PBE) to disclose, on an annual basis, a tabular rate reconciliation using both percentages and currency amounts, broken out into specified categories with certain reconciling items further broken out by nature and jurisdiction to the extent those items exceed a specified threshold. In addition, all entities are required to disclose income taxes paid, net of refunds received disaggregated by federal, state/local, and foreign and by jurisdiction if the amount is at least 5% of total income tax payments, net of refunds received. For PBEs, the new standard is effective for annual periods beginning after December 15, 2024, with early adoption permitted. An entity may apply the amendments in this ASU prospectively by providing the revised disclosures for the period ending December 31, 2025 and continuing to provide the pre-ASU disclosures for the prior periods, or may apply the amendments retrospectively by providing the revised disclosures for all periods presented. We expect this ASU to only impact our disclosures with no impacts to our results of operations, cash flows, and financial condition.
Note 3. Balance Sheet Details
Prepaid and Other Current Assets
Prepaid and other current assets consisted of the following in thousands:
|
|
June 30, 2024 |
|
|
June 30, 2023 |
|
||
Prepaid clinical costs |
|
$ |
|
|
$ |
|
||
Other |
|
|
|
|
|
|
||
Total prepaid and other current assets |
|
$ |
|
|
$ |
|
||
Property and Equipment
Property and equipment consisted of the following, in thousands:
|
|
June 30, 2024 |
|
|
June 30, 2023 |
|
||
Furniture and fixtures |
|
$ |
|
|
$ |
|
||
Equipment |
|
|
|
|
|
|
||
Leasehold improvements |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Less: accumulated depreciation (1) |
|
|
( |
) |
|
|
( |
) |
Property and equipment, net |
|
$ |
|
|
$ |
|
||
_____
(1)
Depreciation expense of property and equipment for the fiscal years ended June 30, 2024 and 2023 are presented in the consolidated statements of cash flows.
Impact of the Agreement (as discussed in Note 9. Leases)
As noted in Note 9. Leases, we agreed to sell our furniture and fixtures to the landlord for $
54
Accrued Liabilities
Accrued liabilities consisted of the following, in thousands:
|
|
|
|
|
|
|
||
|
|
June 30, 2024 |
|
|
June 30, 2023 |
|
||
Accrued pre-clinical and clinical trial expenses |
|
$ |
|
|
$ |
|
||
Accrued compensation and benefits(1) |
|
|
|
|
|
|
||
Accrued legal and professional services |
|
|
|
|
|
|
||
Accrued reimbursement to KKC |
|
|
|
|
|
|
||
Other |
|
|
|
|
|
|
||
Total accrued liabilities |
|
$ |
|
|
$ |
|
||
________
(1)
Note 4. Fair Value Measurements
The carrying amounts of financial instruments such as cash equivalents, short-term investments and accounts payable approximate the related fair values due to the short-term maturities of these instruments. We invest our excess cash in financial instruments which are readily convertible into cash, such as money market funds and U.S. government securities. Cash equivalents and short-term investments are classified as Level 1 as defined by the fair value hierarchy.
In May 2018, we issued warrants in connection with our private placement of shares of common stock, which expired in May 2023. Pursuant to the terms of the warrants, we could have been required to settle the warrants in cash in the event of an acquisition of the company and, as a result, the warrants were required to be measured at fair value and reported as a liability in the consolidated balance sheets. We recorded the fair value of the warrants upon issuance using the Black-Scholes valuation model and are required to revalue the warrants at each reporting date with any changes in fair value recorded on our consolidated statements of operations. The valuation of the warrants is considered under Level 3 of the fair value hierarchy due to the need to use assumptions in the valuation that are both significant to the fair value measurement and unobservable. Inputs used to determine the estimated fair value of the warrant liabilities include the estimated fair value of the underlying stock at the valuation date, the estimated term of the warrants, risk-free interest rates, expected dividends and the expected volatility of the underlying stock. The significant unobservable inputs used in the fair value measurement of the warrant liabilities were the volatility rate and the estimated term of the warrants. Generally, increases or decreases in the fair value of the underlying stock and estimated term would result in a directionally similar impact to the fair value measurement. The changes in the fair value of the Level 3 warrant liability are reflected on the consolidated statements of operations for the fiscal year ended June 30, 2023. During the fiscal year ended June 30, 2023, there were
To calculate the fair value of the warrant liability, the following assumptions were used for the period ended June 30, 2023:
Risk-free interest rate |
|
|
% |
|
Expected life (years) |
|
|
|
|
Expected volatility |
|
|
% |
|
Dividend yield |
|
|
% |
|
Weighted-average grant date fair value |
|
$ |
|
The following table sets forth a summary of changes in the estimated fair value of our Level 3 warrant liability for the fiscal year ended June 30, 2023 (in thousands):
Balance as of June 30, 2022 |
|
$ |
|
|
Change in estimated fair value of liability classified warrants |
|
|
( |
) |
Balance as of June 30, 2023 |
|
$ |
|
Note 5. One-time Employee Termination Benefits
In connection with our joint decision to discontinue development of zandelisib outside of Japan, in December 2022, we announced a realignment of our clinical development efforts that streamlined our organization towards the continued clinical development of our two earlier clinical-stage assets, voruciclib and ME-344. As a result, our Board approved a staggered workforce reduction (the Reduction-in-Force). For the fiscal year ended June 30, 2023, we recorded one-time employee benefits of $
55
and development expense and general and administrative expense, respectively, associated with the termination of
In August 2024, in connection with our Strategic Alternatives announcement, described in Note 1. Description of Business and Basis of Presentation, we commenced a Reduction-in-Force (the 2024 Plan). Including management's contractual 2025 bonuses, we expect to incur charges not to exceed a total of $
The following table summarizes our activity related to one-time employee termination benefits, excluding one-time employee termination benefits recorded subsequent to June 30, 2024, included in accrued liabilities (in thousands):
|
|
One-time Employee Termination Benefits |
|
|
Balance at June 30, 2022 |
|
$ |
|
|
Accrued restructuring |
|
|
|
|
Cash payments |
|
|
( |
) |
Balance at June 30, 2023 |
|
|
|
|
Increase in accrued restructuring |
|
|
|
|
Cash payments |
|
|
( |
) |
Balance at June 30, 2024 |
|
$ |
|
|
Note 6. Commitments and Contingencies
We have contracted with various consultants and third parties to assist us in pre-clinical research and development and clinical trials work for our leading drug compounds. The contracts are terminable at any time, but obligate us to reimburse the providers for any time or costs incurred through the date of termination. We also have employment agreements with certain of our current employees that provide for severance payments and accelerated vesting for share-based awards if their employment is terminated under specified circumstances.
Litigation
From time to time, we may be involved in various lawsuits, legal proceedings, or claims that arise in the ordinary course of business. Management believes there are no claims or actions pending against us as of June 30, 2024 which will have, individually or in the aggregate, a material effect on its business, liquidity, financial position, or results of operations. Litigation, however, is subject to inherent uncertainties, and an adverse result in these or other matters may arise from time to time that may harm our business.
Indemnification
In accordance with our amended and restated memorandum and articles of association, we have indemnification obligations to our officers and directors for certain events or occurrences, subject to certain limits, while they are serving in such capacity. There have been no claims to date, and we have a directors and officers liability insurance policy that may enable it to recover a portion of any amounts paid for future claims.
Presage License Agreement
As discussed in Note 8. Other License Agreements, we are party to a license agreement with Presage under which we may be required to make future payments upon the achievement of certain development, regulatory and commercial milestones, as well as potential future royalties based upon net sales. As of June 30, 2024, we had
Note 7. License Agreements
KKC License, Development and Commercialization Agreement
In April 2020, we entered into the KKC Commercialization Agreement under which we granted to KKC a co-exclusive, sublicensable, payment-bearing license under certain patents and know-how controlled by us to develop and commercialize zandelisib and any pharmaceutical product containing zandelisib for all human indications in the U.S. (the U.S. License), and an exclusive (subject to certain retained rights to perform obligations under the KKC Commercialization Agreement), sublicensable, payment-bearing, license under certain patents and know-how controlled by us to develop and commercialize zandelisib and any
56
pharmaceutical product containing zandelisib for all human indications in countries outside of the U.S. (the Ex-U.S. and the Ex-U.S. License). KKC granted to us a co-exclusive, sublicensable, license under certain patents and know-how controlled by KKC to develop and commercialize zandelisib for all human indications in the U.S., and a co-exclusive, sublicensable, royalty-free, fully paid license under certain patents and know-how controlled by KKC to perform our obligations in the Ex-U.S. under the KKC Commercialization Agreement. KKC paid us an initial payment of $
Prior to the execution of the Termination Agreement, KKC was responsible for the development and commercialization of zandelisib in Ex-U.S. and, subject to certain exceptions, was solely responsible for all costs related thereto. We also provided to KKC certain drug supplies necessary for the development and commercialization of zandelisib in the Ex-U.S., with the understanding that KKC would have assumed responsibility for manufacturing for the Ex-U.S. as soon as practicable.
We assessed the KKC Commercialization Agreement in accordance with Topic 808 and Topic 606 and determined that our obligations comprise the U.S. License, the Ex-U.S. License, and development services (the Development Services). We determined that the KKC Commercialization Agreement is a collaborative arrangement in accordance with Topic 808 that contains multiple units of account, as we and KKC are both active participants in the development and commercialization activities and are exposed to significant risks and rewards that are dependent on commercial success of the activities of the arrangement. The U.S. License is a unit of account under the scope of Topic 808 and is not a deliverable under Topic 606, while the Ex-U.S. License and Development Services performance obligations are under the scope of Topic 606.
As discussed in Note 1. Description of Business and Basis of Presentation, we and KKC jointly decided to discontinue zandelisib development in the U.S. During fiscal year 2023, we updated our assessment of the total transaction price from the KKC Commercialization Agreement to be $
We allocated the transaction price to each unit of account. Variable consideration that relates specifically to our efforts to satisfy specific performance obligations are allocated entirely to those performance obligations. Other components of the transaction price are allocated based on the relative stand-alone selling price, over which management has applied significant judgment. We developed the estimated stand-alone selling price for the licenses using the risk-adjusted net present values of estimated cash flows, and the estimated stand-alone selling price of the development services performance obligations by estimating costs to be incurred, and an appropriate margin, using an income approach.
