
目前FDA官網顯示,恒瑞1月的現場檢查已經進入OAI階段,意味着FDA不滿公司第一階段的回覆和改正措施,做出正式監管決定。不過需要強調的是,恒瑞仍有機會在進一步監管措施前提交整改措施。並且此次現場檢查與雙艾療法出海無關。
近日,恒瑞醫藥收到了美國FDA 483表格,此次FDA檢查涉及公司連雲港地區一處製劑生產場地,檢查時間爲2024年1月8日-16日。根據FDA官網,恒瑞此次的FORM 483掛網時間爲2024年6月4日。
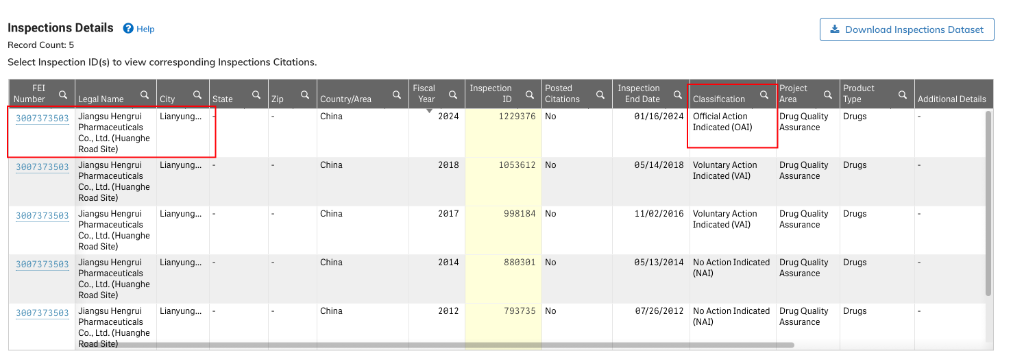
但這份FORM 483已經不是簡單的483第一階段,根據FDA官網數據庫顯示,恒瑞此次的現場檢查已經進入第二階段,OAI(Official Action Indicated)階段。
意味着FDA在審查企業初步回覆和改正措施後,做出了正式監管決定。
對於醫藥製造現場檢查來說,已經進入嚴重階段。
理論上恒瑞還有15天針對OAI進行回覆和整改措施提交。
什麼是FORM 483,OAI意味着什麼?
FDA Form 483
FDA Form 483,也稱爲“檢查觀察表”,是美國食品藥品管理局(FDA)在對藥品、醫療器械、食品等企業進行檢查時,如果發現企業違反了FDA法規,檢查員會使用該表格記錄這些觀察結果。Form 483的目的是通知企業其設施、設備、流程或控制措施存在潛在的違反GMP(良好生產規範)或其他FDA法規的行爲。
主要特點:
- 非最終決定:Form 483只是初步觀察結果,並不代表最終的監管決定。
- 改進機會:企業在收到Form 483後有機會回覆FDA,解釋其觀察結果並提供改進計劃。
- 後續行動:企業的回覆和糾正措施將決定FDA是否會採取進一步的監管行動。
Official Action Indicated (OAI)
Official Action Indicated (OAI)是FDA在審查企業回覆和改正措施後,做出的正式監管決定。OAI表示FDA認爲企業的違規行爲嚴重,需要採取官方監管行動。
主要特點:
- 嚴重違規:OAI狀態表明企業未能充分解決Form 483中指出的問題,存在嚴重違規行爲。
- 監管行動:OAI可能導致FDA採取一系列監管行動,包括警告信(Warning Letter)、進口禁令、產品召回、甚至法律訴訟。
- 整改要求:企業在收到OAI後,通常需要進行更大範圍的整改,並可能面臨進一步的FDA檢查。
關係
Form 483和OAI的關係可以總結如下:
- 初步觀察:FDA檢查員在企業檢查中發現問題,並使用Form 483記錄這些初步觀察結果。
- 企業回覆:企業收到Form 483後,有機會回覆FDA,解釋問題並提供改進計劃。
- 評估和決定:FDA審查企業的回覆和改進措施。如果FDA認爲問題沒有得到充分解決,可能會將企業狀態升級爲OAI。
- 正式行動:在OAI狀態下,FDA可能採取更嚴厲的監管措施,要求企業進行徹底整改,確保符合FDA法規。
因此可見,恒瑞此次1月的現場檢查第一次回覆和整改並沒有得到FDA全面認可,目前已經進入下一步OAI階段。恒瑞將再有一次回覆和整改機會,一旦此次還不能獲得FDA認可,則恒瑞此次現場檢查的工廠將面臨警告信,乃至禁止出口的情況。
這對醫藥公司的質控來說是最嚴厲懲罰,需要公司重視。
作爲全球最嚴監管的FDA,每年在美國境內及境外的現場檢查超過17000次,從FDA官方數據來看,國際檢查數量總體逐年增加,意味着FDA對全球供應鏈的關注和對國際合規性的重視。
此外,OAI分類檢查在最近幾年有所增加,表明嚴重違規行爲的發現增多,可能導致更多的官方行動。
中國藥企要持續提升製造合規,保持競爭優勢
此前,華爾街見聞·見智研究在文章《以藥明康德爲代表的中國CXO產能,會被輕易取代嗎?》的文章中,重點介紹了製造合規對於行業的重要性。相對於印度等國家,中國在高質量產能的合規上已經處於領先。保證合規這也是在目前國內藥企的一個優勢。