The $
Note 8. Other License Agreements
Presage License Agreement
In September 2017, we, as licensee, entered into a license agreement with Presage Biosciences, Inc. (Presage). Under the terms of the license agreement, Presage granted to us exclusive worldwide rights to develop, manufacture and commercialize voruciclib, a clinical-stage, oral and selective CDK inhibitor, and related compounds. In exchange, we paid $
57
BeiGene Collaboration
In October 2018, we entered into a clinical collaboration with BeiGene, Ltd. (BeiGene) to evaluate the safety and efficacy of zandelisib in combination with BeiGene’s zanubrutinib (marketed as Brukinsa®), an inhibitor of Bruton’s tyrosine kinase, for the treatment of patients with B-cell malignancies. Under the terms of the clinical collaboration agreement, we amended our ongoing Phase 1b trial to include evaluation of zandelisib in combination with zanubrutinib in patients with B-cell malignancies. Study costs are being shared equally by the parties, and we agreed to supply zandelisib and BeiGene agreed to supply zanubrutinib. We record the costs reimbursed by BeiGene as a reduction of our research and development expenses. We retained full commercial rights for zandelisib and BeiGene retained full commercial rights for zanubrutinib. With the discontinuation of the zandelisib program outside of Japan, this clinical collaboration was terminated on September 28, 2023. Cost reimbursements recorded as a reduction of research and development costs, in the consolidated statements of operations, during the fiscal years ended June 30, 2024 and 2023 were approximately $
Note 9. Leases
In July 2020, we entered into a lease agreement for approximately
Lease Termination
On June 18, 2024 (the Agreement Date), we entered into a lease termination agreement (Agreement) with our landlord pursuant to which the parties agreed to terminate, as of September 30, 2024, the lease for our existing office space. The original (as amended) scheduled expiration date was
The Agreement was accounted for as a lease modification of the original contract. As a result of the Agreement, we reduced both the remaining ROU asset and lease liability by approximately $
We incurred direct costs of approximately $
The total operating lease costs for the Amended Lease were as follows for the periods presented (in thousands):
|
|
For the Fiscal Year Ended June 30, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Operating lease cost |
|
$ |
|
|
$ |
|
||
Variable lease costs |
|
|
|
|
|
|
||
Total lease costs included in operating expenses |
|
$ |
|
|
$ |
|
||
58
Supplemental cash flow information related to our operating leases was as follows for the periods presented (in thousands):
|
|
For the Fiscal Year Ended June 30, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Cash paid for amount included in the measurement |
|
|
|
|
|
|
||
Operating cash flows from operating leases |
|
$ |
|
|
$ |
|
||
As of June 30, 2024, we have
Note 10. Stockholders’ Equity
Equity Transactions
Shelf Registration Statement
We have a shelf registration statement (February 2024 Self Registration Statement) that permits us to sell, from time to time, up to $
At-The-Market Equity Offering
On February 20, 2024, we entered into a capital on demand sales agreement with JonesTrading Institution Services LLC, pursuant to which we can offer and sell shares having an aggregate offering price of up to $
Rights Agreement
On
Warrants
In May 2023, outstanding warrants to purchase
As of June 30, 2024, we have outstanding warrants to purchase
Description of Capital Stock
Our total authorized share capital is
59
Common Stock
The holders of common stock are entitled to
Preferred Stock
Our Board has the authority to issue up to
Note 11. Share-based Compensation
We use equity-based compensation programs to provide long-term performance incentives for our employees. These incentives consist primarily of stock options and RSUs. In December 2008, we adopted the MEI Pharma, Inc. 2008 Stock Omnibus Equity Compensation Plan (the Omnibus Plan), as amended and restated from time to time, under which
In May 2021, we adopted the 2021 Inducement Plan (Inducement Plan), under which
Total share-based compensation expense for all stock awards consists of the following, in thousands:
|
|
For the Fiscal Year Ended June 30, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Research and development |
|
$ |
|
|
$ |
|
||
General and administrative |
|
|
|
|
|
|
||
Total share-based compensation |
|
$ |
|
|
$ |
|
||
Stock Options
Stock options granted to employees vest
60
A summary of our stock option activity and related data follows:
|
|
Number of |
|
|
Weighted-Average |
|
|
Weighted-Average |
|
|
Aggregate |
|
||||
Outstanding at June 30, 2023 |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
Granted |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
Forfeited |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
Outstanding at June 30, 2024 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
Vested and expected to vest at June 30, 2024 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
As of June 30, 2024, the aggregate intrinsic value of outstanding options is calculated as the difference between the exercise price of the underlying options and the closing price of our common stock of $
Unrecognized compensation expense related to non-vested stock options totaled $
We use a Black-Scholes valuation model to estimate the grant date fair value of stock options. To calculate these fair values, the
|
|
For the Fiscal Year Ended June 30, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Risk-free interest rate |
|
|
% |
|
|
% |
||
Expected life (years) |
|
|
|
|
|
|
||
Volatility |
|
|
% |
|
|
% |
||
Dividend yield |
|
|
% |
|
|
% |
||
Weighted-average grant date fair value |
|
$ |
|
|
$ |
|
||
Restricted Stock Units (RSU)
Each RSU represents the contingent right to receive
Note 12. Income Taxes
Pre-tax loss consists of the following jurisdictions (in thousands):
|
|
For the Fiscal Year Ended June 30, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Domestic |
|
$ |
|
|
$ |
( |
) |
|
Foreign |
|
|
|
|
|
|
||
Pre-tax income (loss) |
|
$ |
|
|
$ |
( |
) |
|
61
The reconciliation of income tax computed at the U.S. federal statutory tax rates to income tax expense is as follows (in thousands):
|
|
For the Fiscal Year Ended June 30, |
|
|||||||||||||
|
|
2024 |
|
|
2023 |
|
||||||||||
|
|
$ |
|
|
% |
|
|
$ |
|
|
% |
|
||||
Tax benefit at U.S. statutory rates |
|
$ |
|
|
|
% |
|
$ |
|
|
|
% |
||||
State tax |
|
|
|
|
|
% |
|
|
|
|
|
% |
||||
Warrant liability costs |
|
|
|
|
|
% |
|
|
|
|
|
% |
||||
Equity compensation |
|
|
|
|
|
% |
|
|
( |
) |
|
|
% |
|||
Change in valuation allowance |
|
|
( |
) |
|
|
( |
)% |
|
|
( |
) |
|
|
( |
)% |
Section 162(m) limitation |
|
|
|
|
|
% |
|
|
|
|
|
% |
||||
Other |
|
|
|
|
|
% |
|
|
( |
) |
|
|
% |
|||
|
|
$ |
|
|
|
% |
|
$ |
|
|
|
% |
||||
Deferred tax liabilities and assets are comprised of the following (in thousands):
|
|
June 30, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Deferred tax assets (liabilities): |
|
|
|
|
|
|
||
Deferred revenue |
|
$ |
|
|
$ |
|
||
Fixed and intangible assets |
|
|
|
|
|
|
||
Capitalization of R&D costs |
|
|
|
|
|
|
||
Share-based payments |
|
|
|
|
|
|
||
Tax losses carried forward |
|
|
|
|
|
|
||
Right-of-use assets |
|
|
( |
) |
|
|
( |
) |
Lease liabilities |
|
|
|
|
|
|
||
Other |
|
|
|
|
|
|
||
Total deferred tax assets |
|
|
|
|
|
|
||
Valuation allowance for deferred tax assets |
|
|
( |
) |
|
|
( |
) |
Net deferred tax assets and liabilities |
|
$ |
|
|
$ |
|
||
We evaluate the recoverability of the deferred tax assets and the amount of the required valuation allowance. Due to the uncertainty surrounding the realization of the tax deductions in future tax returns, we have recorded a valuation allowance against our net deferred tax assets as of June 30, 2024 and 2023. At such time as it is determined that it is more likely than not that the deferred tax assets will be realized, the valuation allowance would be reduced.
We had federal and state net operating loss carryforwards of approximately $
Our ability to utilize our net operating loss carryforwards may be substantially limited due to ownership changes that have occurred or that could occur in the future under Section 382 of the Internal Revenue Code and similar state laws. Through December 31, 2021, we completed a study to analyze whether one or more ownership changes had occurred and determined that two such ownership changes did occur. Although we did not conduct a formal study in 2023, we believe no significant ownership changes occurred subsequent to 2021. While the ownership changes do limit the amount of net operating loss we are able to use each year, all of our net operating losses are expected to be available for utilization prior to expiring.
None of our prior income tax returns have been selected for examination by a major taxing jurisdiction; however, the statutes of limitations for various filings remain open. The oldest filings subject to potential examination for federal and state purposes are 2020 and 2019, respectively. If we utilize a net operating loss related to a closed tax year, the tax year in which the loss was incurred is subject to adjustment up to the amount of the net operating loss.
We have not reduced any tax benefit on our consolidated financial statements due to uncertain tax positions as of June 30, 2024 and we are not aware of any circumstance that would significantly change this result through the end of fiscal year 2024. To the extent we incur income-tax related penalties or interest, we will recognize them as additional income tax expense.
62
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
Disclosure controls and procedures are controls and other procedures of a company that are designed to ensure that information required to be disclosed by the company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officer, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure. As of the end of the period covered by this Annual Report, or June 30, 2024, our management, with the participation of our principal executive officer and principal financial officer, has evaluated the effectiveness of our disclosure controls and procedures as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act as of June 30, 2024. Based on such evaluation, our principal executive officer and principal financial officer have concluded that, as of such date, our disclosure controls and procedures were effective.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act). Internal control over financial reporting is a process designed under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America. Management conducted an assessment of the effectiveness of our internal control over financial reporting based on the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control—Integrated Framework (2013). Based on this assessment, our management concluded that, as of June 30, 2024, our internal control over financial reporting was effective based on those criteria.
This annual report does not include an attestation report of our registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by our registered public accounting firm pursuant to the rules of the Securities and Exchange Commission that permit us to provide only management’s report in this annual report.
Changes in Internal Control over Financial Reporting
There were no changes to our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) that occurred during the quarter ended June 30, 2024, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Inherent Limitations of Internal Controls
Our management, including our Acting Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures or our internal controls over financial reporting will prevent or detect all error and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Due to the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within the company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the controls. The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Over time, controls may become inadequate because of changes in conditions, or the degree of compliance with the policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
Item 9B. Other Information
63
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
Not applicable.
64
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Directors
Set forth below are the names, ages and certain biographical information as of the date of filing of this Annual Report on Form 10-K (Annual Report) regarding our directors.
Name |
|
Age |
|
Positions Held |
|
Expiration of Term |
Nicholas R. Glover, Ph.D. |
|
55 |
|
Director |
|
Fiscal 2025 Annual Meeting of Stockholders |
Frederick W. Driscoll |
|
73 |
|
Director |
|
Fiscal 2025 Annual Meeting of Stockholders |
Thomas C. Reynolds, M.D., Ph.D. |
|
65 |
|
Director |
|
Fiscal 2026 Annual Meeting of Stockholders |
Taheer Datoo |
|
33 |
|
Director |
|
Fiscal 2027 Annual Meeting of Stockholders |
James Flynn |
|
44 |
|
Director |
|
Fiscal 2027 Annual Meeting of Stockholders |
Steven D. Wood |
|
41 |
|
Director |
|
Fiscal 2027 Annual Meeting of Stockholders |
Frederick W. Driscoll, age 73, Chair
Mr. Driscoll has been a director of MEI Pharma since February 2018 and was appointed as chairperson of the board of directors on July 22, 2024. He currently serves on the board of directors of Cue Biopharma, a biotechnology company, Cellectar Biosciences, Inc., a clinical-stage biopharmaceutical company and Calliditas Therapeutics, a commercial-stage biopharmaceutical company. He served as interim Chief Financial Officer at Invivyd, Inc. from September 2022 to May 2023. He served as Chief Financial Officer of Renovacor, Inc., a leading late-stage biotechnology company, from March to June 2022. He served as the Chief Financial Officer of Flexion Therapeutics, Inc., a commercial-stage biopharmaceutical company, from 2013 to 2017 and rejoined in June 2021 as Chief Financial Officer until it was sold to Pacira BioScience. Prior to joining Flexion, he was the Chief Financial Officer at Novavax, Inc. from 2009 to 2013. From 2008 to 2009, Mr. Driscoll served as the Chief Executive Officer at Genelabs Technologies, Inc. and from 2007 to 2008 he served as its Chief Financial Officer. He was also the Chief Executive Officer of OXiGENE, Inc. from 2000 to 2006. Mr. Driscoll also served as the chairman of the board and audit committee chair at OXiGENE and as a member of the audit committee for Cynapsus Therapeutics, Inc. Mr. Driscoll earned a bachelor’s degree in Accounting and Finance from Bentley University.
Nicholas R. Glover, Ph.D., age 55, Director
Dr. Glover has been a director of MEI Pharma since June 2013. He is currently a consultant to the biotech industry. Previously, he served as President and Chief Executive Officer of Sierra Oncology (NASDAQ: SRRA), a drug development company focused on advancing targeted therapeutics for the treatment of patients with cancer, from July 2014 through May 2020. Prior to joining Sierra, he served as President and Chief Executive Officer of YM Biosciences, an oncology drug development company, from November 2010 until its acquisition by Gilead Sciences in February 2013. Previously, Dr. Glover was President and Chief Executive Officer of Viventia Biotech, a biopharmaceutical company involved in the discovery and development of monoclonal antibody-based technologies for the treatment of cancer, which he joined after serving as an investment manager for MDS Capital, a life sciences venture capital firm. Dr. Glover holds a B.Sc. (Hons) in Chemistry from the University of East Anglia, U.K., a M.Sc. in Chemistry from the University of British Columbia, Canada, and a Ph.D. in Chemistry from Simon Fraser University, Canada.
Thomas C. Reynolds, M.D., Ph.D., age 65, Director
Dr. Reynolds has been a director of MEI Pharma since February 2013. He is President of Two Paddles Consulting LLC since December 2013, providing consulting services to biotechnology and pharmaceutical companies. Dr. Reynolds served as an independent director of Trillium Therapeutics Inc. (NASDAQ: TRIL; TSX: TR), an immuno-oncology company, from March 2014 to April 2021. Previously, he served as Chief Medical Officer of Seattle Genetics, a biotechnology company, from March 2007 until his retirement in February 2013. While at Seattle Genetics, he was responsible for building and leading an integrated clinical development, regulatory and medical affairs organization, highlighted by the development and approval of ADCETRIS®. From 2002 to 2007, Dr. Reynolds served at ZymoGenetics (acquired by BMS in 2010), most recently as Vice President, Medical Affairs, where he oversaw the clinical development and regulatory filing of RECOTHROM®. Previously, he was Vice President, Clinical Affairs at Targeted Genetics, and before that was at Somatix Therapy (acquired by Cell Genesys in 1997). Dr. Reynolds received his M.D. and Ph.D. in Biophysics from Stanford University and a B.A. in Chemistry from Dartmouth College.
Taheer Datoo, age 33, Director
Mr. Datoo has been a director of MEI Pharma since October 2023. Mr. Datoo has served in various roles of increasing seniority at Anson Advisors, Inc. (Anson), a hedge fund with a global investment portfolio, since 2016, including most recently as Principal and Portfolio Manager since January 2023, as well as Portfolio Manager from January 2019 to December 2022 and as an Analyst, from April 2016 to December 2019. While employed at Anson, Mr. Datoo has primarily focused on North American smallcap equities,
65
special situations and thematic investing. Prior to joining Anson, Mr. Datoo served as an investment banker for BMO Capital Markets, the capital markets subsidiary of Bank of Montreal (NYSE/TSX: BMO), an international financial services company, from 2013 to 2015. Mr. Datoo earned a B.A. in economics and finance from McGill University.
James Flynn, age 44, Director
Mr. Flynn has been a director of MEI Pharma since October 2023. Mr. Flynn is currently a Managing Member and Portfolio Manager of Nerium Capital LLC, an investment adviser he founded in 2021. Nerium Capital LLC is the General Partner of Nerium Partners LP, a healthcare focused investment partnership. Previously, Mr. Flynn worked as a therapeutics analyst at Aptigon Capital, an investment firm and division of Citadel LLC, a multinational financial services company, from October 2017 to February 2018. Prior to that, Mr. Flynn served in various roles at Amici Capital, LLC, an investment management firm, from 2003 to 2017, including as Healthcare Portfolio Manager, from 2008 to 2017. Earlier in his career, Mr. Flynn worked in the credit research/high yield group at Putnam Investments LLC, an investment management firm, from 2002 to 2003. Mr. Flynn currently serves on the board of directors of ARCA biopharma, Inc. (NASDAQ: ABIO), a clinical-stage biopharmaceutical company, since December 2022, Synlogic, Inc. (NASDAQ: SYBX), a biopharmaceutical company with a focus on rare metabolic disorders, since March 2024, RiceBran Technologies, an innovative specialty ingredients company, since January 2024, and Axiom Health, Inc. (Axiom), a provider of software and big-data solutions to the healthcare industry, since July 2022. He has also served as an advisor to Axiom since August 2020. Mr. Flynn earned a S.B. degree in Management Science with a concentration in Finance and a minor in Economic Science from the Massachusetts Institute of Technology. Mr. Flynn is a Chartered Financial Analyst (CFA) charterholder.
Steven Wood, age 41, Director
Mr. Wood has been a director of MEI Pharma since October 2023. Mr. Wood, CFA, is the founder and Chief Investment Officer of GreenWood Investors, LLC., an investment advisory firm he founded in 2010. Since May 2023, Mr. Wood serves as a board member of Leonardo SpA, an Italian-listed international Aerospace & Defense company, and since May 2019 he serves as a board member of CTT - Correios De Portugal, a publicly listed Iberian logistics company. Prior to founding GreenWood Investors in 2010, Mr. Wood was a research analyst at Carr Securities from 2009 to 2013. Previously, Mr. Wood worked as an investment banking analyst for RBC Capital Markets in the Syndicated and Leveraged Finance group. He began his career with the special situations team at Kellogg Capital Group. Mr. Wood is a Chartered Financial Analyst charterholder. He received a triple degree, Cum Laude, from Tulane University in Economics, Political Economy and International Relations in 2005.
Information about the Board of Directors and its Committees
The Board has responsibility for the overall corporate governance of MEI Pharma. During the fiscal year ended June 30, 2024, a majority of the members of the Board were, and as of the date of this Annual Report are, independent within the meaning of the Nasdaq Stock Market (Nasdaq) rules.
The Board has established an Audit Committee to oversee MEI Pharma’s financial matters, a Compensation Committee to oversee our compensation policies, plans and programs and a Nominating and Governance Committee to assist the Board in nominating board members to be elected by the stockholders at the Annual Meeting, to fill vacancies and newly created directorships, and to evaluate and monitor all matters with respect to governance of MEI Pharma and oversee compliance by MEI Pharma with its legal and regulatory obligations. As of the date of this Annual Report, our schedule of committee members is as follows:
Board Member |
Audit Committee |
Compensation Committee |
Nominating & Governance Committee |
Taheer Datoo* |
|
|
|
Frederick W. Driscoll |
|
|
|
James Flynn |
|
|
|
Nick Glover, PhD. |
|
|
|
Thomas C. Reynolds, MD, PhD. |
|
|
|
Steven D. Wood |
|
|
|
* - Is not an independent member of the Board
 = Committee Member
= Committee Member
 = Committee Chair
= Committee Chair
 = Financial Expert
= Financial Expert
66
Audit Committee
The Audit Committee of the Board has been established in accordance with Section 3(a)(58)(A) of the Securities Exchange Act of 1934, as amended (the Exchange Act). The Audit Committee’s responsibilities include:
Mr. Driscoll has served as Chairman of the Audit Committee since August 29, 2019. The other members of the Audit Committee are Dr. Glover and Mr. Flynn. Mr. Driscoll has been determined by the Board to be an audit committee financial expert as defined by the SEC.
The Board of Directors has determined that each of the Audit Committee members is independent, as defined in accordance with Nasdaq and SEC rules. We have adopted an Audit Committee Charter, which is posted on its website at www.meipharma.com. The Audit Committee met five times during the fiscal year ended June 30, 2024.
Compensation Committee
The Compensation Committee acts on behalf of the Board to fulfill the Board’s responsibilities to:
The Compensation Committee also consults with and considers the recommendations of the chief executive officer with respect to the appropriate level and mix of the various compensation components, focused primarily on the particular goals of applicable executives and employees in a particular year. The Board has adopted a written charter for the Compensation Committee, which is available on our website at www.meipharma.com. Dr. Glover has served as the Chairman of the Compensation Committee since December 16, 2021. The other members of the Compensation Committee are Dr. Reynolds and Mr. Wood. The Board has determined that each member of the Compensation Committee is independent in accordance with applicable Nasdaq and SEC rules. The Compensation Committee met four times during the fiscal year ended June 30, 2024.
67
Nominating and Governance Committee
The Nominating and Governance Committee is responsible for assisting the Board in:
Dr. Reynolds has served as Chairman of the Nominating and Governance Committee since January 2023. The other members of the Nominating and Governance Committee are Mr. Flynn and Mr. Datoo. MEI Pharma’s Nominating and Governance Committee Charter is posted on its website at www.meipharma.com. The Board has determined that Dr. Reynolds and Mr. Flynn are independent members of the Nominating and Governance Committee in accordance with applicable Nasdaq and SEC rules and Mr. Datoo is not considered independent in accordance with applicable Nasdaq and SEC rules. The Nominating Committee met five times during the fiscal year ended June 30, 2024.
Stockholders who would like to propose an independent director candidate for consideration for nomination by the Board at next year’s annual meeting of stockholders may do so by submitting the candidate’s name, resume and biographical information to the attention of Justin J. File, Secretary, MEI Pharma, Inc., 9920 Pacific Heights Blvd, Suite 150, San Diego, California 92121. All stockholder nominations received by the Secretary, which comply with the advance notice provisions of MEI's Amended and Restated Bylaws, will be presented to the Nominating and Governance Committee for the same consideration as individuals identified by the Nominating and Governance Committee through other means.
While we have no minimum qualifications for director nominees, the Nominating and Governance Committee reviews the prospective candidate’s biographical information and assesses each candidate’s independence, diversity, skills and expertise based on a variety of factors, including the following criteria:
Application of these factors requires the exercise of judgment by members of the Nominating and Governance Committee when the Committee makes recommendations to the Board and cannot be measured in a quantitative way. In addition, the Nominating and Governance Committee considers, as one factor among many, the diversity of Board candidates, which may include diversity of skills and experience as well as geographic, gender, age, and ethnic diversity. The Nominating and Governance Committee does not, however, have a formal policy regarding to the consideration of diversity in identifying Board candidates. The Nominating and Governance Committee and the Board generally value the broad business experience and independent business judgment in the health care, life sciences and other fields of each member. Specifically, Mr. Driscoll is qualified for the Board based on his business experience in the pharmaceutical industry, the area of finance and his status as an audit committee financial expert. Dr. Glover is qualified for the Board based on his business experience and his drug development experience in the oncology field. Dr. Reynolds is qualified for the Board based on his medical experience and experience in clinical development and regulatory and medical affairs. Mr. Flynn is qualified for the Board based on his business experience in the pharmaceutical industry and his business development experience. Mr. Datoo is qualified for the Board based on his business development experience. Mr. Wood is qualified for the Board based on his business development experience.
68
In addition, the Nominating and Governance Committee oversees compliance by MEI with its legal and regulatory obligations and periodically reviews our:
Director Independence
The Board has determined the independence of each director in accordance with the elements of independence set forth in the Nasdaq listing standards. Based upon information solicited from each director, the Board has determined that each of Mr. Driscoll, Dr. Glover, Dr. Reynolds, Mr. Flynn and Mr. Wood has no material relationship with MEI Pharma and is independent within the meaning of Nasdaq’s director independence standards as currently in effect. Mr. Datoo is not considered independent within the meaning of Nasdaq’s director independence standards as currently in effect. In making the foregoing determinations, the Board has considered both the objective tests set forth in the Nasdaq independence standards and subjective measures with respect to each director necessary to determine that no relationships exist that would interfere with the exercise of independent judgment by each such director in carrying out responsibilities of a director.
Board Leadership Structure
Mr. Driscoll has served as the Chair of our Board since July 2024. The Board does not have a policy addressing whether the same person should serve as both the Chief Executive Officer and Chair of the Board or if the roles should be separate. Our Board believes that it should have the flexibility to make its determination based upon what it considers to be the appropriate leadership structure for MEI at the time. The Board believes that its current leadership structure is appropriate for MEI at this time.
Board Role in Risk Oversight
Risk is an integral part of the Board and Committee deliberations throughout the year. While the Board has the ultimate oversight responsibility for the risk management process, various committees of the Board also have responsibility for risk management. In particular, the Audit Committee focuses on financial risk, including internal controls, and receives financial risk assessment reports from management. Risks related to the compensation programs are reviewed by the Compensation Committee. The Nominating and Governance Committee exercises oversight of governance risks, including succession planning and legal compliance. The Board is advised by these committees of significant risks and management’s response through periodic updates.
Board Diversity
The table below provides certain information with respect to the composition of our Board. Each of the categories listed in the table has the meaning ascribed to it in Nasdaq Listing Rule 5605(f).