此外,對於市場傳聞的造假一說,從FORM 483的具體內容來看,公司的問題與造假關係不大,主要集中在設備與流程合規上。
具體來看,主要問題包括:無菌保障細節管理及清潔驗證評估細節不充分;文件管理軟件存在漏洞,對廢棄記錄文件銷燬管理不充分;生產個別輔助設備計算機系統不符合21CFR Part11的要求;倉儲空調故障維護不足;故意拖延檢查。
不過,對於恒瑞卡瑞利珠單抗+阿帕替尼的雙艾組合出海來說,此次檢查的生產廠址與雙艾並不在同一地點,沒有影響。
去年君實PD1在美國上市前夕曾收到FORM 483,見智研究在《君實PD-1出海再進一步》中曾對其做出解讀,當時公司合作方收到的3條意見很容易解決,在第一輪迴復FDA後,FDA同意公司整改方案並沒有進一步行動,其特瑞普利單抗後續也順利在美國獲批上市。
因此,恒瑞此次現場檢查雖然進入了OAI階段,但公司仍有一次回覆改進的機會,相信恒瑞也會就此進一步反思,快速進行合規回覆,並在內部查找原因,保證未來不會再出現此類問題。
附FORM 483全文(翻譯+原文)
觀察1
防止聲稱無菌的藥品產品微生物污染的程序未能包括無菌和滅菌過程的充分驗證。具體而言:
A) 產品接觸設備包括(b)(4),塞子鼓,(b)(4)在沒有適當包裹的情況下進行滅菌,並從A級推車轉移到RABS內部時未覆蓋。在填充室(b)(4)內的車間中,A級區域(RABS外部)和B級區域(房間的其餘部分)之間沒有物理分隔。氣流可視化研究未包括A級和B級之間的過渡。
B) 在觀察爲美國市場生產的批次(b)注射劑USP時,我們觀察到添加塞子的例行干預操作,開始時間爲3:58 PM,結束時間爲4:03 PM,但您公司的批次記錄中記錄的開始和結束時間爲4:00 PM至4:01 PM(16:00 - 16:01),這與我們觀察到的干預時間有顯著差異。進一步審查程序PO-101-007《填充崗位操作程序》,生效日期2023年11月30日,發現關於干預開始和結束時間的程序定義不清晰。
C) 您的工藝模擬研究(培養基填充)在您的一般注射車間進行的操作無法確保代表當前的商業生產操作。
D) 關於執行常規和非常規干預的生產操作員的重新資格認證程序不充分。在審查六項培養基填充研究時,我們發現操作員執行了大部分干預,一些操作員執行了幾次干預,而一些操作員在培養基填充研究中沒有執行任何干預,但所有這些操作員都被重新資格認證。因此,無法確保所有參與商業生產操作的操作員都具備在無菌操作過程中執行干預的資格。
觀察2
容器密封系統無法提供足夠的保護,以防止儲存和使用中可能導致藥品產品變質或污染的外部因素。具體而言:
您的容器密封完整性測試用於I(b)注射劑(USP)不充分。測試使用了空瓶而不是填充瓶。微生物挑戰研究沒有正控制。染料滲透研究使用人眼檢測結果“未檢測到”,未指定檢測限。
觀察3
設備和器具未適當清潔,無法防止可能改變藥品產品的安全性、身份、強度、質量或純度的污染。具體而言:
A) 清潔驗證研究未考慮所有設備的最難清潔產品的濃度。此外,某些產品的塞子處理過程包括一個(b)(4)步驟,但未考慮爲清潔驗證的污染物。
B) 清潔驗證的最大週期未必比常規清潔週期更差。
C) 檢測剩餘活性污染物(產品)的擦拭和沖洗樣本包括一些“未檢測到”結果,但報告中未提供檢測限。
觀察4
質量控制單位缺乏批准所有影響藥品產品身份、強度、質量和純度的程序或規範的責任。具體而言:
A) 質量單位缺乏對受控文件(如生產批記錄、驗證協議和報告、變更控制和標準操作程序)的充分控制。在2024年1月8日的檢查過程中,我們觀察到批記錄、驗證報告和變更控制的原始執行頁被丟棄在垃圾桶中。當這些原始記錄與存檔的官方記錄進行比較時,生產數據等信息不一致。
B) 質量單位缺乏對根據程序PR-416《批記錄準備、簽發、審查和歸檔管理程序》(生效日期2023年6月20日)和QS-001《受控文件管理(DMS系統)》(生效日期2023年6月16日)控制的主生產批記錄的充分控制。在檢查期間,發現批記錄通過軟件DMS系統簽發給生產人員進行記錄控制,但生產人員可以通過批記錄上的二維碼條形碼複製批記錄,這在當前檢查中已被證實發生。例如,參見上述關於在檢查過程中在垃圾桶中發現的原始執行/填寫的批記錄頁的信息。
C) 視覺檢查操作員的官方眼科檢查結果未由質量單位審核和管理。
D) 在廢棄區域等待銷燬的注射藥品產品未被妥善保管。在2024年1月8日的檢查過程中,我們觀察到廢棄大樓的(b)(4)門是敞開的且沒有人員在場。這座大樓位於公共人行道和街道的正對面,圍欄約3.5英尺高。根據您的公司,圍欄裝有警報器,以防止私人人員爬過圍欄,但在我們的檢查過程中,發現警報器不工作。
E) 沒有充分的數據完整性計劃,包括由質量保證單位對電子數據進行統計上合理的綜合審查,以確保質量控制實驗室生成的所有色譜和非色譜電子數據的完整性、一致性和準確性。
觀察5
與藥品生產相關且在保留期內的記錄未能在授權檢查時立即提供。具體而言:
在對貴公司的廢棄文件區域進行檢查時,我們要求被帶到丟棄辦公廢物的區域,大約(b)(4)步的距離。我們要求被帶到這個區域時,觀察到至少有一人用手機打電話。完成手頭的任務後,我們被引導繞過建築物外側,通過前門進入公司,這增加了大約(b)(4)小時的步行時間。當我們走向設施內的廢棄垃圾桶時,看到一名女性工作人員快速地將文件放入垃圾桶,周圍還有大約2至3人觀察,其中兩人站在乘客側門前。我們看到其中一人快速地將手從前面移到背後,手中似乎拿着撕碎的文件,隨後注意到車輛下方有一疊約8英寸厚的文件。上述文件內容與觀察4中描述的內容相符。這些事件以及對文件內容的審查延遲了我們檢查的及時有效進行。
觀察6
計算機或相關係統沒有采取適當的控制措施,確保主生產和控制記錄或其他記錄的變更僅由授權人員進行。具體而言:
A) 貴公司的GMP相關計算機系統和設備遍佈各個製造車間,不符合21 CFR Part 11的要求。例如:
貴公司操作的便攜式非活性粒子監測設備用於進行非活性粒子(NVP)計數的測試數據生成,支持一般注射製造操作的環境監測/潔淨室認證活動。設備沒有時間戳審計追蹤、數據管理、報警管理、記錄存檔和檢索功能。我們發現這些設備的設備驗證活動從未由貴公司進行。
B) 貴公司操作的容器完整性測試設備和支撐一般注射製造操作的完整性測試設備沒有時間戳審計追蹤、數據管理、報警管理、記錄存檔和檢索功能。
觀察7
用於儲存藥品的建築物未保持良好的維修狀態。具體而言:
在2024年1月12日對您的倉庫設施(2 - 8°C冰箱)的檢查過程中,我們觀察到倉庫地板上有一灘約100平方英尺的水,已釋放的成品注射藥品產品的托盤直接堆放在這灘水上。此外,我們在另一個空調冷凝器附近看到第二灘水,冷凝器似乎由於鏽蝕和不充分的膠帶固定而泄漏。連接托盤架的金屬梁似乎被腐蝕,並試圖通過增加額外的油漆層覆蓋。此外,我們在冷凝器下方和周圍的地板上看到類似黑黴菌的生長,這些地方距離已釋放的藥品成品不到一英尺。這些藥品包括批次號爲(b)(4)的(b)(4)注射液(用於美國市場),批次號爲(b)(4)的(b)(4)注射液(用於英國市場),準備出口至美國和國際市場。
觀察8
洗滌和廁所設施缺乏冷熱水。具體而言:
在對貴公司注射藥品生產車間(b)(4)的檢查過程中,我們觀察到在進入D級衣物消毒室之前的洗滌設施沒有熱水或溫水。此外,您的車間經理確認整個車間的所有洗手站都沒有溫水或熱水,包括C級和B級區域內的洗滌設施。該車間用於製造無菌注射藥品成品,供應美國市場。
檢查日期 2024年1月8日(星期一),2024年1月9日(星期二),2024年1月10日(星期三),2024年1月11日(星期四),2024年1月12日(星期五),2024年1月15日(星期一),2024年1月16日(星期二)
簽發日期 2024年1月16日
檢查員簽名 Arsen Karapetyan, 調查員, DOC
Qiao Y. Bobo, 部門主任, CDER/OPQ/OPMA