Board Diversity Matrix (As of August 31, 2024) |
||||
Total Number of Directors |
6 |
|||
|
Female |
Male |
Non-Binary |
Did Not Disclose Gender |
Part I: Gender Identity |
|
|
|
|
Directors |
— |
6 |
— |
— |
Part II: Demographic Background |
|
|
|
|
African American or Black |
— |
— |
— |
— |
Alaskan Native or Native American |
— |
— |
— |
— |
Indian or South Asian |
— |
1 |
— |
— |
Hispanic or Latinx |
— |
— |
— |
— |
Native Hawaiian or Pacific Islander |
— |
— |
— |
— |
White |
— |
5 |
— |
— |
Two or More Races or Ethnicities |
— |
— |
— |
— |
LGBTQ+ |
— |
— |
— |
— |
Did Not Disclose Demographic Background |
— |
— |
— |
— |
69
Board Diversity Matrix (As of August 31, 2023) |
||||
Total Number of Directors |
8 |
|||
|
Female |
Male |
Non-Binary |
Did Not Disclose Gender |
Part I: Gender Identity |
|
|
|
|
Directors |
1 |
7 |
— |
— |
Part II: Demographic Background |
|
|
|
|
African American or Black |
— |
— |
— |
— |
Alaskan Native or Native American |
— |
— |
— |
— |
Asian |
— |
1 |
— |
— |
Hispanic or Latinx |
— |
— |
— |
— |
Native Hawaiian or Pacific Islander |
— |
— |
— |
— |
White |
1 |
6 |
— |
— |
Two or More Races or Ethnicities |
— |
— |
— |
— |
LGBTQ+ |
— |
— |
— |
— |
Did Not Disclose Demographic Background |
— |
— |
— |
— |
Anti-Hedging and Pledging Policies
Under our Insider Trading Policy, all directors, officers, employees and consultants of MEI are subject to restrictions on hedging of securities of MEI. These restrictions apply to securities of MEI owned by such persons, regardless of whether such securities were granted by us to such persons as compensatory awards. Our Insider Trading Policy prohibits such persons from engaging in short sales of securities of MEI or in transactions in publicly traded options with respect to our securities. In addition, our Insider Trading Policy permits, but discourages, such persons from holding our securities in a margin account or pledging securities of MEI as collateral for a loan and from entering standing orders with respect to our securities.
Stockholder Communications with the Board of Directors
Our stockholders may communicate with the Board, including non-executive directors or officers, by sending written communications addressed to such person or persons in care of MEI Pharma, Inc., Attention: Secretary, 9920 Pacific Heights Blvd., Suite 150, San Diego, California, 92121. All communications will be compiled by the Secretary and submitted to the addressee. If the Board modifies this process, the revised process will be posted on our website.
Appointment of Directors
Our amended and restated certificate of incorporation and amended and restated by-laws provide that the number of directors will be set by resolution of the board, but shall be between two and nine. We currently have six directors.
Under our amended and restated certificate of incorporation and amended and restated by-laws, directors are to be elected at each annual meeting of stockholders for a term of three years unless the director is removed, retires or the office is vacated earlier. The board is divided into three classes with respect to the term of office, with the terms of office of one class expiring each successive year. This classified board provision could discourage a third-party from making a tender offer for MEI's shares or attempting to obtain control of MEI Pharma. It could also delay stockholders who do not agree with the policies of the Board from removing a majority of the Board for two years.
A director may resign at any time. The resignation is effective upon receipt of notice. Any or all directors may be removed with cause by a resolution of stockholders entitled to vote to elect directors. Vacancies from resignation or removal or expansion of the size of the board may be filled by resolution of a majority of directors then in office or by a sole remaining director, and any director so appointed shall serve for the remainder of the full term of the class of directors in which the vacancy occurred.
Attendance of Directors at Board Meetings and Stockholder Meetings
During the fiscal year ended June 30, 2024, the Board held a total of 24 meetings, and each director attended at least 75% of the total number of meetings of the Board and of the meetings of each committee of the Board on which such director served.
All directors are expected to attend our annual meetings of stockholders. All directors then in the office attended the annual meeting of stockholders held in December 2023.
Code of Ethics
We have adopted a Code of Business and Ethics policy that applies to our directors and employees (including our principal executive officer and our principal financial officer), and have posted the text of our policy on our website (www.meipharma.com), under Investors – Governance Documents. In addition, we intend to promptly disclose (i) the nature of any amendment to the policy that applies to our principal executive officer and principal financial officer and (ii) the nature of any waiver, including an implicit waiver, from a provision of the policy that is granted to one of these specified individuals, the name of such person who is granted the
70
waiver and the date of the waiver on our website in the future. Except as expressly stated herein, information contained on our website is not incorporated by reference herein and shall not be deemed a part of this Annual Report on Form 10-K.
Executive Officers
Our executive officers are appointed by and serve at the discretion of the Board. Set forth below is the name and certain biographical information regarding MEI Pharma’s executive officer as of the date of filing of this Annual Report
Justin J. File, age 54, Acting Chief Executive Officer, Chief Financial Officer and Secretary
Mr. File has been our Acting Chief Executive Officer since August 1, 2024 and our Chief Financial Officer since August 1, 2023. Mr. File has over 30 years of experience in accounting and finance, working in both public and private companies. He has a diverse range of experience, having worked in various industries, including the life sciences industry for the past 17 years. From 2015 to 2023, Mr. File was the Chief Financial Officer and Corporate Secretary of Evofem Biosciences, Inc., a women's health company that developed and commercialized Phexxi®, a nonhormonal contraceptive for women. While at Evofem he helped bring the company public through a reverse merger and was responsible for overseeing corporate finance and accounting, information technology and investor relations. Previously, Mr. File provided executive financial and accounting oversight consulting services to biotechnology companies, and before that led accounting operations and reporting at Sequenom, Inc., a molecular diagnostic company. He additionally served as Treasurer of Sequenom’s diagnostic subsidiary. Before joining industry, Mr. File worked for approximately ten years in public accounting, primarily with Arthur Andersen LLP. Mr. File graduated from Central Washington University with a Bachelor of Science in Accounting and Business Administration. He is a Certified Public Accountant (inactive).
Item 11. Executive Compensation
COMPENSATION DISCUSSION AND ANALYSIS
This Compensation Discussion and Analysis describes the compensation strategy, policies, programs and practices for the named executive officers identified in the Summary Compensation Table. For fiscal year 2024, the named executive officers consist of David M. Urso, Chief Executive Officer and General Counsel, Justin J. File, Chief Financial Officer and Secretary and Richard G. Ghalie, M.D., Chief Medical Officer, to whom we collectively refer in this Compensation Discussion and Analysis as our named executive officers.
Compensation Philosophy and Objectives
We believe that the performance of our executive officers significantly impacts our ability to achieve our corporate goals. We, therefore, place considerable importance on the design and administration of our executive officer compensation program. This program is intended to enhance stockholder value by attracting, motivating and retaining qualified individuals to perform at the highest levels and to contribute to our growth and success. Our executive officer compensation program is designed to provide compensation opportunities that are tied to individual and corporate performance.
Our overall compensation philosophy has been to pay our executive officers an annual base salary and to provide opportunities, through cash and equity incentives, to deliver higher compensation if certain key performance goals are satisfied or exceeded. The primary principles of our fiscal year 2024 compensation strategy were:
The Compensation Committee’s Process
The Compensation Committee acts on behalf of the Board with respect to fulfilling the Board’s responsibilities to oversee our compensation policies, plans and programs and reviewing and determining, as appropriate, the compensation to be paid to executive officers and directors. To achieve this task, the Compensation Committee (i) reviews and approves corporate performance goals and objectives that support and reinforce our long-term strategic goals and compensation plans; (ii) reviews the individual performance of the executive officers; (iii) establishes policies with respect to equity compensation arrangements, timing and pricing of equity awards for newly hired employees, promotions and annual grants for executive and non-executive employees and directors; (iv) reviews regional and industry-wide compensation practices and trends to assess the propriety, adequacy and competitiveness of our executive compensation programs among comparable companies in our industry; (v) reviews and approves the terms of any employment agreements, severance agreements, change-of-control protections and any other compensation arrangements of the executive officers; (vi) performs and considers a compensation risk assessment; and (vii) considers stockholder feedback and Say-on-Pay voting results.
71
With respect to compensation of our Chief Executive Officer, the Compensation Committee evaluates the Chief Executive Officer’s performance in light of relevant performance goals and objectives, taking into account the policies of the Compensation Committee and, with respect to long-term incentive compensation, stockholder return and the results of the most recent stockholder advisory vote on executive compensation. The Compensation Committee reviews and approves (or if appropriate, recommends to the Board for final determination and approval) individual and corporate performance goals and objectives of our other executive officers. The Compensation Committee considers the recommendations of the Chief Executive Officer with respect to the compensation of our other executive officers. The Compensation Committee also makes recommendations to the Board with respect to this Compensation Discussion and Analysis section and recommends that such section be included in any of our annual reports on Form 10-K, registration statements, Proxy statements or information statements.
The Compensation Committee meets at least once a year or more frequently as its members deem necessary or appropriate. Under its charter, the Compensation Committee has the authority, in its sole discretion, to retain or obtain the advice of a compensation consultant, legal counsel or other advisors as the Compensation Committee may determine to assist in the performance of the Compensation Committee’s duties and responsibilities, only after taking into consideration the factors prescribed by the SEC and Nasdaq that bear upon the adviser’s independence.
Setting Executive Compensation
The Compensation Committee considers peer group analysis as a component of its overall executive compensation decision process, but it does not attempt to set executive compensation to a specific benchmark level, or percentile as compared to executive compensation levels at other companies. The Compensation Committee determines the mix of compensation of each executive officer based on its review of such competitive data and an assessment of the individual’s performance. We believe our approach to compensation does not encourage excessive risk-taking by our executives as it is not a market outlier and is based on a typical mix of short- and long-term compensation tied to both internal objectives and to stockholder value.
Our peer group of companies for fiscal year 2024 consisted of 20 similar publicly traded drug development companies, all approved by the Compensation Committee, with input from management and F. W. Cook, our compensation consultant. The peer group is composed of drug development companies with a broadly similar market cap, (median peer market cap at the time the peer data were reviewed by the Committee in June 2023 was $111 million), generally without material revenue from commercial products, and with emphasis on oncology drug development companies, as follows:
Aeglea BioTherapeutics |
Kronos Bio |
BioAtla |
Leap Therapeutics |
Cardiff Oncology |
Molecular Templates |
CEL-SCI Corporation |
Oncternal Therapeutics |
CytomX Therapeutics |
Pieris Pharmaceuticals |
ESSA Pharma |
Rigel Pharmaceuticals |
Fusion Pharma |
Surface Oncology |
G1 Therapeutics |
Syros Pharmaceuticals |
Glycomimetics |
UroGen Pharma Ltd. |
Karyopharm Therapeutics |
Verastem |
The Compensation Committee believes that our base compensation, cash incentives and equity programs reward the achievement of defined corporate goals and objectives. This is critical for ensuring a competitive program that retains our existing executive officers and allows us to hire new executive officers, particularly considering the competitive nature of our industry.
The peer data were used as context for setting fiscal year 2024 executive officer compensation. The Compensation Committee does not set a target benchmark, but in fiscal year 2024 the value of total direct compensation was below the median for the CEO and for all other named executive officers, and Mr. Urso’s total compensation value in fiscal year 2024 as our new CEO was considerably lower than our prior CEO, Dr. Gold’s, compensation value in fiscal year 2023. Further, Mr. Urso was not provided a fiscal year 2024 equity award because it was included in his fiscal year 2023 equity award. The fiscal year 2023 option value for Mr. Urso includes both his regular annual fiscal year 2023 award as COO and General Counsel in July 2022 and a second grant to recognize his promotion to CEO in June 2023, as well as his annual CEO equity award in fiscal year 2024.
Role of Stockholder Say-on-Pay Votes
At our annual meeting of stockholders held in December 2023, approximately 94% of the shares voted at the meeting approved, on an advisory basis, the compensation of our named executive officers. The Compensation Committee considers input from stockholders, its compensation consultant and proxy advisors, when assessing its compensation philosophy and the components of its compensation program, giving further consideration to the level of attainment of corporate goals and to the compensation data of our peer group so that compensation decisions are broadly consistent with market practice.
72
Elements of Compensation
Each executive officer’s compensation has three key elements: (i) base salary, (ii) performance-based cash incentives and (iii) equity-based compensation. These elements of executive compensation are intended to align the interests of our executive officers with those of our stockholders.
Base Salary
Base salaries serve to provide a fixed amount of compensation to our executive officers for successfully fulfilling their responsibilities. We establish base salaries for our executive officers when they join us or upon promotion. Base salaries for executive officers are reviewed and determined by the Compensation Committee annually during the first fiscal quarter, following consultation with our compensation consultant. The Compensation Committee did not increase the salaries of Mr. Urso and Mr. File during fiscal year 2024. The salary of Dr. Ghalie was increased by 5% in fiscal year 2024.
Performance-based Cash Incentives
The Compensation Committee believes that allocating a meaningful amount of our executive officers’ total cash compensation to the achievement of corporate goals and objectives aligns their interests with those of our stockholders. The Compensation Committee establishes annual corporate incentive bonus targets for each of our executive officers, expressed as a percent of base salary. Fiscal year 2024 bonus targets as a percent of base salary were set at the start of the fiscal year at 50% for Mr. Urso, and 40% for Dr. Ghalie and Mr. File. The corporate goals and objectives are generally critical path activities or strategic initiatives designed to move forward our clinical and operating activities and enhance stockholder return.
The goals were as objective as possible in both their definition and their scoring at the end of the period, though scoring goals included a subjective element to recognize the quality of achievements.
The following is a description of the primary corporate goals for fiscal year 2024, which guided the Compensation Committee in determining total compensation.
Description |
Voruciclib |
1) Clinical trial cohort enrollment target |
2) FDA briefing book submission for proposed protocol amendment |
3) Completion of preclinical and clinical data package |
ME-344 |
4) Clinical trial cohort enrollment target |
5) Completion of preclinical and clinical data package |
Financial |
6) Maintain sufficient funds for a minimum cash balance as of fiscal year end |
The Compensation Committee determined we had met 91.5% of target based on achievement of the goals above. The named executive officers were paid 91.5% of their target bonus for the corporate achievement without any individual adjustment.
Equity-based Compensation
The Compensation Committee believes that long-term value creation is achieved through an ownership culture that encourages performance by our executive officers through stock and stock-based awards. This potential reward for stockholder value creation is also key to our retention strategy. Under our Amended and Restated MEI Pharma, Inc. 2008 Omnibus Equity Compensation Plan (the 2008 Equity Plan), we may award incentive and non-qualified stock options, stock appreciation rights, restricted stock, restricted stock units, and performance shares and units. Stock options expire after 10 years, have an exercise price equal to the fair market value at grant, typically vest 25% after one year and in equal monthly installments thereafter over the next 36 months and have a three-month post-termination exercise period. All equity awards to NEOs in fiscal year 2024 were stock options because they are naturally performance-based without any value delivery unless the stock price increases after grant.
The regular annual equity grant cycle occurs at the start of the fiscal year and at the start of fiscal year 2024, we granted to Dr. Ghalie, Chief Medical Officer, options to purchase 30,000 shares of our common stock with an exercise price of $7.01, which was the closing sales price on the date of grant. This option award amount had a grant date fair value that that was less than half the median of the peer companies and was based on MEI's assessment of the relative importance of the executive to the future, the need to retain them, and market data for their position. Mr. Urso was not provided an option award in fiscal year 2024 due to the grant previously provided upon becoming CEO in June 2023 (fiscal year 2023). Mr. File was not provided an option award in fiscal year 2024 because he was granted an award upon being hired in June 2023 (fiscal year 2023). We grant options because they have a natural performance requirement for the stock price to increase after a grant. All options granted before and during 2024 are currently underwater as of the date of the filing of this Annual Report and do not provide any value at our current share price.
73
Executive Benefits and Perquisites
We offer benefit programs to our employees, including named executive officers, which include paid time off, health insurance, a company funded HSA account and a company sponsored 401(k) plan. Our executive officers generally do not receive any supplemental retirement benefits or perquisites and participate in the above listed benefit programs on the same basis as other full-time employees.
Severance and Change in Control Agreements
Each of Mr. Urso’s, Dr. Ghalie’s and Mr. File’s employment agreements provides for certain severance payments upon the applicable employee’s termination by us, other than for cause or by the applicable employee for good reason, as such terms are defined in the respective employment agreement. Upon such a termination of employment, we will: (i) make a payment to the applicable employee in lieu of notice in an amount equal to twelve months of such employee’s base salary (as in effect at the time of such employee’s termination from employment), and (ii) accelerate the vesting of the applicable employee’s options so that such employee will be vested in the same number of shares of common stock subject to the options as if such employee had continued to be employed by us for an additional twelve months. Such payment and additional option vesting will be conditional upon the execution of a customary release of claims in favor of us and our affiliates, in a form prescribed by us. The payment in lieu of notice will be paid to the applicable employee in a single lump sum payment as soon as administratively practicable after the maximum review and revocation period for the release agreement as may be required under applicable law, if any, or such earlier date as determined in our sole discretion, but in no event more than 60 days after the applicable employee’s termination of employment. If their employment had been terminated in accordance with the foregoing provisions on June 30, 2023, Mr. Urso, Dr. Ghalie and Mr. File would have been entitled to payments for 12 months base salary in the amount of $614,000, $503,165, and $450,000, respectively, and payments for fiscal year 2024 bonus funded at 91.5% target of $280,905, $184,158, and $164,700, respectively. Each would be entitled to 12 months of option vesting as follows: Mr. Urso 39,582 shares, Dr. Ghalie, 20,385 shares and Mr. File 13,326 shares. The intrinsic values of the option vesting acceleration on June 30, 2024, would have been nil for all three NEOs because all options were underwater. In the event of a change in control of MEI Pharma, as defined in the 2008 Equity Plan, unless the Compensation Committee of the Board determines otherwise, all options granted to Mr. Urso, Dr. Ghalie and Mr. File will accelerate and become fully exercisable effective upon the date of the change in control. For all three NEOs, there was no intrinsic value of unvested stock options as of June 30, 2024, upon a change in control.
Mr. Urso and Dr. Ghalie’s employment as CEO and CMO, respectively, were terminated by us without cause on August 1, 2024. Mr. Urso and Dr. Ghalie’s severance was paid pursuant to their employment agreements dated June 2, 2023 and January 16, 2024, respectively, with 12 months base salary and accelerated vesting of the portion of their stock options that would have vested over the next 12 months, plus their fiscal year 2025 bonus based on performance and board discretion. Further, a total of 61,646 options accelerated for Mr. Urso and 18,861 for Dr. Ghalie, reflecting the number of shares would have vested during the next 12 months, respectively. Mr. Urso’s and Dr. Ghalie’s vested options may be exercised after separation of service for one year, not to exceed their original term.
Tax and Accounting Considerations
The tax and accounting consequences to us of certain compensation elements are important considerations for the Compensation Committee when evaluating and recommending compensation packages for our executive officers. Generally, the Compensation Committee seeks to balance its objective to create an effective compensation program that attracts, retains and rewards executives in order to maximize the return to stockholders with the need for appropriate tax and accounting consequences of such compensation.
In addition to considering the tax consequences, the Compensation Committee considers the accounting consequences of its decisions, including the impact of expenses being recognized in connection with equity-based awards, in determining the size and form of different equity-based awards.
CEO Pay Ratio
SEC rules require us to disclose the total annual compensation of our principal executive officer for fiscal year 2024, who was David Urso, our President and Chief Executive Officer, the median of the total annual compensation of all employees other than our principal executive officer, as well as their ratio to each other (the CEO Pay Ratio). Total annual compensation for our principal executive officer and for the median of the total annual compensation of all employees is calculated in accordance with SEC rules applicable to the Summary Compensation Table. For fiscal year 2024, these amounts were as follows:
74
In determining the median compensated employee, we chose June 30, 2024, as the determination date. As of this date, we had 27 employees, excluding our principal executive officer. We annualized compensation of employees who were not employed with us for the full fiscal year. In determining our median compensated employee and calculating the CEO Pay Ratio, we did not use any of the exemptions permitted under SEC rules, nor did we rely upon any material assumptions, adjustments or estimates.
We believe that the CEO Pay Ratio set forth above is a reasonable estimate for fiscal year 2024, determined in a manner consistent with SEC rules. The SEC rules for identifying the median compensated employee and calculating the CEO Pay Ratio based on that employee’s total annual compensation permit companies to adopt a variety of methodologies, to apply certain exemptions and to make certain assumptions, adjustments or estimates that reflect their compensation policies. Accordingly, the CEO Pay Ratio may not be comparable to the pay ratios reported by other companies, which may have used different methodologies, assumptions, adjustments or estimates in calculating their pay ratios.
COMPENSATION COMMITTEE REPORT
Our Compensation Committee has reviewed and discussed the Compensation Discussion and Analysis required by Item 402(b) of Regulation S-K and contained within this Proxy Statement with management. Based on such review and discussions, our Compensation Committee recommended to our Board that the Compensation Discussion and Analysis be included as of the date of filing for this Annual Report, by the members of the Compensation Committee of the Board:
Dr. Nicholas R. Glover
Dr. Thomas C. Reynolds
Mr. Steven Wood
This Section is not soliciting material, is not deemed filed with the SEC and is not to be incorporated by reference in any filing of our under the Exchange Act or the Securities Act, other than in our Annual Report on Form 10-K where it shall be deemed to be furnished, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing.
75
EXECUTIVE COMPENSATION
Our Executive Officers
Our named executive officers for the fiscal year ended June 30, 2024, were:
Summary Compensation Table
The table below sets forth for the fiscal years ended June 30, 2024 and 2023, the compensation of our named executive officers.
Name and Principal Position |
|
Fiscal Year |
|
Salary ($) |
|
|
Stock Awards ($)(1) |
|
|
Option Awards ($) (2) |
|
|
Non-Equity Incentive Plan Compensation ($) (3)(4)(5)(6) |
|
|
All Other Compensation ($) |
|
|
Total ($) (6) |
|
||||||
David M. Urso (7) |
|
2024 |
|
$ |
614,000 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
280,905 |
|
|
$ |
— |
|
|
$ |
894,905 |
|
President and Chief Executive Officer |
|
2023 |
|
$ |
541,185 |
|
|
$ |
— |
|
|
$ |
1,352,600 |
|
|
$ |
207,273 |
|
|
$ |
— |
|
|
$ |
2,101,058 |
|
Richard G. Ghalie (9) |
|
2024 |
|
$ |
503,165 |
|
|
$ |
— |
|
|
$ |
159,800 |
|
|
$ |
184,158 |
|
|
$ |
— |
|
|
$ |
847,123 |
|
Chief Medical Officer |
|
2023 |
|
$ |
479,205 |
|
|
$ |
— |
|
|
$ |
218,900 |
|
|
$ |
148,554 |
|
|
$ |
— |
|
|
$ |
846,659 |
|
Justin J. File (8) |
|
2024 |
|
$ |
450,000 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
164,700 |
|
|
$ |
— |
|
|
$ |
614,700 |
|
Chief Financial Officer |
|
2023 |
|
$ |
25,673 |
|
|
$ |
— |
|
|
$ |
295,600 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
321,273 |
|
Employment Agreements
We have entered into written employment agreements with each of the named executive officers, which set forth the terms of their respective employments.
Employment Agreement between David M. Urso and MEI Pharma
On May 31, 2023, the MEI board of directors appointed Mr. Urso as President and Chief Executive Officer of MEI, effective as of June 2, 2023. Mr. Urso was also elected as a member of the MEI board of directors, effective as of June 8, 2023 through August 1,
76
2024. Mr. Urso served as a member of the class of directors whose term was to expire at the 2025 annual meeting of MEI’s stockholders or until his earlier resignation or removal.
In connection with Mr. Urso’s appointment as President and Chief Executive Officer, Mr. Urso and MEI entered into a new employment agreement (the CEO Employment Agreement), effective as of June 2, 2023, that replaced the existing employment agreement dated March 6, 2014, between MEI and Mr. Urso, as amended by Amendment No. 1, dated July 12, 2018. The CEO Employment Agreement provided for an annual base salary of $614,000, with a target annual bonus opportunity of 50% of base salary. Mr. Urso was eligible to participate in MEI’s health, retirement, expense reimbursement and other benefit plans.
The CEO Employment Agreement provided for a grant as of June 2, 2023, of an option to purchase a number of shares of MEI’s common stock, under MEI’s equity compensation plan, equal to 2.5% of MEI’s outstanding shares as of the date of grant, with vesting over a 4-year period, full vesting on a change in control, and other terms and conditions consistent with the CEO Employment Agreement and grants made to other senior executives (the CEO Initial Grant). The exercise price of the CEO Initial Grant was equal to the Nasdaq closing price per share of MEI stock on the date of grant. The CEO Employment Agreement further provided for a stock option grant to be made on the closing date of the Merger, contingent on the consummation of the Merger and subject to Mr. Urso’s being employed by or providing service to MEI or an affiliate at the time of grant (i.e., the closing date of the Merger), that was equal to 2.5% of the outstanding shares of MEI on the closing date of the Merger (calculated immediately after the effective time of the Merger), less the number of MEI shares underlying the CEO Initial Grant; provided, however, that the total number of shares covered by options granted to Mr. Urso in a calendar year shall not exceed 200,000 shares pursuant to the terms of the MEI’s equity compensation plan (the CEO Second Grant). The exercise price of the CEO Second Grant was equal to the Nasdaq closing price per share of MEI stock on the date of grant (the closing date of the Merger). The CEO Second Grant had the same vesting terms as the CEO Initial Grant.
If the full number of options called for pursuant to the CEO Second Grant could not be granted on the closing date of the Merger in 2023 because of the per person share limit under the equity compensation plan, then an option for the number of shares that could not be granted on the closing date would be granted on January 2, 2024 (the Top Off Grant); provided that, if on January 2, 2024, MEI’s equity compensation plan does not have sufficient shares available to make the Top Off Grant, such grant would be made on the first subsequent date on which MEI did have sufficient shares under the equity compensation plan. To receive the Top Off Grant, Mr. Urso must have been employed by or providing services to MEI or an affiliate on the applicable date of grant. The exercise price of the Top Off Grant would be equal to the Nasdaq closing price per share of MEI stock on the date of grant of the Top Off Grant, and the Top Off Grant would have the same vesting terms as the CEO Initial Grant. If the Merger was not consummated, then in certain circumstances a CEO Second Grant (and a Top Off Grant, if applicable) would be made to Mr. Urso according to the terms and conditions of the CEO Employment Agreement. On May 31, 2023, the MEI board of directors approved the CEO Initial Grant, the CEO Second Grant, and the Top Off Grant, as applicable, to be effective on their respective dates of grant.
For 2024 and subsequent years, Mr. Urso would be eligible to receive equity awards on similar terms as other senior executives of MEI. MEI would pay Mr. Urso’s legal fees in connection with negotiation of the CEO Employment Agreement and ancillary agreements, up to $7,500.
Under the CEO Employment Agreement, if Mr. Urso’s employment was terminated by MEI without cause or Mr. Urso resigned for good reason, Mr. Urso would be eligible to receive the following severance benefits if he signed an effective release of claims: (i) lump sum payment equal to 12 months of his base salary, (ii) if he elected COBRA health care continuation coverage, MEI would pay the monthly COBRA premium for 12 months, (iii) payment of a pro-rata annual bonus, if any, for the year of termination, and (iv) accelerated vesting of a portion of Mr. Urso’s outstanding stock options equal to the number of options that would have vested if he had continued to be employed by MEI for 12 months following termination. The CEO Employment Agreement also provided that if, within 3 months before a change in control, MEI terminated Mr. Urso’s employment without cause at the request of the other party to the change in control transaction, or if, upon or within 2 years following a change in control, Mr. Urso’s employment was terminated by MEI without cause or Mr. Urso resigned for good reason, Mr. Urso’s outstanding stock options would fully vest and become exercisable as of his termination date, provided that he signed an effective release.
In the event that Mr. Urso’s employment was terminated due to his death or disability, vesting of a portion of Mr. Urso’s outstanding stock options would accelerate equal to the number of options that would have vested if he had continued to be employed by MEI for 12 months following termination, subject to his execution of an effective release in the event of disability.
Mr. Urso continued to remain subject to his Employee Proprietary Information and Inventions Agreement, dated April 7, 2014.
Employment Agreement between Richard G. Ghalie and MEI Pharma
In connection with Dr. Ghalie’s appointment as Chief Medical Officer, effective May 3, 2021, we entered into an amendment to his Employment Letter, dated February 17, 2016 (as amended, the Ghalie Employment Letter). The Ghalie Employment Letter provided for an annual base salary of $463,000, and a stock option award to purchase 75,000 shares of our common stock. Effective July 1, 2021, pursuant to the terms of the Ghalie Employment Letter, Dr. Ghalie was eligible to earn an annual cash bonus in an
77
amount up to a maximum of 40% of the base salary based on his achievement of milestones established by the Compensation Committee of the Board.
Dr. Ghalie could terminate his employment at any time other than for Good Reason (as defined in the Ghalie Employment Letter), upon providing one (1) month advance notice to us. Dr. Ghalie could terminate his employment with Good Reason by providing us with notice within sixty (60) days of the event giving rise to the Good Reason (and we did not cure the Good Reason event within thirty (30) days after receiving notice). We had the right to terminate the Ghalie Employment Letter with or without Cause (as defined in the Ghalie Employment Letter) at any time. If Dr. Ghalie’s employment was terminated by us without Cause or by Dr. Ghalie for Good Reason, Dr. Ghalie would be entitled to (i) a lump sum payment in an amount equal to twelve (12) months of his base salary and (ii) accelerated vesting of his options such that Dr. Ghalie will be vested in the same number of options as if he had continued to be employed by us for an additional twelve (12) months, subject to his execution and nonrevocation of a release of claims. The Ghalie Employment Letter contained confidentiality provisions.
Employment Agreement between Justin J. File and MEI Pharma
In connection with Mr. File’s appointment as Chief Financial Officer, Mr. File and MEI entered into an employment agreement (the CFO Employment Agreement), effective as of June 12, 2023. The CFO Employment Agreement provides for an annual base salary of $450,000, with a target annual bonus opportunity of 40% of base salary. Mr. File will be eligible to participate in MEI’s health, retirement, expense reimbursement and other benefit plans.
The CFO Employment Agreement provides for a grant as of June 12, 2023 of an option to purchase a number of shares of MEI’s common stock, under MEI’s equity compensation plan, equal to 0.8% of MEI’s outstanding shares as of the date of grant, with vesting over a 4-year period, full vesting on a change in control, and other terms and conditions consistent with the CFO Employment Agreement and grants made to other senior executives. The exercise price of the CFO Initial Grant will be equal to the Nasdaq closing price per share of MEI stock on the date of grant. For 2024 and subsequent years, Mr. File will be eligible to receive equity awards on similar terms as other senior executives of MEI.
Under the CFO Employment Agreement, if Mr. File’s employment is terminated by MEI without cause or Mr. File resigns for good reason, Mr. File will be eligible to receive the following severance benefits if he signs an effective release of claims: (i) lump sum payment equal to 12 months of his base salary, (ii) if he elects COBRA health care continuation coverage, MEI will pay the monthly COBRA premium for 12 months, (iii) payment of a pro-rata annual bonus, if any, for the year of termination, and (iv) accelerated vesting of a portion of Mr. File’s outstanding stock options equal to the number of options that would have vested if he had continued to be employed by MEI for 12 months following termination. The CFO Employment Agreement also provides that if, within 3 months before a change in control, MEI terminates Mr. File’s employment without cause at the request of the other party to the change in control transaction, or if, upon or within 2 years following a change in control, Mr. File’s employment is terminated by MEI without cause or Mr. File resigns for good reason, Mr. File’s outstanding stock options will fully vest and become exercisable as of his termination date, provided that he signs an effective release.
In the event that Mr. File’s employment is terminated due to his death or disability, vesting of a portion of Mr. File’s outstanding stock options will accelerate equal to the number of options that would have vested if he had continued to be employed by MEI for 12 months following termination, subject to his execution of an effective release in the event of disability.
In connection with our announcement on July 22, 2024 to pursue strategic alternatives, Mr. File and MEI entered into an amendment to his employment agreement effective August 1, 2024. Under the amendment, Mr. File was appointed by the Board to serve in the additional capacity of Acting Chief Executive Officer and provided an increase in his annual base salary to $550,000 and an increase in his target annual bonus opportunity to 50% of base salary. In addition, Mr. File is eligible to receive a success fee if the closing cash balance as of the first to occur of (i) December 31, 2024 or (ii) the closing date of a change in control (the Measurement Date) exceeds a specified amount determined by the Board. The success fee will equal 10% of the closing cash balance of the Company in excess of the specified amount as of the Measurement Date and will be paid in a lump sum cash payment on the first to occur of (i) June 30, 2025, or (ii) Mr. File’s termination of employment by the Company without cause, subject to Mr. File remaining employed by the Company through the payment date of the success fee.
78
Grants of Plan Based Awards for Fiscal Year Ended June 30, 2024
|
|
|
|
Estimated Possible Payouts Under Non-Equity Incentive Plan Awards (1) |
|
All Other Stock Awards Number of Shares of Stocks of Units |
|
All Other Option Awards Number of Securities Underlying Options |
|
|
Exercise or Base Price of Option Awards |
|
|
Grant Date Fair Value of Stock and Option Awards |
|
|||||||
Name |
|
Grant Date |
|
Target |
|
|
Maximum |
|
|
|
|
|
|
|
|
|
|
|
||||
David M. Urso |
|
N/A |
|
$ |
307,000 |
|
|
N/A |
|
— |
|
— |
|
|
— |
|
|
— |
|
|||
Richard G. Ghalie, M.D. |
|
September 29, 2023 |
|
$ |
201,266 |
|
|
N/A |
|
— |
|
|
30,000 |
|
|
$ |
7.01 |
|
|
$ |
159,800 |
|
Justin J. File |
|
N/A |
|
$ |
180,000 |
|
|
N/A |
|
— |
|
— |
|
|
— |
|
|
— |
|
|||
(1) The Board established single bonus targets and as disclosed in the Summary Compensation Table, determined to pay out bonuses at 91.5% of the target levels.
Outstanding Equity Awards at June 30, 2024
The following table provides information on all stock options and RSUs held by our named executive officers on June 30, 2024.
|
|
Option Awards |
|
Stock Awards |
|
|||||||||||||||||
|
|
Number of Securities Underlying Unexercised Options (Exercisable) |
|
|
Number of Securities Underlying Unexercised Options (Unexercisable) |
|
|
|
|
Options Exercise Price |
|
|
Option Expiration |
|
Number of Shares or Units of Stock That Have Not Vested |
|
Market Value of Shares or Units of Stock That Have Not Vested |
|
||||
Name |
|
(#) |
|
|
(#) |
|
|
Footnote |
|
($/Share) |
|
|
Date |
|
(#) |
|
($) |
|
||||
David M. Urso |
|
|
41,643 |
|
|
|
124,928 |
|
|
(1), (17) |
|
$ |
7.50 |
|
|
6/2/2033 |
|
— |
|
$ |
— |
|
|
|
|
25,156 |
|
|
|
27,344 |
|
|
(2), (17) |
|
$ |
10.80 |
|
|
7/5/2032 |
|
— |
|
$ |
— |
|
|
|
|
21,875 |
|
|
|
8,125 |
|
|
(3), (17) |
|
$ |
59.00 |
|
|
7/1/2031 |
|
— |
|
$ |
— |
|
|
|
|
25,704 |
|
|
|
546 |
|
|
(4), (17) |
|
$ |
69.80 |
|
|
7/2/2030 |
|
— |
|
$ |
— |
|
|
|
|
17,500 |
|
|
— |
|
|
(5) |
|
$ |
50.40 |
|
|
7/1/2029 |
|
— |
|
$ |
— |
|
|
|
|
|
11,000 |
|
|
— |
|
|
(6) |
|
$ |
85.60 |
|
|
7/12/2028 |
|
— |
|
$ |
— |
|
|
|
|
|
6,500 |
|
|
— |
|
|
(7) |
|
$ |
86.60 |
|
|
6/22/2028 |
|
— |
|
$ |
— |
|
|
|
|
|
6,500 |
|
|
— |
|
|
(8) |
|
$ |
56.60 |
|
|
7/6/2027 |
|
— |
|
$ |
— |
|
|
|
|
|
6,500 |
|
|
— |
|
|
(9) |
|
$ |
27.20 |
|
|
7/28/2026 |
|
— |
|
$ |
— |
|
|
|
|
|
6,375 |
|
|
— |
|
|
(10) |
|
$ |
31.40 |
|
|
7/27/2025 |
|
— |
|
$ |
— |
|
|
Richard G. Ghalie |
|
— |
|
|
|
30,000 |
|
|
(16), (17) |
|
$ |
7.01 |
|
|
9/29/2033 |
|
— |
|
$ |
— |
|
|
|
|
|
13,425 |
|
|
|
14,575 |
|
|
(2), (17) |
|
$ |
10.80 |
|
|
7/5/2032 |
|
— |
|
$ |
— |
|
|
|
|
11,671 |
|
|
|
4,329 |
|
|
(3), (17) |
|
$ |
59.00 |
|
|
7/1/2031 |
|
— |
|
$ |
— |
|
|
|
|
2,892 |
|
|
|
858 |
|
|
(13), (17) |
|
$ |
71.00 |
|
|
5/3/2031 |
|
— |
|
$ |
— |
|
|
|
|
7,344 |
|
|
|
156 |
|
|
(4), (17) |
|
$ |
69.80 |
|
|
7/2/2030 |
|
— |
|
$ |
— |
|
|
|
|
7,500 |
|
|
— |
|
|
(5) |
|
$ |
50.40 |
|
|
7/1/2029 |
|
— |
|
$ |
— |
|
|
|
|
|
6,500 |
|
|
— |
|
|
(6) |
|
$ |
85.60 |
|
|
7/12/2028 |
|
— |
|
$ |
— |
|
|
|
|
|
3,250 |
|
|
— |
|
|
(14) |
|
$ |
57.60 |
|
|
7/7/2027 |
|
— |
|
$ |
— |
|
|
|
|
|
1,250 |
|
|
— |
|
|
(12) |
|
$ |
27.60 |
|
|
7/13/2026 |
|
— |
|
$ |
— |
|
|
|
|
|
6,500 |
|
|
— |
|
|
(11) |
|
$ |
24.20 |
|
|
3/6/2026 |
|
— |
|
$ |
— |
|
|
Justin J. File |
|
|
13,326 |
|
|
|
39,976 |
|
|
(15) |
|
$ |
7.35 |
|
|
6/12/2033 |
|
— |
|
$ |
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
79
Option Exercises and Stock Vested
Mr. Urso, Dr. Ghalie and Mr. File did not exercise any stock options during the fiscal year ended June 30, 2024. 1,000 RSUs vested for Dr. Ghalie during the fiscal year ended June 30, 2023.
Pay Versus Performance
Provided below is our pay versus performance disclosure as required pursuant to Item 402(v) of Regulation S-K promulgated under the Exchange Act. As required by Item 402(v), we have included:
Note: pursuant to Item 402(v)(8), MEI, as a smaller reporting company (SRC), has provided the information required by 402(v) for three years, instead of five years and is not required to provide the disclosure required by 402(v)(2)(iv) or 402(v)(5) with respect to the total shareholder return of any peer group, or our-Selected Measure disclosure required by 402 (v)(2)(vi), or the Tabular List provided pursuant to 402(v)(6).
Given our current pay program, the only difference between the SCT and CAP amounts for our NEOs is the value of equity awards, which for purposes of the SCT is based on the grant date fair value of equity awards granted during the year, and for purposes of CAP is based on the year over year change in the fair value of equity awards that are unvested as of the end of the year, or that vested, or were forfeited during the year.
80
This disclosure has been prepared in accordance with Item 402(v) and does not necessarily reflect value realized by the NEOs from their equity awards. Please refer to our Compensation Discussion and Analysis, above, for a discussion of our executive compensation program objectives and the ways in which we align executive compensation with performance.
Our Most Important Metrics Used for Linking Pay and Performance. As required by Item 402(v), below are the most important metrics linking CAP to performance for fiscal year 2024. Besides stock price, the only financial performance measure the Committee used to link executive compensation to performance in 2024 was Total Cash (total cash, cash equivalents and short-term investments) on the consolidated balance sheets as of June 30, 2024.
Other non-financial performance factors considered when making compensation decisions include clinical milestones, individual performance, scope of responsibility, and an annual assessment of pay competitiveness within the market.
Pay Versus Performance Table. In accordance with Item 402(v) and under rules adopted by the SEC pursuant to the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010, we are providing the tabular disclosure for our Chief Executive Officer (our Principal Executive Officer or PEO) and the average of our NEOs other than the PEO for fiscal years 2022, 2023 and 2024.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Value of Initial Fixed $100 Investment Based on Total Shareholder Return |
|
|
|
|
|
|
|
||||||||||||
Fiscal Year |
|
Summary Compensation Table for Total Current PEO |
|
|
Compensation Actually Paid to Current PEO |
|
|
Summary Compensation Table for Total Former PEO |
|
|
Compensation Actually Paid to Former PEO |
|
|
Average Summary Compensation Table Total for Non-PEO |
|
|
Average Compensation Actually Paid to Non-PEO |
|
|
MEI Total Shareholder Return |
|
Peer Total Shareholder |
|
|
MEI Net Income |
|
|
Company Selected Measure: Total Cash ($Millions)(5) |
|
||||||||||
(a) |
|
(b) |
|
|
(c) |
|
|
(b) |
|
|
(c) |
|
|
(d) |
|
|
(e) |
|
|
(f) |
|
|
|
|
(h) |
|
|
(i) |
|
||||||||||
2024 |
|
$ |
894,905 |
|
|
$ |
233,569 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
730,912 |
|
|
$ |
563,489 |
|
|
$ |
7 |
|
$ |
88 |
|
|
$ |
18 |
|
|
$ |
38 |
|
2023 |
|
$ |
2,101,058 |
|
|
$ |
1,689,893 |
|
|
$ |
2,446,640 |
|
|
$ |
1,853,678 |
|
|
$ |
772,486 |
|
|
$ |
640,229 |
|
|
$ |
12 |
|
$ |
79 |
|
|
$ |
(32 |
) |
|
$ |
101 |
|
2022 |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
2,528,609 |
|
|
$ |
268,643 |
|
|
$ |
1,260,180 |
|
|
$ |
250,830 |
|
|
$ |
21 |
|
$ |
73 |
|
|
$ |
(54 |
) |
|
$ |
153 |
|
Fiscal Year |
|
PEO |
|
Non-PEO NEOs |
2024 |
|
David M. Urso (Current) |
|
Justin J. File and Richard G. Ghalie |
2023 |
|
David M. Urso (Current) |
|
Brian G. Drazba and Richard G. Ghalie |
2022 |
|
Daniel P. Gold |
|
Brian G. Drazba, Richard G. Ghalie and David M. Urso |
81
|
Current PEO (Urso)(i) |
|
|||||||
Fiscal Year |
2022 |
|
2023 |
|
2024 |
|
|||
SCT Total |
$ |
1,744,152 |
|
$ |
2,101,058 |
|
$ |
894,905 |
|
Stock and Option Award Values Reported in SCT for the Covered Year |
|
(1,078,500 |
) |
|
(1,352,600 |
) |
|
— |
|
Fair Value of Outstanding Unvested Stock and Option Awards Granted in the Covered Year |
|
167,280 |
|
|
1,064,015 |
|
|
— |
|
Change in Fair Value of Outstanding Unvested Stock and Option Awards from Prior Years |
|
(508,767 |
) |
|
(70,339 |
) |
|
(501,428 |
) |
Fair Value of Stock and Option Awards Granted in Covered Year that Vested |
|
— |
|
|
— |
|
|
— |
|
Change in Fair Value of Stock and Option Awards from Prior Years that Vested in Covered Year |
|
(127,938 |
) |
|
(52,241 |
) |
|
(159,908 |
) |
Fair Value of Stock and Option Awards Forfeited during the Covered Year |
|
— |
|
|
— |
|
|
— |
|
Compensation Actually Paid |
$ |
196,227 |
|
$ |
1,689,893 |
|
$ |
233,569 |
|
|
|
|
|
|
|
|
|||
|
Former PEO (Gold)(i) |
|
|||||||
Fiscal Year |
2022 |
|
2023 |
|
2024 |
|
|||
SCT Total |
$ |
2,528,609 |
|
$ |
2,446,640 |
|
$ |
— |
|
Stock and Option Award Values Reported in SCT for the Covered Year |
|
(1,536,800 |
) |
|
(584,700 |
) |
|
— |
|
Fair Value of Outstanding Unvested Stock and Option Awards Granted in the Covered Year |
|
238,374 |
|
|
— |
|
|
— |
|
Change in Fair Value of Outstanding Unvested Stock and Option Awards from Prior Years |
|
(747,241 |
) |
|
— |
|
|
— |
|
Fair Value of Stock and Option Awards Granted in Covered Year that Vested |
|
— |
|
|
188,782 |
|
|
— |
|
Change in Fair Value of Stock and Option Awards from Prior Years that Vested in Covered Year |
|
(214,299 |
) |
|
(129,123 |
) |
|
— |
|
Fair Value of Stock and Option Awards Forfeited during the Covered Year |
|
— |
|
|
(67,921 |
) |
|
— |
|
Compensation Actually Paid |
$ |
268,643 |
|
$ |
1,853,678 |
|
$ |
— |
|
|
|
|
|
|
|
|
|||
|
Average Non-PEO (Gold)(i) |
|
|||||||
Fiscal Year |
2022 |
|
2023 |
|
2024 |
|
|||
SCT Total |
$ |
1,260,180 |
|
$ |
772,486 |
|
$ |
730,912 |
|
Stock and Option Award Values Reported in SCT for the Covered Year |
|
(671,067 |
) |
|
(177,850 |
) |
|
(79,900 |
) |
Fair Value of Outstanding Unvested Stock and Option Awards Granted in the Covered Year |
|
104,085 |
|
|
101,680 |
|
|
43,649 |
|
Change in Fair Value of Outstanding Unvested Stock and Option Awards from Prior Years |
|
(349,416 |
) |
|
(31,805 |
) |
|
(100,239 |
) |
Fair Value of Stock and Option Awards Granted in Covered Year that Vested |
|
— |
|
|
— |
|
|
— |
|
Change in Fair Value of Stock and Option Awards from Prior Years that Vested in Covered Year |
|
(92,952 |
) |
|
(24,282 |
) |
|
(30,933 |
) |
Fair Value of Stock and Option Awards Forfeited during the Covered Year |
|
— |
|
|
— |
|
|
— |
|
Compensation Actually Paid |
$ |
250,830 |
|
$ |
640,229 |
|
$ |
563,489 |
|
|
|
|
|
|
|
|
|||
(i) The fair value of options awards used to calculate CAP was determined using the Black-Scholes option pricing model, in accordance with FASB 718 |
|
||||||||
Relationship between CAP and TSR. The chart below reflects the relationship between the PEO and average non-PEO NEO CAP versus our TSR and the Peer Group TSR.

82
Relationship between CAP and GAAP Net Income (Loss). The chart below reflects the relationship between the PEO and average non-PEO NEO CAP and our GAAP Net Income (Loss).

Relationship between CAP and Total Cash (our Selected Measure). The chart below reflects the relationship between the PEO CAP and average non-PEO NEO CAP and our Total Cash.

Compensation of Directors
The following table provides details of the fees paid to our non-executive directors who served on the Board for the fiscal year ended June 30, 2024.
|
|
Fees Earned or Paid in Cash ($) (1) |
|
|
OptionAwards($)(2) |
|
|
Total($) |
|
|||
Name |
|
|
|
|
|
|
|
|
|
|||
Charles V. Baltic III (3) |
|
$ |
96,433 |
|
|
$ |
51,600 |
|
|
$ |
148,033 |
|
Frederick W. Driscoll (4) |
|
|
65,600 |
|
|
|
51,600 |
|
|
|
117,200 |
|
Nicholas R. Glover, Ph.D. (5) |
|
|
71,183 |
|
|
|
51,600 |
|
|
|
122,783 |
|
Thomas C. Reynolds, M.D., Ph.D. (6) |
|
|
63,683 |
|
|
|
51,600 |
|
|
|
115,283 |
|
Daniel P. Gold (7) |
|
|
15,200 |
|
|
|
51,600 |
|
|
|
66,800 |
|
Tamar D. Howson (8) |
|
|
19,367 |
|
|
|
51,600 |
|
|
|
70,967 |
|
Sujay R. Kango (9) |
|
|
19,367 |
|
|
|
51,600 |
|
|
|
70,967 |
|
Taheer Datoo (10) |
|
|
68,060 |
|
|
|
— |
|
|
|
68,060 |
|
James Flynn (11) |
|
|
37,067 |
|
|
|
77,000 |
|
|
|
114,067 |
|
Steven Wood (12) |
|
|
35,400 |
|
|
|
77,000 |
|
|
|
112,400 |
|
83
(1) For the fiscal year ended June 30, 2024, each of our non-executive directors received an annual cash retainer of $45,600. In addition to the annual cash retainer, the chair received additional annual compensation of $35,000, and each Board Committee chair received additional compensation as follows: Audit Committee: $20,000; Compensation Committee: $15,000; Nominating and Governance Committee: $10,000; and Strategic Committee: $10,000. Committee members not receiving compensation as a committee chairperson received additional compensation as follows: Audit Committee: $10,000; Compensation Committee: $7,500; Nominating and Governance Committee: $5,000; and Strategic Committee: $7,000. Such amounts are pro-rated for periods of service less than the full fiscal year.
Indemnification Agreements
We have entered into an indemnification agreement with each of our directors and executive officers. Subject to certain exceptions, the indemnification agreements provide that an indemnitee will be indemnified for all expenses incurred or paid by the indemnitee in connection with a proceeding to which the indemnitee was or is a party, or is threatened to be made a party, by reason of the indemnitee’s status with or service to us or to another entity at our request. In connection with proceedings other than those by or in the right of our company and to which the indemnitee was or is a party, or is threatened to be made a party, by reason of the indemnitee’s status with or service to us or to another entity at our request, the indemnification agreements provide that an indemnitee will also be indemnified for all liabilities incurred or paid by the indemnitee. The indemnification agreements also provide for advancement of expenses incurred by an indemnitee in connection with an indemnifiable claim, subject to reimbursement in certain circumstances.
The rights of each indemnitee are in addition to any other rights provided for under our amended and restated certificate of incorporation, and our amended and restated by-laws, as may be amended from time to time, and under Delaware law.
84
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The following table sets forth information with respect to the beneficial ownership of shares of our common stock as of September 13, 2024 (except as otherwise indicated below) by (i) each person known to beneficially own more than 5% of our common stock, (ii) each of our named executive officers and directors, and (iii) our officers and directors as a group. Beneficial ownership is determined in accordance with the rules and regulations of the SEC. In computing the number of shares beneficially owned by a person and the percentage ownership of that person, shares of common stock subject to options, warrants or restricted stock units, exercisable or convertible on or within sixty (60) days of September 13, 2024, are deemed outstanding. Such shares, however, are not deemed outstanding for the purposes of computing the percentage ownership of any other person. The percentage of beneficial ownership described below is based on 6,662,857 shares of common stock outstanding, plus adjustments to the number of shares of common stock outstanding as described above, as of September 13, 2024.
Name and Address of Beneficial Owner |
|
Amount & Nature of Beneficial Ownership |
|
|
Percentage of Shares Beneficially Owned |
|
|
||
Anson Funds Management LP (1) |
|
|
1,093,188 |
|
|
|
16.40 |
|
% |
Cable Car Capital LLC (2) |
|
|
611,440 |
|
|
|
9.20 |
|
% |
The Vanguard Group (3) |
|
|
389,807 |
|
|
|
5.85 |
|
% |
Directors and Named Executive Officers |
|
|
|
|
|
|
|
||
Justin J. File (4) |
|
|
18,878 |
|
|
* |
|
|
|
Frederick W. Driscoll (5) |
|
|
26,916 |
|
|
* |
|
|
|
Nicholas R. Glover, Ph.D. (6) |
|
|
28,375 |
|
|
* |
|
|
|
Thomas C. Reynolds, M.D., Ph.D. (7) |
|
|
28,875 |
|
|
* |
|
|
|
Taheer Datoo (8) |
|
|
- |
|
|
* |
|
|
|
James Flynn (9) |
|
|
17,378 |
|
|
* |
|
|
|
Steven Wood (10) |
|
|
86,570 |
|
|
|
1.30 |
|
% |
All Current Directors and Executive Officers as a Group (7 People) |
|
3.05 |
|
% |
|||||
85
Delinquent Section 16(a) Reports
Section 16(a) of the Securities Exchange Act of 1934, as amended, requires our Directors and certain of our officers to file reports of holdings and transactions in our equity with the SEC. Based on our records and other information, we believe that in fiscal year 2024 our Directors and our officers who were subject to Section 16(a) met all applicable filing requirements, except as follows: due to an inadvertent administrative error, each member of the Board - Kango Sujay, Thomas Reynolds, Tannar Howson, Daniel Gold, Nicholas Glover, Frederick Driscoll and Charles Baltic III - and our Chief Medical Officer, Richard Ghalie, filed a late Form 4 on October 12, 2023 to report equity grants issued on September 29, 2023.
Item 13. Certain Relationships and Related Transactions, and Director Independence
There were no related party transactions required to be disclosed pursuant to Item 404 of the Regulation S-K during the two years ended June 30, 2024.
Item 14. Principal Accountant Fees and Services.
The Audit Committee has selected Deloitte & Touche LLP (Deloitte) as independent registered public accounting firm to audit the financial statements of MEI for the fiscal year ending June 30, 2025. The Board is submitting the appointment of Deloitte to the stockholders for ratification as a matter of good corporate practice.
Representatives of Deloitte are expected to attend the Annual Meeting. The Deloitte representatives will have an opportunity to make a statement at the meeting and are expected to be available to respond to appropriate questions.
Fees Paid to Independent Registered Public Accounting Firm
Our independent registered public accounting firm is Deloitte & Touche LLP (Deloitte) Auditor Firm ID: 34.
BDO USA, P.C. (BDO) served as the independent registered public accounting firm for us for the period from January 18, 2011 through the fiscal year ended June 30, 2023 and the subsequent interim period ended September 30, 2023. On December 19, 2023 our Audit Committee approved the change in our independent registered public accounting firm effective December 26, 2023 to Deloitte.
The following table represents the aggregate fees from our principal accounting firm, Deloitte for the fiscal year ended June 30, 2024 and our former principal accounting firm, BDO, for the fiscal year ended June 30, 2023.
|
|
Deloitte |
|
|
BDO |
|
||
|
|
June 30, 2024 |
|
|
June 30, 2023 |
|
||
Audit Fees (1) |
|
$ |
528,525 |
|
|
$ |
806,100 |
|
Audit-Related Fees |
|
|
— |
|
|
|
— |
|
Tax Fees (2) |
|
|
— |
|
|
|
76,580 |
|
All Other Fees |
|
|
— |
|
|
|
— |
|
Total Fees |
|
$ |
528,525 |
|
|
$ |
882,680 |
|
Pre-Approval Policies and Procedures
The Audit Committee has adopted a policy and procedure for pre-approving all audit and non-audit services to be performed by our independent auditors. The policy requires pre-approval of all services rendered by our independent auditors either as part of the Audit Committee’s approval of the scope of the engagement of the independent auditors or on a case-by-case basis.
86
PART IV
Item 15. Exhibits, Financial Statement Schedules
Reference is made to the Consolidated Financial Statements under Item 8. Consolidated Financial Statements and Supplementary Data in Part II hereof.
2. Financial Statement Schedules
The Financial Statement Schedules have been omitted either because they are not required or because the information has been included in the consolidated financial statements or the notes thereto included in this Annual Report on Form 10-K.
3. Exhibits
Exhibit Index
|
|
|
|
Incorporated by Reference Herein |
||||||
Exhibit Number |
|
Description |
|
Schedule/ Form |
|
File No. |
|
Exhibit |
|
Filing Date |
2.1 |
|
|
8-K |
|
000-50484 |
|
2.1 |
|
February 23, 2023 |
|
3.1 |
|
|
8-K |
|
000-50484 |
|
3.1 |
|
April 14, 2023 |
|
3.2 |
|
Sixth Amended and Restated Bylaws of MEI Pharma, Inc. adopted as of December 18, 2023 |
|
8-K |
|
001-41827 |
|
3.1 |
|
December 22, 2023 |
3.3 |
|
Certificate of Designation of Series A Convertible Preferred Stock of Marshall Edwards, Inc. |
|
8-K |
|
000-50484 |
|
3.1 |
|
May 11, 2011 |
3.4 |
|
Certificate of Designation of Series B Preferred Stock of Marshall Edwards, Inc. |
|
8-K |
|
000-50484 |
|
10.1 |
|
March 18, 2011 |
3.6 |
|
Certification of Designation of Series A Junior Participating Preferred Stock |
|
8-K |
|
000-50484 |
|
3.1 |
|
October 3, 2023 |
4.1 |
|
|
S-1 |
|
333-109129 |
|
4.1 |
|
October 31, 2023 |
|
4.2 |
|
|
8-K |
|
000-50484 |
|
10.1 |
|
May 16, 2018 |
|
4.3 |
|
|
10-K |
|
000-50484 |
|
4.3 |
|
September 9, 2020 |
|
4.4 |
|
|
10-K |
|
000-50484 |
|
4.4 |
|
September 26, 2023 |
|
4.5 |
|
|
8-K |
|
000-50484 |
|
4.1 |
|
October 3, 2023 |
|
10.1† |
|
Amended and Restated 2008 Stock Omnibus Equity Compensation Plan (December 2023) |
|
10-Q |
|
001-41827 |
|
10.1 |
|
May 9, 2024 |
10.2† |
|
Employment letter dated February 1, 2017, between MEI Pharma, Inc. and Brian G. Drazba |
|
8-K |
|
000-50484 |
|
10.1 |
|
April 3, 2017 |
10.3† |
|
Employment letter dated February 17, 2016, between MEI Pharma, Inc. and Richard G. Ghalie |
|
10-K |
|
000-50484 |
|
10.5 |
|
September 2, 2021 |
10.4† |
|
|
10-K |
|
000-50484 |
|
10.6 |
|
September 2, 2021 |
|
10.5† |
|
|
8-K |
|
000-50484 |
|
10.1 |
|
August 29, 2011 |
|
10.6** |
|
|
10-Q |
|
000-50484 |
|
10.1 |
|
November 8, 2017 |
87
88
|
|
Chapter 63 of Title 18 of the United States Code (18 U.S.C 1350). |
|
|
|
|
|
|
|
|
97* |
|
MEI Clawback Policy |
|
|
|
|
|
|
|
|
101INS |
|
Inline XBRL Instance Document – the instance document does not appear in the Interactive Data File because XBRL tags are embedded within the Inline XBRL document. |
|
|
|
|
|
|
|
|
104 |
|
Cover Page Interactive Data File – the cover page interactive data file does not appear in the Interactive Data File because its XBRL tags are embedded within the XBRL document. |
|
|
|
|
|
|
|
|
* |
|
Filed herewith |
** |
|
Portions of this exhibit have been redacted pursuant to a confidential treatment request filed with the Securities and Exchange Commission. |
+ |
|
Portions of this exhibit have been omitted in accordance with Item 601(a)(6) and Item 601(b)(10) of Regulation S-K because such information (i) is not material and (ii) is the type that the registrant treats as private or confidential. |
† |
|
Each marked exhibit is a management contract or a compensatory plan, contract or arrangement in which a director or executive officer of the registrant participates or has participated. |
Item 16. Form 10-K Summary
Not applicable.
89
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused report to be signed on its behalf by the undersigned, thereunto duly authorized, on September 19, 2024.
|
|
|
MEI PHARMA, INC. |
||
|
|
|
By: |
|
/s/ Justin J. File |
|
|
Justin J. File |
|
|
Acting Chief Executive Officer, |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Signatures |
|
|
By: |
|
/s/ Justin J. File |
|
|
|
Justin J. File Acting Chief Executive Officer, Chief Financial Officer and Secretary (Principal Executive Officer, Principal Financial and Accounting Officer) September 19, 2024 |
|
|
|
|
|
By: |
|
/s/ Thomas C. Reynolds |
|
|
|
Thomas C. Reynolds Director September 19, 2024 |
|
|
|
|
|
By: |
|
/s/ Nicholas R. Glover |
|
|
|
Nicholas R. Glover Director September 19, 2024 |
|
|
|
|
|
By: |
|
/s/ Steven Wood |
|
|
|
Steven Wood Director September 19, 2024 |
|
|
|
|
|
By: |
|
/s/ Frederick W. Driscoll |
|
|
|
Frederick W. Driscoll Director September 19, 2024 |
|
|
|
|
|
By: |
|
/s/ James Flynn |
|
|
|
James Flynn Director September 19, 2024 |
|
|
|
|
|
By: |
|
/s/ Taheer Datoo |
|
|
|
Taheer Datoo Director September 19, 2024 |
|
90









